
Chromatin Structure and “DNA Sequence View”: The Role of Satellite DNA in Ectopic Pairing of the Drosophila X Polytene Chromosome

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
- DNA sequences of the chromosome sections (A) are taken one by one, searching for short single-stranded fragments (k-mers) of a given length that are the same as fragments of the other sections (B). For each pair of sections (A-B), FMF is calculated as the total number of matching fragments for both DNA chains. To increase matching specificity, short DNA repeats (microsatellites) can be excluded at this stage.
- For each section A, the rho value of the Spearman correlation between FEC and FMF is computed. Both specific and unspecific correlations are considered (FEC and FMF values correspond to the same or different section pairs, respectively).
- For all sections A, the average rho values (R) and the proportion of statistically significant FEC-FMF correlations (P) are calculated at different fragment lengths, for different Drosophila strains, and statistically analyzed.
- For each A-B pair, the list of the matching fragments is generated and ordered according to their numbers of occurrence. This lets us reveal short DNA sequences that specifically impact FEC-FMF correlation.
2. Results
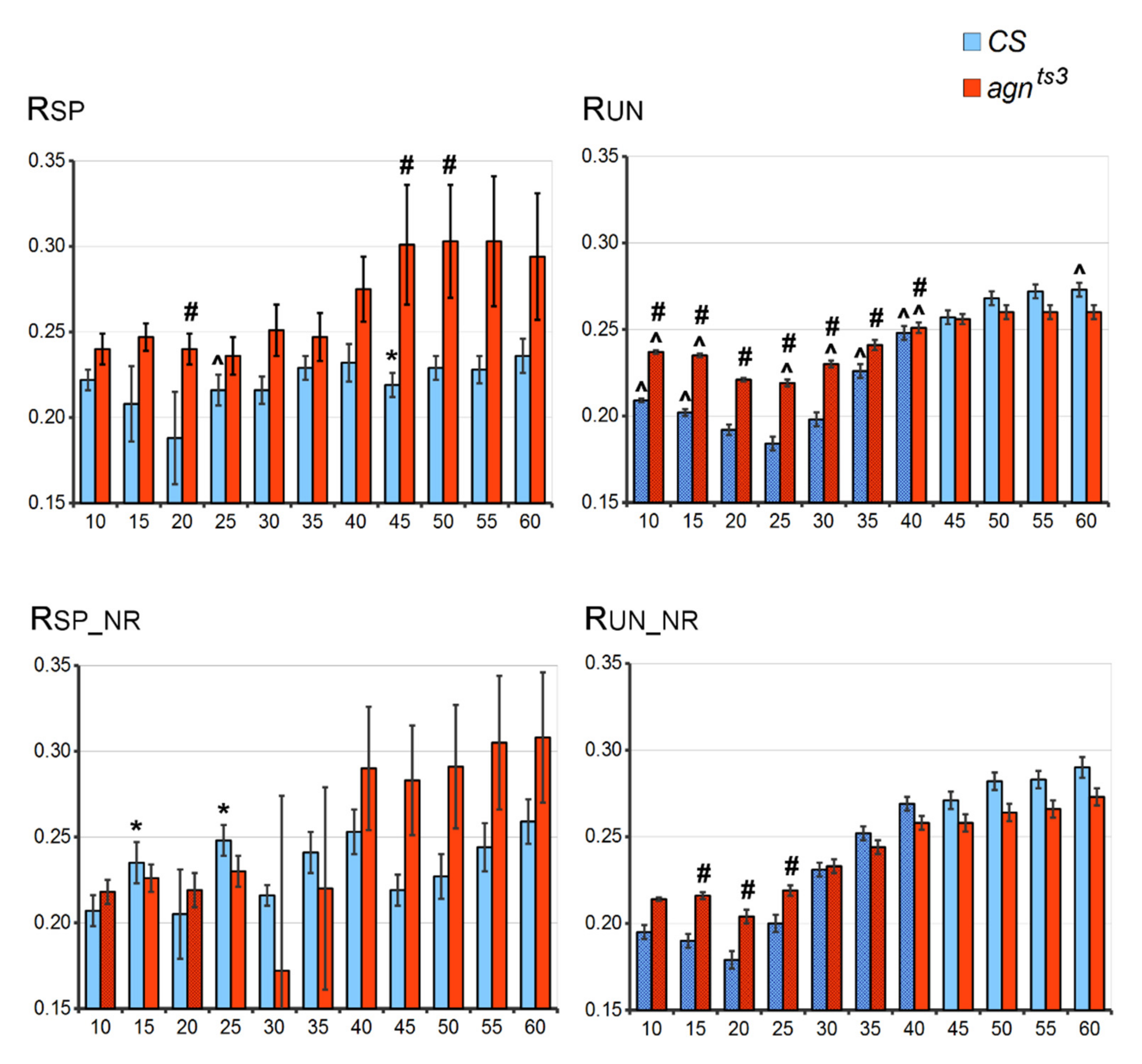
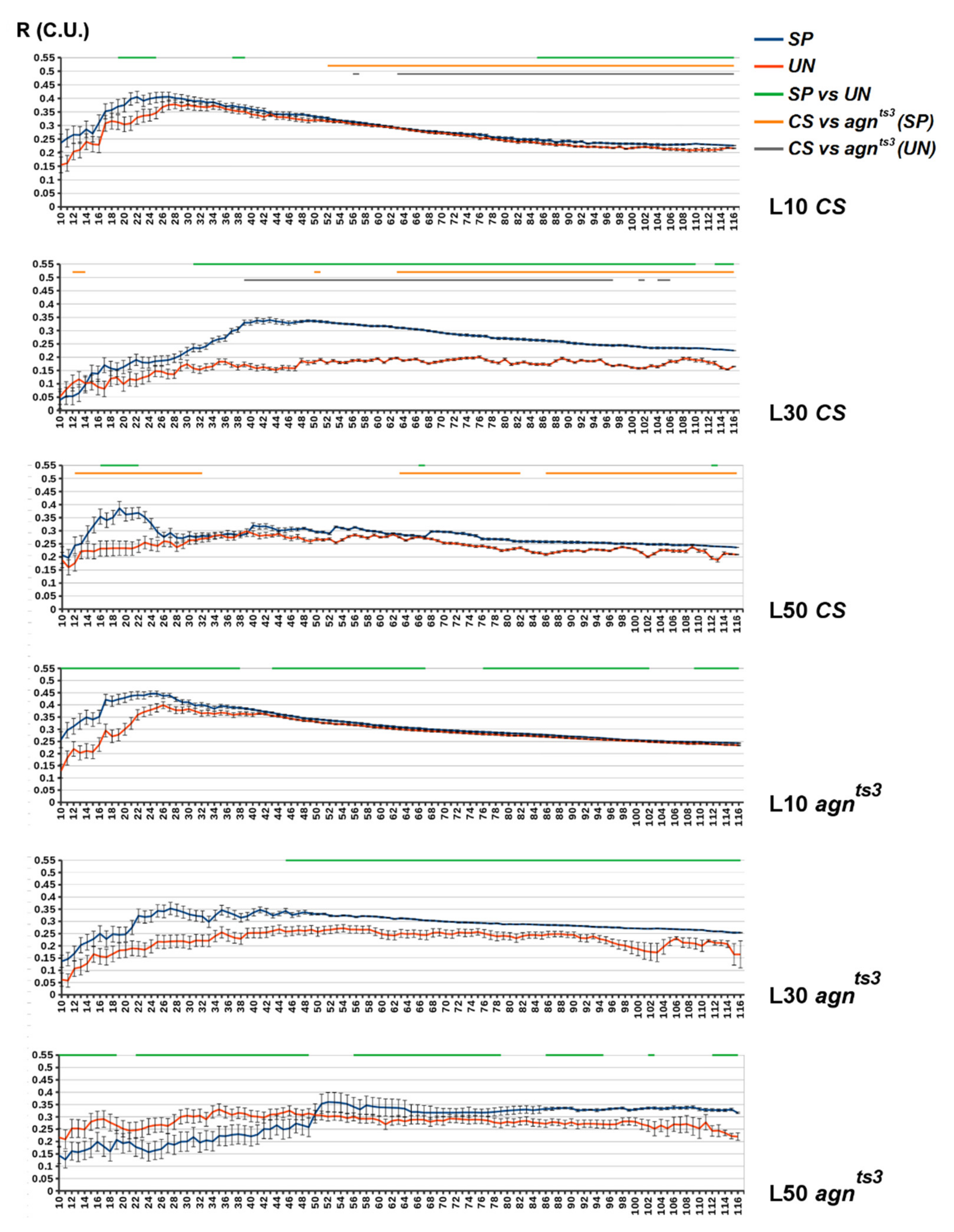
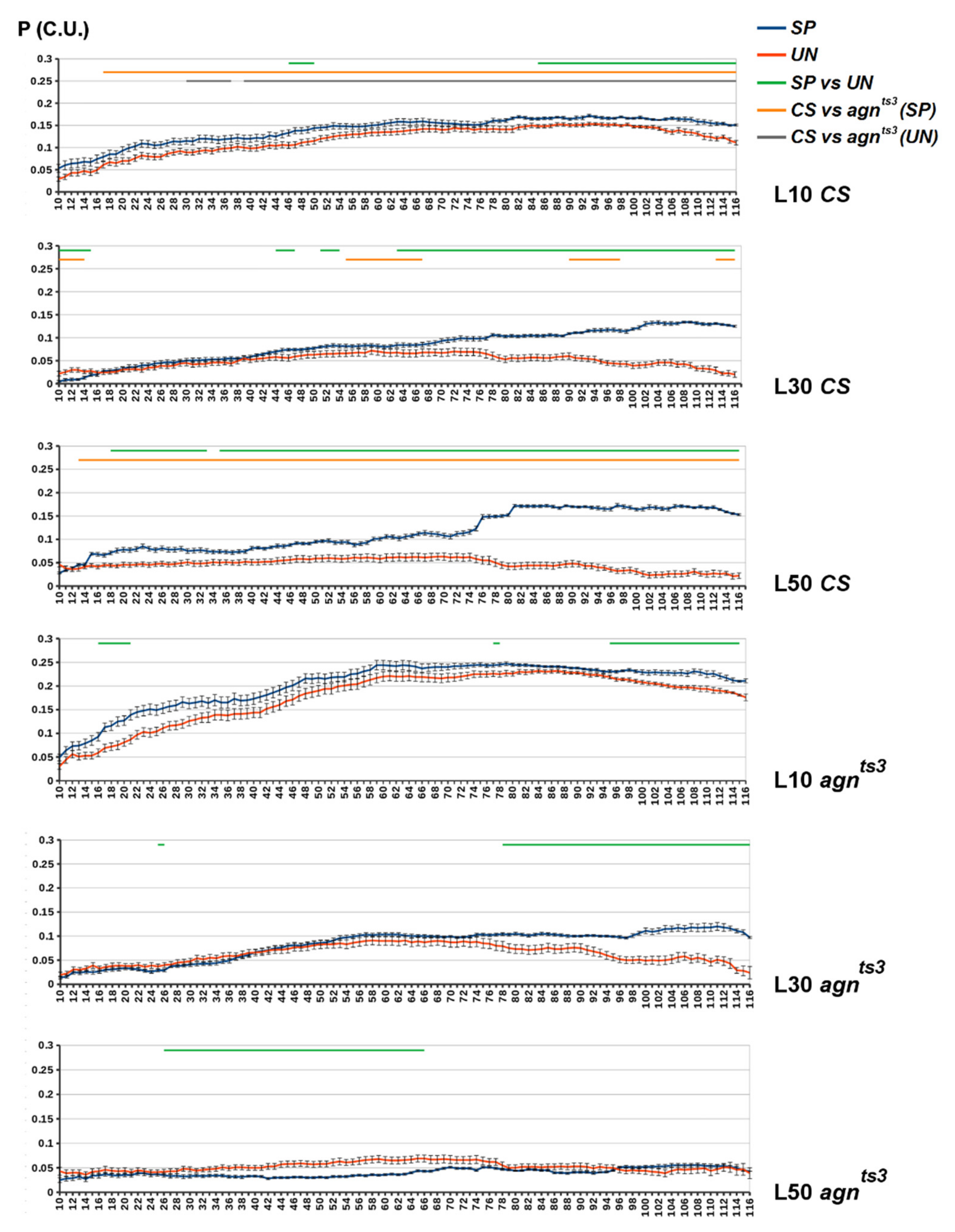
2.1. FEC-FMF Correlations for the Whole X Chromosome
2.2. FEC-FMF Correlation for Chromosomal Regions of the Different Length
2.3. Sections Prone to Ectopic Pairing
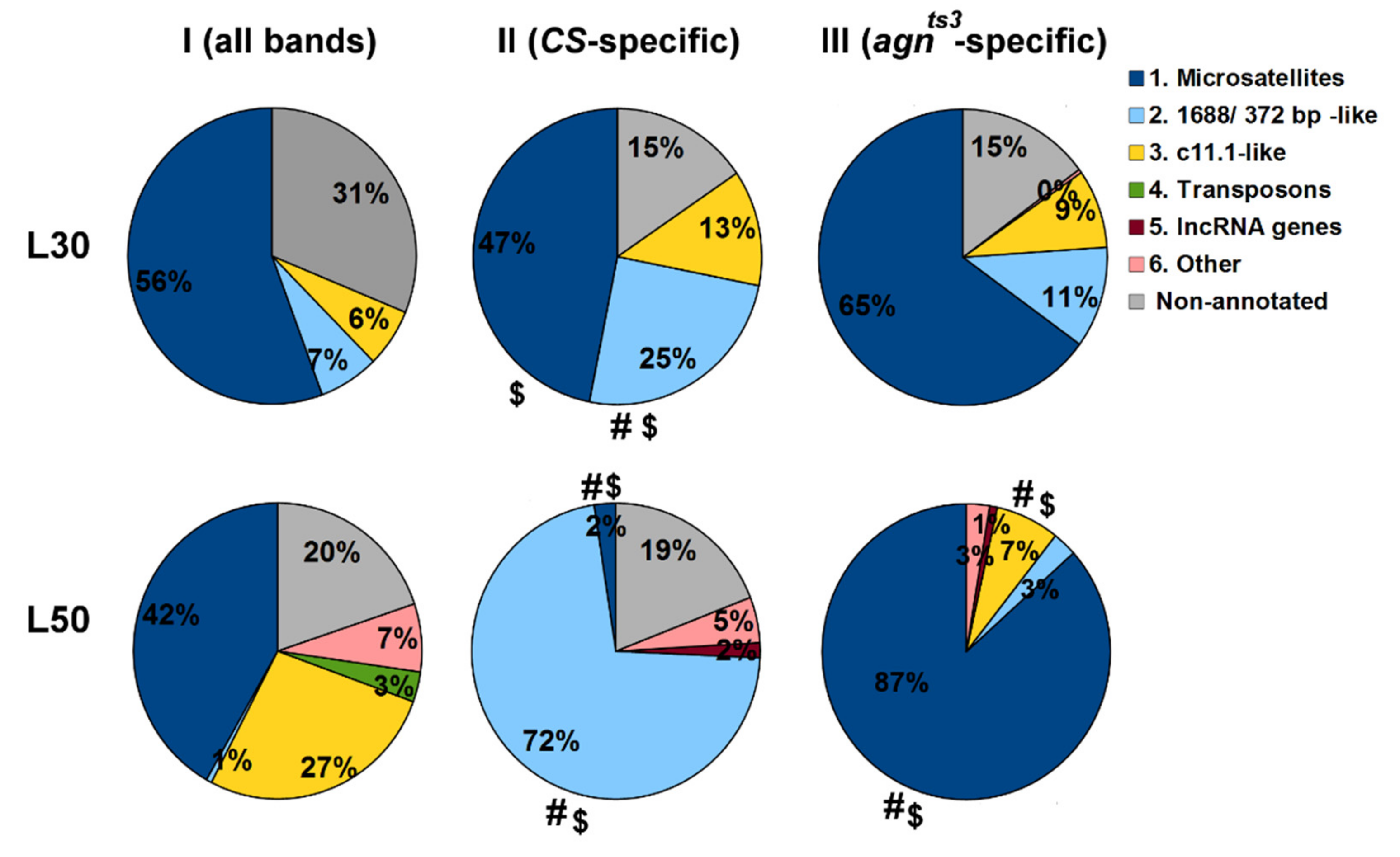
2.4. The Biological Nature of Sequences Making Impact into Ectopic Pairing
3. Discussion
4. Materials and Methods
4.1. FEC Matrices
4.2. Bioinformatics Analysis
- Sequential selection (with a step of 1 nt) of short DNA fragments of a given length (L) from one specific section of the X chromosome.
- Search of the section fragments, as well as the complimentary fragments, within all the other sections of the X chromosome, using Aho-Corasick algorithm.
- For each X chromosome section (B) except A: calculation of normalized frequency of all fragments matching for A and B (FMF(A-B)). The average FMF for all chunks of 10 kb length is equal to 1. The list of the localized fragments is saved for each B.
- Stages 1–3. are repeated for all X chromosome sections.
- 5.
- Specific correlation computation:
- For each section A: calculation of FEC(A)-FMF(A) Spearman rank correlation coefficient (rho; p < 0.05, n = 119).
- Calculation of the average rho value (R) and proportion of statistically significant FEC-FMF cases (P). P is calculated as follows: P = n (p < 0.05)/total n of estimations for which Spearman correlation data were obtained.
- 6.
- Unspecific correlation computation: For all different sections A and B: calculation of FEC(A)-FMF(B) Spearman rank correlation rho and p. R and P values are calculated as in 5b.
- 7.
- Stages 1–6. are repeated at different fragment lengths (L) (10–60 nt, with a step of 5 nt).
- 8.
- Excluding DNA microsatellites: Stages 1–7 are repeated with fragment samplings excluding fragments that contain DNA repeats. By default, a repeat contains identical elements in a row: four nucleotides or three dinucleotides or two trinucleotides.
- 9.
- Estimation of section proximity effect. By default, the distance (D) between the boundary sections varies from 10 to 116, and only fragments of a specific L (10, 30, 50 nt) are used to compute FEC-FMF correlations. The procedure is performed as follows:
- For each D: a sequential selection of the X chromosome zones (Z) of D length, with a step of 1 section (e.g., for D = 30, 90 different Z are selected, starting from 1 (Z1, or 1A–5F) and up to 90 (Z90, or 15F–20F)). The section notations A–F are equivalent to 1–6, so Z90 of D30 is also denoted as 156–206.
- For each Z(D): specific and unspecific R and P calculation, as described in Stages 5–8, taking into account only the sections within Z. Currently, analysis of unspecific correlations is time consuming, taking up about 3 h for each L. So some cases (e.g., with specific L values or excluded repeats) may be omitted to speed up the processes. For each D, R(D) and P(D) values constitute samplings for further statistical analysis. The sampling size n is equal to the number of Z(D): n = 120–D.
4.3. Statistical Analysis
- For results obtained at Stages 5–8.: R are compared using a two-sided Mann–Whitney U-test, P are compared using a Chi-square test for two sample proportion comparisons. The parameters of analysis are automatically varied: strain (CS/agnts3); type of correlation (specific/unspecific); repeats exclusion (“no”/“yes”); chromosome regions (with/without division into sections); type of analysis (comparison of data obtained for different Ls using the same parameters/comparison of data obtained for the same L using different parameters).
- For results obtained at Stage 9.: R and P and compared using a two-sided Mann–Whitney U-test. Samplings obtained for different Ds and Ls are analyzed independently. The parameters of analysis are automatically varied: strain (CS/agnts3); type of correlation (specific/unspecific); repeats exclusion (“no”/“yes”).
4.4. BLAST Analysis of Fragments Contributing to FMF
- For the given L values (here, L30 or L50), the full list of fragments of all sections making contributions to FMF are generated and arranged according to the number of fragment occurrences (NO) > 10, starting from the maximum NO. If the NO is equal for different fragments, only the first fragment is chosen.
- The same procedure is performed for a set of sections showing statistically significant FEC-FMF correlations for the given strain and L value (see Stage 5.).
- The biological nature of the first 50 fragments in each list is revealed using NCBI Blast (http://blast.ncbi.nlm.nih.gov; accessed on 20 February 2021): BLASTN, database—Nucleotide collection, species—Drosophila melanogaster, max target sequences—100, other parameters—by default.
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Croft, J.A.; Bridger, J.M.; Boyle, S.; Perry, P.; Teague, P.; Bickmore, W.A. Differences in the Localization and Morphology of Chromosomes in the Human Nucleus. J. Cell Biol. 1999, 145, 1119–1131. [Google Scholar] [CrossRef] [Green Version]
- Ranade, D.; Koul, S.; Thompson, J.; Prasad, K.B.; Sengupta, K. Chromosomal Aneuploidies Induced upon Lamin B2 Depletion Are Mislocalized in the Interphase Nucleus. Chromosoma 2017, 126, 223–244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goetze, S.; Mateos-Langerak, J.; Gierman, H.J.; de Leeuw, W.; Giromus, O.; Indemans, M.H.G.; Koster, J.; Ondrej, V.; Versteeg, R.; van Driel, R. The Three-Dimensional Structure of Human Interphase Chromosomes Is Related to the Transcriptome Map. Mol. Cell. Biol. 2007, 27, 4475–4487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osborne, C.S.; Chakalova, L.; Mitchell, J.A.; Horton, A.; Wood, A.L.; Bolland, D.J.; Corcoran, A.E.; Fraser, P. Myc Dynamically and Preferentially Relocates to a Transcription Factory Occupied by Igh. PLoS Biol. 2007, 5, e192. [Google Scholar] [CrossRef] [PubMed]
- Branco, M.R.; Pombo, A. Intermingling of Chromosome Territories in Interphase Suggests Role in Translocations and Transcription-Dependent Associations. PLoS Biol. 2006, 4, e138. [Google Scholar] [CrossRef] [Green Version]
- Kumaran, R.I.; Thakar, R.; Spector, D.L. Chromatin Dynamics and Gene Positioning. Cell 2008, 132, 929–934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osborne, C.S.; Chakalova, L.; Brown, K.E.; Carter, D.; Horton, A.; Debrand, E.; Goyenechea, B.; Mitchell, J.A.; Lopes, S.; Reik, W.; et al. Active Genes Dynamically Colocalize to Shared Sites of Ongoing Transcription. Nat. Genet. 2004, 36, 1065–1071. [Google Scholar] [CrossRef] [Green Version]
- Zhimulev, I.F.; Belyaeva, E.S.; Vatolina, T.Y.; Demakov, S.A. Banding Patterns in Drosophila Melanogaster Polytene Chromosomes Correlate with DNA-Binding Protein Occupancy. Bioessays 2012, 34, 498–508. [Google Scholar] [CrossRef] [PubMed]
- Zhimulev, I.F. Polytene Chromosomes, Heterochromatin, and Position Effect Variegation. Adv. Genet. 1998, 37, 1–566. [Google Scholar] [CrossRef] [PubMed]
- Kolesnikova, T.D. Banding Pattern of Polytene Chromosomes as a Representation of Universal Principles of Chromatin Organization into Topological Domains. Biochemistry 2018, 83, 338–349. [Google Scholar] [CrossRef]
- Zhimulev, I.F.; Zykova, T.Y.; Goncharov, F.P.; Khoroshko, V.A.; Demakova, O.V.; Semeshin, V.F.; Pokholkova, G.V.; Boldyreva, L.V.; Demidova, D.S.; Babenko, V.N.; et al. Genetic Organization of Interphase Chromosome Bands and Interbands in Drosophila Melanogaster. PLoS ONE 2014, 9, e101631. [Google Scholar] [CrossRef] [Green Version]
- Kolesnikova, T.D.; Goncharov, F.P.; Zhimulev, I.F. Similarity in Replication Timing between Polytene and Diploid Cells Is Associated with the Organization of the Drosophila Genome. PLoS ONE 2018, 13, e0195207. [Google Scholar] [CrossRef]
- Hochstrasser, M.; Mathog, D.; Gruenbaum, Y.; Saumweber, H.; Sedat, J.W. Spatial Organization of Chromosomes in the Salivary Gland Nuclei of Drosophila Melanogaster. J. Cell Biol. 1986, 102, 112–123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Wit, E.; de Laat, W. A Decade of 3C Technologies: Insights into Nuclear Organization. Genes Dev. 2012, 26, 11–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eagen, K.P.; Hartl, T.A.; Kornberg, R.D. Stable Chromosome Condensation Revealed by Chromosome Conformation Capture. Cell 2015, 163, 934–946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sexton, T.; Yaffe, E.; Kenigsberg, E.; Bantignies, F.; Leblanc, B.; Hoichman, M.; Parrinello, H.; Tanay, A.; Cavalli, G. Three-Dimensional Folding and Functional Organization Principles of the Drosophila Genome. Cell 2012, 148, 458–472. [Google Scholar] [CrossRef] [Green Version]
- Belyaeva, E.S.; Demakov, S.A.; Pokholkova, G.V.; Alekseyenko, A.A.; Kolesnikova, T.D.; Zhimulev, I.F. DNA Underreplication in Intercalary Heterochromatin Regions in Polytene Chromosomes of Drosophila Melanogaster Correlates with the Formation of Partial Chromosomal Aberrations and Ectopic Pairing. Chromosoma 2006, 115, 355–366. [Google Scholar] [CrossRef] [PubMed]
- Nikitina, E.; Medvedeva, A.; Zakharov, G.; Savvateeva-Popova, E. Williams Syndrome as a Model for Elucidation of the Pathway Genes—the Brain—Cognitive Functions: Genetics and Epigenetics. Acta Nat. 2014, 6, 9–22. [Google Scholar] [CrossRef]
- Nikitina, E.; Medvedeva, A.; Zakharov, G.; Savvateeva-Popova, E. The Drosophila Agnostic Locus: Involvement in the Formation of Cognitive Defects in Williams Syndrome. Acta Nat. 2014, 6, 53–61. [Google Scholar] [CrossRef]
- Manetti, F. LIM Kinases Are Attractive Targets with Many Macromolecular Partners and Only a Few Small Molecule Regulators. Med. Res. Rev. 2012, 32, 968–998. [Google Scholar] [CrossRef]
- Savvateeva-Popova, E.V.; Zhuravlev, A.V.; Brázda, V.; Zakharov, G.A.; Kaminskaya, A.N.; Medvedeva, A.V.; Nikitina, E.A.; Tokmatcheva, E.V.; Dolgaya, J.F.; Kulikova, D.A.; et al. Drosophila Model for the Analysis of Genesis of LIM-Kinase 1-Dependent Williams-Beuren Syndrome Cognitive Phenotypes: INDELs, Transposable Elements of the Tc1/Mariner Superfamily and MicroRNAs. Front. Genet. 2017, 8, 123. [Google Scholar] [CrossRef]
- Medvedeva, A.; Molotkov, D.; Nikitina, E.; Popov, A.; Karagodin, D.; Baricheva, E.; Savvateeva-Popova, E. Systemic Regulation of Genetic and Cytogenetic Processes by a Signal Cascade of Actin Remodeling: Locus Agnostic in Drosophila. Russ. J. Genet. 2008, 44, 669–681. [Google Scholar] [CrossRef]
- Zhuravlev, A.; Zakharov, G. Homology Segment Analysis. Available online: https://bitbucket.org/beneor/homology-segment-analysis/src/master/ (accessed on 30 July 2021).
- Medvedeva, A.; Tokmatcheva, E.; Kaminskaya, A.; Vasileva, S.; Nikitina, E.; Zhuravlev, A.; Zakharov, G.; Zatsepina, O.; Savvateeva-Popova, E. Parent-of-Origin Effects on Nuclear Chromatin Organization and Behavior in Drosophila Model for Williams-Beuren Syndrome. Vavilovskii Zhurnal Genet. Selektsii 2021, in press. [Google Scholar]
- Waring, G.L.; Pollack, J.C. Cloning and Characterization of a Dispersed, Multicopy, X Chromosome Sequence in Drosophila Melanogaster. Proc. Natl. Acad. Sci. USA 1987, 84, 2843–2847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Lima, L.G.; Hanlon, S.L.; Gerton, J.L. Origins and Evolutionary Patterns of the 1.688 Satellite DNA Family in Drosophila Phylogeny. G3 Genes Genomes Genet. 2020, 10, 4129–4146. [Google Scholar] [CrossRef]
- Menon, D.U.; Coarfa, C.; Xiao, W.; Gunaratne, P.H.; Meller, V.H. SiRNAs from an X-Linked Satellite Repeat Promote X-Chromosome Recognition in Drosophila Melanogaster. Proc. Natl. Acad. Sci. USA 2014, 111, 16460–16465. [Google Scholar] [CrossRef] [Green Version]
- Rošić;, S.; Köhler, F.; Erhardt, S. Repetitive Centromeric Satellite RNA Is Essential for Kinetochore Formation and Cell Division. J. Cell Biol. 2014, 207, 335–349. [Google Scholar] [CrossRef] [Green Version]
- Akoglu, H. User’s guide to correlation coefficients. Turk. J. Emerg. Med. 2018, 18, 91–93. [Google Scholar] [CrossRef]
- Savvateeva-Popova, E.; Peresleni, A.; Scharagina, L.; Medvedeva, A.; Korochkina, S.; Grigorieva, I.; Dyuzhikova, N.; Popov, A.; Baricheva, E.; Karagodin, D.; et al. Architecture of the X Chromosome, Expression of LIM Kinase 1, and Recombination in the Agnostic Mutants of Drosophila: A Model for Human Williams Syndrome. Russ. J. Genet. 2004, 40, 605–624. [Google Scholar] [CrossRef]
- Zhimulev, I.; Semeshin, V.; Kulichkov, V.; Belyaeva, E. Intercalary Heterochromatin in Drosophila. Chromosoma 1982, 87, 197–228. [Google Scholar] [CrossRef]
- Shatskikh, A.S.; Kotov, A.A.; Adashev, V.E.; Bazylev, S.S.; Olenina, L.V. Functional Significance of Satellite DNAs: Insights From Drosophila. Front. Cell Dev. Biol. 2020, 8, 312. [Google Scholar] [CrossRef] [PubMed]
- Lohe, A.R.; Brutlag, D.L. Multiplicity of Satellite DNA Sequences in Drosophila Melanogaster. Proc. Natl. Acad. Sci. USA 1986, 83, 696–700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jagannathan, M.; Warsinger-Pepe, N.; Watase, G.J.; Yamashita, Y.M. Comparative Analysis of Satellite DNA in the Drosophila Melanogaster Species Complex. G3 Genes Genomes Genet. 2017, 7, 693–704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DiBartolomeis, S.M.; Tartof, K.D.; Jackson, F.R. A Superfamily of Drosophila Satellite Related (SR) DNA Repeats Restricted to the X Chromosome Euchromatin. Nucleic Acids Res. 1992, 20, 1113–1116. [Google Scholar] [CrossRef] [Green Version]
- Savvateeva-Popova, E.; Peresleny, A.; Scharagina, L.; Tokmacheva, E.; Medvedeva, A.; Kamyshev, N.; Popov, A.; Ozersky, P.; Baricheva, E.; Karagodin, D.; et al. Complex Study of Drosophila Mutants in the Agnostic Locus: A Model for Coupling Chromosomal Architecture and Cognitive Functions. J. Evol. Biochem. Physiol. 2002, 38, 706–733. [Google Scholar] [CrossRef]
- Savvateeva, E. Genetic Control of Second Messenger Systems and Their Role in Learning. Usp. Sovr. Genet. 1991, 17, 33–99. [Google Scholar]
- Rath, U.; Ding, Y.; Deng, H.; Qi, H.; Bao, X.; Zhang, W.; Girton, J.; Johansen, J.; Johansen, K. The Chromodomain Protein, Chromator, Interacts with JIL-1 Kinase and Regulates the Structure of Drosophila Polytene Chromosomes. J. Cell. Sci. 2006, 119, 2332–2341. [Google Scholar] [CrossRef] [Green Version]
- Sage, B.T.; Csink, A.K. Heterochromatic Self-Association, a Determinant of Nuclear Organization, Does Not Require Sequence Homology in Drosophila. Genetics 2003, 165, 1183–1193. [Google Scholar] [CrossRef]
- Nikitina, E.A.; Medvedeva, A.V.; Gerasimenko, M.S.; Pronikov, V.S.; Surma, S.V.; Shchegolev, B.F.; Savvateeva-Popova, E.V. A Weakened Geomagnetic Field: Effects on Genomic Transcriptiln Activity, Learning, and Memory in Drosophila Melanogaster. Neurosci. Behav. Phys. 2018, 48, 796–803. [Google Scholar] [CrossRef]
- Bourassa, M.W.; Ratan, R.R. The Interplay between MicroRNAs and Histone Deacetylases in Neurological Diseases. Neurochem. Int. 2014, 77, 33–39. [Google Scholar] [CrossRef] [Green Version]
- Klages-Mundt, N.L.; Kumar, A.; Zhang, Y.; Kapoor, P.; Shen, X. The Nature of Actin-Family Proteins in Chromatin-Modifying Complexes. Front. Genet. 2018, 9, 398. [Google Scholar] [CrossRef]
- Davis, R.L.; Zhong, Y. The Biology of Forgetting-A Perspective. Neuron 2017, 95, 490–503. [Google Scholar] [CrossRef]
- Seum, C.; Delattre, M.; Spierer, A.; Spierer, P. Ectopic HP1 Promotes Chromosome Loops and Variegated Silencing in Drosophila. EMBO J. 2001, 20, 812–818. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lachner, M.; O’Carroll, D.; Rea, S.; Mechtler, K.; Jenuwein, T. Methylation of Histone H3 Lysine 9 Creates a Binding Site for HP1 Proteins. Nature 2001, 410, 116–120. [Google Scholar] [CrossRef] [PubMed]
- Usakin, L.; Abad, J.; Vagin, V.V.; de Pablos, B.; Villasante, A.; Gvozdev, V.A. Transcription of the 1.688 Satellite DNA Family Is under the Control of RNA Interference Machinery in Drosophila Melanogaster Ovaries. Genetics 2007, 176, 1343–1349. [Google Scholar] [CrossRef] [Green Version]
- Tartof, K.D.; Hobbs, C.; Jones, M. A Structural Basis for Variegating Position Effects. Cell 1984, 37, 869–878. [Google Scholar] [CrossRef]
- Menon, D.U.; Meller, V.H. Identification of the Drosophila X Chromosome: The Long and Short of It. RNA Biol. 2015, 12, 1088–1093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zerbino, D.R.; Achuthan, P.; Akanni, W.; Amode, M.R.; Barrell, D.; Bhai, J.; Billis, K.; Cummins, C.; Gall, A.; Girón, C.G.; et al. Ensembl 2018. Nucleic Acids Res. 2018, 46, D754–D761. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhuravlev, A.V.; Zakharov, G.A.; Anufrieva, E.V.; Medvedeva, A.V.; Nikitina, E.A.; Savvateeva-Popova, E.V. Chromatin Structure and “DNA Sequence View”: The Role of Satellite DNA in Ectopic Pairing of the Drosophila X Polytene Chromosome. Int. J. Mol. Sci. 2021, 22, 8713. https://doi.org/10.3390/ijms22168713
Zhuravlev AV, Zakharov GA, Anufrieva EV, Medvedeva AV, Nikitina EA, Savvateeva-Popova EV. Chromatin Structure and “DNA Sequence View”: The Role of Satellite DNA in Ectopic Pairing of the Drosophila X Polytene Chromosome. International Journal of Molecular Sciences. 2021; 22(16):8713. https://doi.org/10.3390/ijms22168713
Chicago/Turabian StyleZhuravlev, Aleksandr V., Gennadii A. Zakharov, Ekaterina V. Anufrieva, Anna V. Medvedeva, Ekaterina A. Nikitina, and Elena V. Savvateeva-Popova. 2021. "Chromatin Structure and “DNA Sequence View”: The Role of Satellite DNA in Ectopic Pairing of the Drosophila X Polytene Chromosome" International Journal of Molecular Sciences 22, no. 16: 8713. https://doi.org/10.3390/ijms22168713
APA StyleZhuravlev, A. V., Zakharov, G. A., Anufrieva, E. V., Medvedeva, A. V., Nikitina, E. A., & Savvateeva-Popova, E. V. (2021). Chromatin Structure and “DNA Sequence View”: The Role of Satellite DNA in Ectopic Pairing of the Drosophila X Polytene Chromosome. International Journal of Molecular Sciences, 22(16), 8713. https://doi.org/10.3390/ijms22168713