
The Proteasome Inhibitor Bortezomib Induces Apoptosis and Activation in Gel-Filtered Human Platelets

 , , and
, , and 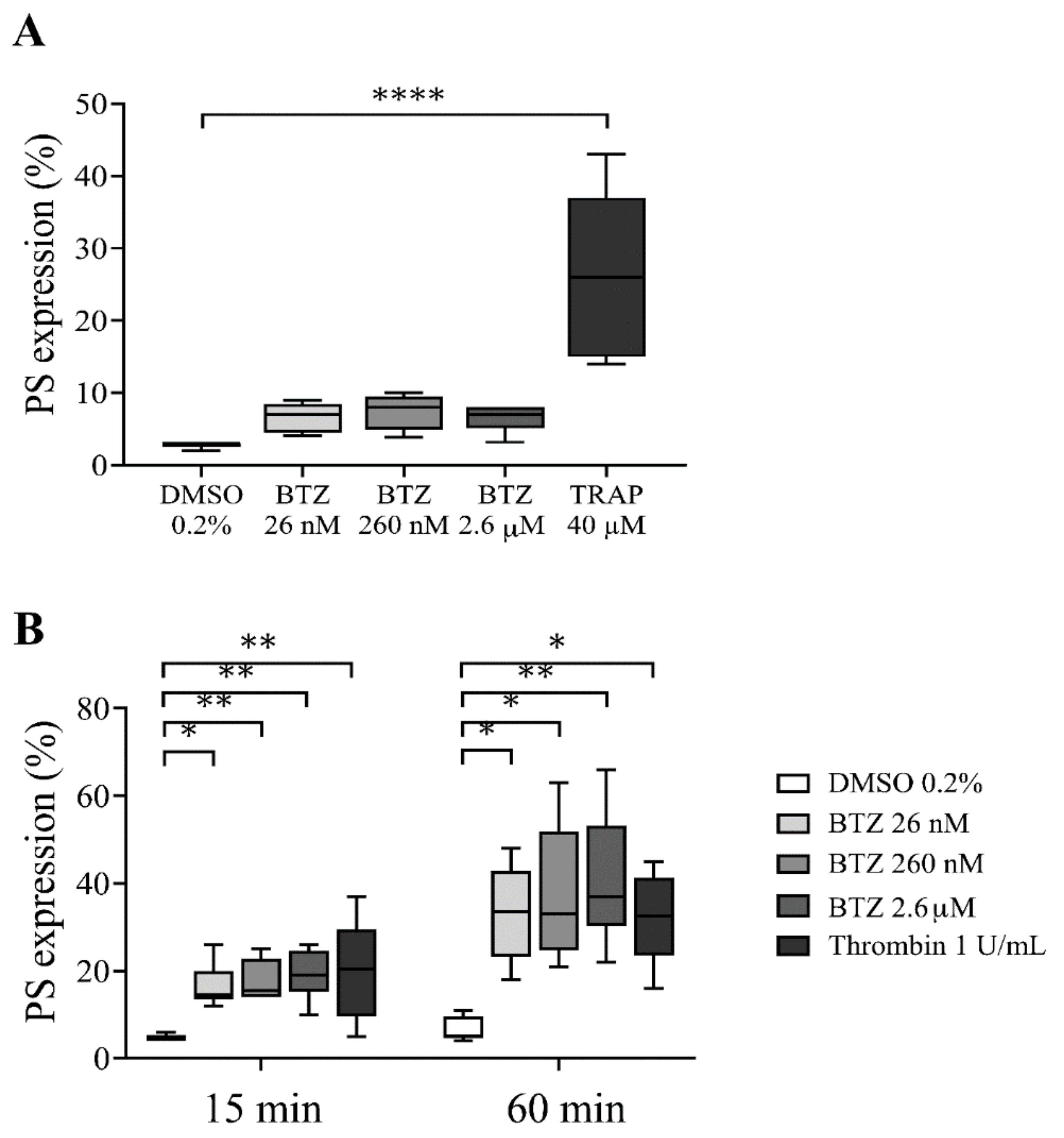{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
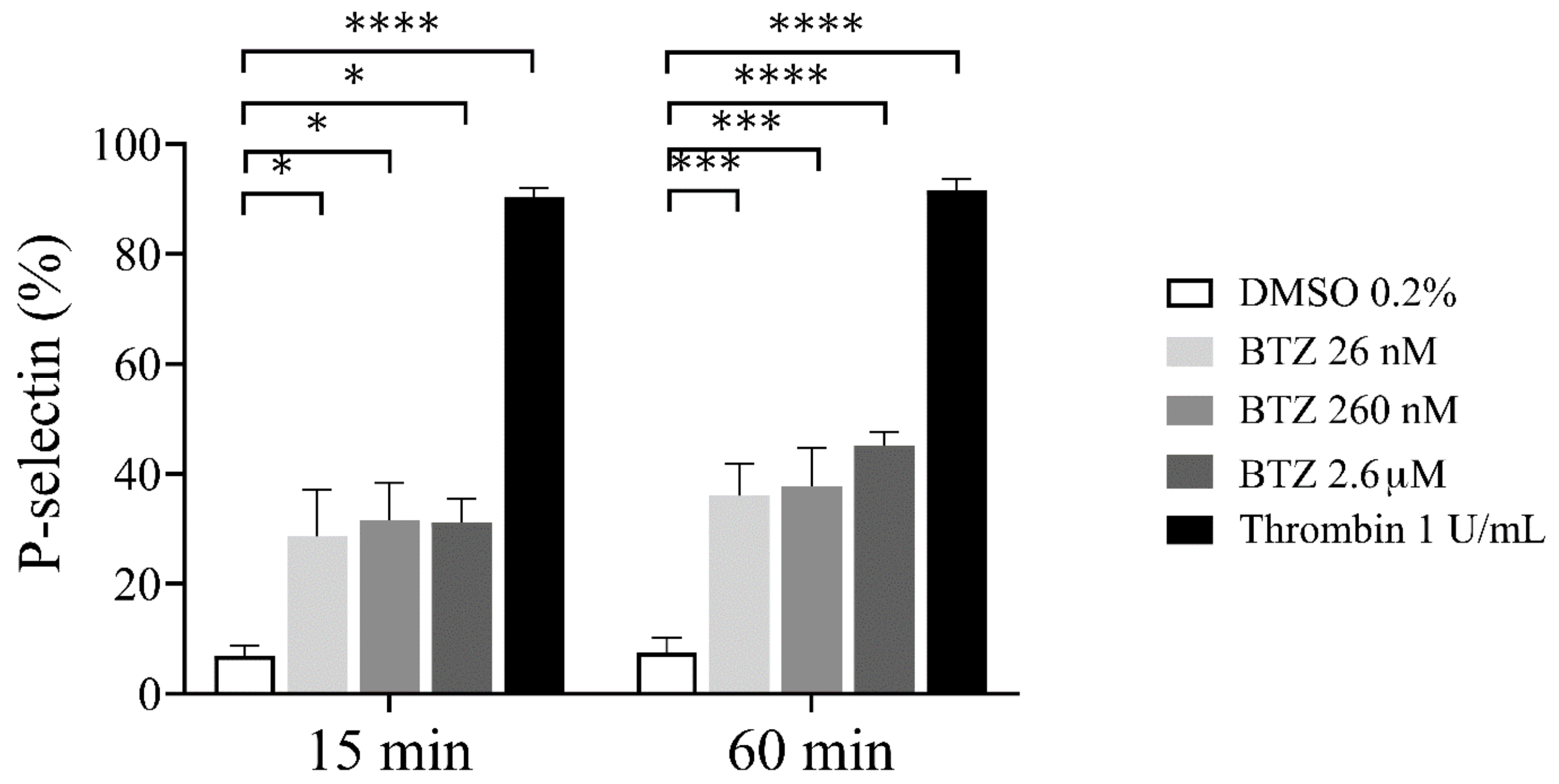
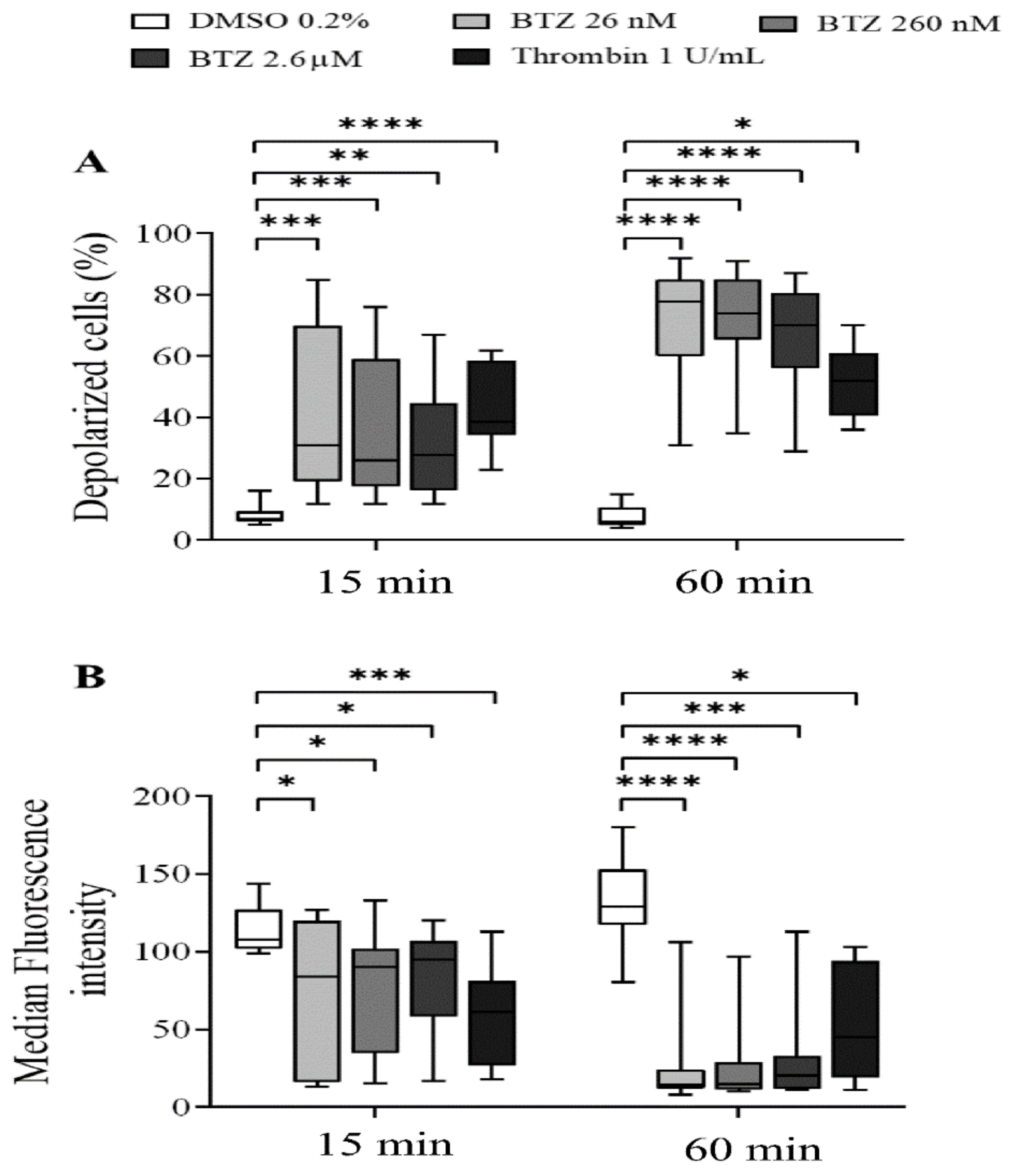
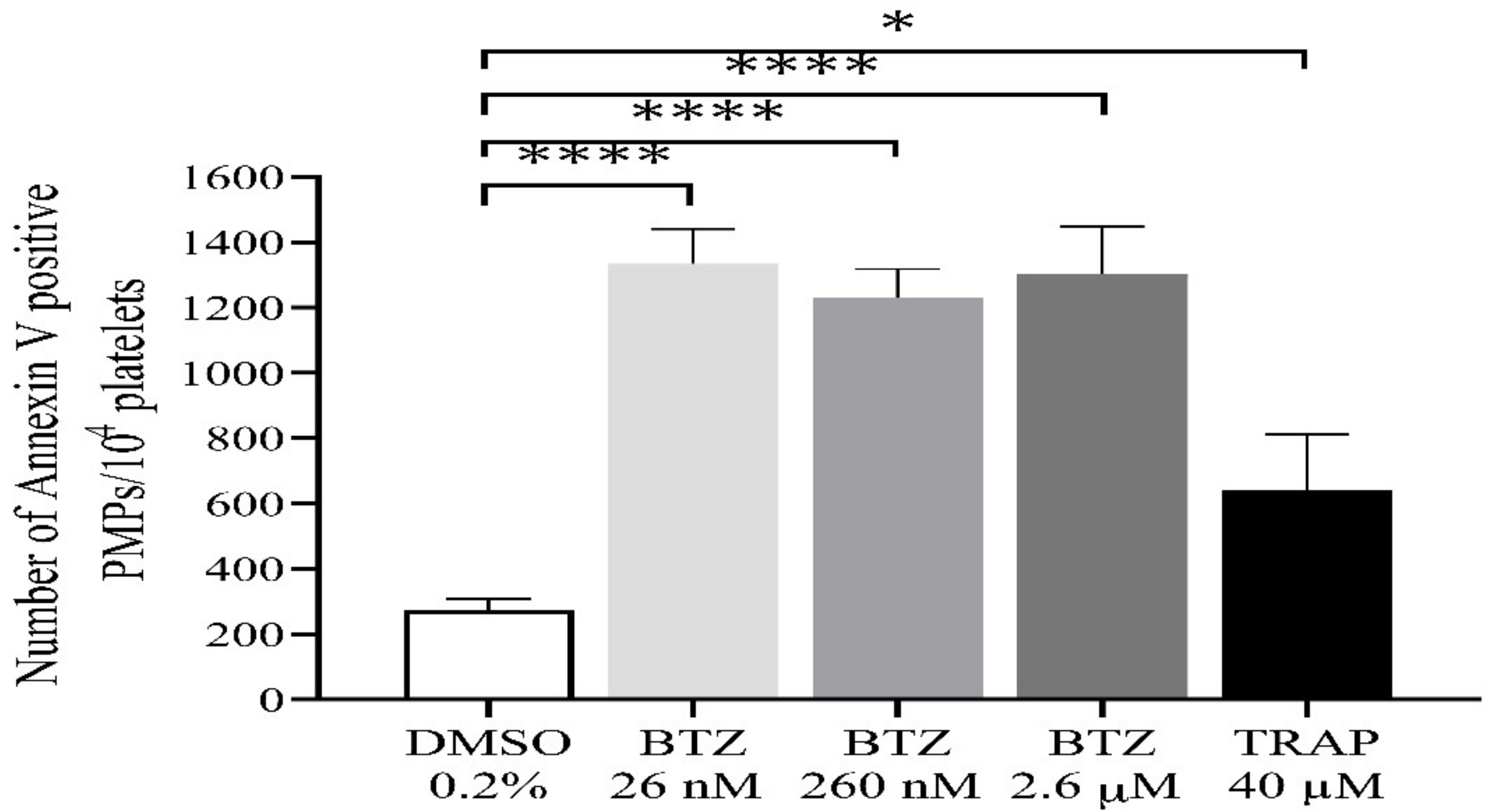
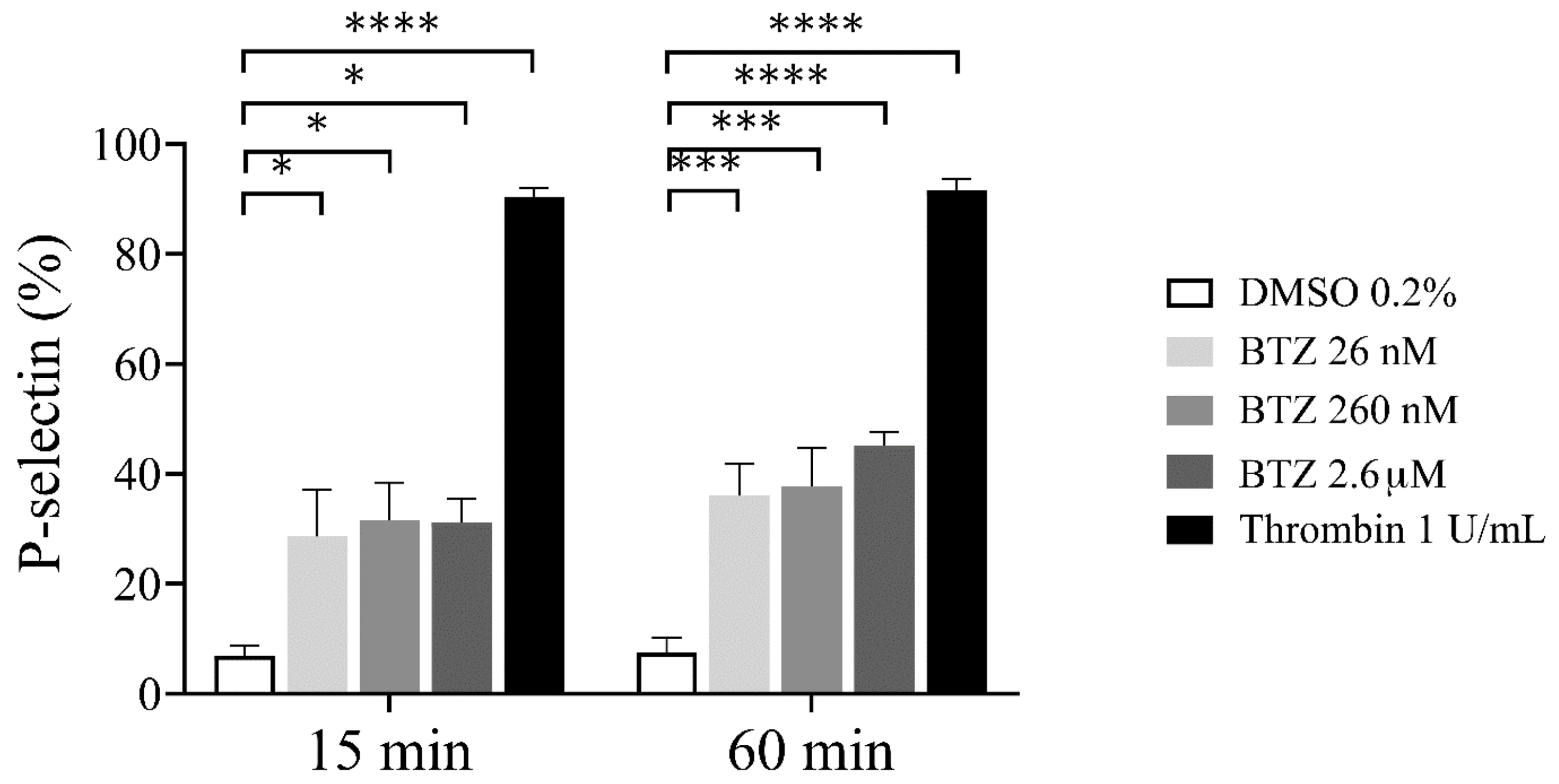
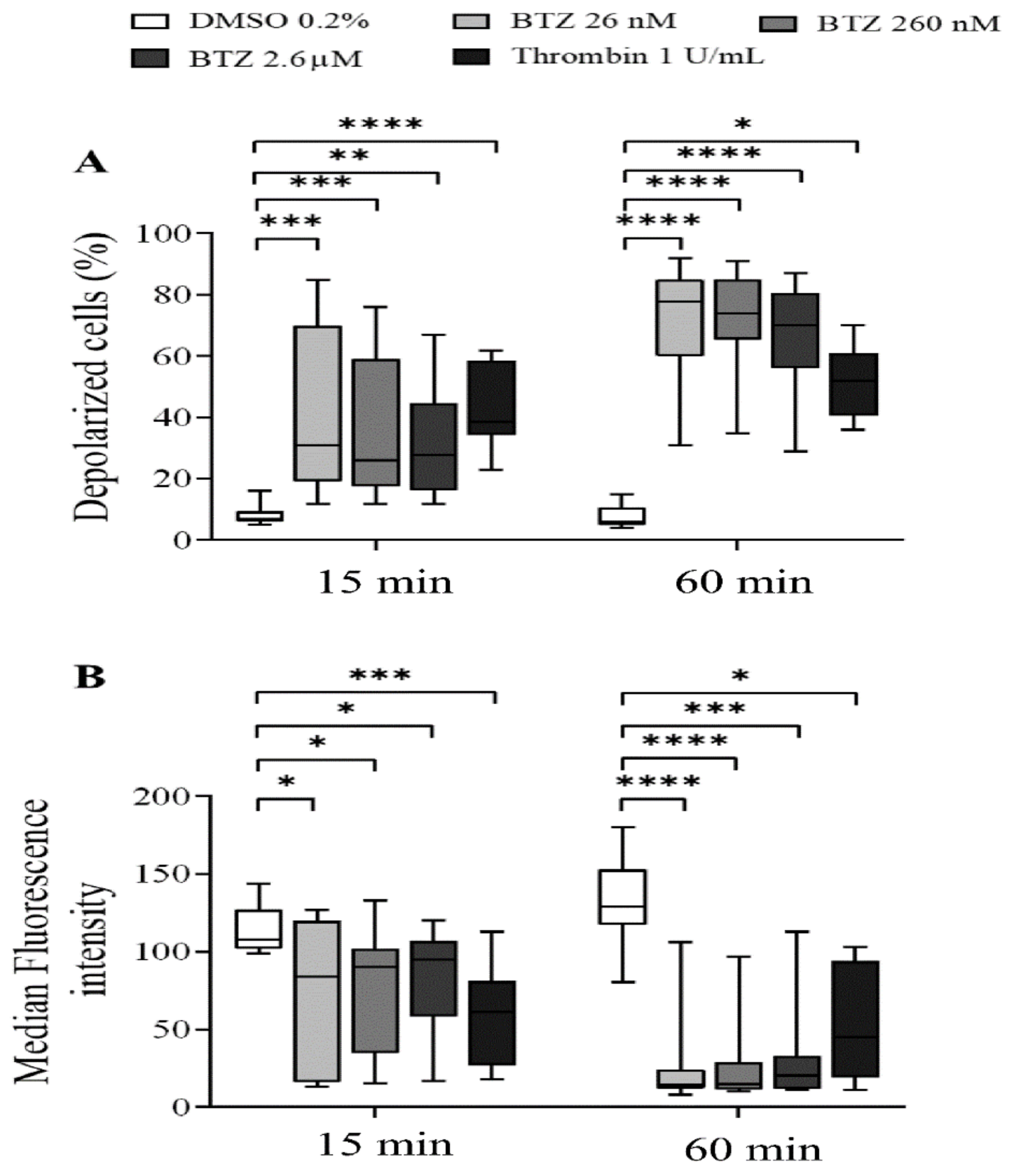
2.1. Platelet Activation and Apoptosis Studies with BTZ
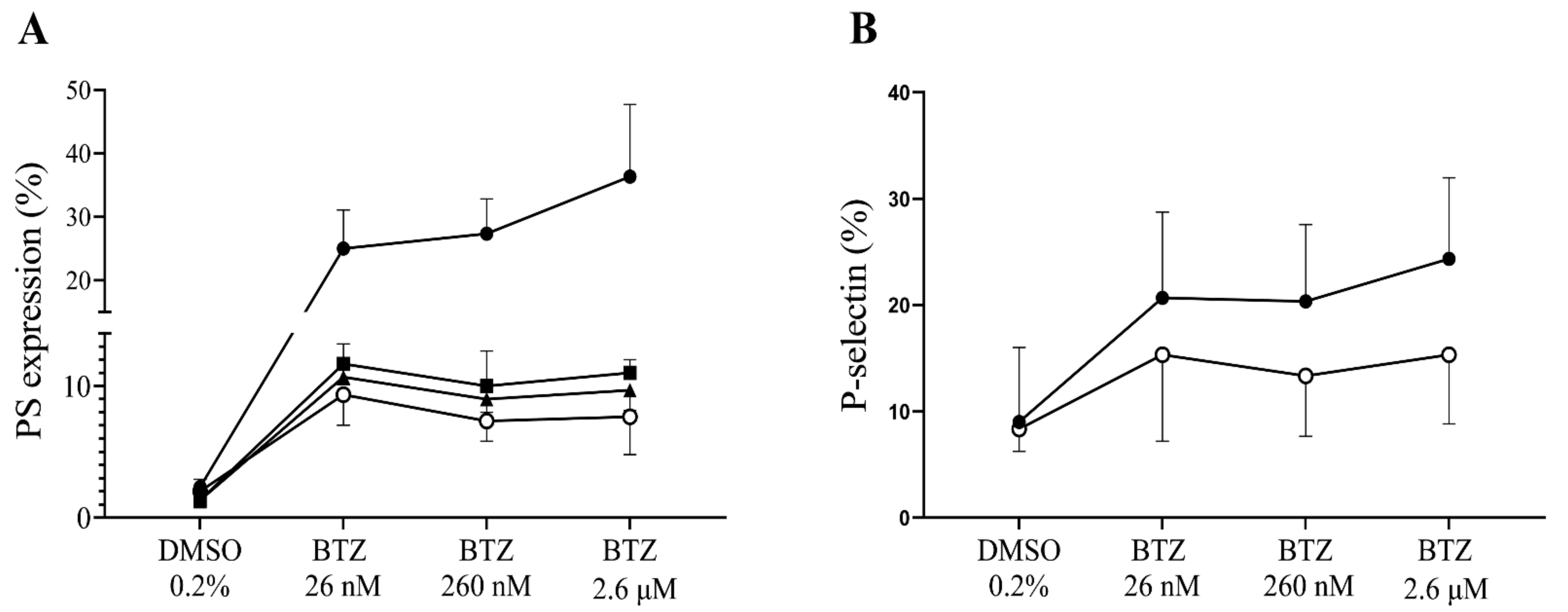
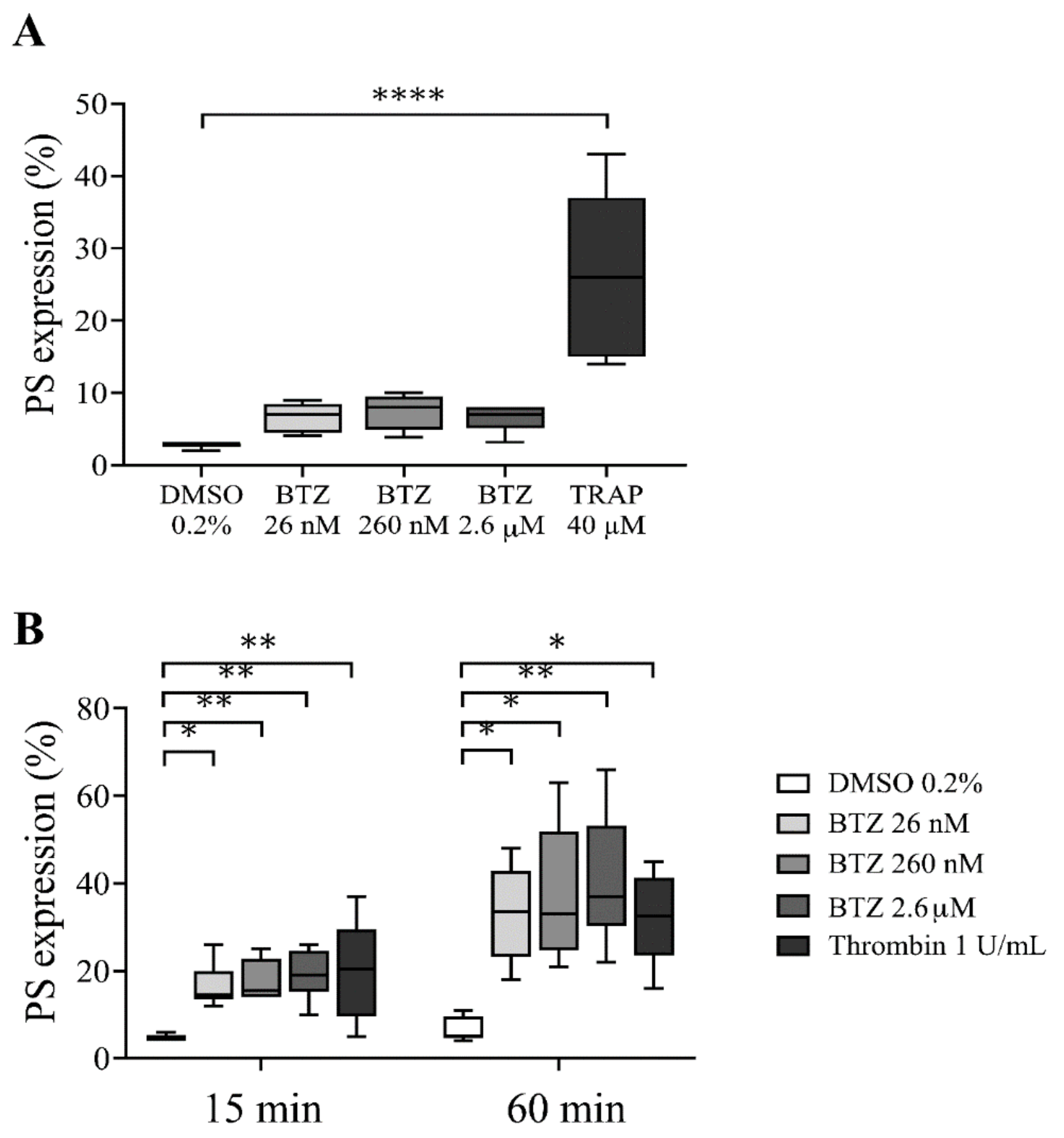
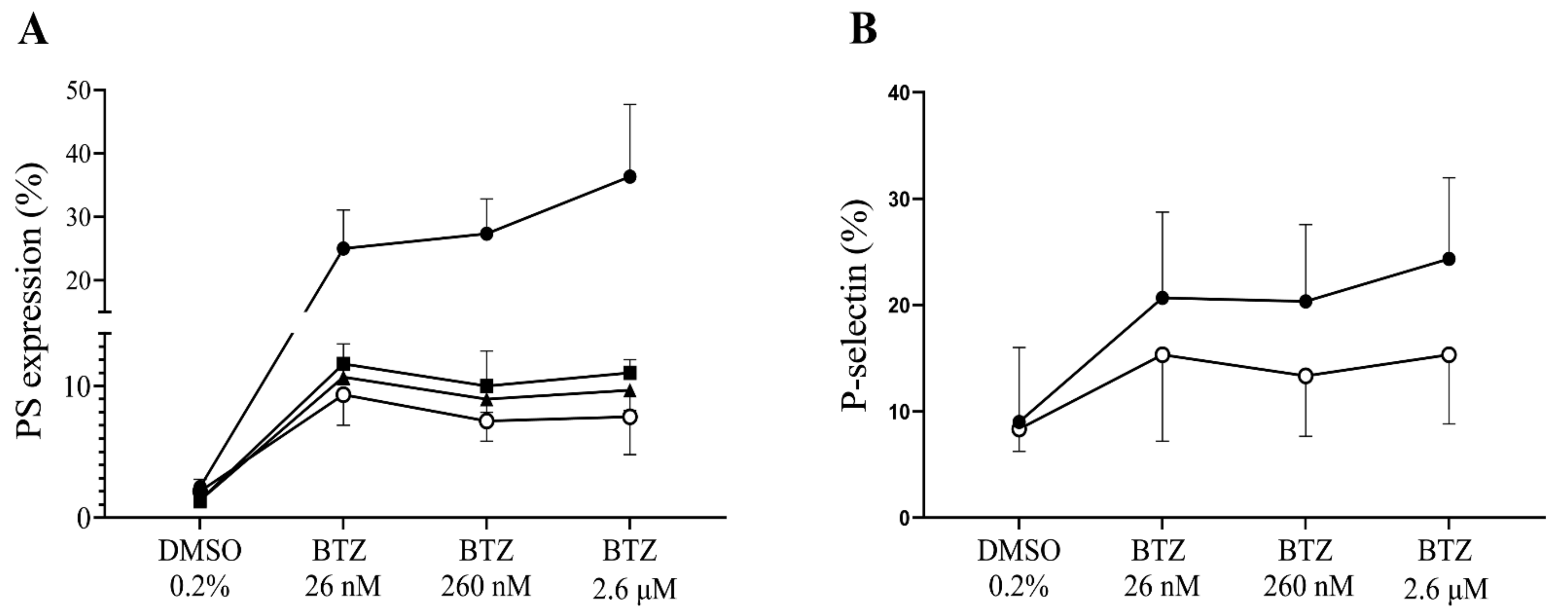
2.2. Functional Assays for Enhanced Phosphatidylserine (PS) Expression
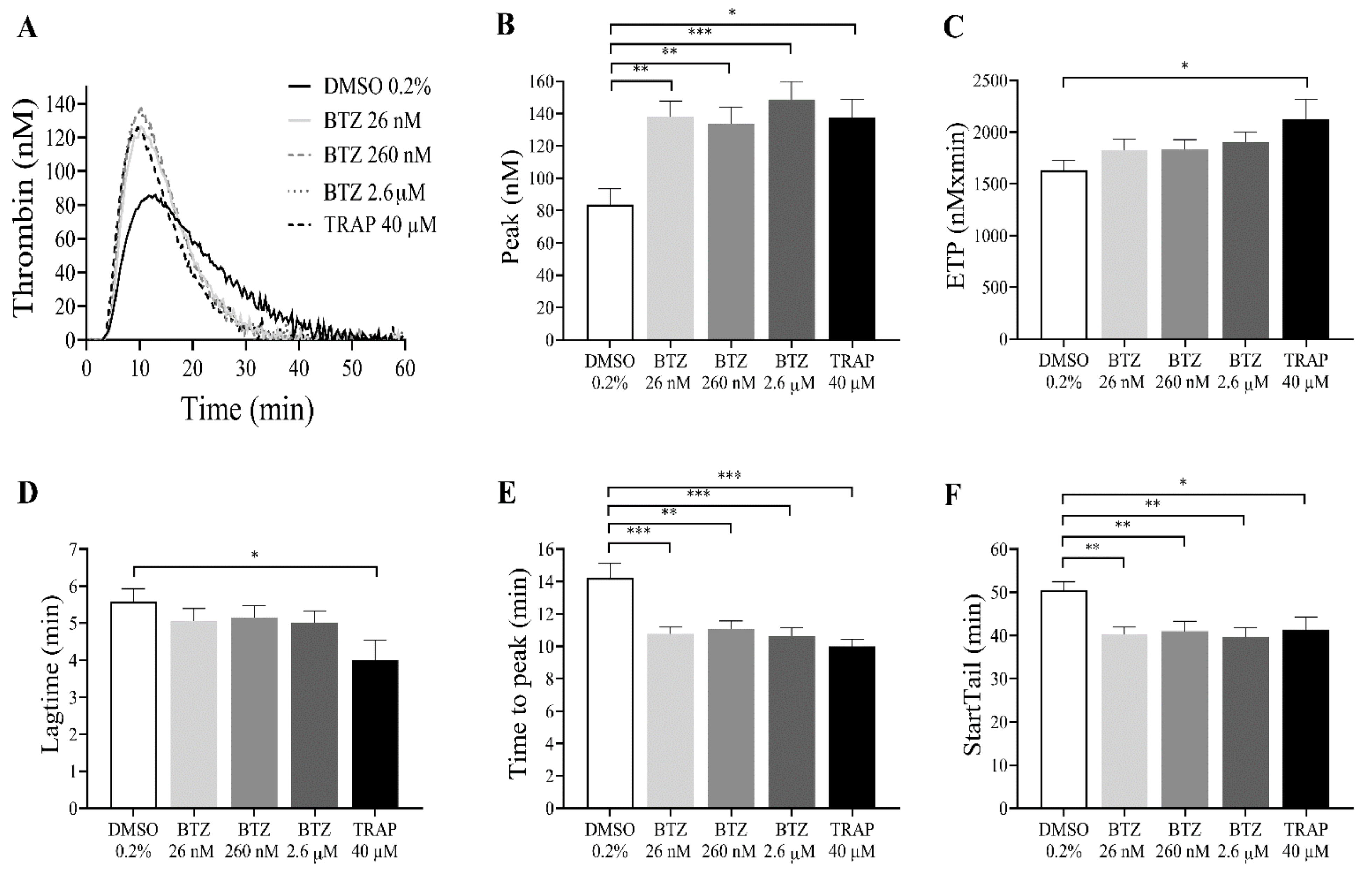
2.3. Plasma Proteins Neutralize the Effect of BTZ on Human Platelets In Vitro
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Blood Collection
4.3. Plasma Preparation
4.4. Isolation of Platelets by Gel-Filtration Chromatography
4.4.1. PS Expression in Platelets and PMPs
4.4.2. P-Selectin Expression
4.4.3. Mitochondrial Inner Membrane Potential (Δψm) Depolarization
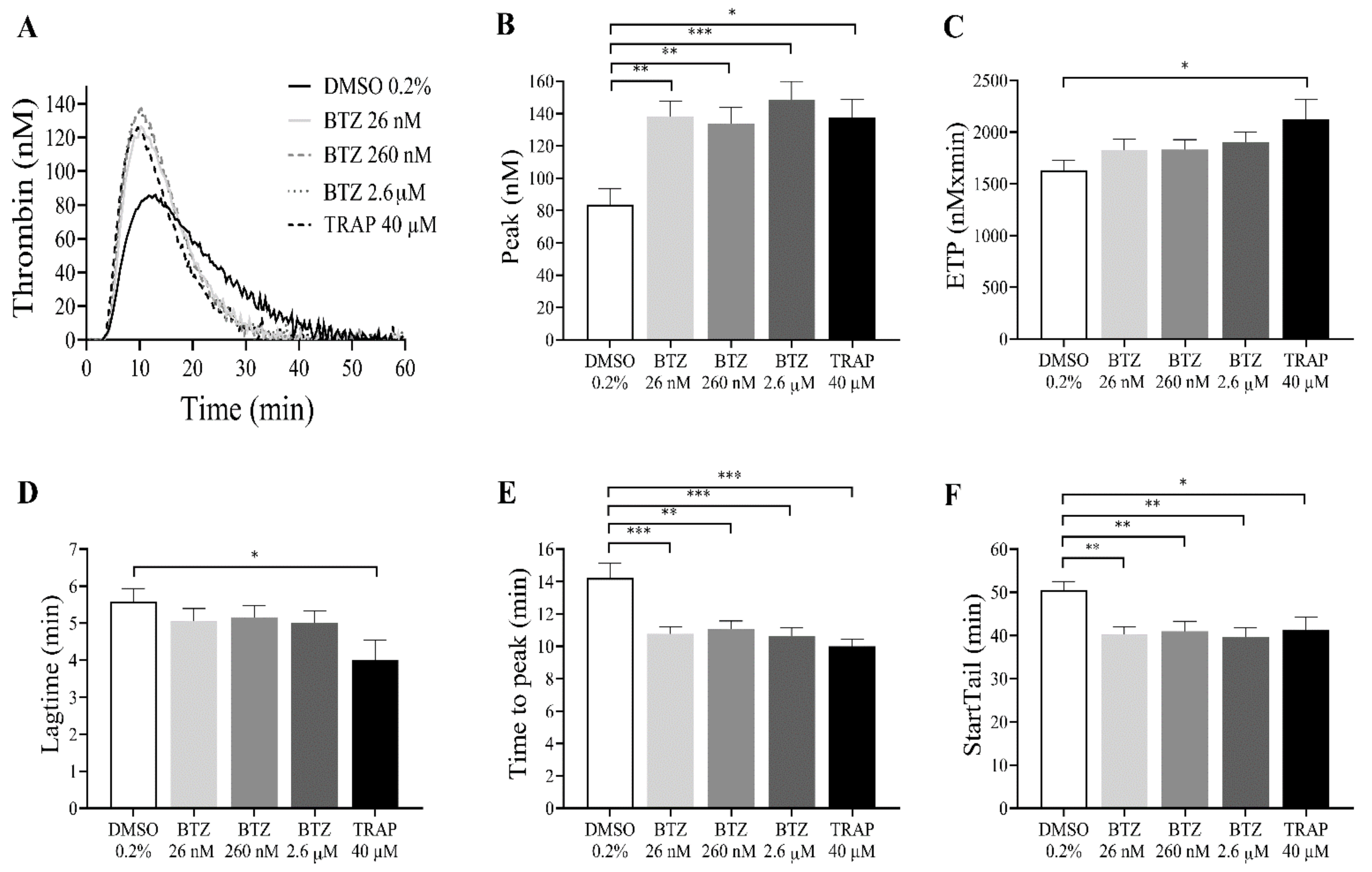
4.5. Thrombin Generation Assay
4.6. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lata, S.; Mishra, R.; Banerjea, A.C. Proteasomal degradation machinery: Favorite target of HIV-1 proteins. Front. Microbiol. 2018, 9, 1–17. [Google Scholar] [CrossRef]
- Guedes, R.A.; Serra, P.; Salvador, J.A.R.; Guedes, R.C. Computational approaches for the discovery of human proteasome inhibitors: An overview. Molecules 2016, 21, 927. [Google Scholar] [CrossRef] [Green Version]
- Thibaudeau, T.A.; Smith, D.M. A practical review of proteasome pharmacology. Pharmacol. Rev. 2019, 71, 170–197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanaka, K. The proteasome: Overview of structure and functions. Proc. Japan Acad. Ser. B Phys. Biol. Sci. 2009, 85, 12–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kish-Trier, E.; Hill, C.P. Structural biology of the proteasome. Annu. Rev. Biophys. 2013, 42, 29–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Groll, M.; Bajorek, M.; Köhler, A.; Moroder, L.; Rubin, D.M.; Huber, R.; Glickman, M.H.; Finley, D. A gated channel into the proteasome core particle. Nat. Struct. Biol. 2000, 7, 1062–1067. [Google Scholar] [CrossRef]
- Groll, M.; Heinemeyer, W.; Jäger, S.; Ullrich, T.; Bochtler, M.; Wolf, D.H.; Huber, R. The catalytic sites of 20S proteasomes and their role in subunit maturation: A mutational and crystallographic study. Proc. Natl. Acad. Sci. USA 1999, 96, 10976–10983. [Google Scholar] [CrossRef] [Green Version]
- Crawford, L.J.; Walker, B.; Irvine, A.E. Proteasome inhibitors in cancer therapy. J. Cell Commun. Signal. 2011, 5, 101–110. [Google Scholar] [CrossRef] [Green Version]
- Latonen, L.; Moore, H.M.; Bai, B.; Jäämaa, S.; Laiho, M. Proteasome inhibitors induce nucleolar aggregation of proteasome target proteins and polyadenylated RNA by altering ubiquitin availability. Oncogene 2011, 30, 790–805. [Google Scholar] [CrossRef] [Green Version]
- Obeng, E.A.; Carlson, L.M.; Gutman, D.M.; Harrington, W.J.; Lee, K.P.; Boise, L.H. Proteasome inhibitors induce a terminal unfolded protein response in multiple myeloma cells. Blood 2006, 107, 4907–4916. [Google Scholar] [CrossRef] [Green Version]
- Jung, T.; Catalgol, B.; Grune, T. The proteasomal system. Mol. Aspects Med. 2009, 30, 191–296. [Google Scholar] [CrossRef] [PubMed]
- Park, J.E.; Miller, Z.; Jun, Y.; Lee, W.; Kim, K.B. Next-generation proteasome inhibitors for cancer therapy. Transl. Res. 2018, 198, 1–16. [Google Scholar] [CrossRef]
- Fricker, L.D. Proteasome Inhibitor Drugs. Annu. Rev. Pharmacol. Toxicol. 2020, 60, 457–476. [Google Scholar] [CrossRef] [Green Version]
- Ito, S. Proteasome inhibitors for the treatment of multiple myeloma. Cancers 2020, 12, 265. [Google Scholar] [CrossRef] [Green Version]
- Gandolfi, S.; Laubach, J.P.; Hideshima, T.; Chauhan, D.; Anderson, K.C.; Richardson, P.G. The proteasome and proteasome inhibitors in multiple myeloma. Cancer Metastasis Rev. 2017, 36, 561–584. [Google Scholar] [CrossRef]
- Sherman, D.J.; Li, J. Proteasome inhibitors: Harnessing proteostasis to combat disease. Molecules 2020, 25, 671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, D.; Frezza, M.; Schmitt, S.; Kanwar, J.; Dou, Q.P. Bortezomib as the First Proteasome Inhibitor Anticancer Drug: Current Status and Future Perspectives. Curr. Cancer Drug Targets 2011, 11, 239–253. [Google Scholar] [CrossRef] [Green Version]
- Tundo, G.R.; Sbardella, D.; Santoro, A.M.; Coletta, A.; Oddone, F.; Grasso, G.; Milardi, D.; Lacal, P.M.; Marini, S.; Purrello, R.; et al. The proteasome as a druggable target with multiple therapeutic potentialities: Cutting and non-cutting edges. Pharmacol. Ther. 2020, 213, 107579. [Google Scholar] [CrossRef] [PubMed]
- Manasanch, E.E.; Orlowski, R.Z. Proteasome inhibitors in cancer therapy. Nat. Rev. Clin. Oncol. 2017, 14, 417–433. [Google Scholar] [CrossRef] [PubMed]
- Mofers, A.; Selvaraju, K.; Gubat, J.; D’Arcy, P.; Linder, S. Identification of proteasome inhibitors using analysis of gene expression profiles. Eur. J. Pharmacol. 2020, 889, 173709. [Google Scholar] [CrossRef]
- Robak, P.; Robak, T. Bortezomib for the Treatment of Hematologic Malignancies: 15 Years Later. Drugs R D 2019, 19, 73–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sevim, E.; Willis, R.; Erkan, D. Is there a role for immunosuppression in antiphospholipid syndrome? Hematology 2019, 2019, 426–432. [Google Scholar] [CrossRef]
- El-Kadiry, A.E.-H.; Merhi, Y. The Role of the Proteasome in Platelet Function. Int. J. Mol. Sci. 2021, 22, 3999. [Google Scholar] [CrossRef]
- Nayak, M.K.; Kumar, K.; Dash, D. Regulation of proteasome activity in activated human platelets. Cell Calcium 2011, 49, 226–232. [Google Scholar] [CrossRef]
- Ostrowska, H.; Ostrowska, J.K.; Worowski, K.; Radziwon, P. Human platelet 20S proteasome: Inhibition of its chymotrypsin-like activity and identification of the proteasome activator PA28. A preliminary report. Platelets 2003, 14, 151–157. [Google Scholar] [CrossRef]
- Klockenbusch, C.; Walsh, G.M.; Brown, L.M.; Hoffman, M.D.; Ignatchenko, V.; Kislinger, T.; Kast, J. Global proteome analysis identifies active immunoproteasome subunits in human platelets. Mol. Cell. Proteom. 2014, 13, 3308–3319. [Google Scholar] [CrossRef] [Green Version]
- Shi, D.S.; Smith, M.C.P.; Campbell, R.A.; Zimmerman, P.W.; Franks, Z.B.; Kraemer, B.F.; Machlus, K.R.; Ling, J.; Kamba, P.; Schwertz, H.; et al. Proteasome function is required for platelet production. J. Clin. Investig. 2014, 124, 3757–3766. [Google Scholar] [CrossRef]
- Nayak, M.K.; Kulkarni, P.P.; Dash, D. Regulatory role of proteasome in determination of platelet life span. J. Biol. Chem. 2013, 288, 6826–6834. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lonial, S.; Waller, E.K.; Richardson, P.G.; Jagannath, S.; Orlowski, R.Z.; Giver, C.R.; Jaye, D.L.; Francis, D.; Giusti, S.; Torre, C.; et al. Risk factors and kinetics of thrombocytopenia associated with bortezomib for relapsed, refractory multiple myeloma. Blood 2005, 106, 3777–3784. [Google Scholar] [CrossRef] [PubMed]
- Murai, K.; Kowata, S.; Shimoyama, T.; Yashima-Abo, A.; Fujishima, Y.; Ito, S.; Ishida, Y. Bortezomib induces thrombocytopenia by the inhibition of proplatelet formation of megakaryocytes. Eur. J. Haematol. 2014, 93, 290–296. [Google Scholar] [CrossRef] [PubMed]
- Koessler, J.; Etzel, J.; Weber, K.; Boeck, M.; Kobsar, A. Evaluation of dose-dependent effects of the proteasome inhibitor bortezomib in human platelets. Eur. J. Pharmacol. 2016, 791, 99–104. [Google Scholar] [CrossRef] [PubMed]
- Koessler, J.; Schuepferling, A.; Klingler, P.; Koessler, A.; Weber, K.; Boeck, M.; Kobsar, A. The role of proteasome activity for activating and inhibitory signalling in human platelets. Cell. Signal. 2019, 62, 109351. [Google Scholar] [CrossRef]
- Klingler, P.; Niklaus, M.; Koessler, J.; Weber, K.; Koessler, A.; Boeck, M.; Kobsar, A. Influence of long-term proteasome inhibition on platelet responsiveness mediated by bortezomib. Vascul. Pharmacol. 2021, 138, 106830. [Google Scholar] [CrossRef] [PubMed]
- Tan, C.R.C.; Abdul-Majeed, S.; Cael, B.; Barta, S.K. Clinical Pharmacokinetics and Pharmacodynamics of Bortezomib. Clin. Pharmacokinet. 2018, 58, 1–12. [Google Scholar] [CrossRef]
- Curran, M.P.; McKeage, K. Bortezomib: A review of its use in patients with multiple myeloma. Drugs 2009, 69, 859–888. [Google Scholar] [CrossRef]
- Bross, P.F.; Kane, R.; Farrell, A.T.; Abraham, S.; Benson, K.; Brower, M.E.; Bradley, S.; Gobburu, J.V.; Goheer, A.; Lee, S.L.; et al. Approval summary for bortezomib for injection in the treatment of multiple myeloma. Clin. Cancer Res. 2004, 10, 3954–3964. [Google Scholar] [CrossRef] [Green Version]
- Antwi-Baffour, S.; Adjei, J.; Aryeh, C.; Kyeremeh, R.; Kyei, F.; Seidu, M.A. Understanding the biosynthesis of platelets-derived extracellular vesicles. Immun. Inflamm. Dis. 2015, 3, 133–140. [Google Scholar] [CrossRef]
- Takano, K.; Asazuma, N.; Satoh, K.; Yatomi, Y.; Ozaki, Y. Collagen-induced generation of platelet-derived microparticles in whole blood is dependent on ADP released from red blood cells and calcium ions. Platelets 2004, 15, 223–229. [Google Scholar] [CrossRef]
- Pinto, V.; Bergantim, R.; Caires, H.R.; Seca, H.; Guimarães, J.E.; Vasconcelos, M.H. Multiple myeloma: Available therapies and causes of drug resistance. Cancers 2020, 12, 407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Almond, J.B.; Cohen, G.M. The proteasome: A novel target for cancer chemotherapy. Leukemia 2002, 16, 433–443. [Google Scholar] [CrossRef] [Green Version]
- Arpinati, M.; Chirumbolo, G.; Nicolini, B.; Agostinelli, C.; Rondelli, D. Selective apoptosis of monocytes and monocyte-derived DCs induced by bortezomib (Velcade). Bone Marrow Transplant. 2009, 43, 253–259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kholmukhamedov, A.; Jobe, S. Mitochondria and cell death. Thromb. Haemost. 2017, 117, 2207–2208. [Google Scholar]
- Libourel, E.J.; Sonneveld, P.; Van Der Holt, B.; De Maat, M.P.M.; Leebeek, F.W.G. High incidence of arterial thrombosis in young patients treated for multiple myeloma: Results of a prospective cohort study. Blood 2010, 116, 22–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mateos, M.V. Management of treatment-related adverse events in patients with multiple myeloma. Cancer Treat. Rev. 2010, 36, 24–32. [Google Scholar] [CrossRef]
- Yui, J.C.; Van Keer, J.; Weiss, B.M.; Waxman, A.J.; Palmer, M.B.; D’Agati, V.D.; Kastritis, E.; Dimopoulos, M.A.; Vij, R.; Bansal, D.; et al. Proteasome inhibitor associated thrombotic microangiopathy. Am. J. Hematol. 2016, 91, E348–E352. [Google Scholar] [CrossRef] [PubMed]
- Nechipurenko, D.Y.; Receveur, N.; Yakimenko, A.O.; Shepelyuk, T.O.; Yakusheva, A.A.; Kerimov, R.R.; Obydennyy, S.I.; Eckly, A.; Léon, C.; Gachet, C.; et al. Clot Contraction Drives the Translocation of Procoagulant Platelets to Thrombus Surface. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 37–47. [Google Scholar] [CrossRef]
- Alberio, L.; Ravanat, C.; Hechler, B.; Mangin, P.H.; Lanza, F.; Gachet, C. Delayed-onset of procoagulant signalling revealed by kinetic analysis of COAT platelet formation. Thromb. Haemost. 2017, 117, 1101–1114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kholmukhamedov, A.; Janecke, R.; Choo, H.J.; Jobe, S.M. The mitochondrial calcium uniporter regulates procoagulant platelet formation. J. Thromb. Haemost. 2018, 16, 2315–2321. [Google Scholar] [CrossRef] [Green Version]
- Guo, L.; Tong, D.; Yu, M.; Zhang, Y.; Li, T.; Wang, C.; Zhou, P.; Jin, J.; Li, B.; Liu, Y.; et al. Phosphatidylserine-exposing cells contribute to the hypercoagulable state in patients with multiple myeloma. Int. J. Oncol. 2018, 52, 1981–1990. [Google Scholar] [CrossRef] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ghansah, H.; Debreceni, I.B.; Fejes, Z.; Nagy, B., Jr.; Kappelmayer, J. The Proteasome Inhibitor Bortezomib Induces Apoptosis and Activation in Gel-Filtered Human Platelets. Int. J. Mol. Sci. 2021, 22, 8955. https://doi.org/10.3390/ijms22168955
Ghansah H, Debreceni IB, Fejes Z, Nagy B Jr., Kappelmayer J. The Proteasome Inhibitor Bortezomib Induces Apoptosis and Activation in Gel-Filtered Human Platelets. International Journal of Molecular Sciences. 2021; 22(16):8955. https://doi.org/10.3390/ijms22168955
Chicago/Turabian StyleGhansah, Harriet, Ildikó Beke Debreceni, Zsolt Fejes, Béla Nagy, Jr., and János Kappelmayer. 2021. "The Proteasome Inhibitor Bortezomib Induces Apoptosis and Activation in Gel-Filtered Human Platelets" International Journal of Molecular Sciences 22, no. 16: 8955. https://doi.org/10.3390/ijms22168955
APA StyleGhansah, H., Debreceni, I. B., Fejes, Z., Nagy, B., Jr., & Kappelmayer, J. (2021). The Proteasome Inhibitor Bortezomib Induces Apoptosis and Activation in Gel-Filtered Human Platelets. International Journal of Molecular Sciences, 22(16), 8955. https://doi.org/10.3390/ijms22168955