
Mechanisms of Binding Specificity among bHLH Transcription Factors



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Variability and Complexity of Transcription Factor Regulatory Activity
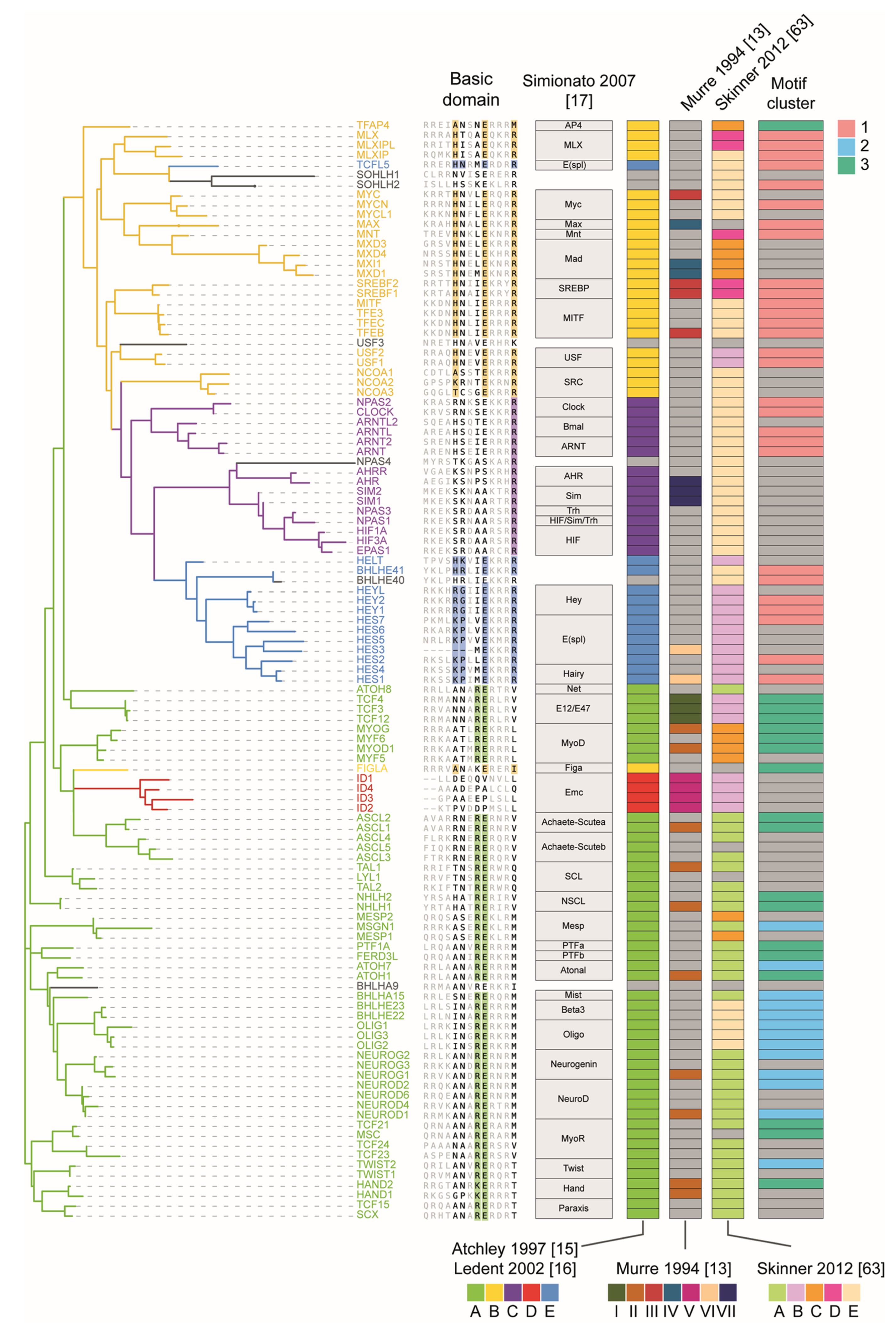
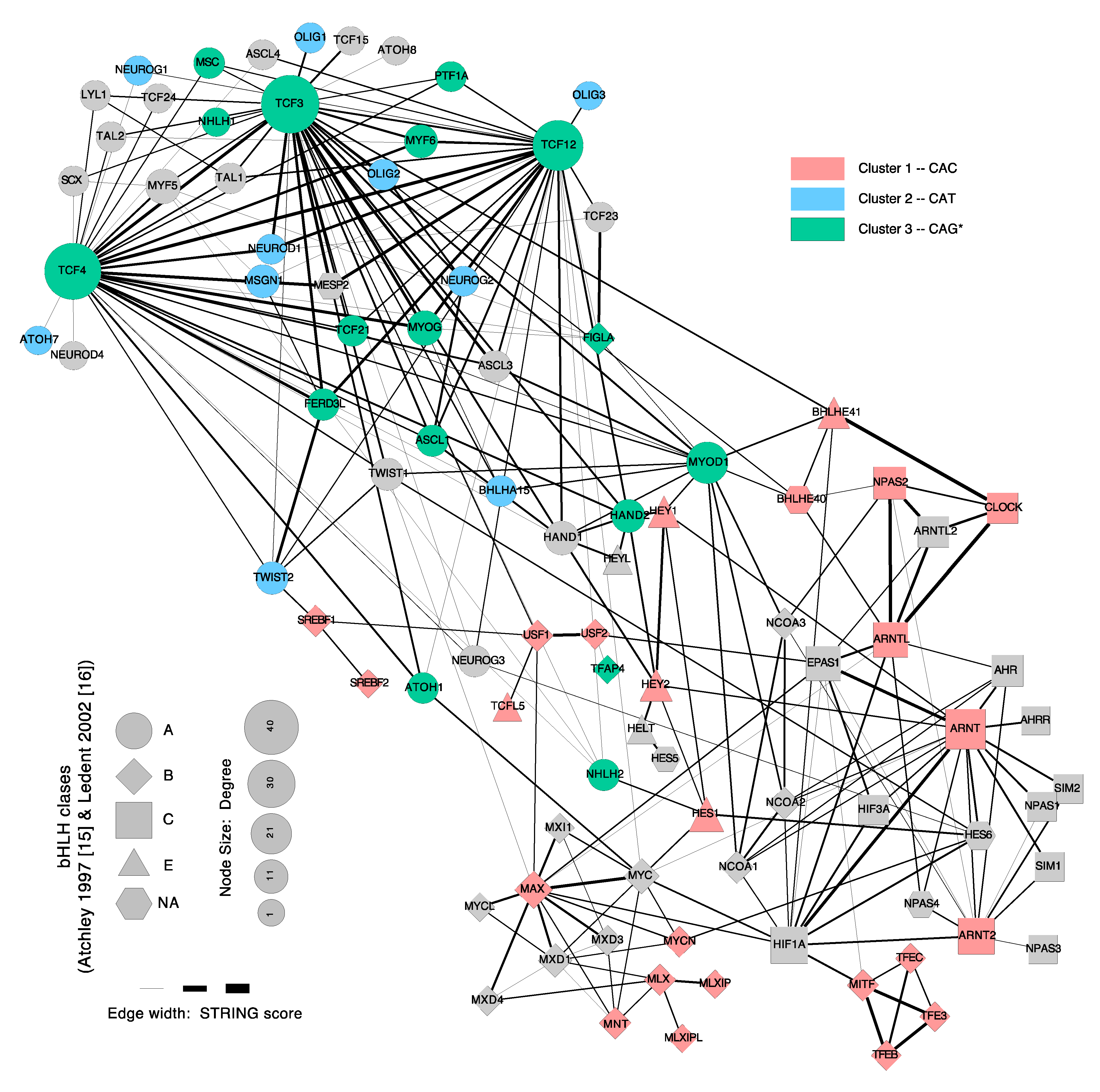
3. The bHLH Family of Transcription Factors
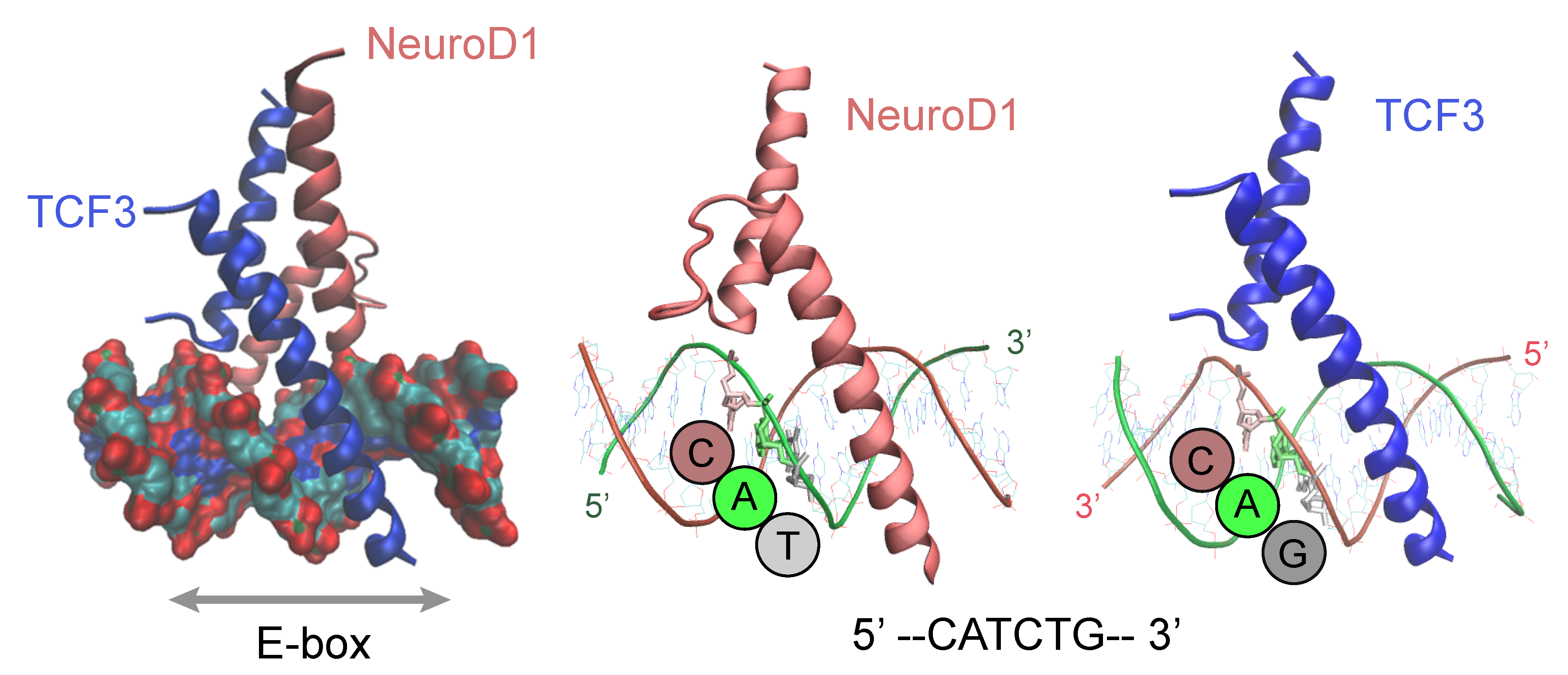
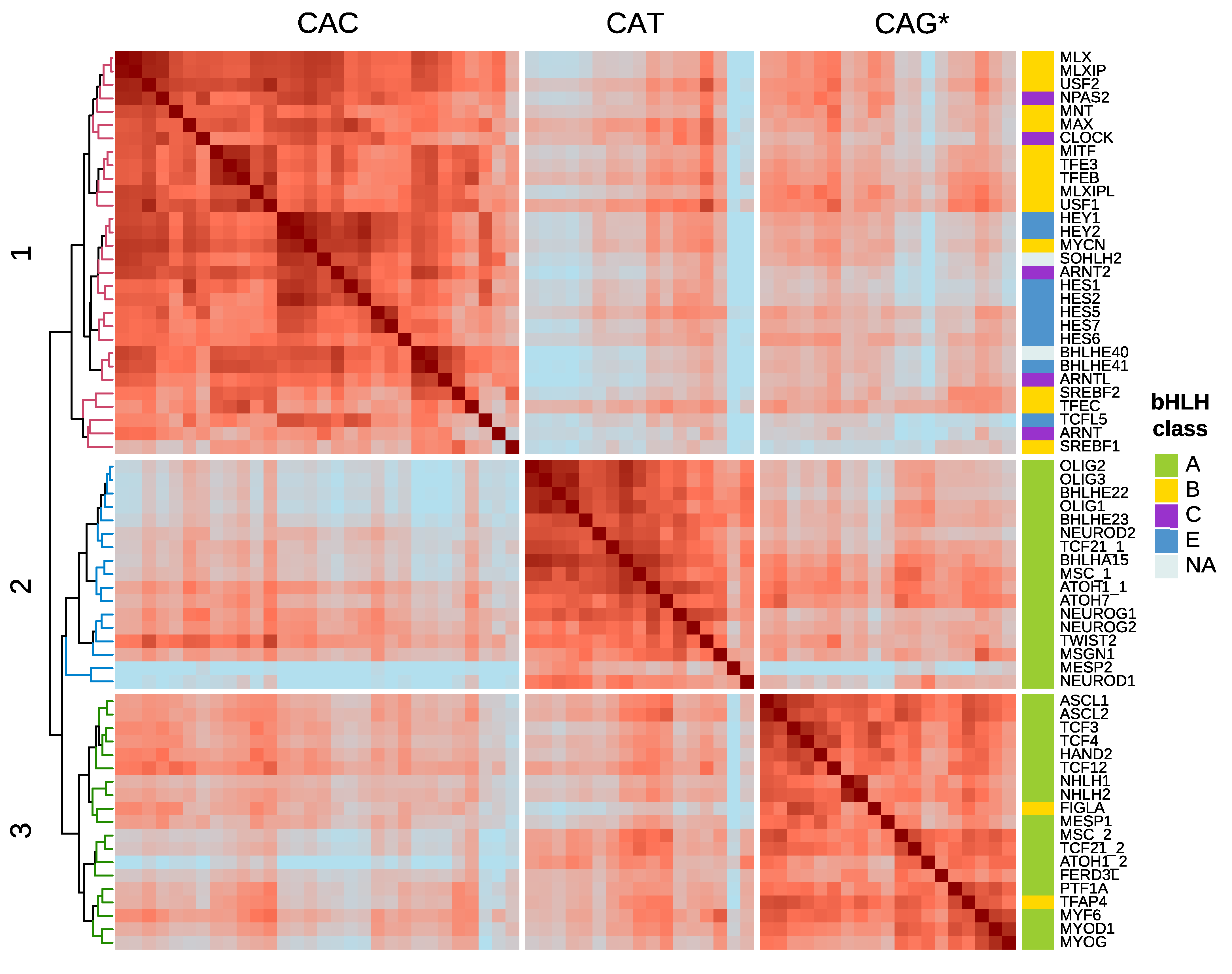
4. DNA-Motif Preferences
5. Dimerization
6. Cooperative Binding with Other Transcription-Factors
7. Chromatin Accessibility and Pioneer Factors
8. DNA Modifications
9. Shape
10. Binding to Non-B DNA
11. Expression Levels
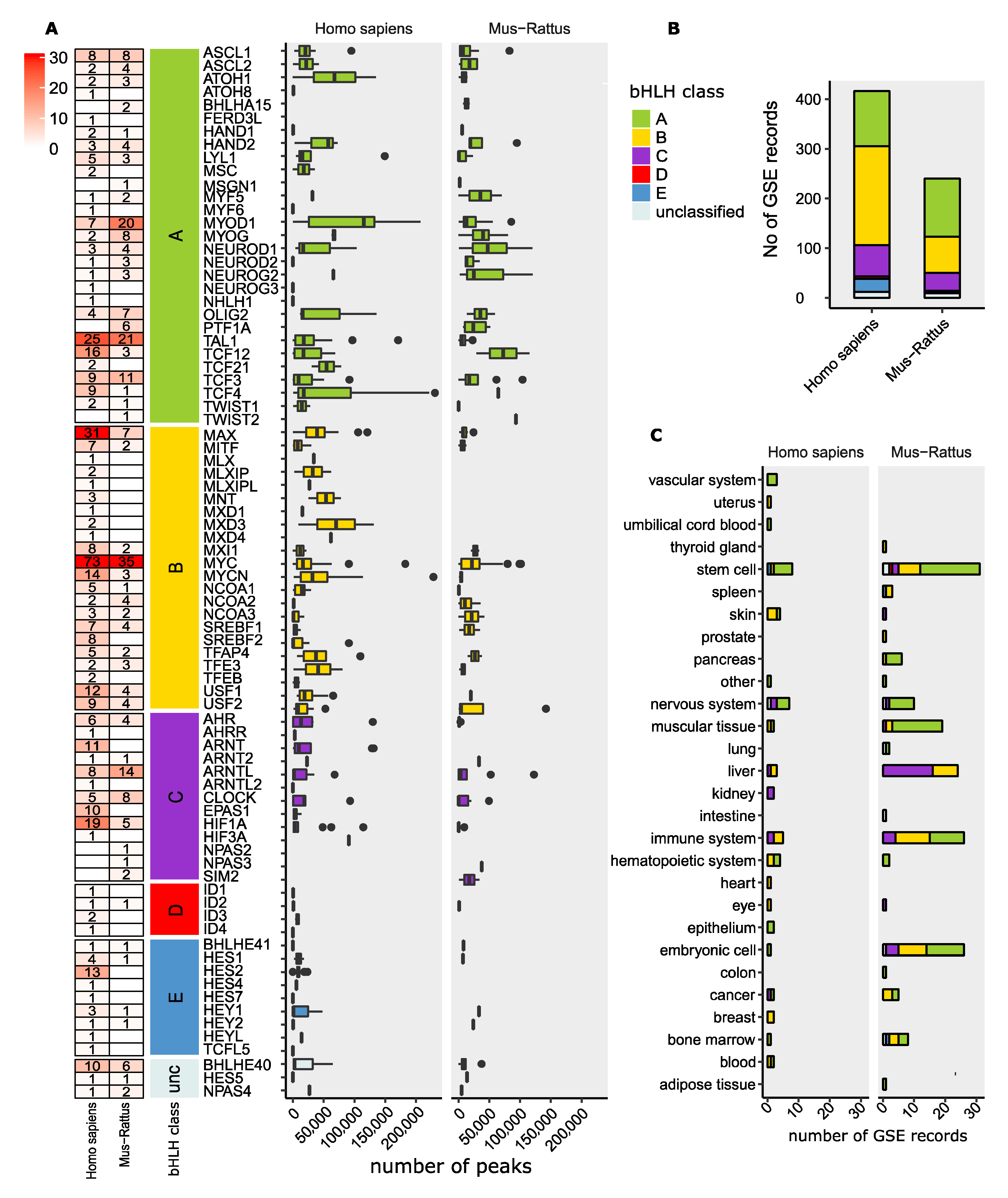
12. ChIP-Seq
13. Conclusions and Future Directions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Lambert, S.A.; Jolma, A.; Campitelli, L.F.; Das, P.K.; Yin, Y.; Albu, M.; Chen, X.; Taipale, J.; Hughes, T.R.; Weirauch, M.T. The Human Transcription Factors. Cell 2018, 172, 650–665. [Google Scholar] [CrossRef]
- Thurman, R.E.; Rynes, E.; Humbert, R.; Vierstra, J.; Maurano, M.T.; Haugen, E.; Sheffield, N.C.; Stergachis, A.B.; Wang, H.; Vernot, B.; et al. The accessible chromatin landscape of the human genome. Nature 2012, 489, 75–82. [Google Scholar] [CrossRef] [PubMed]
- Vierstra, J.; Rynes, E.; Sandstrom, R.; Zhang, M.; Canfield, T.; Scott Hansen, R.; Stehling-Sun, S.; Sabo, P.J.; Byron, R.; Humbert, R.; et al. Mouse regulatory DNA landscapes reveal global principles of cis-regulatory evolution. Science 2014, 346, 1007–1012. [Google Scholar] [CrossRef] [PubMed]
- Maurano, M.T.; Haugen, E.; Sandstrom, R.; Vierstra, J.; Shafer, A.; Kaul, R.; Stamatoyannopoulos, J.A. Large-scale identification of sequence variants influencing human transcription factor occupancy in vivo. Nat. Genet. 2015, 47, 1393–1401. [Google Scholar] [CrossRef] [PubMed]
- Atak, Z.K.; Taskiran, I.I.; Demeulemeester, J.; Flerin, C.; Mauduit, D.; Minnoye, L.; Hulselmans, G.; Christiaens, V.; Ghanem, G.E.; Wouters, J.; et al. Interpretation of allele-specific chromatin accessibility using cell state–aware deep learning. Genome Res. 2021, 31, 1082–1096. [Google Scholar] [CrossRef] [PubMed]
- Kasowski, M.; Grubert, F.; Heffelfinger, C.; Hariharan, M.; Asabere, A.; Waszak, S.M.; Habegger, L.; Rozowsky, J.; Shi, M.; Urban, A.E.; et al. Variation in transcription factor binding among humans. Science 2010, 328, 232–235. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Qiu, Y.; Ribeiro dos Santos, A.M.; Yin, Y.; Li, Y.E.; Vinckier, N.; Nariai, N.; Benaglio, P.; Raman, A.; Li, X.; et al. Systematic analysis of binding of transcription factors to noncoding variants. Nature 2021, 591, 147–151. [Google Scholar] [CrossRef]
- Gronau, I.; Arbiza, L.; Mohammed, J.; Siepel, A. Inference of natural selection from interspersed genomic elements based on polymorphism and divergence. Mol. Biol. Evol. 2013, 30, 1159–1171. [Google Scholar] [CrossRef] [PubMed]
- Samee, M.A.H.; Bruneau, B.G.; Pollard, K.S. A De Novo Shape Motif Discovery Algorithm Reveals Preferences of Transcription Factors for DNA Shape Beyond Sequence Motifs. Cell Syst. 2019, 8, 27–42.e6. [Google Scholar] [CrossRef]
- Rubinstein, M.; Souza, F.S.J. de Evolution of Transcriptional Enhancers and Animal Diversity. Philos. Trans. Royal Soc. B Biol. Sci. 2013, 368, 20130017. [Google Scholar] [CrossRef]
- Klemm, S.L.; Shipony, Z.; Greenleaf, W.J. Chromatin accessibility and the regulatory epigenome. Nat. Rev. Genet. 2019, 20, 207–220. [Google Scholar] [CrossRef]
- Kribelbauer, J.F.; Rastogi, C.; Bussemaker, H.J.; Mann, R.S. Low-Affinity Binding Sites and the Transcription Factor Specificity Paradox in Eukaryotes. Annu Rev Cell Dev Bi 2019, 35, 1–23. [Google Scholar] [CrossRef]
- Murre, C.; Bain, G.; van Dijk, M.A.; Engel, I.; Furnari, B.A.; Massari, M.E.; Matthews, J.R.; Quong, M.W.; Rivera, R.R.; Stuiver, M.H. Structure and function of helix-loop-helix proteins. BBA Gene Struct. Expr. 1994, 1218, 129–135. [Google Scholar] [CrossRef]
- Massari, M.E.; Murre, C. Helix-Loop-Helix Proteins: Regulators of Transcription in Eucaryotic Organisms. Mol. Cell. Biol. 2000, 20, 429–440. [Google Scholar] [CrossRef]
- Atchley, W.R.; Fitch, W.M. A natural classification of the basic helix-loop-helix class of transcription factors. Proc. Natl. Acad. Sci. USA 1997, 94, 5172–5176. [Google Scholar] [CrossRef]
- Ledent, V.; Paquet, O.; Vervoort, M. Phylogenetic analysis of the human basic helix-loop-helix proteins. Genome Biol. 2002, 3, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Simionato, E.; Ledent, V.; Richards, G.; Thomas-Chollier, M.; Kerner, P.; Coornaert, D.; Degnan, B.M.; Vervoort, M. Origin and diversification of the basic helix-loop-helix gene family in metazoans: Insights from comparative genomics. BMC Evol. Biol. 2007, 7, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Heim, M.A.; Jakoby, M.; Werber, M.; Martin, C.; Weisshaar, B.; Bailey, P.C. The basic helix-loop-helix transcription factor family in plants: A genome-wide study of protein structure and functional diversity. Mol. Biol. Evol. 2003, 20, 735–747. [Google Scholar] [CrossRef]
- Dennis, D.J.; Han, S.; Schuurmans, C. bHLH transcription factors in neural development, disease, and reprogramming. Brain Res. 2019, 1705, 48–65. [Google Scholar] [CrossRef]
- Block, N.E.; Miller, J.B. Expression of MRF4, a myogenic helix-loop-helix protein, produces multiple changes in the myogenic program of BC3H-1 cells. Mol. Cell. Biol. 1992, 12, 2484–2492. [Google Scholar] [CrossRef] [PubMed]
- Dezan, C.; Meierhans, D.; Künne, A.G.E.; Allemann, R.K. Acquisition of myogenic specificity through replacement of one amino acid of MASH-1 and introduction of an additional α-helical turn. Biol. Chem. 1999, 380, 705–710. [Google Scholar] [CrossRef]
- Fujisawa-Sehara, A.; Nabeshima, Y.; Komiya, T.; Uetsuki, T.; Asakura, A.; Nabeshima, Y.I. Differential trans-activation of muscle-specific regulatory elements including the mysosin light chain box by chicken MyoD, myogenin, and MRF4. J. Biol. Chem. 1992, 267, 10031–10038. [Google Scholar] [CrossRef]
- Lu, J.; Webb, R.; Richardson, J.A.; Olson, E.N. MyoR: A muscle-restricted basic helix-loop-helix transcription factor that antagonizes the actions of MyoD. Proc. Natl. Acad. Sci. USA 1999, 96, 552–557. [Google Scholar] [CrossRef] [PubMed]
- Penn, B.H.; Bergstrom, D.A.; Dilworth, F.J.; Bengal, E.; Tapscott, S.J. A MyoD-generated feed-forward circuit temporally patterns gene expression dining skeletal muscle differentiation. Genes Dev. 2004, 18, 2348–2353. [Google Scholar] [CrossRef] [PubMed]
- Weintraub, H.; Dwarki, V.J.; Verma, I.; Davis, R.; Hollenberg, S.; Snider, L.; Lassar, A.; Tapscott, S.J. Muscle-specific transcriptional activation by MyoD. Genes Dev. 1991, 5, 1377–1386. [Google Scholar] [CrossRef] [PubMed]
- Porcher, C.; Liao, E.C.; Fujiwara, Y.; Zon, L.I.; Orkin, S.H. Specification of hematopoietic and vascular development by the bHLH transcription factor SCL without direct DNA binding. Development 1999, 126, 4603–4615. [Google Scholar] [CrossRef]
- Shivdasanl, R.A.; Mayer, E.L.; Orkin, S.H. Absence of blood formation in mice lacking the T-cell leukaemia oncoprotein tal-1/SCL. Nature 1995, 373, 432–434. [Google Scholar] [CrossRef] [PubMed]
- Larigot, L.; Juricek, L.; Dairou, J.; Coumoul, X. AhR signaling pathways and regulatory functions. Biochim. Open 2018, 7, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, K.; Kataoka, N. Regulation of gene expression under hypoxic conditions. Int. J. Mol. Sci. 2019, 20, 3278. [Google Scholar] [CrossRef]
- McDonald, M.J.; Rosbash, M.; Emery, P. Wild-Type Circadian Rhythmicity Is Dependent on Closely Spaced E Boxes in the Drosophila timelessPromoter. Mol. Cell. Biol. 2001, 21, 1207–1217. [Google Scholar] [CrossRef][Green Version]
- Nakahata, Y.; Yoshida, M.; Takano, A.; Soma, H.; Yamamoto, T.; Yasuda, A.; Nakatsu, T.; Takumi, T. A direct repeat of E-box-like elements is required for cell-autonomous circadian rhythm of clock genes. BMC Mol. Biol. 2008, 9, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Sato, F.; Kawamoto, T.; Fujimoto, K.; Noshiro, M.; Honda, K.K.; Honma, S.; Honma, K.I.; Kato, Y. Functional analysis of the basic helix-loop-helix transcription factor DEC1 in circadian regulation: Interaction with BMAL1. Eur. J. Biochem. 2004, 271, 4409–4419. [Google Scholar] [CrossRef] [PubMed]
- Carroll, P.A.; Freie, B.W.; Mathsyaraja, H.; Eisenman, R.N. The MYC transcription factor network: Balancing metabolism, proliferation and oncogenesis. Front. Med. 2018, 12, 412–425. [Google Scholar] [CrossRef] [PubMed]
- Aguilar-Rodríguez, J.; Peel, L.; Stella, M.; Wagner, A.; Payne, J.L. The architecture of an empirical genotype-phenotype map. Evolution (N. Y.) 2018, 72, 1242–1260. [Google Scholar] [CrossRef]
- Liu, Z.; Venkatesh, S.S.; Maley, C.C. Sequence space coverage, entropy of genomes and the potential to detect non-human DNA in human samples. BMC Genom. 2008, 9, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Ellenberger, T.; Fass, D.; Arnaud, M.; Harrison, S.C. Crystal structure of transcription factor E47: E-box recognition by a basic region helix-loop-helix dimer. Genes Dev. 1994, 8, 970–980. [Google Scholar] [CrossRef] [PubMed]
- Ferré-D’Amaré, A.R.; Prendergast, G.C.; Ziff, E.B.; Burley, S.K. Recognition by Max of its cognate DNA through a dimeric b/HLH/Z domain. Nature 1993, 363, 38–45. [Google Scholar] [CrossRef] [PubMed]
- Ferre-D’Amare, A.R.; Pognonec, P.; Roeder, R.G.; Burley, S.K. Structure and function of the b/HLH/Z domain of USF. EMBO J. 1994, 13, 180–189. [Google Scholar] [CrossRef]
- Ma, P.C.M.; Rould, M.A.; Weintraub, H.; Pabo, C.O. Crystal structure of MyoD bHLH domain-DNA complex: Perspectives on DNA recognition and implications for transcriptional activation. Cell 1994, 77, 451–459. [Google Scholar] [CrossRef]
- Longo, A.; Guanga, G.P.; Rose, R.B. Crystal structure of E47-NeuroD1/Beta2 bHLH domain-DNA complex: Heterodimer selectivity and DNA recognition. Biochemistry 2008, 47, 218–229. [Google Scholar] [CrossRef]
- Murre, C.; McCaw, P.S.; Vaessin, H.; Caudy, M.; Jan, L.Y.; Jan, Y.N.; Cabrera, C.V.; Buskin, J.N.; Hauschka, S.D.; Lassar, A.B.; et al. Interactions between heterologous helix-loop-helix proteins generate complexes that bind specifically to a common DNA sequence. Cell 1989, 58, 537–544. [Google Scholar] [CrossRef]
- Blackwell, T.K.; Weintraub, H. Differences and similarities in DNA-binding preferences of MyoD and E2A protein complexes revealed by binding site selection. Science 1990, 250, 1104–1110. [Google Scholar] [CrossRef] [PubMed]
- Akazawa, C.; Sasai, Y.; Nakanishi, S.; Kageyama, R. Molecular characterization of a rat negative regulator with a basic helix- loop-helix structure predominantly expressed in the developing nervous system. J. Biol. Chem. 1992, 267, 21879–21885. [Google Scholar] [CrossRef]
- Blackwell, T.K.; Huang, J.; Ma, A.; Kretzner, L.; Alt, F.W.; Eisenman, R.N.; Weintraub, H. Binding of myc proteins to canonical and noncanonical DNA sequences. Mol. Cell. Biol. 1993, 13, 5216–5224. [Google Scholar] [CrossRef]
- Yokoyama, C.; Wang, X.; Briggs, M.R.; Admon, A.; Wu, J.; Hua, X.; Goldstein, J.L.; Brown, M.S. SREBP-1, a basic-helix-loop-helix-leucine zipper protein that controls transcription of the low density lipoprotein receptor gene. Cell 1993, 75, 187–197. [Google Scholar] [CrossRef]
- Ishibashi, M.; Sasai, Y.; Nakanishi, S.; Kageyama, R. Molecular characterization of HES-2, a mammalian helix-loop-helix factor structurally related to Drosophila hairy and Enhancer of split. Eur. J. Biochem. 1993, 215, 645–652. [Google Scholar] [CrossRef]
- Fisher, F.; Crouch, D.H.; Jayaraman, P.S.; Clark, W.; Gillespie, D.A.F.; Goding, C.R. Transcription activation by Myc and Max: Flanking sequences target activation to a subset of CACGTG motifs in vivo. EMBO J. 1993, 12, 5075–5082. [Google Scholar] [CrossRef] [PubMed]
- Whitelaw, M.; Pongratz, I.; Wilhelmsson, A.; Gustafsson, J.A.; Poellinger, L. Ligand-dependent recruitment of the Arnt coregulator determines DNA recognition by the dioxin receptor. Mol. Cell. Biol. 1993, 13, 2504–2514. [Google Scholar] [CrossRef]
- Tontonoz, P.; Kim, J.B.; Graves, R.A.; Spiegelman, B.M. ADD1: A novel helix-loop-helix transcription factor associated with adipocyte determination and differentiation. Mol. Cell. Biol. 1993, 13, 4753–4759. [Google Scholar] [CrossRef]
- Takebayashi, K.; Sasai, Y.; Sakai, Y.; Watanabe, T.; Nakanishi, S.; Kageyama, R. Structure, chromosomal locus, and promoter analysis of the gene encoding the mouse helix-loop-helix factor HES-1. Negative autoregulation through the multiple N box elements. J. Biol. Chem. 1994, 269, 5150–5156. [Google Scholar] [CrossRef]
- Kim, J.B.; Spotts, G.D.; Halvorsen, Y.D.; Shih, H.M.; Ellenberger, T.; Towle, H.C.; Spiegelman, B.M. Dual DNA binding specificity of ADD1/SREBP1 controlled by a single amino acid in the basic helix-loop-helix domain. Mol. Cell. Biol. 1995, 15, 2582–2588. [Google Scholar] [CrossRef]
- Robert, L.D.; Pei-Feng, C.; Lassar, B.A.; Harold, W. The MyoD DNA binding domain contains a recognition code for muscle-specific gene activation. Cell 1990, 60, 733–746. [Google Scholar]
- Halazonetis, T.D. Determination of the c-MYC DNA-binding site. Proc. Natl. Acad. Sci. 1991, 88, 6162–6166. [Google Scholar] [CrossRef] [PubMed]
- Blackwood, E.M.; Eisenman, R.N. Max: A helix-loop-helix zipper protein that forms a sequence-specific DNA-binding complex with Myc. Science 1991, 251, 1211–1217. [Google Scholar] [CrossRef] [PubMed]
- Kerkhoff, E.; Bister, K.; Klempnauer, K.H. Sequence-specific DNA binding by Myc proteins. Proc. Natl. Acad. Sci. USA 1991, 88, 4323–4327. [Google Scholar] [CrossRef] [PubMed]
- Wright, W.E.; Binder, M.; Funk, W. Cyclic amplification and selection of targets (CASTing) for the myogenin consensus binding site. Mol. Cell. Biol. 1991, 11, 4104–4110. [Google Scholar] [CrossRef] [PubMed]
- Fisher, F.; Goding, C.R. Single amino acid substitutions alter helix-loop-helix protein specificity for bases flanking the core CANNTG motif. EMBO J. 1992, 11, 4103–4109. [Google Scholar] [CrossRef]
- Sasai, Y.; Kageyama, R.; Tagawa, Y.; Shigemoto, R.; Nakanishi, S. Two mammalian helix-loop-helix factors structurally related to Drosophila hairy and Enhancer of split. Genes Dev. 1992, 6, 2620–2634. [Google Scholar] [CrossRef]
- Reyes, H.; Reisz-Porszasz, S.; Hankinson, O. Identification of the Ah receptor nuclear translocator protein (Arnt) as a component of the DNA binding form of the Ah receptor. Science 1992, 256, 1193–1195. [Google Scholar] [CrossRef]
- Yin, Y.; Morgunova, E.; Jolma, A.; Kaasinen, E.; Sahu, B.; Khund-Sayeed, S.; Das, P.K.; Kivioja, T.; Dave, K.; Zhong, F.; et al. Impact of cytosine methylation on DNA binding specificities of human transcription factors. Science 2017, 356. [Google Scholar] [CrossRef]
- Jolma, A.; Yan, J.; Whitington, T.; Toivonen, J.; Nitta, K.R.; Rastas, P.; Morgunova, E.; Enge, M.; Taipale, M.; Wei, G.; et al. DNA-binding specificities of human transcription factors. Cell 2013, 152, 327–339. [Google Scholar] [CrossRef] [PubMed]
- Grove, C.A.; De Masi, F.; Barrasa, M.I.; Newburger, D.E.; Alkema, M.J.; Bulyk, M.L.; Walhout, A.J.M. A Multiparameter Network Reveals Extensive Divergence between C. elegans bHLH Transcription Factors. Cell 2009, 138, 314–327. [Google Scholar] [CrossRef]
- Skinner, M.K.; Rawls, A.; Wilson-Rawls, J.; Roalson, E.H. Basic helix-loop-helix transcription factor gene family. Differentiation 2011, 80, 1–8. [Google Scholar] [CrossRef] [PubMed]
- De Masi, F.; Grove, C.A.; Vedenko, A.; Alibés, A.; Gisselbrecht, S.S.; Serrano, L.; Bulyk, M.L.; Walhout, A.J.M. Using a structural and logics systems approach to infer bHLH-DNA binding specificity determinants. Nucleic Acids Res. 2011, 39, 4553–4563. [Google Scholar] [CrossRef] [PubMed]
- Bouard, C.; Terreux, R.; Honorat, M.; Manship, B.; Ansieau, S.; Vigneron, A.M.; Puisieux, A.; Payen, L. Deciphering the molecular mechanisms underlying the binding of the TWIST1/E12 complex to regulatory E-box sequences. Nucleic Acids Res. 2016, 44, 5470–5489. [Google Scholar] [CrossRef][Green Version]
- Pellanda, P.; Dalsass, M.; Filipuzzi, M.; Loffreda, A.; Verrecchia, A.; Castillo Cano, V.; Thabussot, H.; Doni, M.; Morelli, M.J.; Soucek, L.; et al. Integrated requirement of non-specific and sequence-specific DNA binding in Myc-driven transcription. EMBO J. 2021, 40, 1–17. [Google Scholar] [CrossRef]
- Dang, C.V.; Dolde, C.; Gillison, M.L.; Kato, G.J. Discrimination between related DNA sites by a single amino acid residue of Myc-related basic-helix-loop-helix proteins (DNA-protein interaction/transcription). Genetics 1992, 89, 599–602. [Google Scholar]
- Swanson, H.I.; Chan, W.K.; Bradfield, C.A. DNA binding specificities and pairing rules of the Ah receptor, ARNT, and SIM proteins. J. Biol. Chem. 1995, 270, 26292–26302. [Google Scholar] [CrossRef]
- Atchley, W.R.; Zhao, J. Molecular architecture of the DNA-binding region and its relationship to classification of basic helix-loop-helix proteins. Mol. Biol. Evol. 2007, 24, 192–202. [Google Scholar] [CrossRef][Green Version]
- Firulli, B.A.; Redick, B.A.; Conway, S.J.; Firulli, A.B. Mutations within helix I of twist1 result in distinct limb defects and variation of DNA binding affinities. J. Biol. Chem. 2007, 282, 27536–27546. [Google Scholar] [CrossRef]
- Ozdemir, A.; Fisher-Aylor, K.I.; Pepke, S.; Samanta, M.; Dunipace, L.; McCue, K.; Zeng, L.; Ogawa, N.; Wold, B.J.; Stathopoulos, A. High resolution mapping of Twist to DNA in Drosophila embryos: Efficient functional analysis and evolutionary conservation. Genome Res. 2011, 21, 566–577. [Google Scholar] [CrossRef]
- Kophengnavong, T.; Michnowicz, J.E.; Blackwell, T.K. Establishment of Distinct MyoD, E2A, and Twist DNA Binding Specificities by Different Basic Region-DNA Conformations. Mol. Cell. Biol. 2000, 20, 261–272. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Van Doren, M.; Bailey, A.M.; Esnayra, J.; Ede, K.; Posakony, J.W. Negative regulation of proneural gene activity: Hairy is a direct transcriptional repressor of achaete. Genes Dev. 1994, 8, 2729–2742. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, T.; Toumoto, A.; Ihara, K.; Shimizu, M.; Kyogoku, Y.; Ogawa, N.; Oshima, Y.; Hakoshima, T. Crystal structure of PHO4 bHLH domain-DNA complex: Flanking base recognition. EMBO J. 1997, 16, 4689–4697. [Google Scholar] [CrossRef] [PubMed]
- Brownlie, P.; Ceska, T.A.; Lamers, M.; Romier, C.; Stier, G.; Teo, H.; Suck, D. The crystal structure of an intact human Max-DNA complex: New insights into mechanisms of transcriptional control. Structure 1997, 5, 509–520. [Google Scholar] [CrossRef]
- Sato, F.; Bhawal, U.K.; Kawamoto, T.; Fujimoto, K.; Imaizumi, T.; Imanaka, T.; Kondo, J.; Koyanagi, S.; Noshiro, M.; Yoshida, H.; et al. Basic-helix-loop-helix (bHLH) transcription factor DEC2 negatively regulates vascular endothelial growth factor expression. Genes Cells 2008, 13, 131–144. [Google Scholar] [CrossRef] [PubMed]
- Kimura, H.; Weisz, A.; Ogura, T.; Hitomi, Y.; Kurashima, Y.; Hashimoto, K.; D’Acquisto, F.; Makuuchi, M.; Esumi, H. Identification of hypoxia-inducible factor 1 ancillary sequence and its function in vascular endothelial growth factor gene induction by hypoxia and nitric oxide. J. Biol. Chem. 2001, 276, 2292–2298. [Google Scholar] [CrossRef] [PubMed]
- Murre, C. Helix–loop–helix proteins and the advent of cellular diversity: 30 years of discovery. Genes Dev. 2019, 33, 6–25. [Google Scholar] [CrossRef]
- Tietze, K.; Oellers, N.; Knust, E. Enhancer of splitD, a dominant mutation of Drosophila, and its use in the study of functional domains of a helix-loop-helix protein. Proc. Natl. Acad. Sci. USA 1992, 89, 6152–6156. [Google Scholar] [CrossRef]
- Bessho, Y.; Miyoshi, G.; Sakata, R.; Kageyama, R. Hes7: A bHLH-type repressor gene regulated by Notch and expressed in the presomitic mesoderm. Genes Cells 2001, 6, 175–185. [Google Scholar] [CrossRef] [PubMed]
- Hwang, B.; Lee, J.H.; Bang, D. Single-cell RNA sequencing technologies and bioinformatics pipelines. Exp. Mol. Med. 2018, 50, 1–14. [Google Scholar] [CrossRef]
- Iso, T.; Chung, G.; Hamamori, Y.; Kedes, L. HERP1 is a cell type-specific primary target of Notch. J. Biol. Chem. 2002, 277, 6598–6607. [Google Scholar] [CrossRef]
- Firulli, A.B. A HANDful of questions: The molecular biology of the heart and neural crest derivatives (HAND)-subclass of basic helix-loop-helix transcription factors. Gene 2003, 312, 27–40. [Google Scholar] [CrossRef]
- Knöfler, M.; Meinhardt, G.; Bauer, S.; Loregger, T.; Vasicek, R.; Bloor, D.J.; Kimber, S.J.; Husslein, P. Human Hand1 basic helix-loop-helix (bHLH) protein: Extra-embryonic expression pattern, interaction partners and identification of its transcriptional repressor domains. Biochem. J. 2002, 361, 641–651. [Google Scholar] [CrossRef] [PubMed]
- Benezra, R.; Davis, R.L.; Lockshon, D.; Turner, D.L.; Weintraub, H. The protein Id: A negative regulator of helix-loop-helix DNA binding proteins. Cell 1990, 61, 49–59. [Google Scholar] [CrossRef]
- Wendt, H.; Thomas, R.M.; Ellenberger, T. DNA-mediated Folding and Assembly of MyoD-E47 Heterodimers. J. Biol. Chem. 1998, 273, 5735–5743. [Google Scholar] [CrossRef]
- Powell, L.M.; Jarman, A.P. Context dependence of proneural bHLH proteins. Curr. Opin. Genet. Dev. 2008, 18, 411–417. [Google Scholar] [CrossRef]
- Chien, C.T.; Hsiao, C.D.; Jan, L.Y.; Jan, Y.N. Neuronal type information encoded in the basic-helix-loop-helix domain of proneural genes. Proc. Natl. Acad. Sci. USA 1996, 93, 13239–13244. [Google Scholar] [CrossRef]
- Guo, J.; Li, T.; Schipper, J.; Nilson, K.A.; Fordjour, F.K.; Cooper, J.J.; Gordân, R.; Price, D.H. Sequence specificity incompletely defines the genome-wide occupancy of Myc. Genome Biol. 2014, 15, 482. [Google Scholar] [CrossRef]
- Hejna, M.; Moon, W.M.; Cheng, J.; Kawakami, A.; Fisher, D.E.; Song, J.S. Local genomic features predict the distinct and overlapping binding patterns of the bHLH-Zip family oncoproteins MITF and MYC-MAX. Pigment Cell Melanoma Res. 2019, 32, 500–509. [Google Scholar] [CrossRef] [PubMed]
- Jennings, B.H.; Tyler, D.M.; Bray, S.J. Target Specificities of DrosophilaEnhancer of split Basic Helix-Loop-Helix Proteins. Mol. Cell. Biol. 1999, 19, 4600–4610. [Google Scholar] [CrossRef] [PubMed]
- MacQuarrie, K.L.; Yao, Z.; Fong, A.P.; Diede, S.J.; Rudzinski, E.R.; Hawkins, D.S.; Tapscott, S.J. Comparison of Genome-Wide Binding of MyoD in Normal Human Myogenic Cells and Rhabdomyosarcomas Identifies Regional and Local Suppression of Promyogenic Transcription Factors. Mol. Cell. Biol. 2013, 33, 773–784. [Google Scholar] [CrossRef][Green Version]
- Maerkl, S.J.; Quake, S.R. A systems approach to measuring the binding energy landscapes of transcription factors. Science 2007, 315, 233–237. [Google Scholar] [CrossRef] [PubMed]
- Nair, S.K.; Burley, S.K. X-Ray Structures of Myc-Max and Mad-Max Recognizing DNA. Cell 2003, 112, 193–205. [Google Scholar] [CrossRef]
- Wang, J.; Zhuang, J.; Iyer, S.; Lin, X.Y.; Whitfield, T.W.; Greven, M.C.; Pierce, B.G.; Dong, X.; Kundaje, A.; Cheng, Y.; et al. Sequence features and chromatin structure around the genomic regions bound by 119 human transcription factors. Genome Res. 2012, 22, 1798–1812. [Google Scholar] [CrossRef] [PubMed]
- Beltran, A.C.; Dawson, P.E.; Gottesfeld, J.M. Role of DNA sequence in the binding specificity of synthetic basic-helix-loop-helix domains. ChemBioChem 2005, 6, 104–113. [Google Scholar] [CrossRef]
- MacQuarrie, K.L.; Yao, Z.; Fong, A.P.; Tapscott, S.J. Genome-wide binding of the basic helix-loop-helix myogenic inhibitor musculin has substantial overlap with MyoD: Implications for buffering activity. Skelet. Muscle 2013, 3, 1–10. [Google Scholar] [CrossRef]
- Soufi, A.; Garcia, M.F.; Jaroszewicz, A.; Osman, N.; Pellegrini, M.; Zaret, K.S. Pioneer transcription factors target partial DNA motifs on nucleosomes to initiate reprogramming. Cell 2015, 161, 555–568. [Google Scholar] [CrossRef]
- Casey, B.H.; Kollipara, R.K.; Pozo, K.; Johnson, J.E. Intrinsic DNA binding properties demonstrated for lineage-specifying basic helix-loop-helix transcription factors. Genome Res. 2018, 28, 484–496. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Potluri, N.; Lu, J.; Kim, Y.; Rastinejad, F. Structural integration in hypoxia-inducible factors. Nature 2015, 524, 303–308. [Google Scholar] [CrossRef] [PubMed]
- Murakami, M.; Kataoka, K.; Fukuhara, S.; Nakagawa, O.; Kurihara, H. Akt-dependent phosphorylation negatively regulates the transcriptional activity of dHAND by inhibiting the DNA binding activity. Eur. J. Biochem. 2004, 271, 3330–3339. [Google Scholar] [CrossRef] [PubMed]
- Takebayashi, K.; Takahashi, S.; Yokota, C.; Tsuda, H.; Nakanishi, S.; Asashima, M.; Kageyama, R. Conversion of ectoderm into a neural fate by ATH-3, a vertebrate basic helix-loop-helix gene homologous to Drosophila proneural gene atonal. EMBO J. 1997, 16, 384–395. [Google Scholar] [CrossRef]
- Ström, A.; Castella, P.; Rockwood, J.; Wagner, J.; Caudy, M. Mediation of NGF signaling by post-translational inhibition of HES-1, a basic helix-loop-helix repressor of neuronal differentiation. Genes Dev. 1997, 11, 3168–3181. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Li, L.; Zhou, J.; James, G.; Heller-Harrison, R.; Czech, M.P.; Olson, E.N. FGF inactivates myogenic helix-loop-helix proteins through phosphorylation of a conserved protein kinase C site in their DNA-binding domains. Cell 1992, 71, 1181–1194. [Google Scholar] [CrossRef]
- Fan, X.; Waardenberg, A.J.; Demuth, M.; Osteil, P.; Sun, J.Q.J.; Loebel, D.A.F.; Graham, M.; Tam, P.P.L. TWIST1 Homodimers and Heterodimers Orchestrate Lineage- Specific Differentiation. Mol. Cell. Biol. 2020, 40, 1–20. [Google Scholar] [CrossRef]
- Quan, X.J.; Yuan, L.; Tiberi, L.; Claeys, A.; De Geest, N.; Yan, J.; Van Der Kant, R.; Xie, W.R.; Klisch, T.J.; Shymkowitz, J.; et al. Post-translational Control of the Temporal Dynamics of Transcription Factor Activity Regulates Neurogenesis. Cell 2016, 164, 460–475. [Google Scholar] [CrossRef]
- Berberich, S.J.; Cole, M.D. Casein kinase II inhibits the DNA-binding activity of Max homodimers but not Myc/Max heterodimers. Genes Dev. 1992, 6, 166–176. [Google Scholar] [CrossRef] [PubMed]
- Huang, N.; Chelliah, Y.; Shan, Y.; Taylor, C.A.; Yoo, S.H.; Partch, C.; Green, C.B.; Zhang, H.; Takahashi, J.S. Crystal structure of the heterodimeric CLOCK:BMAL1 transcriptional activator complex. Science 2012, 337, 189–194. [Google Scholar] [CrossRef] [PubMed]
- Taelman, V.; Van Wayenbergh, R.; Sölter, M.; Pichon, B.; Pieler, T.; Christophe, D.; Bellefroid, E.J. Sequences downstream of the bHLH domain of the Xenopus hairy-related transcription factor-1 act as an extended dimerization domain that contributes to the selection of the partners. Dev. Biol. 2004, 276, 47–63. [Google Scholar] [CrossRef]
- Fischer, A.; Gessler, M. Delta-Notch-and then? Protein interactions and proposed modes of repression by Hes and Hey bHLH factors. Nucleic Acids Res. 2007, 35, 4583–4596. [Google Scholar] [CrossRef]
- Jones, N. Transcriptional regulation by dimerization: Two sides to an incestuous relationship. Cell 1990, 61, 9–11. [Google Scholar] [CrossRef]
- Kadesch, T. Consequences of heteromeric interactions among helix-loop-helix proteins. Cell Growth Differ. 1993, 4, 49–55. [Google Scholar]
- Le Dréau, G.; Escalona, R.; Fueyo, R.; Herrera, A.; Martínez, J.D.; Usieto, S.; Menendez, A.; Pons, S.; Martinez-Balbas, M.A.; Marti, E. E proteins sharpen neurogenesis by modulating proneural bHLH transcription factors’ activity in an E-box-dependent manner. Elife 2018, 7, 1–29. [Google Scholar] [CrossRef]
- Ohsako, S.; Hyer, J.; Panganiban, G.; Oliver, I.; Caudy, M. Hairy function as a DNA-binding helix-loop-helix repressor of Drosophila sensory organ formation. Genes Dev. 1994, 8, 2743–2755. [Google Scholar] [CrossRef]
- Wang, Z.; Wu, Y.; Li, L.; Su, X.D. Intermolecular recognition revealed by the complex structure of human CLOCK-BMAL1 basic helix-loop-helix domains with E-box DNA. Cell Res. 2013, 23, 213–224. [Google Scholar] [CrossRef]
- Lusska, A.; Shen, E.; Whitlock, J.P. Protein-DNA interactions at a dioxin-responsive enhancer. Analysis of six bona fide DNA-binding sites for the liganded Ah receptor. J. Biol. Chem. 1993, 268, 6575–6580. [Google Scholar] [CrossRef]
- Kinoshita, K.; Kikuchi, Y.; Sasakura, Y.; Suzuki, M.; Fujii-Kuriyama, Y.; Sogawa, K. Altered DNA binding specificity of Arnt by selection of partner bHLH-PAS proteins. Nucleic Acids Res. 2004, 32, 3169–3179. [Google Scholar] [CrossRef][Green Version]
- Siggers, T.; Gordân, R. Protein-DNA binding: Complexities and multi-protein codes. Nucleic Acids Res. 2014, 42, 2099–2111. [Google Scholar] [CrossRef] [PubMed]
- Párraga, A.; Bellsolell, L.; Ferré-D’Amaré, A.R.; Burley, S.K. Co-crystal structure of sterol regulatory element binding protein 1a at 2.3 Å resolution. Structure 1998, 6, 661–672. [Google Scholar] [CrossRef]
- Jensen, L.J.; Kuhn, M.; Stark, M.; Chaffron, S.; Creevey, C.; Muller, J.; Doerks, T.; Julien, P.; Roth, A.; Simonovic, M.; et al. STRING 8—A global view on proteins and their functional interactions in 630 organisms. Nucleic Acids Res. 2009, 37, 412–416. [Google Scholar] [CrossRef] [PubMed]
- Mitsui, K.; Shirakata, M.; Paterson, B.M. Phosphorylation inhibits the DNA-binding activity of MyoD homodimers but not MyoD-E12 heterodimers. J. Biol. Chem. 1993, 268, 24415–24420. [Google Scholar] [CrossRef]
- Li, S.; Mattar, P.; Zinyk, D.; Singh, K.; Chaturvedi, C.P.; Kovach, C.; Dixit, R.; Kurrasch, D.M.; Ma, Y.C.; Chan, J.A.; et al. GSK3 temporally regulates Neurogenin 2 proneural activity in the neocortex. J. Neurosci. 2012, 32, 7791–7805. [Google Scholar] [CrossRef]
- Kalousi, A.; Mylonis, I.; Politou, A.S.; Chachami, G.; Paraskeva, E.; Simos, G. Casein kinase 1 regulates human hypoxia-inducible factor HIF-1. J. Cell Sci. 2010, 123, 2976–2986. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Paes de Faria, J.; Andrew, P.; Nitarska, J.; Richardson, W.D. Phosphorylation Regulates OLIG2 Cofactor Choice and the Motor Neuron-Oligodendrocyte Fate Switch. Neuron 2011, 69, 918–929. [Google Scholar] [CrossRef] [PubMed]
- El Omari, K.; Hoosdally, S.J.; Tuladhar, K.; Karia, D.; Hall-Ponselé, E.; Platonova, O.; Vyas, P.; Patient, R.; Porcher, C.; Mancini, E.J. Structural Basis for LMO2-Driven Recruitment of the SCL: E47bHLH Heterodimer to Hematopoietic-Specific Transcriptional Targets. Cell Rep. 2013, 4, 135–147. [Google Scholar] [CrossRef]
- Spicer, D.B.; Rhee, J.; Cheung, W.L.; Lassar, A.B. Inhibition of Myogenic bHLH and MEF2 Transcription Factors by the bHLH Protein Twist. Science 1996, 272, 1476–1480. [Google Scholar] [CrossRef] [PubMed]
- Langlands, K.; Yin, X.; Anand, G.; Prochownik, E.V. Differential interactions of Id proteins with basic-helix-loop-helix transcription factors. J. Biol. Chem. 1997, 272, 19785–19793. [Google Scholar] [CrossRef] [PubMed]
- Jen, Y.; Weintraub, H.; Benezra, R. Overexpression of Id protein inhibits the muscle differentiation program: In vivo association of Id with E2A proteins. Genes Dev. 1992, 6, 1466–1479. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.H.; Copeland, N.G.; Jenkins, N.A.; Baltimore, D. Id proteins Id1 and Id2 selectively inhibit DNA binding by one class of helix-loop-helix proteins. Mol. Cell. Biol. 1991, 11, 5603–5611. [Google Scholar] [CrossRef]
- Cochrane, S.W.; Zhao, Y.; Welner, R.S.; Sun, X.H. Balance between Id and E proteins regulates myeloid-versus-lymphoid lineage decisions. Blood 2009, 113, 1016–1026. [Google Scholar] [CrossRef][Green Version]
- Rivera, R.; Murre, C. The regulation and function of the Id proteins in lymphocyte development. Oncogene 2001, 20, 8308–8316. [Google Scholar] [CrossRef] [PubMed]
- Davis, R.L.; Turner, D.L. Vertebrate hairy and Enhancer of split related proteins: Transcriptional repressors regulating cellular differentiation and embryonic patterning. Oncogene 2001, 20, 8342–8357. [Google Scholar] [CrossRef] [PubMed]
- Azmi, S.; Ozog, A.; Taneja, R. Sharp-1/DEC2 inhibits skeletal muscle differentiation through repression of myogenic transcription factors. J. Biol. Chem. 2004, 279, 52643–52652. [Google Scholar] [CrossRef] [PubMed]
- Dhar, M.; Taneja, R. Cross-regulatory interaction between Stra13 and USF results in functional antagonism. Oncogene 2001, 20, 4750–4756. [Google Scholar] [CrossRef] [PubMed]
- Ejarque, M.; Altirriba, J.; Gomis, R.; Gasa, R. Characterization of the transcriptional activity of the basic helix-loop-helix (bHLH) transcription factor Atoh8. Biochim. Biophys. Acta Gene Regul. Mech. 2013, 1829, 1175–1183. [Google Scholar] [CrossRef] [PubMed]
- Lemercier, C.; To, R.Q.; Carrasco, R.A.; Konieczny, S.F. The basic helix-loop-helix transcription factor Mist1 functions as a transcriptional repressor of MyoD. EMBO J. 1998, 17, 1412–1422. [Google Scholar] [CrossRef]
- Castanon, I.; Von Stetina, S.; Kass, J.; Baylies, M.K. Dimerization partners determine the activity of the Twist bHLH protein during Drosophila mesoderm development. Development 2001, 128, 3145–3159. [Google Scholar] [CrossRef]
- Allevato, M.; Bolotin, E.; Grossman, M.; Mane-padros, D.; Sladek, M.; Martinez, E. Sequence-specific DNA binding by MYC/MAX to low-affinity non-E-box motifs. PLoS ONE 2017, 7, e0180147. [Google Scholar] [CrossRef]
- Matter-Sadzinski, L.; Puzianowska-Kuznicka, M.; Hernandez, J.; Ballivet, M.; Matter, J.M. A bHLH transcriptional network regulating the specification of retinal ganglion cells. Development 2005, 132, 3907–3921. [Google Scholar] [CrossRef]
- Hernandez, J.; Matter-Sadzinski, L.; Skowronska-Krawczyk, D.; Chiodini, F.; Alliod, C.; Ballivet, M.; Matter, J.M. Highly conserved sequences mediate the dynamic interplay of basic helix-loop-helix proteins regulating retinogenesis. J. Biol. Chem. 2007, 282, 37894–37905. [Google Scholar] [CrossRef]
- Sharma, N.; Pollina, E.A.; Nagy, M.A.; Yap, E.L.; DiBiase, F.A.; Hrvatin, S.; Hu, L.; Lin, C.; Greenberg, M.E. ARNT2 Tunes Activity-Dependent Gene Expression through NCoR2-Mediated Repression and NPAS4-Mediated Activation. Neuron 2019, 102, 390–406.e9. [Google Scholar] [CrossRef]
- Wolf, E.; Lin, C.Y.; Eilers, M.; Levens, D.L. Taming of the beast: Shaping Myc-dependent amplification. Trends Cell Biol. 2015, 25, 241–248. [Google Scholar] [CrossRef] [PubMed]
- Soleimani, V.D.; Yin, H.; Jahani-Asl, A.; Ming, H.; Kockx, C.E.M.; van Ijcken, W.F.J.; Grosveld, F.; Rudnicki, M.A. Snail Regulates MyoD Binding-Site Occupancy to Direct Enhancer Switching and Differentiation-Specific Transcription in Myogenesis. Mol. Cell 2012, 47, 457–468. [Google Scholar] [CrossRef]
- Tanoue, S.; Fujimoto, K.; Myung, J.; Hatanaka, F.; Kato, Y.; Takumi, T. DEC2-E4BP4 heterodimer represses the transcriptional enhancer activity of the EE element in the Per2 promoter. Front. Neurol. 2015, 6, 166. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Pognonec, P.; Boulukos, K.E.; Aperlo, C.; Fujimoto, M.; Ariga, H.; Nomoto, A.; Kato, H. Cross-family interaction between the bHLHZip USF and bZip Fra1 proteins results in down-regulation of AP1 activity. Oncogene 1997, 14, 2091–2098. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Bengal, E.; Ransone, L.; Scharfmann, R.; Dwarki, V.J.; Tapscott, S.J.; Weintraub, H.; Verma, I.M. Functional antagonism between c-Jun and MyoD proteins: A direct physical association. Cell 1992, 68, 507–519. [Google Scholar] [CrossRef]
- Peukert, K.; Staller, P.; Schneider, A.; Carmichael, G.; Hänel, F.; Eilers, M. An alternative pathway for gene regulation by Myc. EMBO J. 1997, 16, 5672–5686. [Google Scholar] [CrossRef] [PubMed]
- Liu, A.; Li, J.; Marin-Husstege, M.; Kageyama, R.; Fan, Y.; Gelinas, C.; Casaccia-Bonnefil, P. A molecular insight of Hes5-dependent inhibition of myelin gene expression: Old partners and new players. EMBO J. 2006, 25, 4833–4842. [Google Scholar] [CrossRef] [PubMed]
- Planque, N.; Leconte, L.; Coquelle, F.M.; Martin, P.; Saulet, S. Specific Pax-6/Microphthalmia Transcription Factor Interactions Involve Their DNA-binding Domains and Inhibit Transcriptional Properties of Both Proteins. J. Biol. Chem. 2001, 276, 29330–29337. [Google Scholar] [CrossRef]
- Brennan, T.J.; Chakraborty, T.; Olson, E.N. Mutagenesis of the myogenin basic region identifies an ancient protein motif critical for activation of myogenesis. Proc. Natl. Acad. Sci. USA 1991, 88, 5675–5679. [Google Scholar] [CrossRef]
- Molkentin, J.D.; Olson, E.N. Combinatorial control of muscle development by basic helix-loop-helix and MADS-box transcription factors. Proc. Natl. Acad. Sci. USA, 1996; 93, 9366–9373. [Google Scholar]
- Quan, X.J.; Denayer, T.; Yan, J.; Jafar-Nejad, H.; Philippi, A.; Lichtarge, O.; Vleminckx, K.; Hassan, B.A. Evolution of neural precursor selection: Functional divergence of proneural proteins. Development 2004, 131, 1679–1689. [Google Scholar] [CrossRef]
- Skowronska-Krawczyk, D.; Ballivet, M.; Dynlacht, B.D.; Matter, J.M. Highly specific interactions between bHLH transcription factors and chromatin during retina development. Development 2004, 131, 4447–4454. [Google Scholar] [CrossRef] [PubMed]
- Weintraub, H.; Genetta, T.; Kadesch, T. Tissue-specific gene activation by MyoD: Determination of specificity by cis-acting repression elements. Genes Dev. 1994, 8, 2203–2211. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Heidt, A.B.; Rojas, A.; Harris, I.S.; Black, B.L. Determinants of Myogenic Specificity within MyoD Are Required for Noncanonical E Box Binding. Mol. Cell. Biol. 2007, 27, 5910–5920. [Google Scholar] [CrossRef]
- Molkentin, J.D.; Black, B.L.; Martin, J.F.; Olson, E.N. Cooperative activation of muscle gene expression by MEF2 and myogenic bHLH proteins. Cell 1995, 83, 1125–1136. [Google Scholar] [CrossRef]
- Lai, H.C.; Meredith, D.M.; Johnson, J.E. bHLH Factors in Neurogenesis and Neuronal Subtype Specification; Elsevier Inc.: Amsterdam, The Netherlands, 2013; ISBN 9780123972651. [Google Scholar]
- Nakada, Y.; Hunsaker, T.L.; Henke, R.M.; Johnson, J.E. Distinct domains within Mash1 and Math1 are required for function in neuronal differentiation versus neuronal cell-type specification. Development 2004, 131, 1319–1330. [Google Scholar] [CrossRef]
- Ohneda, K.; Mirmira, R.G.; Wang, J.; Johnson, J.D.; German, M.S. The Homeodomain of PDX-1 Mediates Multiple Protein-Protein Interactions in the Formation of a Transcriptional Activation Complex on the Insulin Promoter. Mol. Cell. Biol. 2000, 20, 900–911. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Berkes, C.A.; Bergstrom, D.A.; Penn, B.H.; Seaver, K.J.; Knoepfler, P.S.; Tapscott, S.J. Pbx marks genes for activation by MyoD indicating a role for a homeodomain protein in establishing myogenic potential. Mol. Cell 2004, 14, 465–477. [Google Scholar] [CrossRef]
- Fong, A.P.; Yao, Z.; Zhong, J.W.; Johnson, N.M.; Farr, G.H.; Maves, L.; Tapscott, S.J. Conversion of MyoD to a neurogenic factor: Binding site specificity determines lineage. Cell Rep. 2015, 10, 1937–1946. [Google Scholar] [CrossRef]
- Lee, Q.Y.; Mall, M.; Chanda, S.; Zhou, B.; Sharma, K.S.; Schaukowitch, K.; Adrian-Segarra, J.M.; Grieder, S.D.; Kareta, M.S.; Wapinski, O.L.; et al. Pro-neuronal activity of Myod1 due to promiscuous binding to neuronal genes. Nat. Cell Biol. 2020, 22, 401–411. [Google Scholar] [CrossRef] [PubMed]
- Beres, T.M.; Masui, T.; Swift, G.H.; Shi, L.; Henke, R.M.; MacDonald, R.J. PTF1 Is an Organ-Specific and Notch-Independent Basic Helix-Loop-Helix Complex Containing the Mammalian Suppressor of Hairless (RBP-J) or Its Paralogue, RBP-L. Mol. Cell. Biol. 2006, 26, 117–130. [Google Scholar] [CrossRef]
- Glick, E.; Leshkowitz, D.; Walker, M.D. Transcription Factor BETA2 Acts Cooperatively with E2A and PDX1 to Activate the Insulin Gene Promoter. J. Biol. Chem. 2000, 275, 2199–2204. [Google Scholar] [CrossRef]
- Hori, K.; Cholewa-Waclaw, J.; Nakada, Y.; Glasgow, S.M.; Masui, T.; Henke, R.M.; Wildner, H.; Martarelli, B.; Beres, T.M.; Epstein, J.A.; et al. A nonclassical bHLH-Rbpj transcription factor complex is required for specification of GABAergic neurons independent of Notch signaling. Genes Dev. 2008, 22, 166–178. [Google Scholar] [CrossRef]
- Allen, R.D.; Kim, H.K.; Sarafova, S.D.; Siu, G. Negative Regulation of CD4 Gene Expression by a HES-1–c-Myb Complex. Mol. Cell. Biol. 2001, 21, 3071–3082. [Google Scholar] [CrossRef]
- Roy, A.L.; Carruthers, C.; Gutjahr, T.; Robert, B. Direct role for Myc in transcription initiation mediated by interactions with TFII-I. Nature 1993, 365, 359–361. [Google Scholar] [CrossRef]
- Roy, A.L.; Du, H.; Gregor, P.D.; Novina, C.D.; Martinez, E.; Roeder, R.G. Cloning of an inr- and E-box-binding protein, TFII-I, that interacts physically and functionally with USF1. EMBO J. 1997, 16, 7091–7104. [Google Scholar] [CrossRef]
- Sieweke, M.H.; Tekotte, H.; Jarosch, U.; Graf, T. Cooperative interaction of Ets-1 with USF-1 required for HIV-1 enhancer activity in T cells. EMBO J. 1998, 17, 1728–1739. [Google Scholar] [CrossRef]
- Yang, M.H.; Hsu, D.S.S.; Wang, H.W.; Wang, H.J.; Lan, H.Y.; Yang, W.H.; Huang, C.H.; Kao, S.Y.; Tzeng, C.H.; Tai, S.K.; et al. Bmi1 is essential in Twist1-induced epithelial-mesenchymal transition. Nat. Cell Biol. 2010, 12, 982–992. [Google Scholar] [CrossRef]
- Blaiseau, P.L.; Thomas, D. Multiple transcriptional activation complexes tether the yeast activator Met4 to DNA. EMBO J. 1998, 17, 6327–6336. [Google Scholar] [CrossRef] [PubMed]
- Kuras, L.; Cherest, H.; Surdin-Kerjan, Y.; Thomas, D. A heteromeric complex containing the centromere binding factor 1 and two basic leucine zipper factors, Met4 and Met28, mediates the transcription activation of yeast sulfur metabolism. EMBO J. 1996, 15, 2519–2529. [Google Scholar] [CrossRef] [PubMed]
- Siggers, T.; Duyzend, M.H.; Reddy, J.; Khan, S.; Bulyk, M.L. Non-DNA-binding cofactors enhance DNA-binding specificity of a transcriptional regulatory complex. Mol. Syst. Biol. 2011, 7, 1–14. [Google Scholar] [CrossRef]
- Castro, D.S.; Skowronska-Krawczyk, D.; Armant, O.; Donaldson, I.J.; Parras, C.; Hunt, C.; Critchley, J.A.; Nguyen, L.; Gossler, A.; Göttgens, B.; et al. Proneural bHLH and Brn Proteins Coregulate a Neurogenic Program through Cooperative Binding to a Conserved DNA Motif. Dev. Cell 2006, 11, 831–844. [Google Scholar] [CrossRef]
- Lorenzin, F.; Benary, U.; Baluapuri, A.; Walz, S.; Jung, L.A.; von Eyss, B.; Kisker, C.; Wolf, J.; Eilers, M.; Wolf, E. Different promoter affinities account for specificity in MYC-dependent gene regulation. Elife 2016, 5, 1–35. [Google Scholar] [CrossRef] [PubMed]
- Lécuyer, E.; Herblot, S.; Saint-Denis, M.; Martin, R.; Glenn Begley, C.; Porcher, C.; Orkin, S.H.; Hoang, T. The SCL complex regulates c-kit expression in hematopoietic cells through functional interaction with Sp1. Blood 2002, 100, 2430–2440. [Google Scholar] [CrossRef] [PubMed]
- Kassouf, M.T.; Hughes, J.R.; Taylor, S.; McGowan, S.J.; Soneji, S.; Green, A.L.; Vyas, P.; Porcher, C. Genome-wide identification of TAL1’s functional targets: Insights into its mechanisms of action in primary erythroid cells. Genome Res. 2010, 20, 1064–1083. [Google Scholar] [CrossRef]
- Osada, H.; Grutz, G.; Axelson, H.; Forster, A.; Rabbitts, T.H. Association of erythroid transcription factors: Complexes involving the LIM protein RBTN2 and the zinc-finger protein GATA1. Proc. Natl. Acad. Sci. USA 1995, 92, 9585–9589. [Google Scholar] [CrossRef] [PubMed]
- Wadman, I.A.; Osada, H.; Grütz, G.G.; Agulnick, A.D.; Westphal, H.; Forster, A.; Rabbitts, T.H. The LIM-only protein Lmo2 is a bridging molecule assembling an erythroid, DNA-binding complex which includes the TAL1, E47, GATA-1 and Ldb1/NLI proteins. EMBO J. 1997, 16, 3145–3157. [Google Scholar] [CrossRef]
- Ono, Y.; Fukuhara, N.; Yoshie, O. TAL1 and LIM-Only Proteins Synergistically Induce Retinaldehyde Dehydrogenase 2 Expression in T-Cell Acute Lymphoblastic Leukemia by Acting as Cofactors for GATA3. Mol. Cell. Biol. 1998, 18, 6939–6950. [Google Scholar] [CrossRef]
- Han, G.C.; Vinayachandran, V.; Bataille, A.R.; Park, B.; Chan-salis, K.Y.; Keller, C.A.; Long, M.; Mahony, S.; Hardison, R.C.; Pugh, B.F. Genome-wide organization of GATA1 and TAL1 determined at high resolution. Mol. Cell. Biology 2016, 36, 157–172. [Google Scholar] [CrossRef]
- Soler, E.; Andrieu-Soler, C.; De Boer, E.; Bryne, J.C.; Thongjuea, S.; Stadhouders, R.; Palstra, R.J.; Stevens, M.; Kockx, C.; Van Ijcken, W.; et al. The genome-wide dynamics of the binding of Ldb1 complexes during erythroid differentiation. Genes Dev. 2010, 24, 277–289. [Google Scholar] [CrossRef]
- Chang, A.T.; Liu, Y.; Ayyanathan, K.; Benner, C.; Jiang, Y.; Prokop, J.W.; Paz, H.; Wang, D.; Li, H.R.; Fu, X.D.; et al. An evolutionarily conserved DNA architecture determines target specificity of the TWIST family bHLH transcription factors. Genes Dev. 2015, 29, 603–616. [Google Scholar] [CrossRef] [PubMed]
- Fong, A.P.; Yao, Z.; Zhong, J.W.; Cao, Y.; Ruzzo, W.L.; Gentleman, R.C.; Tapscott, S.J. Genetic and Epigenetic Determinants of Neurogenesis and Myogenesis. Dev. Cell 2012, 22, 721–735. [Google Scholar] [CrossRef] [PubMed]
- Weintraub, H.; Davis, R.; Lockshon, D.; Lassar, A. MyoD binds cooperatively to two sites in a target enhancer sequence: Occupancy of two sites is required for activation. Proc. Natl. Acad. Sci. USA 1990, 87, 5623–5627. [Google Scholar] [CrossRef]
- Shively, C.A.; Liu, J.; Chen, X.; Loell, K.; Mitra, R.D. Homotypic cooperativity and collective binding are determinants of bHLH specificity and function. Proc. Natl. Acad. Sci. USA 2019, 116, 16143–16152. [Google Scholar] [CrossRef]
- Walhout, A.J.M.; Gubbels, J.M.; Bernards, R.; Van Der Vliet, P.C.; Timmers, H.T.M. C-Myc/Max heterodimers bind cooperatively to the E-box sequences located in the first intron of the rat ornithine decarboxylase (ODC) gene. Nucleic Acids Res. 1997, 25, 1493–1501. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Sham, Y.Y.; Walters, K.J.; Towle, H.C. A critical role for the loop region of the basic helix-loop-helix/ leucine zipper protein Mlx in DNA binding and glucose-regulated transcription. Nucleic Acids Res. 2007, 35, 35–44. [Google Scholar] [CrossRef]
- Seo, S.; Lim, J.W.; Yellajoshyula, D.; Chang, L.W.; Kroll, K.L. Neurogenin and NeuroD direct transcriptional targets and their regulatory enhancers. EMBO J. 2007, 26, 5093–5108. [Google Scholar] [CrossRef]
- Wilkinson, G.; Dennis, D.; Schuurmans, C. Proneural genes in neocortical development. Neuroscience 2013, 253, 256–273. [Google Scholar] [CrossRef]
- Gotea, V.; Visel, A.; Westlund, J.M.; Nobrega, M.A.; Pennacchio, L.A.; Ovcharenko, I. Homotypic clusters of transcription factor binding sites are a key component of human promoters and enhancers. Genome Res. 2010, 20, 565–577. [Google Scholar] [CrossRef]
- Ezer, D.; Zabet, N.R.; Adryan, B. Homotypic clusters of transcription factor binding sites: A model system for understanding the physical mechanics of gene expression. Comput. Struct. Biotechnol. J. 2014, 10, 63–69. [Google Scholar] [CrossRef]
- Lee, S.K.; Pfaff, S.L. Synchronization of neurogenesis and motor neuron specification by direct coupling of bHLH and homeodomain transcription factors. Neuron 2003, 38, 731–745. [Google Scholar] [CrossRef]
- Thaler, J.P.; Lee, S.K.; Jurata, L.W.; Gill, G.N.; Pfaff, S.L. LIM factor Lhx3 contributes to the specification of motor neuron and interneuron identity through cell-type-specific protein-protein interactions. Cell 2002, 110, 237–249. [Google Scholar] [CrossRef]
- Ma, Y.C.; Song, M.R.; Park, J.P.; Henry Ho, H.Y.; Hu, L.; Kurtev, M.V.; Zieg, J.; Ma, Q.; Pfaff, S.L.; Greenberg, M.E. Regulation of Motor Neuron Specification by Phosphorylation of Neurogenin 2. Neuron 2008, 58, 65–77. [Google Scholar] [CrossRef] [PubMed]
- Desbarats, L.; Gaubatz, S.; Eilers, M. Discrimination between different E-box-binding proteins at an endogenous target gene of c-myc. Genes Dev. 1996, 10, 447–460. [Google Scholar] [CrossRef] [PubMed]
- Genetta, T.; Ruezinsky, D.; Kadesch, T. Displacement of an E-box-binding repressor by basic helix-loop-helix proteins: Implications for B-cell specificity of the immunoglobulin heavy-chain enhancer. Mol. Cell. Biol. 1994, 14, 6153–6163. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Poulin, G.; Lebel, M.; Chamberland, M.; Paradis, F.W.; Drouin, J. Specific Protein-Protein Interaction between Basic Helix-Loop-Helix Transcription Factors and Homeoproteins of the Pitx Family. Mol. Cell. Biol. 2000, 20, 4826–4837. [Google Scholar] [CrossRef] [PubMed]
- Bertrand, N.; Castro, D.S.; Guillemot, F. Proneural genes and the specification of neural cell types. Nat. Rev. Neurosci. 2002, 3, 517–530. [Google Scholar] [CrossRef] [PubMed]
- Ramain, P.; Khechumian, R.; Khechumian, K.; Arbogast, N.; Ackermann, C.; Heitzler, P. Interactions between chip and the achaete/scute-daughterless heterodimers are required for Pannier-driven proneural patterning. Mol. Cell 2000, 6, 781–790. [Google Scholar] [CrossRef]
- Cheol, Y.H.; Gong, E.Y.; Kim, K.; Ji, H.S.; Ko, H.M.; Hyun, J.L.; Choi, H.S.; Lee, K. Modulation of the expression and transactivation of androgen receptor by the basic helix-loop-helix transcription factor pod-1 through recruitment of histone deacetylase 1. Mol. Endocrinol. 2005, 19, 2245–2257. [Google Scholar] [CrossRef]
- Curtis, A.M.; Seo, S.B.; Westgate, E.J.; Rudic, R.D.; Smyth, E.M.; Chakravarti, D.; FitzGerald, G.A.; McNamara, P. Histone Acetyltransferase-dependent Chromatin Remodeling and the Vascular Clock. J. Biol. Chem. 2004, 279, 7091–7097. [Google Scholar] [CrossRef] [PubMed]
- Hamamori, Y.; Wu, H.Y.; Sartorelli, V.; Kedes, L. The basic domain of myogenic basic helix-loop-helix (bHLH) proteins is the novel target for direct inhibition by another bHLH protein, Twist. Mol. Cell. Biol. 1997, 17, 6563–6573. [Google Scholar] [CrossRef]
- Belandia, B.; Powell, S.M.; García-Pedrero, J.M.; Walker, M.M.; Bevan, C.L.; Parker, M.G. Hey1, a Mediator of Notch Signaling, Is an Androgen Receptor Corepressor. Mol. Cell. Biol. 2005, 25, 1425–1436. [Google Scholar] [CrossRef]
- King, I.N.; Kathiriya, I.S.; Murakami, M.; Nakagawa, M.; Gardner, K.A.; Srivastava, D.; Nakagawa, O. Hrt and Hes negatively regulate Notch signaling through interactions with RBP-Jκ. Biochem. Biophys. Res. Commun. 2006, 345, 446–452. [Google Scholar] [CrossRef]
- Cho, Y.; Noshiro, M.; Choi, M.; Morita, K.; Kawamoto, T.; Fujimoto, K.; Kato, Y.; Makishima, M. The basic helix-loop-helix proteins differentiated embryo chondrocyte (DEC) 1 and DEC2 function as corepressors of retinoid X receptors. Mol. Pharmacol. 2009, 76, 1360–1369. [Google Scholar] [CrossRef] [PubMed]
- Gulbagci, N.T.; Li, L.; Ling, B.; Gopinadhan, S.; Walsh, M.; Rossner, M.; Nave, K.A.; Taneja, R. SHARP1/DEC2 inhibits adipogenic differentiation by regulating the activity of C/EBP. EMBO Rep. 2009, 10, 79–86. [Google Scholar] [CrossRef] [PubMed]
- Honma, S.; Kawamoto, T.; Takagi, Y.; Fujimoto, K.; Sato, F.; Noshiro, M.; Kato, Y.; Honma, K.I. Dec1 and Dec2 are regulators of the mammalian molecular clock. Nature 2002, 419, 841–844. [Google Scholar] [CrossRef] [PubMed]
- Dai, Y.S.; Cserjesi, P.; Markham, B.E.; Molkentin, J.D. The transcription factors GATA4 and dHAND physically interact to synergistically activate cardiac gene expression through a p300-dependent mechanism. J. Biol. Chem. 2002, 277, 24390–24398. [Google Scholar] [CrossRef]
- McLarren, K.W.; Lo, R.; Grbavec, D.; Thirunavukkarasu, K.; Karsenty, G.; Stifani, S. The mammalian basic helix loop helix protein HES-1 binds to and modulates the transactivating function of the runt-related factor Cbfa1. J. Biol. Chem. 2000, 275, 530–538. [Google Scholar] [CrossRef]
- Zang, M.X.; Li, Y.; Xue, L.X.; Jia, H.T.; Jing, H. Cooperative activation of atrial naturetic peptide promoter by dHAND and MEF2C. J. Cell. Biochem. 2004, 93, 1255–1266. [Google Scholar] [CrossRef]
- Kamakura, S.; Oishi, K.; Yoshimatsu, T.; Nakafuku, M.; Masuyama, N.; Gotoh, Y. Hes binding to STAT3 mediates crosstalk between Notch and JAK-STAT signalling. Nat. Cell Biol. 2004, 6, 547–554. [Google Scholar] [CrossRef]
- Cruickshank, M.N.; Dods, J.; Taylor, R.L.; Karimi, M.; Fenwick, E.J.; Quail, E.A.; Rea, A.J.; Holers, V.M.; Abraham, L.J.; Ulgiati, D. Analysis of tandem E-box motifs within human Complement receptor 2 (CR2/CD21) promoter reveals cell specific roles for RP58, E2A, USF and localized chromatin accessibility. Int. J. Biochem. Cell Biol. 2015, 64, 107–119. [Google Scholar] [CrossRef]
- Zhou, X.; O’Shea, E.K. Integrated Approaches Reveal Determinants of Genome-wide Binding and Function of the Transcription Factor Pho4. Mol. Cell 2011, 42, 826–836. [Google Scholar] [CrossRef]
- Kindrick, J.D.; Mole, D.R. Hypoxic regulation of gene transcription and chromatin: Cause and effect. Int. J. Mol. Sci. 2020, 21, 8320. [Google Scholar] [CrossRef]
- Guccione, E.; Martinato, F.; Finocchiaro, G.; Luzi, L.; Tizzoni, L.; Dall’ Olio, V.; Zardo, G.; Nervi, C.; Bernard, L.; Amati, B. Myc-binding-site recognition in the human genome is determined by chromatin context. Nat. Cell Biol. 2006, 8, 764–770. [Google Scholar] [CrossRef]
- Wapinski, O.L.; Vierbuchen, T.; Qu, K.; Lee, Q.Y.; Chanda, S.; Fuentes, D.R.; Giresi, P.G.; Ng, Y.H.; Marro, S.; Neff, N.F.; et al. XHierarchical mechanisms for direct reprogramming of fibroblasts to neurons. Cell 2013, 155, 621. [Google Scholar] [CrossRef]
- Park, N.I.; Guilhamon, P.; Desai, K.; McAdam, R.F.; Langille, E.; O’Connor, M.; Lan, X.; Whetstone, H.; Coutinho, F.J.; Vanner, R.J.; et al. ASCL1 Reorganizes Chromatin to Direct Neuronal Fate and Suppress Tumorigenicity of Glioblastoma Stem Cells. Cell Stem Cell 2017, 21, 209–224.e7. [Google Scholar] [CrossRef]
- Raposo, A.A.S.F.; Vasconcelos, F.F.; Drechsel, D.; Marie, C.; Johnston, C.; Dolle, D.; Bithell, A.; Gillotin, S.; van den Berg, D.L.C.; Ettwiller, L.; et al. Ascl1 coordinately regulates gene expression and the chromatin landscape during neurogenesis. Cell Rep. 2015, 10, 1544–1556. [Google Scholar] [CrossRef] [PubMed]
- Pataskar, A.; Jung, J.; Smialowski, P.; Noack, F.; Calegari, F.; Straub, T.; Tiwari, V.K. NeuroD1 reprograms chromatin and transcription factor landscapes to induce the neuronal program. EMBO J. 2016, 35, 24–45. [Google Scholar] [CrossRef] [PubMed]
- Guillemot, F.; Hassan, B.A. Beyond proneural: Emerging functions and regulations of proneural proteins. Curr. Opin. Neurobiol. 2017, 42, 93–101. [Google Scholar] [CrossRef]
- Smith, D.K.; Yang, J.; Liu, M.L.; Zhang, C.L. Small Molecules Modulate Chromatin Accessibility to Promote NEUROG2-Mediated Fibroblast-to-Neuron Reprogramming. Stem Cell Reports 2016, 7, 955–969. [Google Scholar] [CrossRef] [PubMed]
- De la Serna, I.L.; Ohkawa, Y.; Berkes, C.A.; Bergstrom, D.A.; Dacwag, C.S.; Tapscott, S.J.; Imbalzano, A.N. MyoD Targets Chromatin Remodeling Complexes to the Myogenin Locus Prior to Forming a Stable DNA-Bound Complex. Mol. Cell. Biol. 2005, 25, 3997–4009. [Google Scholar] [CrossRef]
- Knoepfler, P.S.; Bergstrom, D.A.; Uetsuki, T.; Dac-Korytko, L.; Sun, Y.H.; Wright, W.E.; Tapscott, S.J.; Kamps, M.P. A conserved motif N-terminal to the DNA-binding domains of myogenic bHLH transcription factors mediates cooperative DNA binding with Pbx-Meis1/Prep1. Nucleic Acids Res. 1999, 27, 3752–3761. [Google Scholar] [CrossRef]
- Maves, L.; Waskiewicz, A.J.; Paul, B.; Cao, Y.; Tyler, A.; Moens, C.B.; Tapscott, S.J. Pbx homeodomain proteins direct Myod activity to promote fast-muscle differentiation. Development 2007, 134, 3371–3382. [Google Scholar] [CrossRef]
- Ali, F.; Hindley, C.; McDowell, G.; Deibler, R.; Jones, A.; Kirschner, M.; Guillemot, F.; Philpott, A. Cell cycle-regulated multi-site phosphorylation of neurogenin 2 coordinates cell cycling with differentiation during neurogenesis. Development 2011, 138, 4267–4277. [Google Scholar] [CrossRef] [PubMed]
- Ali, F.R.; Cheng, K.; Kirwan, P.; Metcalfe, S.; Livesey, F.J.; Barker, R.A.; Philpott, A. The phosphorylation status of Ascl1 is a key determinant of neuronal differentiation and maturation in vivo and in vitro. Dev. 2014, 141, 2216–2224. [Google Scholar] [CrossRef]
- Liu, Y.; MacDonald, R.J.; Swift, G.H. DNA Binding and Transcriptional Activation by a PDX1·PBX1b· MEIS2b Trimer and Cooperation with a Pancreas-specific Basic Helix-Loop-Helix Complex. J. Biol. Chem. 2001, 276, 17985–17993. [Google Scholar] [CrossRef]
- Meredith, D.M.; Borromeo, M.D.; Deering, T.G.; Casey, B.H.; Savage, T.K.; Mayer, P.R.; Hoang, C.; Tung, K.-C.; Kumar, M.; Shen, C.; et al. Program Specificity for Ptf1a in Pancreas versus Neural Tube Development Correlates with Distinct Collaborating Cofactors and Chromatin Accessibility. Mol. Cell. Biol. 2013, 33, 3166–3179. [Google Scholar] [CrossRef] [PubMed]
- Ito, S.; Shen, L.; Dai, Q.; Wu, S.C.; Collins, L.B.; Swenberg, J.A.; He, C.; Zhang, Y. Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science 2011, 333, 1300–1303. [Google Scholar] [CrossRef]
- Perini, G.; Diolaiti, D.; Porro, A.; Della Valle, G. In vivo transcriptional regulation of N-Myc target genes is controlled by E-box methylation. Proc. Natl. Acad. Sci. USA 2005, 102, 12117–12122. [Google Scholar] [CrossRef] [PubMed]
- Prendergast, G.C.; Lawe, D.; Ziff, E.B. Association of Myn, the murine homolog of Max, with c-Myc stimulates methylation-sensitive DNA binding and ras cotransformation. Cell 1991, 65, 395–407. [Google Scholar] [CrossRef]
- Wang, D.; Hashimoto, H.; Zhang, X.; Barwick, B.G.; Lonial, S.; Boise, L.H.; Vertino, P.M.; Cheng, X. MAX is an epigenetic sensor of 5-carboxylcytosine and is altered in multiple myeloma. Nucleic Acids Res. 2017, 45, 2396–2407. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Zhang, X.; Blumenthal, R.M.; Cheng, X. Detection of DNA Modifications by Sequence-Specific Transcription Factors. J. Mol. Biol. 2020, 432, 1661–1673. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Horton, J.R.; Li, J.; Huang, Y.; Zhang, X.; Blumenthal, R.M.; Cheng, X. Structural basis for preferential binding of human TCF4 to DNA containing 5-carboxylcytosine. Nucleic Acids Res. 2019, 47, 8375–8387. [Google Scholar] [CrossRef] [PubMed]
- Golla, J.P.; Zhao, J.; Mann, I.K.; Sayeed, S.K.; Mandal, A.; Rose, R.B.; Vinson, C. Carboxylation of cytosine (5caC) in the CG dinucleotide in the E-box motif (CGCAG|GTG) increases binding of the Tcf3|Ascl1 helix-loop-helix heterodimer 10-fold. Biochem. Biophys. Res. Commun. 2014, 449, 248–255. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Zhou, T.; Dror, I.; Mathelier, A.; Wasserman, W.W.; Gordân, R.; Rohs, R. TFBSshape: A motif database for DNA shape features of transcription factor binding sites. Nucleic Acids Res. 2014, 42, 148–155. [Google Scholar] [CrossRef] [PubMed]
- Aydin, B.; Kakumanu, A.; Rossillo, M.; Moreno-estellés, M.; Garipler, G.; Ringstad, N.; Flames, N.; Mahony, S.; Mazzoni, E.O. Proneural factors Ascl1 and Neurog2 contribute to neuronal subtype identities by establishing distinct chromatin landscapes. Nat. Neurosci. 2019, 22, 897–908. [Google Scholar] [CrossRef] [PubMed]
- Gordân, R.; Shen, N.; Dror, I.; Zhou, T.; Horton, J.; Rohs, R.; Bulyk, M.L. Genomic Regions Flanking E-Box Binding Sites Influence DNA Binding Specificity of bHLH Transcription Factors through DNA Shape. Cell Rep. 2013, 3, 1093–1104. [Google Scholar] [CrossRef] [PubMed]
- Huppert, J.L.; Balasubramanian, S. G-quadruplexes in promoters throughout the human genome. Nucleic Acids Res. 2007, 35, 406–413. [Google Scholar] [CrossRef]
- Huppert, J.L.; Bugaut, A.; Kumari, S.; Balasubramanian, S. G-quadruplexes: The beginning and end of UTRs. Nucleic Acids Res. 2008, 36, 6260–6268. [Google Scholar] [CrossRef]
- Huppert, J.L.; Balasubramanian, S. Prevalence of quadruplexes in the human genome. Nucleic Acids Res. 2005, 33, 2908–2916. [Google Scholar] [CrossRef]
- Etzioni, S.; Yafe, A.; Khateb, S.; Weisman-Shomer, P.; Bengal, E.; Fry, M. Homodimeric MyoD preferentially binds tetraplex structures of regulatory sequences of muscle-specific genes. J. Biol. Chem. 2005, 280, 26805–26812. [Google Scholar] [CrossRef]
- Shklover, J.; Etzioni, S.; Weisman-Shomer, P.; Yafe, A.; Bengal, E.; Fry, M. MyoD uses overlapping but distinct elements to bind E-box and tetraplex structures of regulatory sequences of muscle-specific genes. Nucleic Acids Res. 2007, 35, 7087–7095. [Google Scholar] [CrossRef]
- Walsh, K.; Gualberto, A. MyoD binds to the guanine tetrad nucleic acid structure. J. Biol. Chem. 1992, 267, 13714–13718. [Google Scholar] [CrossRef]
- Yafe, A.; Etzioni, S.; Weisman-Shomer, P.; Fry, M. Formation and properties of hairpin and tetraplex structures of guanine-rich regulatory sequences of muscle-specific genes. Nucleic Acids Res. 2005, 33, 2887–2900. [Google Scholar] [CrossRef] [PubMed]
- Yafe, A.; Shklover, J.; Weisman-Shomer, P.; Bengal, E.; Fry, M. Differential binding of quadruplex structures of muscle-specific genes regulatory sequences by MyoD, MRF4 and myogenin. Nucleic Acids Res. 2008, 36, 3916–3925. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Shklover, J.; Weisman-Shomer, P.; Yafe, A.; Michael, F. Quadruplex structures of muscle gene promoter sequences enhance in vivo MyoD-dependent gene expression. Nucleic Acids Res. 2010, 38, 2369–2377. [Google Scholar] [CrossRef] [PubMed]
- Bormuth, I.; Yan, K.; Yonemasu, T.; Gummert, M.; Zhang, M.; Wichert, S.; Grishina, O.; Pieper, A.; Zhang, W.; Goebbels, S.; et al. Neuronal basic helix-loop-helix proteins neurod2/6 regulate cortical commissure formation before midline interactions. J. Neurosci. 2013, 33, 641–651. [Google Scholar] [CrossRef] [PubMed]
- Conerly, M.L.; Yao, Z.; Zhong, J.W.; Groudine, M.; Tapscott, S.J. Distinct Activities of Myf5 and MyoD Indicate Separate Roles in Skeletal Muscle Lineage Specification and Differentiation. Dev. Cell 2016, 36, 375–385. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Song, X.; Ma, Y.; Liu, J.; Yang, D.; Yan, B. DNA binding, but not interaction with Bmal1, is responsible for DEC1-mediated transcription regulation of the circadian gene mPer1. Biochem. J. 2004, 382, 895–904. [Google Scholar] [CrossRef]
- Jiang, J.; Levine, M. Binding affinities and cooperative interactions with bHLH activators delimit threshold responses to the dorsal gradient morphogen. Cell 1993, 72, 741–752. [Google Scholar] [CrossRef]
- Mall, M.; Kareta, M.S.; Chanda, S.; Ahlenius, H.; Perotti, N.; Zhou, B.; Grieder, S.D.; Ge, X.; Drake, S.; Euong Ang, C.; et al. Myt1l safeguards neuronal identity by actively repressing many non-neuronal fates. Nature 2017, 544, 245–249. [Google Scholar] [CrossRef] [PubMed]
- Postigo, A.A.; Dean, D.C. ZEB, a vertebrate homolog of Drosophila Zfh-1, is a negative regulator of muscle differentiation. EMBO J. 1997, 16, 3935–3943. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, P.C.; Frank, S.R.; Wang, L.; Schroeder, M.; Liu, S.; Greene, J.; Cocito, A.; Amati, B. Genomic targets of the human c-Myc protein. Genes Dev. 2003, 17, 1115–1129. [Google Scholar] [CrossRef] [PubMed]
- Sessa, A.; Ciabatti, E.; Drechsel, D.; Massimino, L.; Colasante, G.; Giannelli, S.; Satoh, T.; Akira, S.; Guillemot, F.; Broccoli, V. The Tbr2 Molecular Network Controls Cortical Neuronal Differentiation Through Complementary Genetic and Epigenetic Pathways. Cereb. Cortex 2017, 27, 3378–3396. [Google Scholar] [CrossRef]
- Huang, J.; Blackwell, T.K.; Kedes, L.; Weintraub, H. Differences between MyoD DNA binding and activation site requirements revealed by functional random sequence selection. Mol. Cell. Biol. 1996, 16, 3893–3900. [Google Scholar] [CrossRef]
- Costa, A.; Powell, L.M.; Soufi, A.; Lowell, S.; Jarman, A.P. Atoh1 is repurposed from neuronal to hair cell determinant by Gfi1 acting as a coactivator without redistributing Atoh1’s genomic binding sites. bioRxiv 2019, 1–25. [Google Scholar] [CrossRef]
- Domcke, S.; Hill, A.J.; Daza, R.M.; Cao, J.; Day, D.R.O.; Pliner, H.A.; Aldinger, K.A.; Pokholok, D.; Zhang, F.; Milbank, J.H.; et al. A human cell atlas of fetal chromatin accessibility. Science 2020, 809. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Yao, Z.; Sarkar, D.; Lawrence, M.; Sanchez, G.J.; Parker, M.H.; MacQuarrie, K.L.; Davison, J.; Morgan, M.T.; Ruzzo, W.L.; et al. Genome-wide MyoD Binding in Skeletal Muscle Cells: A Potential for Broad Cellular Reprogramming. Dev. Cell 2010, 18, 662–674. [Google Scholar] [CrossRef] [PubMed]
- Borromeo, M.D.; Meredith, D.M.; Castro, D.S.; Chang, J.C.; Tung, K.C.; Guillemot, F.; Johnson, J.E. A transcription factor network specifying inhibitory versus excitatory neurons in the dorsal spinal cord. Dev. 2014, 141, 2803–2812. [Google Scholar] [CrossRef]
- Hahn, M.A.; Jin, S.G.; Li, A.X.; Liu, J.; Huang, Z.; Wu, X.; Kim, B.W.; Johnson, J.; Bilbao, A.D.V.; Tao, S.; et al. Reprogramming of DNA methylation at NEUROD2-bound sequences during cortical neuron differentiation. Sci. Adv. 2019, 5, 1–14. [Google Scholar] [CrossRef]
- Palii, C.G.; Perez-Iratxeta, C.; Yao, Z.; Cao, Y.; Dai, F.; Davison, J.; Atkins, H.; Allan, D.; Dilworth, F.J.; Gentleman, R.; et al. Differential genomic targeting of the transcription factor TAL1 in alternate haematopoietic lineages. EMBO J. 2011, 30, 494–509. [Google Scholar] [CrossRef] [PubMed]
- Yevshin, I.; Sharipov, R.; Valeev, T.; Kel, A.; Kolpakov, F. GTRD: A database of transcription factor binding sites identified by ChIP-seq experiments. Nucleic Acids Res. 2017, 45, D61–D67. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
de Martin, X.; Sodaei, R.; Santpere, G. Mechanisms of Binding Specificity among bHLH Transcription Factors. Int. J. Mol. Sci. 2021, 22, 9150. https://doi.org/10.3390/ijms22179150
de Martin X, Sodaei R, Santpere G. Mechanisms of Binding Specificity among bHLH Transcription Factors. International Journal of Molecular Sciences. 2021; 22(17):9150. https://doi.org/10.3390/ijms22179150
Chicago/Turabian Stylede Martin, Xabier, Reza Sodaei, and Gabriel Santpere. 2021. "Mechanisms of Binding Specificity among bHLH Transcription Factors" International Journal of Molecular Sciences 22, no. 17: 9150. https://doi.org/10.3390/ijms22179150
APA Stylede Martin, X., Sodaei, R., & Santpere, G. (2021). Mechanisms of Binding Specificity among bHLH Transcription Factors. International Journal of Molecular Sciences, 22(17), 9150. https://doi.org/10.3390/ijms22179150