
Mitochondria-Induced Immune Response as a Trigger for Neurodegeneration: A Pathogen from Within



Abstract
:1. Mitochondria in the Eukaryotic Cell: A Double-Edged Sword
2. mtDNA and the Immune System: Keeping Your Friend Close and Your Enemy Closer
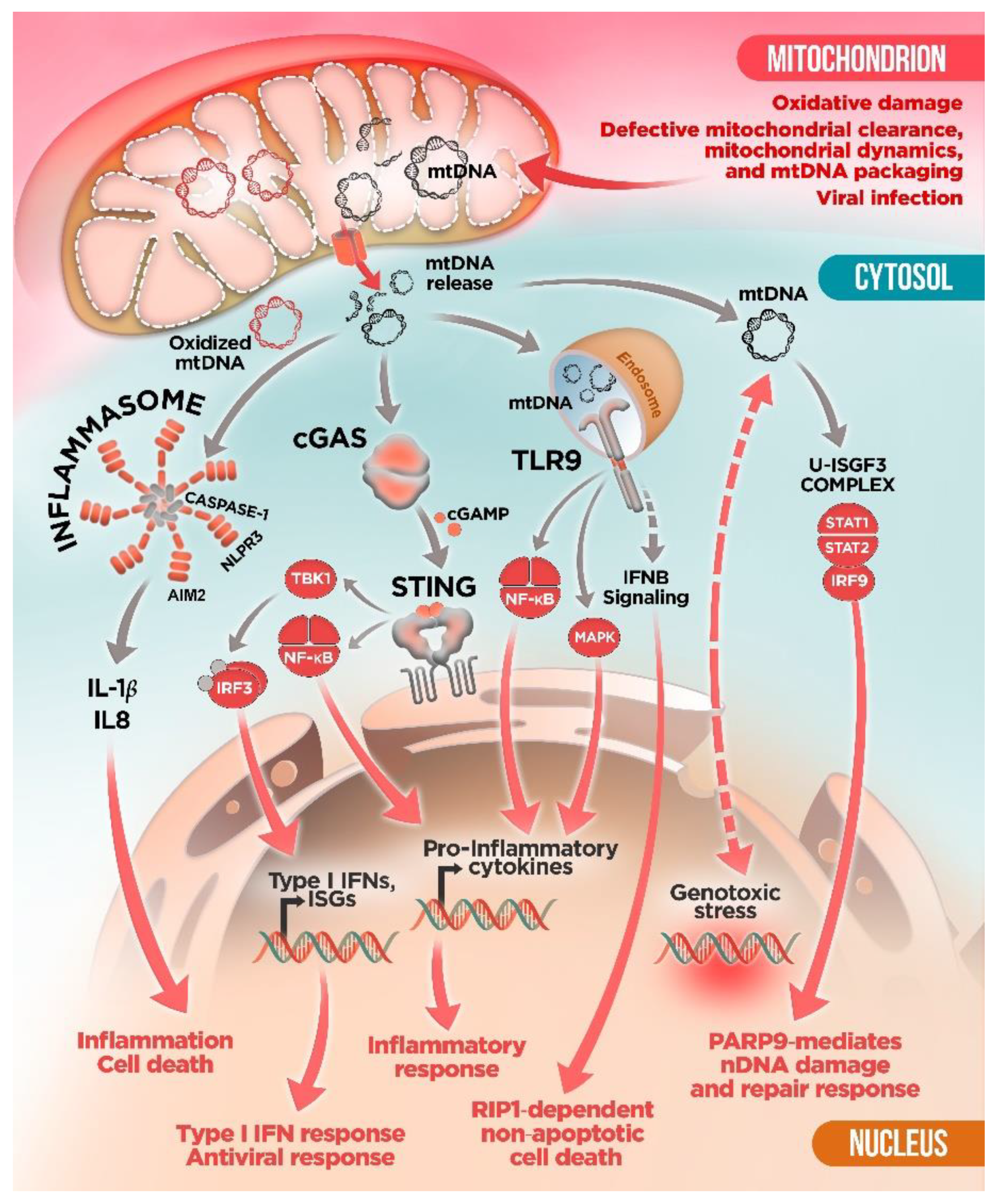
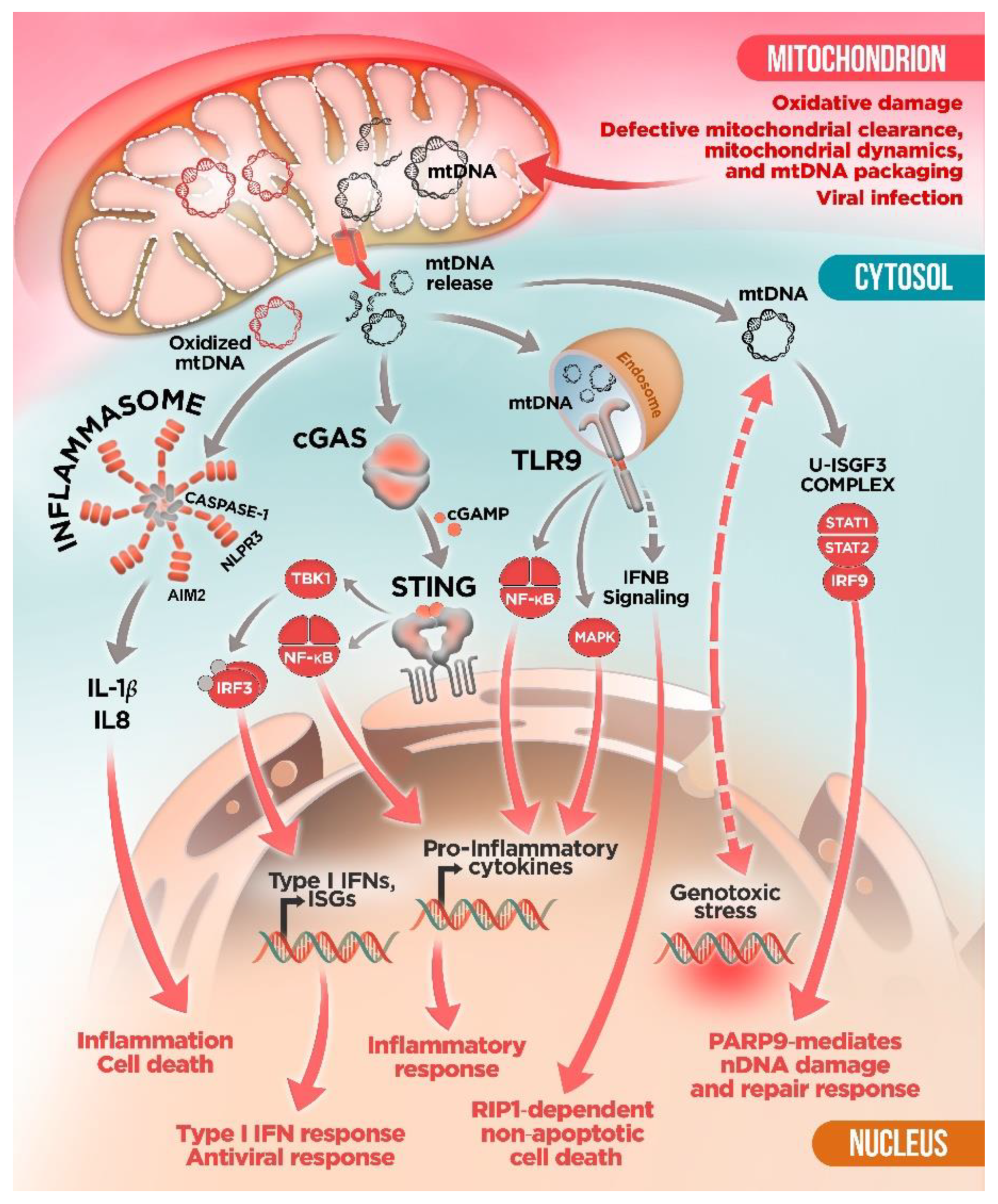
3. Innate Immune Sensors and mtDNA: To Detect and to “Protect”?
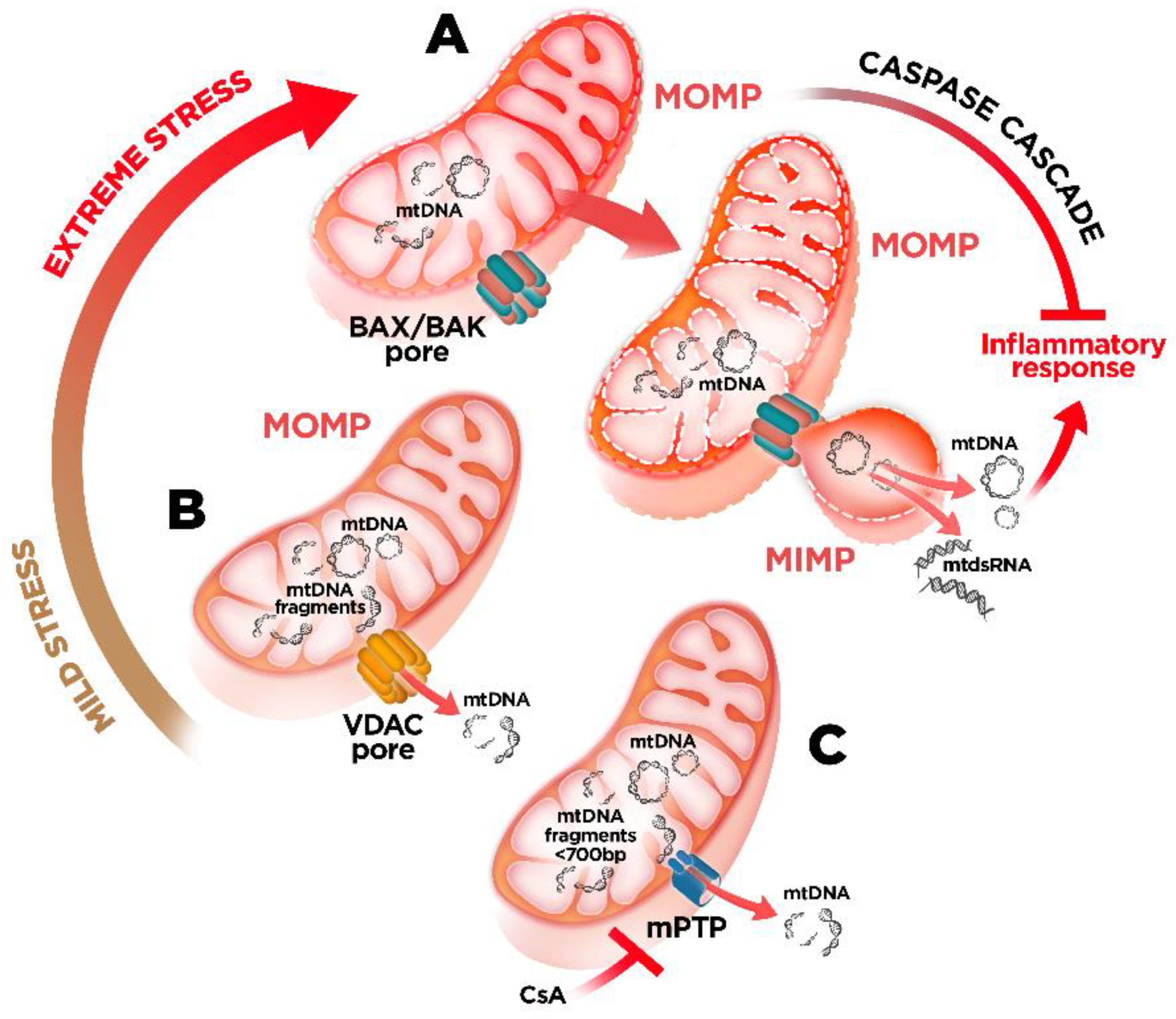
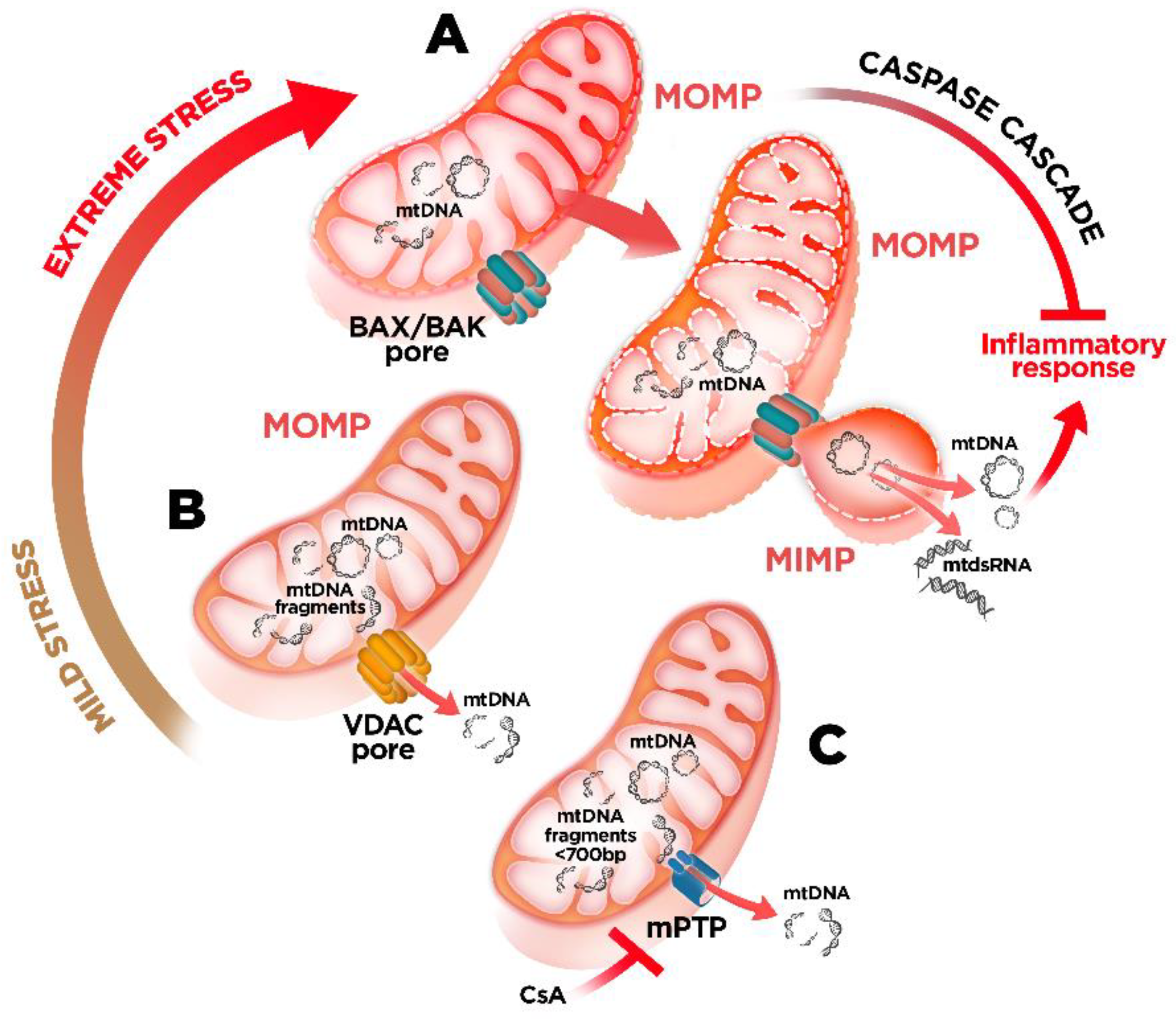
4. mtDNA Mitochondrial Escape: A Double-Membrane Prison Break
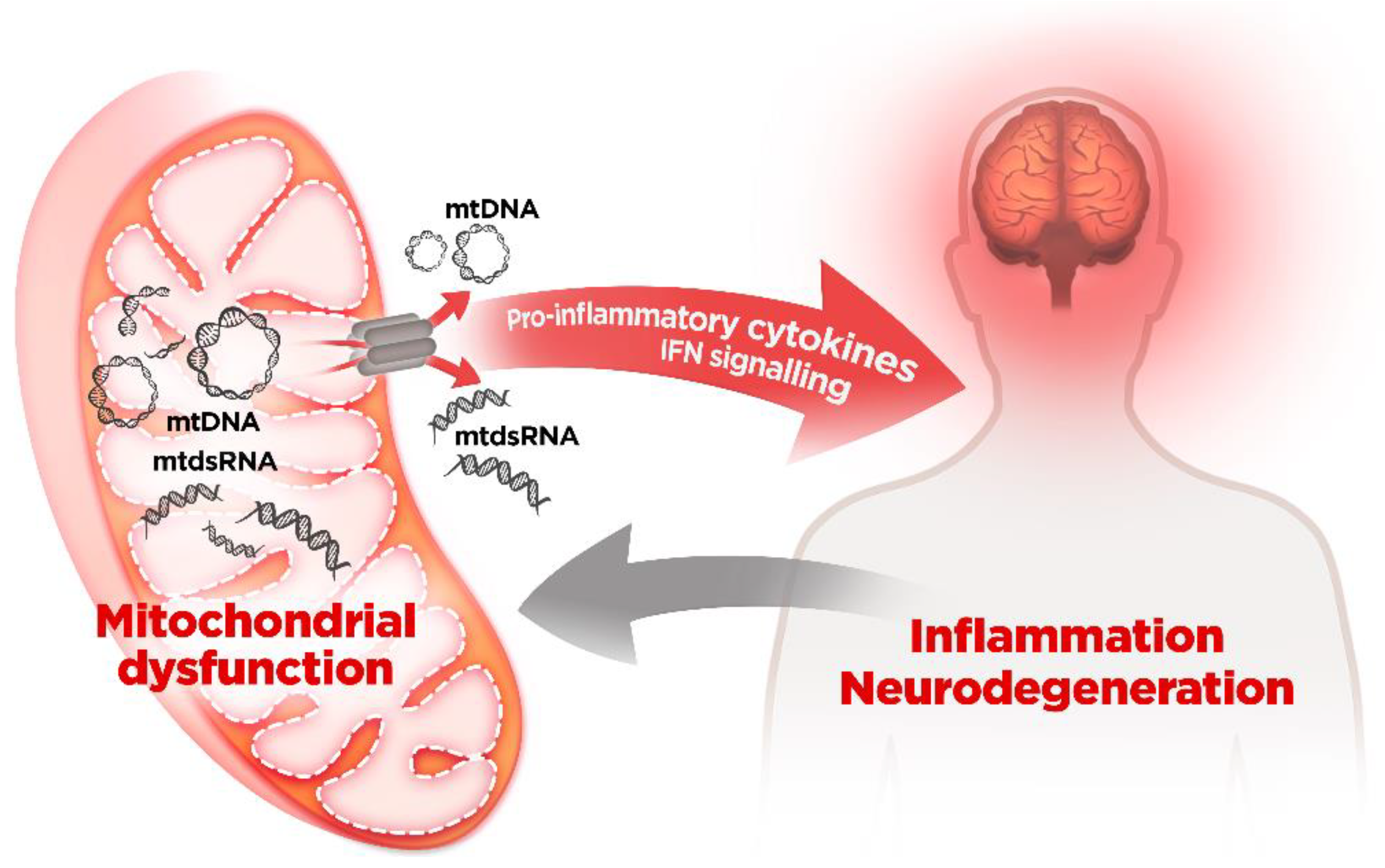
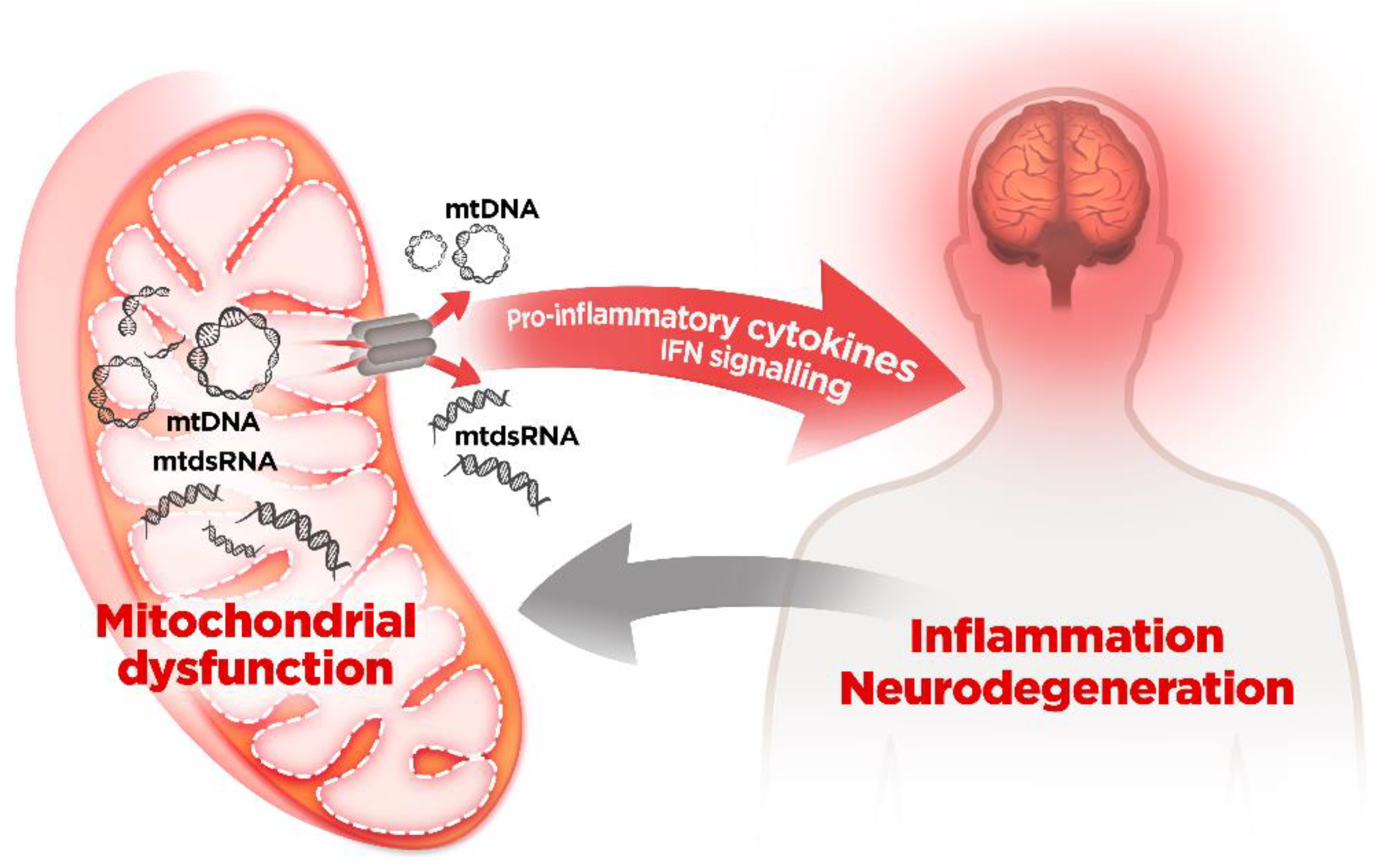
5. Mitochondrial Dysfunction and mtDNA Release: A Crossline between Health and Disease
mtDNA and Neurodegenerative Diseases
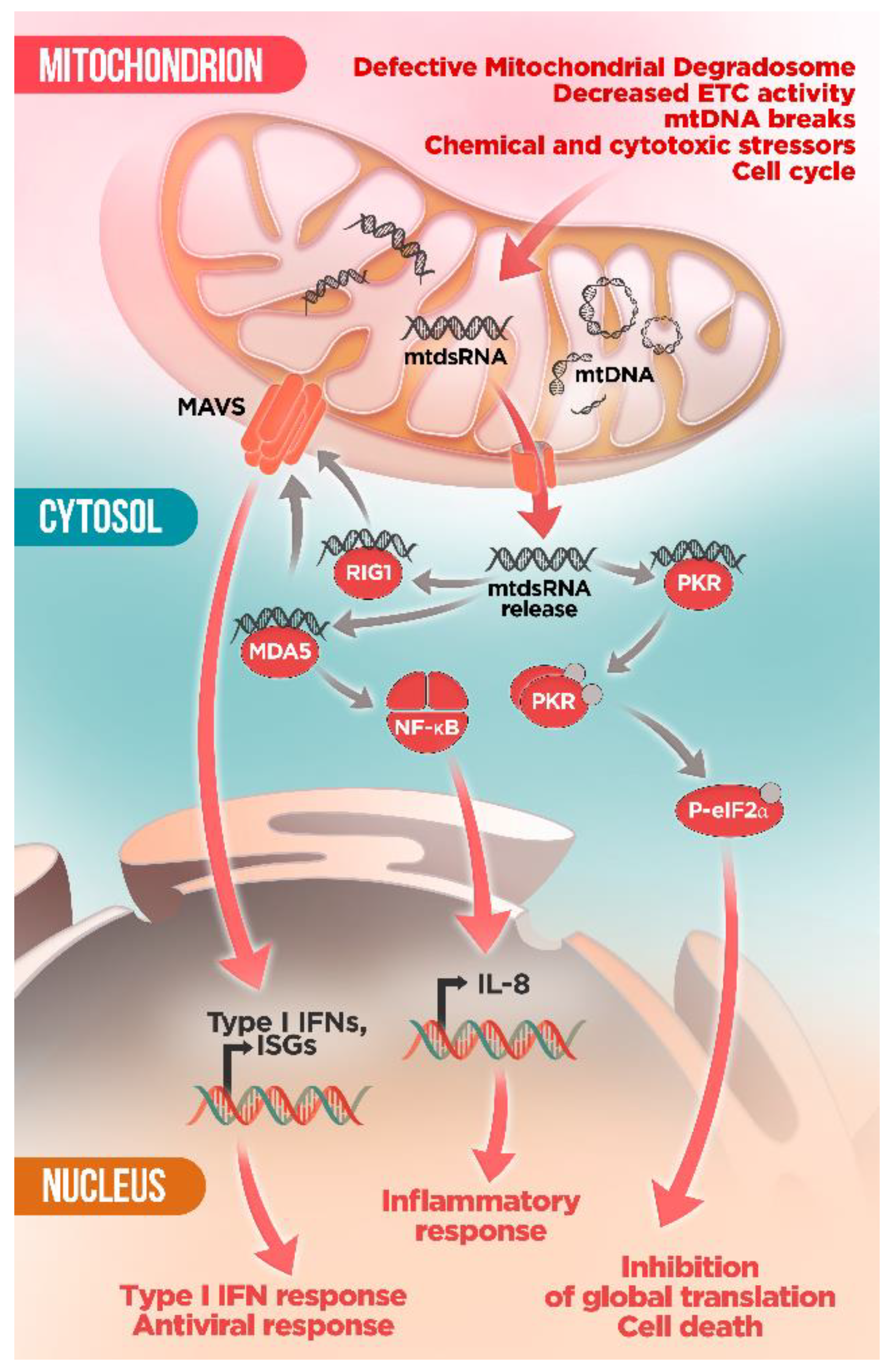
6. Mitochondrial RNA: A New Villain in Town
7. Concluding Remarks: PAMP-Ing the Cell up
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AD | Alzheimer’s Disease |
| AIM2 | Absent In Melanoma 2 |
| ALS | Amyotrophic Lateral Sclerosis |
| ALS | Amyotrophic Lateral Sclerosis |
| CARD | Caspase Recruitment Domain |
| cGAMP | Cyclic GMP–AMP |
| cGAS–STING | Cyclic GMP/AMP Synthase–Stimulator Of Interferon Genes |
| CLPP | Caseinolytic Mitochondrial Matrix Peptidase Proteolytic Subunit |
| CsA | Cyclosporine A |
| CyPD | Cyclophilin D |
| DAMPS | Damage-Associated Molecular Patterns |
| ETC | Electron Transport Chain |
| GSDMD | Gasdermin D |
| HD | Huntington’s Disease |
| IFN | Interferon |
| IKKε | IKB Kinase-ε |
| IMM | Inner Mitochondrial Membrane |
| IRF3 | Interferon Regulatory Factor 3 |
| ISG | Interferon-Stimulated Genes |
| LPS | Ligand Lipopolysaccharide |
| MAPK | Mitogen-Activated Protein Kinases |
| MAVS | Antiviral-Signaling Protein |
| MD | Mitochondrial Disease |
| MDA5 | Melanoma Differentiation Associated Gene 5 |
| MIMP | Mitochondrial Inner Membrane Permeabilization |
| MOMP | Mitochondrial Outer Membrane Permeabilization |
| MPTP | Mitochondrial Permeability Transition Pore |
| mt- | Mitochondrial |
| mtDNA | Mitochondrial DNA |
| mtdsRNA | Double-Stranded Mitochondrial RNA |
| mtRNA | Mitochondrial RNA |
| nDNA | Nuclear DNA |
| NLRPs | Nucleotide-Binding Oligomerization Domain (NOD)-Like Receptors Cyclic |
| NRF2 | Nuclear factor erythroid 2–related factor 2 |
| OMM | Outer Mitochondrial Membrane |
| OXPHOS | Oxidative Phosphorylation |
| PAMPS | Pathogen-Associated Molecular Patterns |
| PD | Parkinson’s Disease |
| PKR | Protein Kinase RNA-Activated |
| PMD | Primary Mitochondrial Disorders |
| PNPase | Polynucleotide Phosphorylase |
| pPKR | Phosphorylated PKR |
| PRRs | Pattern-Recognition Receptors |
| RA | Rheumatoid Arthritis |
| RIG-I | Retinoic Acid Inducible Protein I |
| ROS | Reactive Oxygen Species |
| SLE | Systemic Lupus Erythematosus |
| TBK1 | TANK-Binding Kinase 1 |
| TDP-43 | TAR DNA Binding Protein 43 |
| TFAM | Transcription Factor A |
| TLRs | Toll-Like Receptors |
| VDAC | Voltage Dependent Anion Channel |
References
- Friedman, J.; Nunnari, J. Mitochondrial form and function. Nat. Cell Biol. 2014, 505, 335–343. [Google Scholar] [CrossRef] [Green Version]
- Vafai, S.B.; Mootha, V.K. Mitochondrial disorders as windows into an ancient organelle. Nat. Cell Biol. 2012, 491, 374–383. [Google Scholar] [CrossRef]
- Schaefer, A.; Taylor, R.W.; Turnbull, D.M.; Chinnery, P.F. The epidemiology of mitochondrial disorders—Past, present and future. Biochim. Biophys. Acta Bioenerg. 2004, 1659, 115–120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lechuga-Vieco, A.V.; Justo-Méndez, R.; Enríquez, J.A. Not all mitochondrial DNAs are made equal and the nucleus knows it. IUBMB Life 2020, 73, 511–529. [Google Scholar] [CrossRef] [PubMed]
- Garone, C.; Viscomi, C. Towards a therapy for mitochondrial disease: An update. Biochem. Soc. Trans. 2018, 46, 1247–1261. [Google Scholar] [CrossRef] [Green Version]
- Niyazov, D.M.; Kahler, S.G.; Frye, R.E. Primary mitochondrial disease and secondary mitochondrial dysfunction: Importance of distinction for diagnosis and treatment. Mol. Syndr. 2016, 7, 122–137. [Google Scholar] [CrossRef] [Green Version]
- Roubicek, D.A.; de Souza-Pinto, N.C. Mitochondria and mitochondrial DNA as relevant targets for environmental contaminants. Toxicology 2017, 391, 100–108. [Google Scholar] [CrossRef]
- Consolini, A.E.; Ragone, M.I.; Bonazzola, P.; Colareda, G.A. Mitochondrial bioenergetics during ischemia and reperfusion. In Mitochondrial Dynamics in Cardiovascular Medicine; Santulli, G., Ed.; Springer: Cham, Switzerland, 2017; pp. 141–167. ISBN 978-3-319-55330-6. [Google Scholar]
- Chouchani, E.T.; Pell, V.R.; Gaude, E.; Aksentijevic, D.; Sundier, S.Y.; Robb, E.L.; Logan, A.; Nadtochiy, S.M.; Ord, E.; Smith, A.; et al. Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nat. Cell Biol. 2014, 515, 431–435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abarca-Rojano, E.; Rosas-Medina, P.; Zamudio-Cortez, P.; Mondragon-Flores, R.; Sanchez-Garcia, F.J. Mycobacterium tuberculosis virulence correlates with mitochondrial cytochrome c release in infected macrophages. Scand. J. Immunol. 2003, 58, 419–427. [Google Scholar] [CrossRef] [Green Version]
- Aguirre, S.; Luthra, P.; Sanchez-Aparicio, M.T.; Maestre, A.M.; Patel, J.; Lamothe, F.; Fredericks, A.C.; Tripathi, S.; Zhu, T.; Pintado-Silva, J.; et al. Dengue virus NS2B protein targets cGAS for degradation and prevents mitochondrial DNA sensing during infection. Nat. Microbiol. 2017, 2, 1–11. [Google Scholar] [CrossRef] [PubMed]
- López-Armada, M.J.; Riveiro-Naveira, R.R.; Vaamonde-García, C.; Valcarcel-Ares, M.N. Mitochondrial dysfunction and the inflammatory response. Mitochondrion 2013, 13, 106–118. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.T.; Beal, M.F. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nat. Cell Biol. 2006, 443, 787–795. [Google Scholar] [CrossRef]
- Trushina, E.; McMurray, C. Oxidative stress and mitochondrial dysfunction in neurodegenerative diseases. Neuroscience 2007, 145, 1233–1248. [Google Scholar] [CrossRef]
- Manji, H.K.; Kato, T.; Di Prospero, N.A.; Ness, S.; Beal, M.F.; Krams, M.; Chen, G. Impaired mitochondrial function in psychiatric disorders. Nat. Rev. Neurosci. 2012, 13, 293–307. [Google Scholar] [CrossRef]
- Rossignol, D.A.; Frye, R.E. Mitochondrial dysfunction in autism spectrum disorders: A systematic review and meta-analysis. Mol. Psychiatry 2011, 17, 290–314. [Google Scholar] [CrossRef] [Green Version]
- Nasrallah, C.M.; Horvath, T.L. Mitochondrial dynamics in the central regulation of metabolism. Nat. Rev. Endocrinol. 2014, 10, 650–658. [Google Scholar] [CrossRef]
- Srinivasan, S.; Guha, M.; Kashina, A.; Avadhani, N.G. Mitochondrial dysfunction and mitochondrial dynamics—The cancer connection. Biochim. Biophys. Acta Bioenerg. 2017, 1858, 602–614. [Google Scholar] [CrossRef]
- Dubinsky, J.M. Heterogeneity of nervous system mitochondria: Location, location, location! Exp. Neurol. 2009, 218, 293–307. [Google Scholar] [CrossRef]
- Mottis, A.; Herzig, S.; Auwerx, J. Mitocellular communication: Shaping health and disease. Science 2019, 366, 827–832. [Google Scholar] [CrossRef] [PubMed]
- Youle, R.J. Mitochondria—Striking a balance between host and endosymbiont. Science 2019, 365, eaaw9855. [Google Scholar] [CrossRef] [PubMed]
- West, A.P.; Khoury-Hanold, W.; Staron, M.; Tal, M.; Pineda, C.M.; Lang, S.M.; Bestwick, M.; Duguay, B.A.; Raimundo, N.; MacDuff, D.A.; et al. Mitochondrial DNA stress primes the antiviral innate immune response. Nat. Cell Biol. 2015, 520, 553–557. [Google Scholar] [CrossRef] [Green Version]
- Dhir, A.; Dhir, S.; Borowski, L.; Jimenez, L.; Teitell, M.; Rötig, A.; Crow, Y.J.; Rice, G.I.; Duffy, D.; Tamby, C.; et al. Mitochondrial double-stranded RNA triggers antiviral signalling in humans. Nat. Cell Biol. 2018, 560, 238–242. [Google Scholar] [CrossRef]
- Thornton, B.; Cohen, B.; Copeland, W.; Maria, B.L. Mitochondrial disease. J. Child Neurol. 2014, 29, 1179–1207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barends, M.; Verschuren, L.; Morava, E.; Nesbitt, V.; Turnbull, D.; McFarland, R. Causes of death in adults with mitochondrial disease. In JIMD Reports; Zschocke, J., Gibson, K.M., Brown, G., Morava-Kozicz, E., Peters, V., Eds.; Springer: Berlin, Germany, 2015; Volume 26, pp. 103–113. [Google Scholar]
- Kapnick, S.M.; Pacheco, S.E.; McGuire, P.J. The emerging role of immune dysfunction in mitochondrial diseases as a paradigm for understanding immunometabolism. Metabolism 2018, 81, 97–112. [Google Scholar] [CrossRef] [PubMed]
- Gustafsson, C.M.; Falkenberg, M.; Larsson, N.-G. Maintenance and expression of mammalian mitochondrial DNA. Annu. Rev. Biochem. 2016, 85, 133–160. [Google Scholar] [CrossRef] [PubMed]
- D’Erchia, A.M.; Atlante, A.; Gadaleta, G.; Pavesi, G.; Chiara, M.; De Virgilio, C.; Manzari, C.; Mastropasqua, F.; Prazzoli, G.M.; Picardi, E.; et al. Tissue-specific mtDNA abundance from exome data and its correlation with mitochondrial transcription, mass and respiratory activity. Mitochondrion 2015, 20, 13–21. [Google Scholar] [CrossRef]
- Liu, B.; Du, Q.; Chen, L.; Fu, G.; Li, S.; Fu, L.; Zhang, X.; Ma, C.; Bin, C. CpG methylation patterns of human mitochondrial DNA. Sci. Rep. 2016, 6, 23421. [Google Scholar] [CrossRef] [Green Version]
- Van Der Wijst, M.G.P.; Van Tilburg, A.Y.; Ruiters, M.H.J.; Rots, M.G. Experimental mitochondria-targeted DNA methylation identifies GpC methylation, not CpG methylation, as potential regulator of mitochondrial gene expression. Sci. Rep. 2017, 7, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Patil, V.; Cuenin, C.; Chung, F.; Rodríguez-Aguilera, J.R.; Fernandez-Jimenez, N.; Romero-Garmendia, I.; Bilbao, J.R.; Cahais, V.; Rothwell, J.; Herceg, Z. Human mitochondrial DNA is extensively methylated in a non-CpG context. Nucleic Acids Res. 2019, 47, 10072–10085. [Google Scholar] [CrossRef] [Green Version]
- Janeway, C.A.; Medzhitov, R. Innate immune recognition. Annu. Rev. Immunol. 2002, 20, 197–216. [Google Scholar] [CrossRef] [Green Version]
- Takeuchi, O.; Akira, S. Pattern recognition receptors and inflammation. Cell 2010, 140, 805–820. [Google Scholar] [CrossRef] [Green Version]
- Gong, T.; Liu, L.; Jiang, W.; Zhou, R. DAMP-sensing receptors in sterile inflammation and inflammatory diseases. Nat. Rev. Immunol. 2019, 20, 95–112. [Google Scholar] [CrossRef]
- West, A.P.; Shadel, G.S. Mitochondrial DNA in innate immune responses and inflammatory pathology. Nat. Rev. Immunol. 2017, 17, 363–375. [Google Scholar] [CrossRef] [PubMed]
- Riley, J.S.; Tait, S.W. Mitochondrial DNA in inflammation and immunity. EMBO Rep. 2020, 21, e49799. [Google Scholar] [CrossRef]
- Yuk, J.-M.; Silwal, P.; Jo, E.-K. Inflammasome and mitophagy connection in health and disease. Int. J. Mol. Sci. 2020, 21, 4714. [Google Scholar] [CrossRef]
- Kelley, N.; Jeltema, D.; Duan, Y.; He, Y. The NLRP3 Inflammasome: An overview of mechanisms of activation and regulation. Int. J. Mol. Sci. 2019, 20, 3328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, D.; Liwinski, T.; Elinav, E. Inflammasome activation and regulation: Toward a better understanding of complex mechanisms. Cell Discov. 2020, 6, 1–22. [Google Scholar] [CrossRef]
- Nakahira, K.; Haspel, J.A.; Rathinam, V.A.K.; Lee, S.-J.; Dolinay, T.; Lam, H.C.; Englert, J.; Rabinovitch, M.; Cernadas, M.; Kim, H.P.; et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat. Immunol. 2010, 12, 222–230. [Google Scholar] [CrossRef] [Green Version]
- Zhou, R.; Yazdi, A.S.; Menu, P.; Tschopp, J. A role for mitochondria in NLRP3 inflammasome activation. Nat. Cell Biol. 2010, 469, 221–225. [Google Scholar] [CrossRef]
- Shimada, K.; Crother, T.; Karlin, J.; Dagvadorj, J.; Chiba, N.; Chen, S.; Ramanujan, V.K.; Wolf, A.J.; Vergnes, L.; Ojcius, D.; et al. Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity 2012, 36, 401–414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhong, Z.; Liang, S.; Sanchez-Lopez, E.; He, F.; Shalapour, S.; Lin, X.-J.; Wong, J.; Ding, S.; Seki, E.; Schnabl, B.; et al. New mitochondrial DNA synthesis enables NLRP3 inflammasome activation. Nat. Cell Biol. 2018, 560, 198–203. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Shen, Y.; Jin, K.; Wen, Z.; Cao, W.; Wu, B.; Wen, R.; Tian, L.; Berry, G.J.; Goronzy, J.J.; et al. The DNA repair nuclease MRE11A functions as a mitochondrial protector and prevents T cell pyroptosis and tissue inflammation. Cell Metab. 2019, 30, 477–492. [Google Scholar] [CrossRef]
- Kawai, T.; Akira, S. Signaling to NF-κB by toll-like receptors. Trends Mol. Med. 2007, 13, 460–469. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Itagaki, K.; Hauser, C. mitochondrial DNA is released by shock and activates neutrophils via P38 map kinase. Shock 2010, 34, 55–59. [Google Scholar] [CrossRef] [PubMed]
- Oka, T.; Hikoso, S.; Yamaguchi, O.; Taneike, M.; Takeda, T.; Tamai, T.; Oyabu, J.; Murakawa, T.; Nakayama, H.; Nishida, K.; et al. Mitochondrial DNA that escapes from autophagy causes inflammation and heart failure. Nat. Cell Biol. 2012, 485, 251–255. [Google Scholar] [CrossRef]
- Zhang, J.-Z.; Liu, Z.; Liu, J.; Ren, J.-X.; Sun, T.-S. Mitochondrial DNA induces inflammation and increases TLR9/NF-κB expression in lung tissue. Int. J. Mol. Med. 2014, 33, 817–824. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Martinez, I.; Santoro, N.; Chen, Y.; Hoque, R.; Ouyang, X.; Caprio, S.; Shlomchik, M.J.; Coffman, R.L.; Candia, A.; Mehal, W.Z. Hepatocyte mitochondrial DNA drives nonalcoholic steatohepatitis by activation of TLR9. J. Clin. Investig. 2016, 126, 859–864. [Google Scholar] [CrossRef] [Green Version]
- Rodríguez-Nuevo, A.; Díaz-Ramos, A.; Noguera, E.; Díaz-Sáez, F.; Duran, X.; Muñoz, J.P.; Romero, M.; Plana, N.; Sebastián, D.; Tezze, C.; et al. Mitochondrial DNA and TLR9 drive muscle inflammation upon Opa1 deficiency. EMBO J. 2018, 37, e96553. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.-H.; Davidson, S.; Harapas, C.R.; Hilton, J.B.; Mlodzianoski, M.J.; Laohamonthonkul, P.; Louis, C.; Low, R.R.J.; Moecking, J.; De Nardo, D.; et al. TDP-43 triggers mitochondrial DNA release via mPTP to activate cGAS/STING in ALS. Cell 2020, 183, 636–649. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.S.; Hong, Z.; Wu, W.; Xiong, S.; Gao, X.; Rehman, J.; Malik, A.B. mtDNA activates cGAS signaling and suppresses the YAP-mediated endothelial cell proliferation program to promote inflammatory injury. Immunity 2020, 52, 475–486. [Google Scholar] [CrossRef]
- Maekawa, H.; Inoue, T.; Ouchi, H.; Jao, T.-M.; Inoue, R.; Nishi, H.; Fujii, R.; Ishidate, F.; Tanaka, T.; Tanaka, Y.; et al. Mitochondrial damage causes inflammation via cGAS-STING signaling in acute kidney injury. Cell Rep. 2019, 29, 1261–1273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bai, J.; Cervantes, C.; Liu, J.; He, S.; Zhou, H.; Zhang, B.; Cai, H.; Yin, D.; Hu, D.; Li, Z.; et al. DsbA-L prevents obesity-induced inflammation and insulin resistance by suppressing the mtDNA release-activated cGAS-cGAMP-STING pathway. Proc. Natl. Acad. Sci. USA 2017, 114, 12196–12201. [Google Scholar] [CrossRef] [Green Version]
- Torres-Odio, S.; Lei, Y.; Gispert, S.; Maletzko, A.; Key, J.; Menissy, S.S.; Wittig, I.; Auburger, G.; West, A.P. Loss of mitochondrial protease CLPP activates type I IFN responses through the mitochondrial DNA–cGAS–STING signaling axis. J. Immunol. 2021, ji2001016. [Google Scholar] [CrossRef]
- Hopfner, K.-P.; Hornung, V. Molecular mechanisms and cellular functions of cGAS–STING signalling. Nat. Rev. Mol. Cell Biol. 2020, 21, 501–521. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Oeck, S.; West, A.P.; Mangalhara, K.C.; Sainz, A.G.; Newman, L.; Zhang, X.-O.; Wu, L.; Yan, Q.; Bosenberg, M.; et al. Mitochondrial DNA stress signalling protects the nuclear genome. Nat. Metab. 2019, 1, 1209–1218. [Google Scholar] [CrossRef] [PubMed]
- Lei, Y.; Martinez, C.G.; Torres-Odio, S.; Bell, S.L.; Birdwell, C.E.; Bryant, J.D.; Tong, C.W.; Watson, R.O.; West, L.C.; West, A.P. Elevated type I interferon responses potentiate metabolic dysfunction, inflammation, and accelerated aging in mtDNA mutator mice. Sci. Adv. 2021, 7, eabe7548. [Google Scholar] [CrossRef] [PubMed]
- White, M.J.; McArthur, K.; Metcalf, D.; Lane, R.M.; Cambier, J.; Herold, M.; van Delft, M.; Bedoui, S.; Lessene, G.; Ritchie, M.; et al. Apoptotic caspases suppress mtDNA-induced STING-mediated type I IFN production. Cell 2014, 159, 1549–1562. [Google Scholar] [CrossRef] [Green Version]
- Rongvaux, A.; Jackson, R.; Harman, C.C.; Li, T.; West, A.P.; De Zoete, M.R.; Wu, Y.; Yordy, B.; Lakhani, S.; Kuan, C.-Y.; et al. Apoptotic caspases prevent the induction of type I interferons by mitochondrial DNA. Cell 2014, 159, 1563–1577. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Gupta, R.; Blanco, L.P.; Yang, S.; Shteinfer-Kuzmine, A.; Wang, K.; Zhu, J.; Yoon, H.E.; Wang, X.; Kerkhofs, M.; et al. VDAC oligomers form mitochondrial pores to release mtDNA fragments and promote lupus-like disease. Science 2019, 366, 1531–1536. [Google Scholar] [CrossRef]
- Patrushev, M.; Kasymov, V.; Patrusheva, V.; Ushakova, T.; Gogvadze, V.; Gaziev, A. Mitochondrial permeability transition triggers the release of mtDNA fragments. Cell. Mol. Life Sci. 2004, 61, 3100–3103. [Google Scholar] [CrossRef]
- Tait, S.; Green, D. Mitochondria and cell death: Outer membrane permeabilization and beyond. Nat. Rev. Mol. Cell Biol. 2010, 11, 621–632. [Google Scholar] [CrossRef]
- Ichim, G.; Lopez, J.; Ahmed, S.U.; Muthalagu, N.; Giampazolias, E.; Delgado, M.E.; Haller, M.; Riley, J.; Mason, S.; Athineos, D.; et al. Limited mitochondrial permeabilization causes DNA damage and genomic instability in the absence of cell death. Mol. Cell 2015, 57, 860–872. [Google Scholar] [CrossRef] [Green Version]
- Giampazolias, E.; Zunino, B.; Dhayade, S.; Bock, F.; Cloix, C.; Cao, K.; Roca, A.; Lopez, J.; Ichim, G.; Proïcs, E.; et al. Mitochondrial permeabilization engages NF-κB-dependent anti-tumour activity under caspase deficiency. Nat. Cell Biol. 2017, 19, 1116–1129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riley, J.S.; Quarato, G.; Cloix, C.; Lopez, J.; O’Prey, J.; Pearson, M.; Chapman, J.; Sesaki, H.; Carlin, L.M.; Passos, J.F.; et al. Mitochondrial inner membrane permeabilisation enables mt DNA release during apoptosis. EMBO J. 2018, 37. [Google Scholar] [CrossRef]
- McArthur, K.; Whitehead, L.W.; Heddleston, J.M.; Li, L.; Padman, B.S.; Oorschot, V.; Geoghegan, N.D.; Chappaz, S.; Davidson, S.; Chin, H.S.; et al. BAK/BAX macropores facilitate mitochondrial herniation and mtDNA efflux during apoptosis. Science 2018, 359, eaao6047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saito, Y.; Hikita, H.; Nozaki, Y.; Kai, Y.; Makino, Y.; Nakabori, T.; Tanaka, S.; Yamada, R.; Shigekawa, M.; Kodama, T.; et al. DNase II activated by the mitochondrial apoptotic pathway regulates RIP1-dependent non-apoptotic hepatocyte death via the TLR9/IFN-β signaling pathway. Cell Death Differ. 2018, 26, 470–486. [Google Scholar] [CrossRef] [PubMed]
- He, W.-R.; Cao, L.-B.; Yang, Y.-L.; Hua, D.; Hu, M.-M.; Shu, H.-B. VRK2 is involved in the innate antiviral response by promoting mitostress-induced mtDNA release. Cell. Mol. Immunol. 2021, 18, 1186–1196. [Google Scholar] [CrossRef] [PubMed]
- Krauskopf, A.; Eriksson, O.; Craigen, W.J.; Forte, M.A.; Bernardi, P. Properties of the permeability transition in VDAC1 −/− mitochondria. Biochim. Biophys. Acta Bioenerg. 2006, 1757, 590–595. [Google Scholar] [CrossRef]
- Baines, C.; Kaiser, R.A.; Sheiko, T.; Craigen, W.J.; Molkentin, J.D. Voltage-dependent anion channels are dispensable for mitochondrial-dependent cell death. Nat. Cell Biol. 2007, 9, 550–555. [Google Scholar] [CrossRef]
- Bauer, M.K.; Schubert, A.; Rocks, O.; Grimm, S. Adenine nucleotide translocase-1, a component of the permeability transition pore, can dominantly induce apoptosis. J. Cell Biol. 1999, 147, 1493–1502. [Google Scholar] [CrossRef]
- Kokoszka, J.E.; Waymire, K.G.; Levy, S.; Sligh, J.E.; Cai, J.; Jones, D.P.; MacGregor, G.R.; Wallace, D.C. The ADP/ATP translocator is not essential for the mitochondrial permeability transition pore. Nat. Cell Biol. 2004, 427, 461–465. [Google Scholar] [CrossRef]
- Bernardi, P.; Lippe, G. Channel formation by F-ATP synthase and the permeability transition pore: An update. Curr. Opin. Physiol. 2017, 3, 1–5. [Google Scholar] [CrossRef]
- He, J.; Ford, H.C.; Carroll, J.; Ding, S.; Fearnley, I.M.; Walker, J.E. Persistence of the mitochondrial permeability transition in the absence of subunit c of human ATP synthase. Proc. Natl. Acad. Sci. USA 2017, 114, 3409–3414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- García, N.; Chávez, E. Mitochondrial DNA fragments released through the permeability transition pore correspond to specific gene size. Life Sci. 2007, 81, 1160–1166. [Google Scholar] [CrossRef] [PubMed]
- Carroll, E.C.; Jin, L.; Mori, A.; Muñoz-Wolf, N.; Oleszycka, E.; Moran, H.B.; Mansouri, S.; McEntee, C.; Lambe, E.; Agger, E.M.; et al. The vaccine adjuvant chitosan promotes cellular immunity via DNA sensor cGAS-STING-dependent induction of type I interferons. Immunity 2016, 44, 597–608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bueter, C.L.; Lee, C.K.; Rathinam, V.A.; Healy, G.J.; Taron, C.H.; Specht, C.A.; Levitz, S.M. Chitosan but not chitin activates the inflammasome by a mechanism dependent upon phagocytosis. J. Biol. Chem. 2011, 286, 35447–35455. [Google Scholar] [CrossRef] [Green Version]
- Kerur, N.; Fukuda, S.; Banerjee, D.; Kim, Y.; Fu, D.; Apicella, I.; Varshney, A.; Yasuma, R.; Fowler, B.J.; Baghdasaryan, E.; et al. cGAS drives noncanonical-inflammasome activation in age-related macular degeneration. Nat. Med. 2017, 24, 50–61. [Google Scholar] [CrossRef]
- Liu, R.; Xu, F.; Bi, S.; Zhao, X.; Jia, B.; Cen, Y. Mitochondrial DNA-induced inflammatory responses and lung injury in thermal injury murine model: Protective effect of cyclosporine-A. J. Burn Care Res. 2019, 40, 355–360. [Google Scholar] [CrossRef]
- Yu, Y.; Liu, Y.; An, W.; Song, J.; Zhang, Y.; Zhao, X. STING-mediated inflammation in Kupffer cells contributes to progression of nonalcoholic steatohepatitis. J. Clin. Investig. 2018, 129, 546–555. [Google Scholar] [CrossRef]
- Cunningham, C. Microglia and neurodegeneration: The role of systemic inflammation. Glia 2012, 61, 71–90. [Google Scholar] [CrossRef]
- Moehlman, A.T.; Youle, R.J. Mitochondrial quality control and restraining innate immunity. Annu. Rev. Cell Dev. Biol. 2020, 36, 265–289. [Google Scholar] [CrossRef]
- Pickrell, A.; Youle, R.J. The roles of PINK1, parkin, and mitochondrial fidelity in Parkinson’s Disease. Neuron 2015, 85, 257–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sliter, D.A.; Martinez, J.; Hao, L.; Chen, X.; Sun, N.; Fischer, T.D.; Burman, J.L.; Li, Y.; Zhang, Z.; Narendra, D.P.; et al. Parkin and PINK1 mitigate STING-induced inflammation. Nat. Cell Biol. 2018, 561, 258–262. [Google Scholar] [CrossRef] [PubMed]
- Matsui, H.; Ito, J.; Matsui, N.; Uechi, T.; Onodera, O.; Kakita, A. Cytosolic dsDNA of mitochondrial origin induces cytotoxicity and neurodegeneration in cellular and zebrafish models of Parkinson’s disease. Nat. Commun. 2021, 12, 1–12. [Google Scholar] [CrossRef]
- Jauhari, A.; Baranov, S.V.; Suofu, Y.; Kim, J.; Singh, T.; Yablonska, S.; Li, F.; Wang, X.; Oberly, P.; Minnigh, M.B.; et al. Melatonin inhibits cytosolic mitochondrial DNA–induced neuroinflammatory signaling in accelerated aging and neurodegeneration. J. Clin. Investig. 2020, 130, 3124–3136. [Google Scholar] [CrossRef] [Green Version]
- Sharma, M.; Rajendrarao, S.; Shahani, N.; Ramírez-Jarquín, U.N.; Subramaniam, S. Cyclic GMP-AMP synthase promotes the inflammatory and autophagy responses in Huntington disease. Proc. Natl. Acad. Sci. USA 2020, 117, 15989–15999. [Google Scholar] [CrossRef]
- Lee, H.; Fenster, R.J.; Pineda, S.S.; Gibbs, W.S.; Mohammadi, S.; Davila-Velderrain, J.; Garcia, F.J.; Therrien, M.; Novis, H.S.; Gao, F.; et al. Cell type-specific transcriptomics reveals that mutant Huntingtin leads to mitochondrial RNA release and neuronal innate immune activation. Neuron 2020, 107, 891–908. [Google Scholar] [CrossRef]
- Borsche, M.; König, I.R.; Delcambre, S.; Petrucci, S.; Balck, A.; Brüggemann, N.; Zimprich, A.; Wasner, K.; Pereira, S.L.; Avenali, M.; et al. Mitochondrial damage-associated inflammation highlights biomarkers in PRKN/PINK1 parkinsonism. Brain 2020, 143, 3041–3051. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.J.; Andreazza, S.; Whitworth, A.J. The STING pathway does not contribute to behavioural or mitochondrial phenotypes in Drosophila Pink1/parkin or mtDNA mutator models. Sci. Rep. 2020, 10, 1–10. [Google Scholar] [CrossRef]
- Gispert, S.; Parganlija, D.; Klinkenberg, M.; Dröse, S.; Wittig, I.; Mittelbronn, M.; Grzmil, P.; Koob, S.; Hamann, A.; Walter, M.; et al. Loss of mitochondrial peptidase Clpp leads to infertility, hearing loss plus growth retardation via accumulation of CLPX, mtDNA and inflammatory factors. Hum. Mol. Genet. 2013, 22, 4871–4887. [Google Scholar] [CrossRef] [Green Version]
- Sprenger, H.; Wani, G.; Hesseling, A.; König, T.; Patron, M.; MacVicar, T.; Ahola, S.; Wai, T.; Barth, E.; Rugarli, E.I.; et al. Loss of the mitochondrial i-AAA protease YME1L leads to ocular dysfunction and spinal axonopathy. EMBO Mol. Med. 2018, 11, e9288. [Google Scholar] [CrossRef]
- Sprenger, H.-G.; MacVicar, T.; Bahat, A.; Fiedler, K.U.; Hermans, S.; Ehrentraut, D.; Ried, K.; Milenkovic, D.; Bonekamp, N.; Larsson, N.-G.; et al. Cellular pyrimidine imbalance triggers mitochondrial DNA–dependent innate immunity. Nat. Metab. 2021, 3, 636–650. [Google Scholar] [CrossRef] [PubMed]
- Neumann, M. Molecular neuropathology of TDP-43 proteinopathies. Int. J. Mol. Sci. 2009, 10, 232–246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCauley, M.E.; O’Rourke, J.G.; Yáñez, A.; Markman, J.L.; Ho, R.; Wang, X.; Chen, S.; Lall, D.; Jin, M.; Muhammad, A.K.M.G.; et al. C9orf72 in myeloid cells suppresses STING-induced inflammation. Nat. Cell Biol. 2020, 585, 96–101. [Google Scholar] [CrossRef]
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef] [Green Version]
- Jang, J.Y.; Blum, A.; Liu, J.; Finkel, T. The role of mitochondria in aging. J. Clin. Investig. 2018, 128, 3662–3670. [Google Scholar] [CrossRef] [Green Version]
- Picca, A.; Lezza, A.M.S.; Leeuwenburgh, C.; Pesce, V.; Calvani, R.; Landi, F.; Bernabei, R.; Marzetti, E. Fueling inflamm-aging through mitochondrial dysfunction: Mechanisms and molecular targets. Int. J. Mol. Sci. 2017, 18, 933. [Google Scholar] [CrossRef]
- Arnoult, D.; Soares, F.; Tattoli, I.; Girardin, S.E. Mitochondria in innate immunity. EMBO Rep. 2011, 12, 901–910. [Google Scholar] [CrossRef] [Green Version]
- West, A.P.; Shadel, G.S.; Ghosh, S. Mitochondria in innate immune responses. Nat. Rev. Immunol. 2011, 11, 389–402. [Google Scholar] [CrossRef] [Green Version]
- Zevini, A.; Olagnier, D.; Hiscott, J. Crosstalk between cytoplasmic RIG-I and STING sensing pathways. Trends Immunol. 2017, 38, 194–205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, Y.; Park, J.; Kim, S.; Kim, M.; Kang, M.-G.; Kwak, C.; Kang, M.; Kim, B.; Rhee, H.-W.; Kim, V.N. PKR senses nuclear and mitochondrial signals by interacting with endogenous double-stranded RNAs. Mol. Cell 2018, 71, 1051–1063. [Google Scholar] [CrossRef] [Green Version]
- Karikó, K.; Buckstein, M.; Ni, H.; Weissman, D. Suppression of RNA recognition by toll-like receptors: The impact of nucleoside modification and the evolutionary origin of RNA. Immunity 2005, 23, 165–175. [Google Scholar] [CrossRef] [Green Version]
- Borowski, L.; Dziembowski, A.; Hejnowicz, M.S.; Stepien, P.P.; Szczesny, R.J. Human mitochondrial RNA decay mediated by PNPase-hSuv3 complex takes place in distinct foci. Nucleic Acids Res. 2012, 41, 1223–1240. [Google Scholar] [CrossRef] [PubMed]
- Pajak, A.; Laine, I.; Clemente, P.; El-Fissi, N.; Schober, F.A.; Maffezzini, C.; Garrido, J.C.; Wibom, R.; Filograna, R.; Dhir, A.; et al. Defects of mitochondrial RNA turnover lead to the accumulation of double-stranded RNA in vivo. PLoS Genet. 2019, 15, e1008240. [Google Scholar] [CrossRef] [Green Version]
- Arnaiz, E.; Miar, A.; Dias, A.G.; Prasad, N.; Schulze, U.; Waithe, D.; Rehwinkel, J.; Harris, A.L. Hypoxia regulates endogenous double-stranded RNA production via reduced mitochondrial DNA transcription. bioRxiv 2020. [Google Scholar] [CrossRef]
- Tigano, M.; Vargas, D.C.; Tremblay-Belzile, S.; Fu, Y.; Sfeir, A. Nuclear sensing of breaks in mitochondrial DNA enhances immune surveillance. Nat. Cell Biol. 2021, 591, 477–481. [Google Scholar] [CrossRef]
- Zhou, X.; Backman, L.J.; Danielson, P. Activation of NF-κB signaling via cytosolic mitochondrial RNA sensing in kerotocytes with mitochondrial DNA common deletion. Sci. Rep. 2021, 11, 1–13. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pathology | Mitochondial Nucleic Acid Involvement |
|---|---|
| Parkinson’s Disease (PD) | Cytosolic mtDNA accumulation associated with type I IFN responses and PD phenotypes in vitro and in vivo [85,86]. Circulating cell-free mtDNA and increased mtDNA in postmortem brain tissues from PD patients [86]. |
| Perrault syndrome | mtDNA instability and escape into the cytosol drives cGAS–STING–IFN-I signaling in cells lacking CLPP [55]. |
| Amyotrophic lateral sclerosis (ALS) | mtDNA-mediated cGAS–STING pathway activation leads to induction of type I IFNs and inflammatory cytokines in TDP-43 accumulating cells [51]. |
| Huntington’s disease (HD) | Upregulation of cGAS–STING expression in the striatum of a mouse model and HD human patient’s tissue [87,88].mtDNA release in R6/2 mouse model of HD [87].Upregulation of mtRNA in post-mortem brains from HD patients and mouse models [89]. |
| Leigh Syndrome | mtdsRNA accumulation in fibroblasts from patients PNTP1 Mutations and upregulation of ISGs and markers of immune response in peripheral blood [23]. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Luna-Sánchez, M.; Bianchi, P.; Quintana, A. Mitochondria-Induced Immune Response as a Trigger for Neurodegeneration: A Pathogen from Within. Int. J. Mol. Sci. 2021, 22, 8523. https://doi.org/10.3390/ijms22168523
Luna-Sánchez M, Bianchi P, Quintana A. Mitochondria-Induced Immune Response as a Trigger for Neurodegeneration: A Pathogen from Within. International Journal of Molecular Sciences. 2021; 22(16):8523. https://doi.org/10.3390/ijms22168523
Chicago/Turabian StyleLuna-Sánchez, Marta, Patrizia Bianchi, and Albert Quintana. 2021. "Mitochondria-Induced Immune Response as a Trigger for Neurodegeneration: A Pathogen from Within" International Journal of Molecular Sciences 22, no. 16: 8523. https://doi.org/10.3390/ijms22168523
APA StyleLuna-Sánchez, M., Bianchi, P., & Quintana, A. (2021). Mitochondria-Induced Immune Response as a Trigger for Neurodegeneration: A Pathogen from Within. International Journal of Molecular Sciences, 22(16), 8523. https://doi.org/10.3390/ijms22168523