
MiR-126-3p Is Dynamically Regulated in Endothelial-to-Mesenchymal Transition during Fibrosis

 , , and
, , and 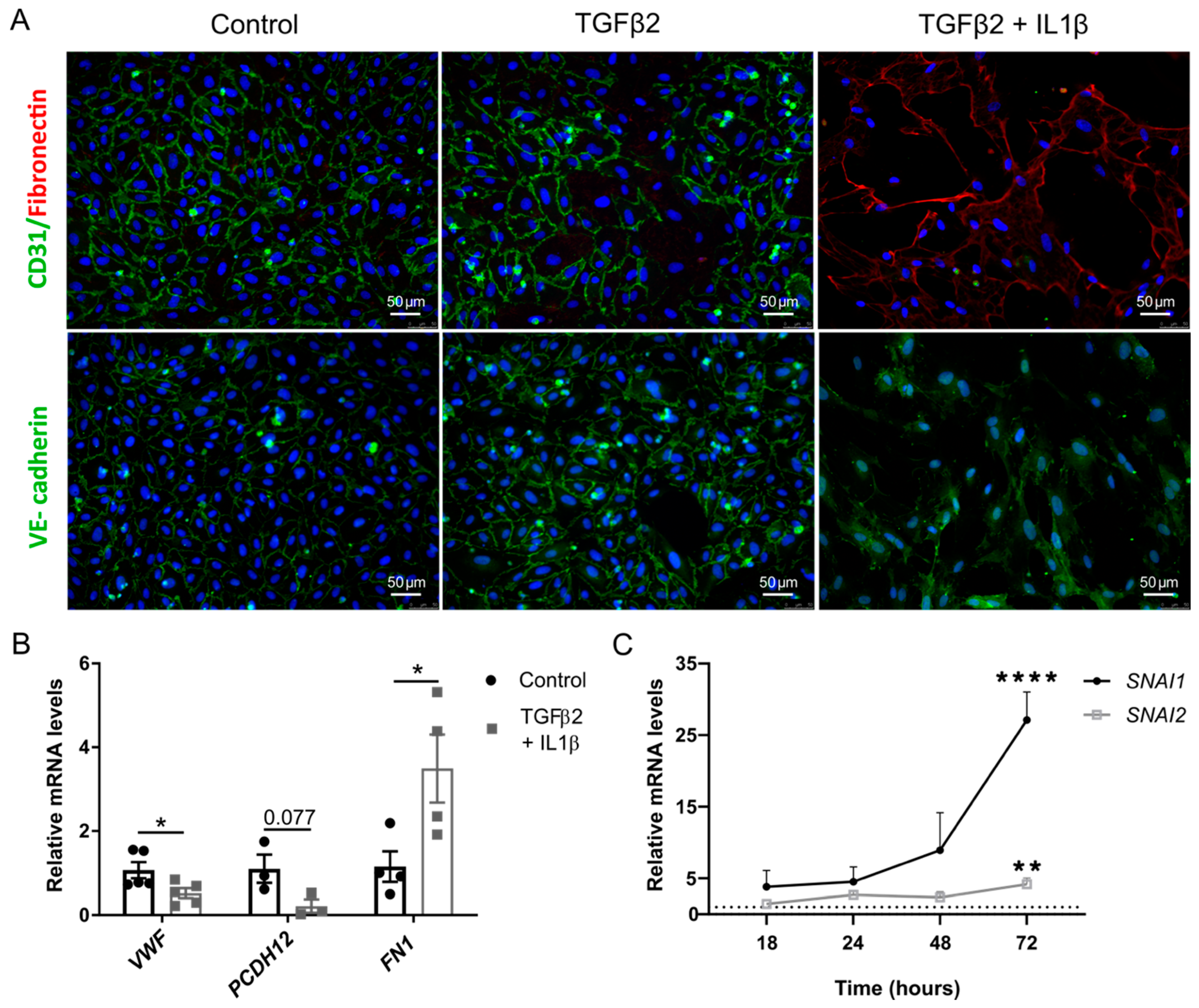{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. TGFβ2 and IL1β Treatment Induces EndMT in Primary Endothelial Cells
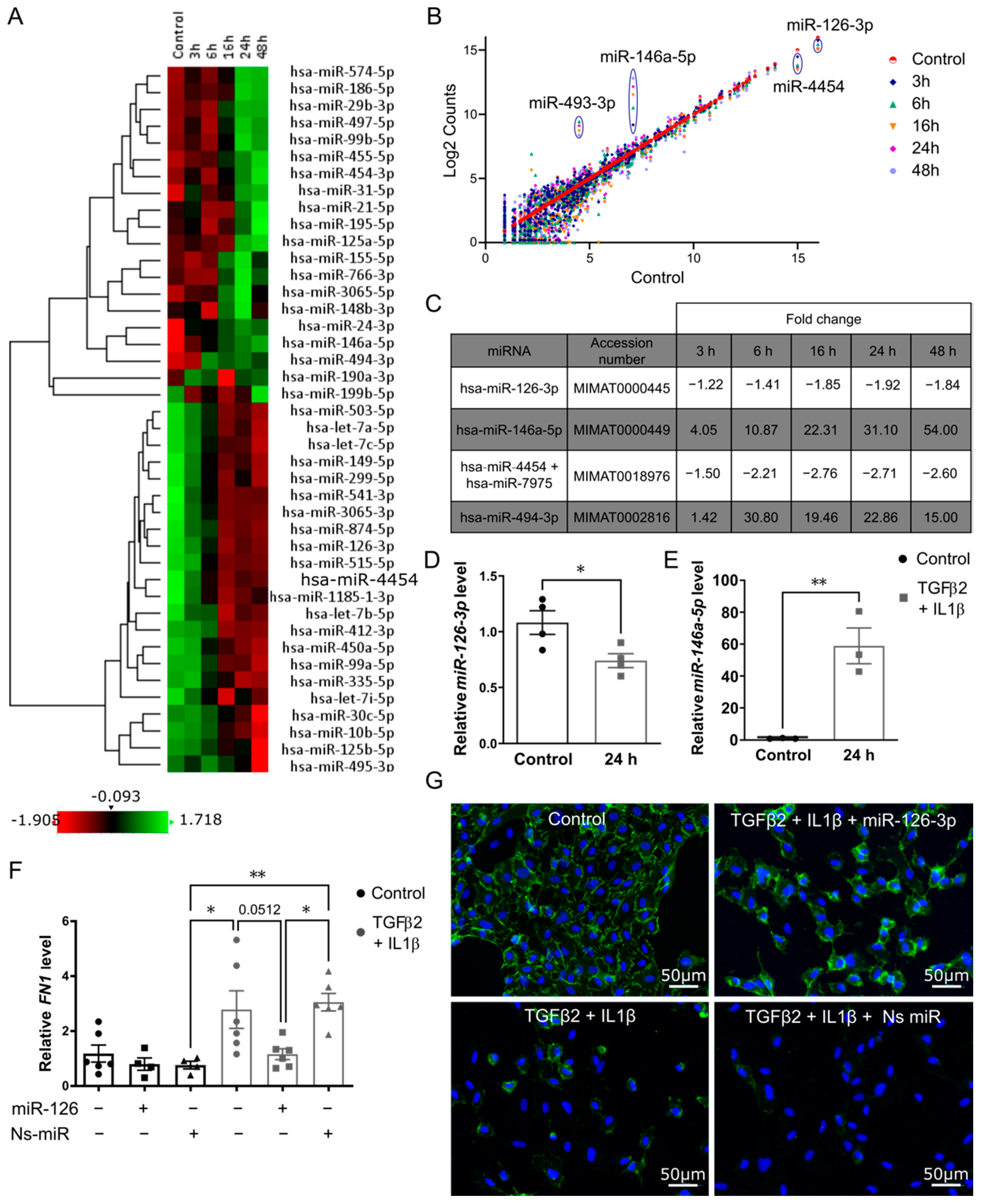
2.2. Changes in miRNA Expression in Primary Endothelial Cells Undergoing EndMT
2.3. Over-Expression of miR126-3p Stabilizes Endothelial Phenotype
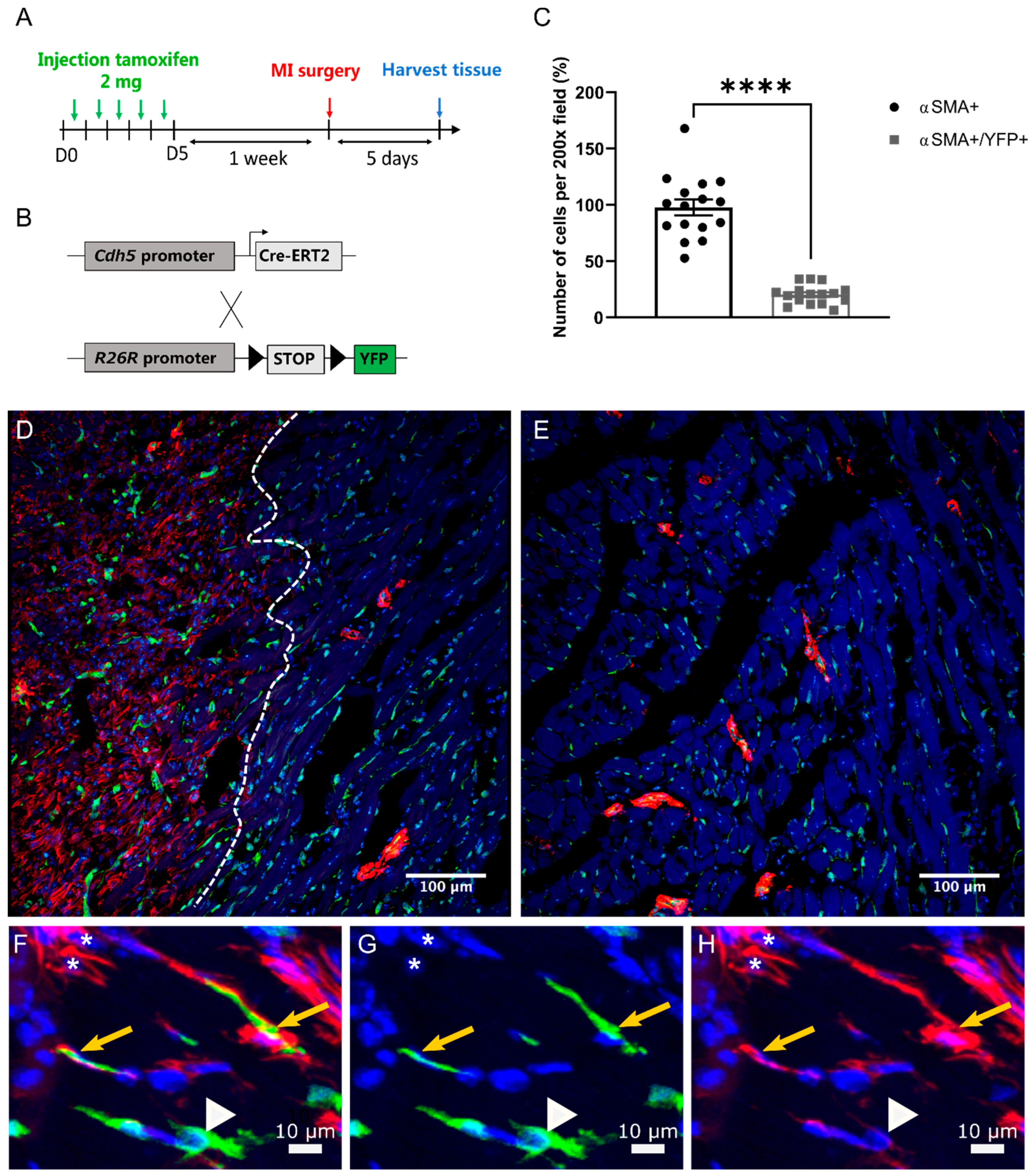
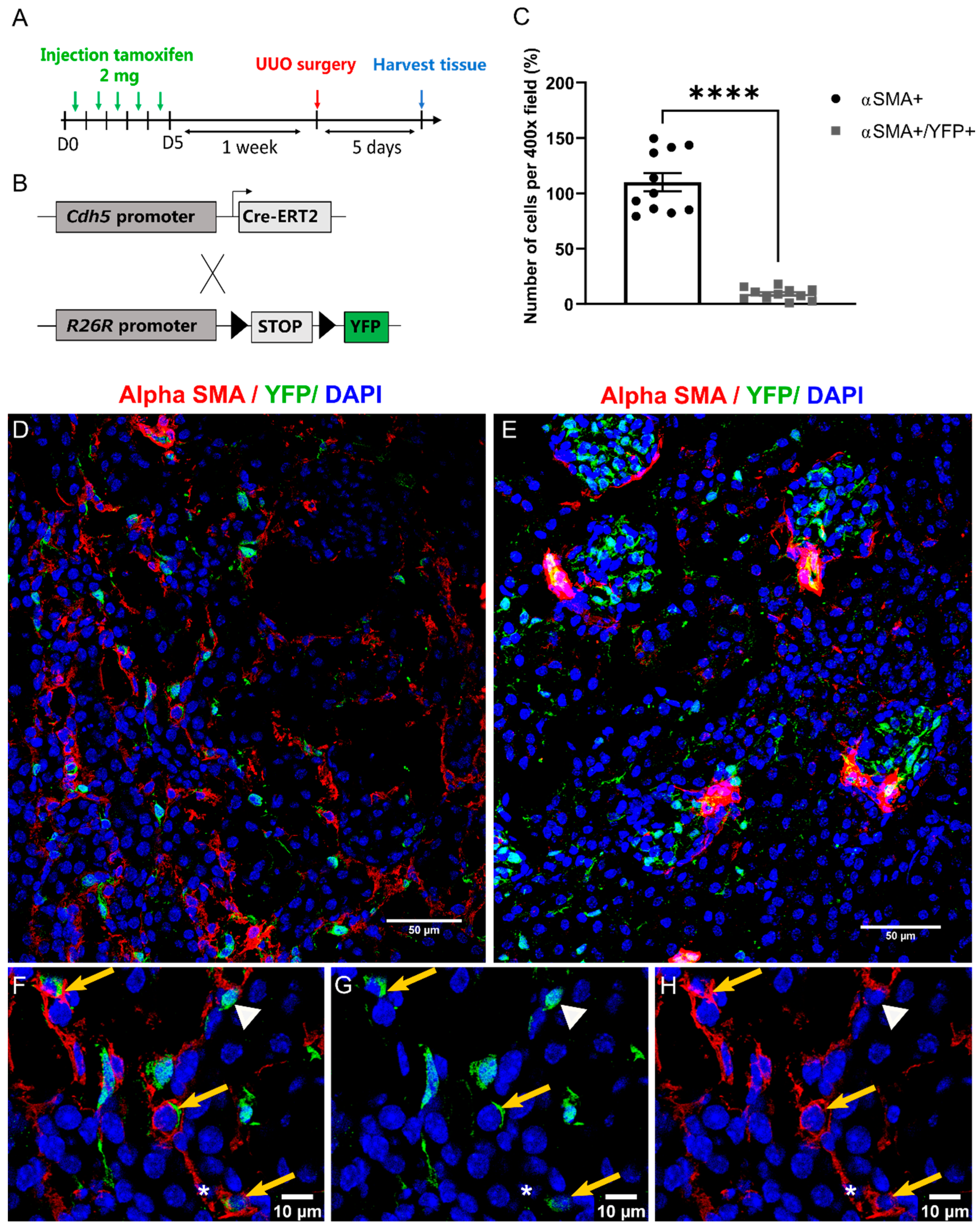
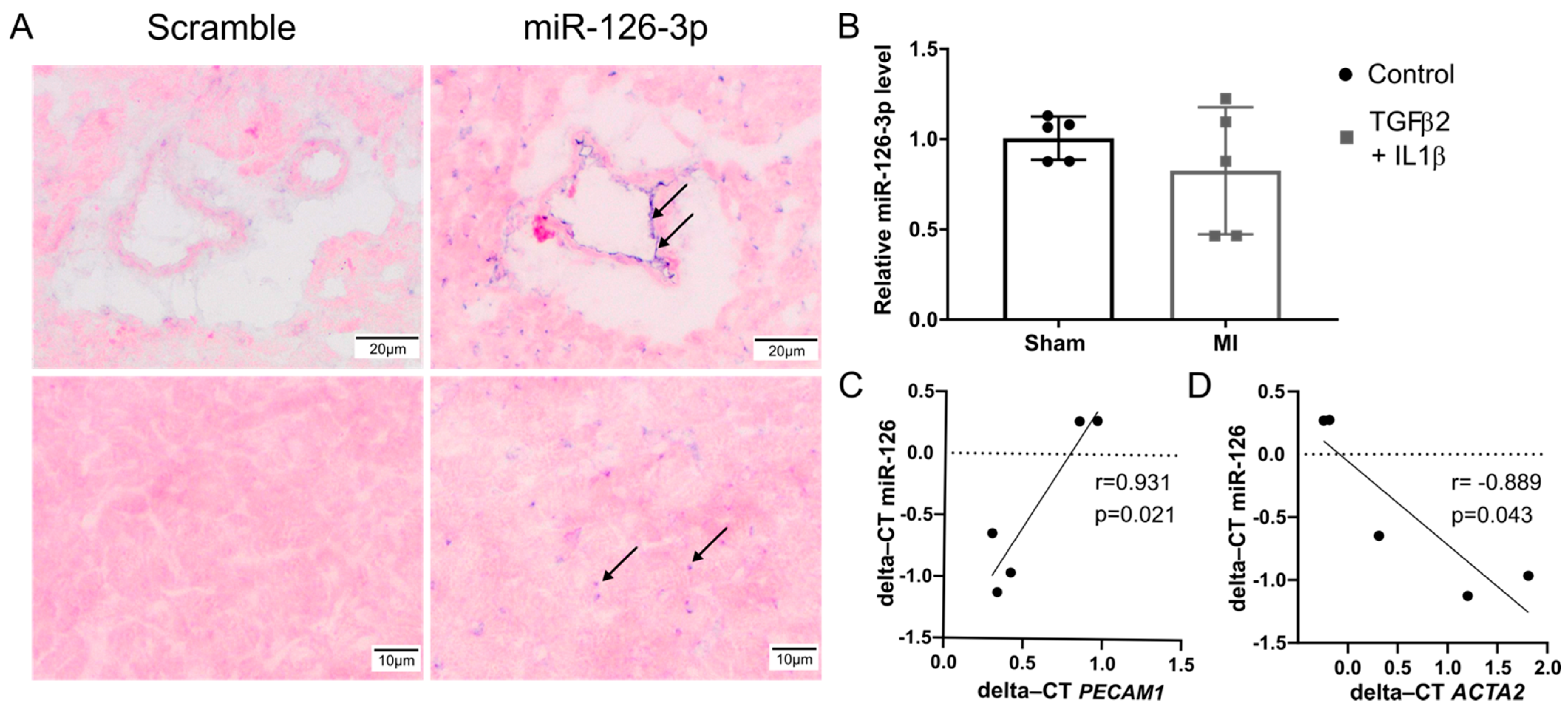
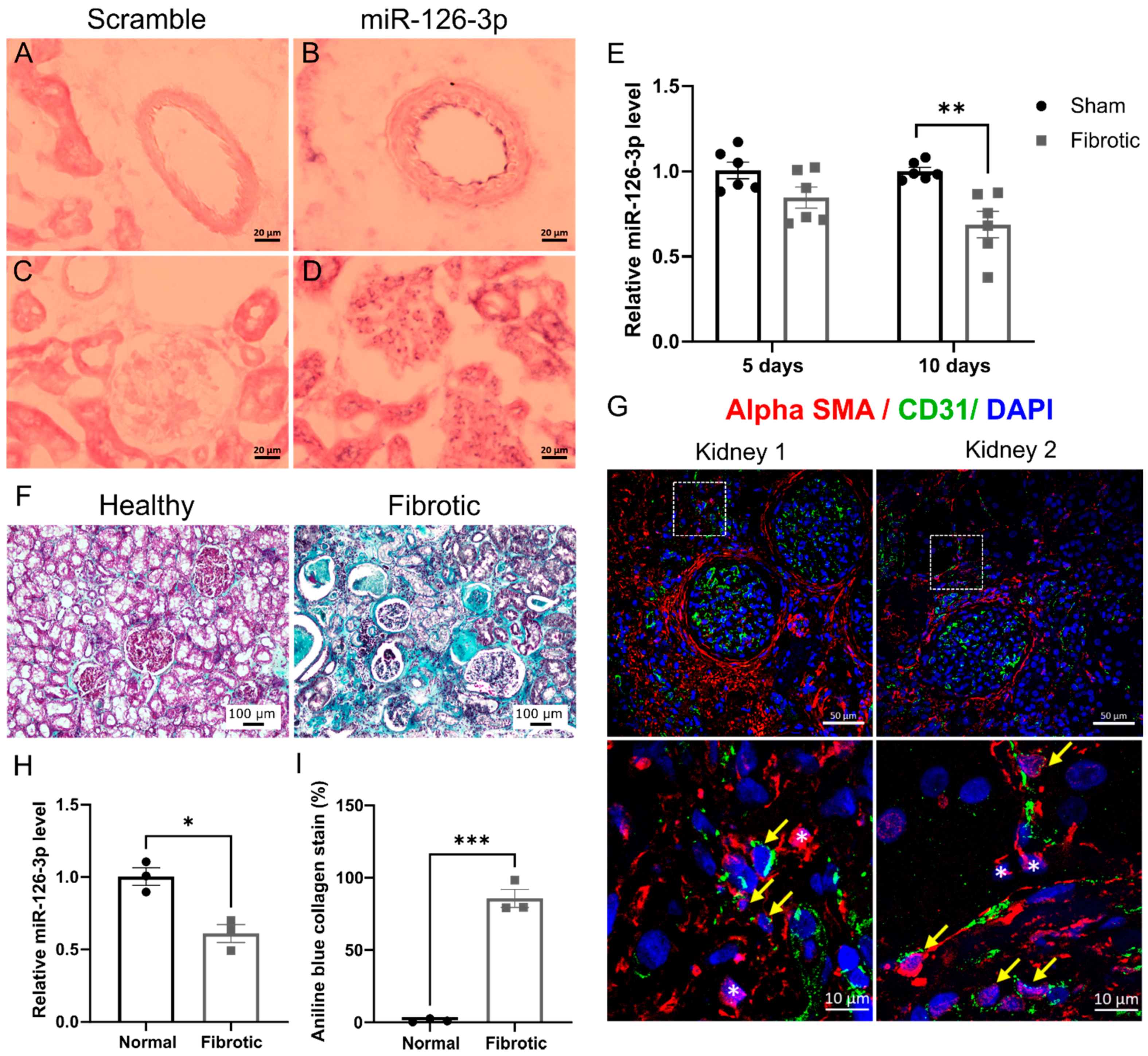
2.4. EndMT Occurs in Cardiac and Renal Fibrosis
2.5. Expression of miR-126-3p in Normal Cardiac Tissue
Expression of miR-126-3p in Normal Renal Tissue
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Animals
4.3. Animal Procedures
4.4. Human Kidney Tissue
4.5. Immunofluorescent Cytostaining
4.6. Immunofluorescent Tissue Staining
4.7. Real-Time Polymerase Chain Reaction
4.8. MiRNA Profile
4.9. In-Situ Hybridisation
4.10. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bon, D.; Chatauret, N.; Giraud, S.; Thuillier, R.; Favreau, F.; Hauet, T. New strategies to optimize kidney recovery and preservation in transplantation. Nat. Rev. Nephrol. 2012, 8, 339–347. [Google Scholar] [CrossRef] [PubMed]
- Burton, C.M.; Iversen, M.; Milman, N.; Zemtsovski, M.; Carlsen, J.; Steinbrüchel, D.; Mortensen, J.; Andersen, C.B. Outcome of lung transplanted patients with primary graft dysfunction. Eur. J. Cardiothorac. Surg. 2007, 31, 75–82. [Google Scholar] [CrossRef] [PubMed]
- Yarlagadda, S.G.; Coca, S.G.; Formica, R.N.; Poggio, E.D.; Parikh, C.R. Association between delayed graft function and allograft and patient survival: A systematic review and meta-analysis. Nephrol. Dial. Transplant. 2009, 24, 1039–1047. [Google Scholar] [CrossRef] [PubMed]
- Situmorang, G.R.; Sheerin, N.S. Ischaemia reperfusion injury: Mechanisms of progression to chronic graft dysfunction. Pediatr. Nephrol. 2019, 34, 951–963. [Google Scholar] [CrossRef] [PubMed]
- Thuillier, R.; Favreau, F.; Celhay, O.; MacChi, L.; Milin, S.; Hauet, T. Thrombin inhibition during kidney ischemia-reperfusion reduces chronic graft inflammation and tubular atrophy. Transplantation 2010, 90, 612–621. [Google Scholar] [CrossRef]
- Wynn, T. Cellular and molecular mechanisms of fibrosis. J. Pathol. 2008, 214, 199. [Google Scholar] [CrossRef]
- Sun, Y.B.Y.; Qu, X.; Caruana, G.; Li, J. The origin of renal fibroblasts/myofibroblasts and the signals that trigger fibrosis. Differentiation 2016, 92, 102–107. [Google Scholar] [CrossRef]
- Maleszewska, M.; Moonen, J.R.; Huijkman, N.; van de Sluis, B.; Krenning, G.; Harmsen, M.C. IL-1beta and TGFbeta2 synergistically induce endothelial to mesenchymal transition in an NFkappaB-dependent manner. Immunobiology 2013, 218, 443–454. [Google Scholar] [CrossRef] [PubMed]
- Bijkerk, R.; de Bruin, R.G.; van Solingen, C.; Duijs, J.M.G.J.; Kobayashi, K.; van der Veer, E.P.; ten Dijke, P.; Rabelink, T.J.; Goumans, M.J.; van Zonneveld, A.J. MicroRNA-155 Functions as a negative regulator of RhoA signaling in TGF-β-induced endothelial to mesenchymal transition. MicroRNA 2012, 1, 2–10. [Google Scholar] [CrossRef] [PubMed]
- Zeisberg, E.M.; Tarnavski, O.; Zeisberg, M.; Dorfman, A.L.; McMullen, J.R.; Gustafsson, E.; Chandraker, A.; Yuan, X.; Pu, W.T.; Roberts, A.B.; et al. Endothelial-to-mesenchymal transition contributes to cardiac fibrosis. Nat. Med. 2007, 13, 952–961. [Google Scholar] [CrossRef] [PubMed]
- LeBleu, V.S.; Taduri, G.; O’Connell, J.; Teng, Y.; Cooke, V.G.; Woda, C.; Sugimoto, H.; Kalluri, R. Origin and function of myofibroblasts in kidney fibrosis. Nat. Med. 2013, 19, 1047–1053. [Google Scholar] [CrossRef]
- Choi, S.-H.; Hong, Z.-Y.; Nam, J.-K.; Lee, H.-J.; Jang, J.; Yoo, R.J.; Lee, Y.J.; Lee, C.Y.; Kim, K.H.; Park, S.; et al. A hypoxia-induced vascular endothelial-to-mesenchymal transition in development of radiation-induced pulmonary fibrosis. Clin. Cancer Res. 2015, 21, 3716–3726. [Google Scholar] [CrossRef]
- Rieder, F.; Kessler, S.P.; West, G.A.; Bhilocha, S.; de la Motte, C.; Sadler, T.M.; Gopalan, B.; Stylianou, E.; Fiocchi, C. Inflammation-induced endothelial-to-mesenchymal transition: A novel mechanism of intestinal fibrosis. Am. J. Pathol. 2011, 179, 2660–2673. [Google Scholar] [CrossRef]
- Kumarswamy, R.; Volkmann, I.; Jazbutyte, V.; Dangwal, S.; Park, D.-H.; Thum, T. Transforming growth factor-induced endothelial-to-mesenchymal transition is partly mediated by MicroRNA-21. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 361–369. [Google Scholar] [CrossRef]
- Kanasaki, K.; Shi, S.; Kanasaki, M.; He, J.; Nagai, T.; Nakamura, Y.; Ishigaki, Y.; Kitada, M.; Srivastava, S.P.; Koya, D. Linagliptin-mediated DPP-4 inhibition ameliorates kidney fibrosis in streptozotocin-induced diabetic mice by inhibiting endothelial-to-mesenchymal transition in a therapeutic regimen. Diabetes 2014, 63, 2120–2131. [Google Scholar] [CrossRef] [PubMed]
- Kwon, O.-S.; Kim, K.-T.; Lee, E.; Kim, M.; Choi, S.-H.; Li, H.; Fornace, A.J.; Cho, J.-H.; Lee, Y.-S.; Lee, J.-S.; et al. Induction of MiR-21 by stereotactic body radiotherapy contributes to the pulmonary fibrotic response. PLoS ONE 2016, 11, e0154942. [Google Scholar] [CrossRef] [PubMed]
- He, M.; Chen, Z.; Martin, M.; Zhang, J.; Sangwung, P.; Woo, B.; Tremoulet, A.H.; Shimizu, C.; Jain, M.K.; Burns, J.C.; et al. MiR-483 targeting of CTGF suppresses endothelial-to-mesenchymal transitionnovelty and significance. Circ. Res. 2017, 120, 354–365. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Aurora, A.B.; Johnson, B.A.; Qi, X.; McAnally, J.; Hill, J.A.; Richardson, J.A.; Bassel-Duby, R.; Olson, E.N. The endothelial-specific MicroRNA MiR-126 governs vascular integrity and angiogenesis. Dev. Cell 2008, 15, 261–271. [Google Scholar] [CrossRef]
- Fish, J.E.; Santoro, M.M.; Morton, S.U.; Yu, S.; Yeh, R.-F.; Wythe, J.D.; Ivey, K.N.; Bruneau, B.G.; Stainier, D.Y.R.; Srivastava, D. MiR-126 regulates angiogenic signaling and vascular integrity. Dev. Cell 2008, 15, 272–284. [Google Scholar] [CrossRef] [PubMed]
- Medici, D.; Potenta, S.; Kalluri, R. Transforming growth factor-beta2 promotes snail-mediated endothelial-mesenchymal transition through convergence of smad-dependent and smad-independent signalling. Biochem. J. 2011, 437, 515–520. [Google Scholar] [CrossRef]
- Cooley, B.C.; Nevado, J.; Mellad, J.; Yang, D.; Hilaire, C.S.; Negro, A.; Fang, F.; Chen, G.; San, H.; Walts, A.D.; et al. TGF-β signaling mediates endothelial-to-mesenchymal transition (EndMT) during vein graft remodeling. Sci. Transl. Med. 2014, 6, 227ra34. [Google Scholar] [CrossRef]
- Xu, X.; Tan, X.; Tampe, B.; Sanchez, E.; Zeisberg, M.; Zeisberg, E.M. Snail is a direct target of hypoxia-inducible factor 1α (HIF1α) in hypoxia-induced endothelial to mesenchymal transition of human coronary endothelial cells. J. Biol. Chem. 2015, 290, 16653–16664. [Google Scholar] [CrossRef]
- Harris, T.A.; Yamakuchi, M.; Ferlito, M.; Mendell, J.T.; Lowenstein, C.J. MicroRNA-126 regulates endothelial expression of vascular cell adhesion molecule 1. Proc. Natl. Acad. Sci. USA 2008, 105, 1516–1521. [Google Scholar] [CrossRef] [PubMed]
- Qu, Q.; Bing, W.; Meng, X.; Xi, J.; Bai, X.; Liu, Q.; Guo, Y.; Zhao, X.; Bi, Y. Upregulation of MiR-126-3p promotes human saphenous vein endothelial cell proliferation in vitro and prevents vein graft neointimal formation ex vivo and in vivo. Oncotarget 2017, 8, 106790–106806. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Zhang, Z.; Zhang, D.Y.; Zhu, J.; Zhang, T.; Wang, C. MicroRNA 126 inhibits the transition of endothelial progenitor cells to mesenchymal cells via the PIK3R2- PI3K/Akt signalling pathway. PLoS ONE 2013, 8, e83294. [Google Scholar] [CrossRef] [PubMed]
- Moore-Morris, T.; Guimarães-Camboa, N.; Banerjee, I.; Zambon, A.C.; Kisseleva, T.; Velayoudon, A.; Stallcup, W.B.; Gu, Y.; Dalton, N.D.; Cedenilla, M.; et al. Resident fibroblast lineages mediate pressure overload–induced cardiac fibrosis. J. Clin. Investig. 2014, 124, 2921–2934. [Google Scholar] [CrossRef] [PubMed]
- Curci, C.; Castellano, G.; Stasi, A.; Divella, C.; Loverre, A.; Gigante, M.; Simone, S.; Cariello, M.; Montinaro, V.; Lucarelli, G.; et al. Endothelial-to-mesenchymal transition and renal fibrosis in ischaemia/reperfusion injury are mediated by complement anaphylatoxins and Akt pathway. Nephrol. Dial. Transplant. 2014, 29, 799–808. [Google Scholar] [CrossRef] [PubMed]
- Xu-Dubois, Y.; Ahmadpoor, P.; Brocheriou, I.; Louis, K.; Arzouk Snanoudj, N.; Rouvier, P.; Taupin, J.; Corchia, A.; Galichon, P.; Barrou, B.; et al. Microvasculature partial endothelial mesenchymal transition in early post-transplant biopsy with acute tubular necrosis identifies poor recovery renal allografts. Am. J. Transplant. 2020, 20, 2400–2412. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Shiesh, S.C.; Lee, G.B.; Chen, C. Two-step magnetic bead-based (2MBB) techniques for immunocapture of extracellular vesicles and quantification of MicroRNAs for cardiovascular diseases: A pilot study. PLoS ONE 2020, 15, e0229610. [Google Scholar] [CrossRef] [PubMed]
- Long, G.; Wang, F.; Duan, Q.; Chen, F.; Yang, S.; Gong, W.; Wang, Y.; Chen, C.; Wang, D.W. Human circulating MicroRNA-1 and MicroRNA-126 as potential novel indicators for acute myocardial infarction. Int. J. Biol. Sci. 2012, 8, 811–818. [Google Scholar] [CrossRef] [PubMed]
- Lovisa, S.; Fletcher-Sananikone, E.; Sugimoto, H.; Hensel, J.; Lahiri, S.; Hertig, A.; Taduri, G.; Lawson, E.; Dewar, R.; Revuelta, I.; et al. Endothelial-to-mesenchymal transition compromises vascular integrity to induce myc-mediated metabolic reprogramming in kidney fibrosis. Sci. Signal. 2020, 13, eaaz2597. [Google Scholar] [CrossRef] [PubMed]
- Jordan, N.P.; Nicholson, M.L.; Hosgood, S.A. MicroRNA-126-3p is downregulated in human kidneys in a model of reperfusion injury. Kidney Int. Rep. 2020, 5, 2357–2360. [Google Scholar] [CrossRef] [PubMed]
- Bijkerk, R.; Van Solingen, C.; De Boer, H.C.; Van Der Pol, P.; Khairoun, M.; De Bruin, R.G.; Van Oeveren-Rietdijk, A.M.; Lievers, E.; Schlagwein, N.; Van Gijlswijk, D.J.; et al. Hematopoietic MicroRNA-126 protects against renal ischemia/reperfusion injury by promoting vascular integrity. J. Am. Soc. Nephrol. 2014, 25, 1710–1722. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Nakayama, M.; Pitulescu, M.E.; Schmidt, T.S.; Bochenek, M.L.; Sakakibara, A.; Adams, S.; Davy, A.; Deutsch, U.; Lüthi, U.; et al. Ephrin-B2 controls VEGF-induced angiogenesis and lymphangiogenesis. Nature 2010, 465, 483–486. [Google Scholar] [CrossRef] [PubMed]
- Srinivas, S.; Watanabe, T.; Lin, C.S.; William, C.M.; Tanabe, Y.; Jessell, T.M.; Costantini, F. Cre reporter strains produced by targeted insertion of EYFP and ECFP into the ROSA26 locus. BMC Dev. Biol. 2001, 1, 4. [Google Scholar] [CrossRef] [PubMed]
- Redgrave, R.E.; Tual-Chalot, S.; Davison, B.J.; Singh, E.; Hall, D.; Amirrasouli, M.M.; Gilchrist, D.; Medvinsky, A.; Arthur, H.M. Cardiosphere-derived cells require endoglin for paracrine-mediated angiogenesis. Stem Cell Rep. 2017, 8, 1287–1298. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jordan, N.P.; Tingle, S.J.; Shuttleworth, V.G.; Cooke, K.; Redgrave, R.E.; Singh, E.; Glover, E.K.; Ahmad Tajuddin, H.B.; Kirby, J.A.; Arthur, H.M.; et al. MiR-126-3p Is Dynamically Regulated in Endothelial-to-Mesenchymal Transition during Fibrosis. Int. J. Mol. Sci. 2021, 22, 8629. https://doi.org/10.3390/ijms22168629
Jordan NP, Tingle SJ, Shuttleworth VG, Cooke K, Redgrave RE, Singh E, Glover EK, Ahmad Tajuddin HB, Kirby JA, Arthur HM, et al. MiR-126-3p Is Dynamically Regulated in Endothelial-to-Mesenchymal Transition during Fibrosis. International Journal of Molecular Sciences. 2021; 22(16):8629. https://doi.org/10.3390/ijms22168629
Chicago/Turabian StyleJordan, Nina P., Samuel J. Tingle, Victoria G. Shuttleworth, Katie Cooke, Rachael E. Redgrave, Esha Singh, Emily K. Glover, Hafiza B. Ahmad Tajuddin, John A. Kirby, Helen M. Arthur, and et al. 2021. "MiR-126-3p Is Dynamically Regulated in Endothelial-to-Mesenchymal Transition during Fibrosis" International Journal of Molecular Sciences 22, no. 16: 8629. https://doi.org/10.3390/ijms22168629
APA StyleJordan, N. P., Tingle, S. J., Shuttleworth, V. G., Cooke, K., Redgrave, R. E., Singh, E., Glover, E. K., Ahmad Tajuddin, H. B., Kirby, J. A., Arthur, H. M., Ward, C., Sheerin, N. S., & Ali, S. (2021). MiR-126-3p Is Dynamically Regulated in Endothelial-to-Mesenchymal Transition during Fibrosis. International Journal of Molecular Sciences, 22(16), 8629. https://doi.org/10.3390/ijms22168629