
Modelling Mitochondrial Disease in Human Pluripotent Stem Cells: What Have We Learned?

 , and
, and
Abstract
1. Introduction
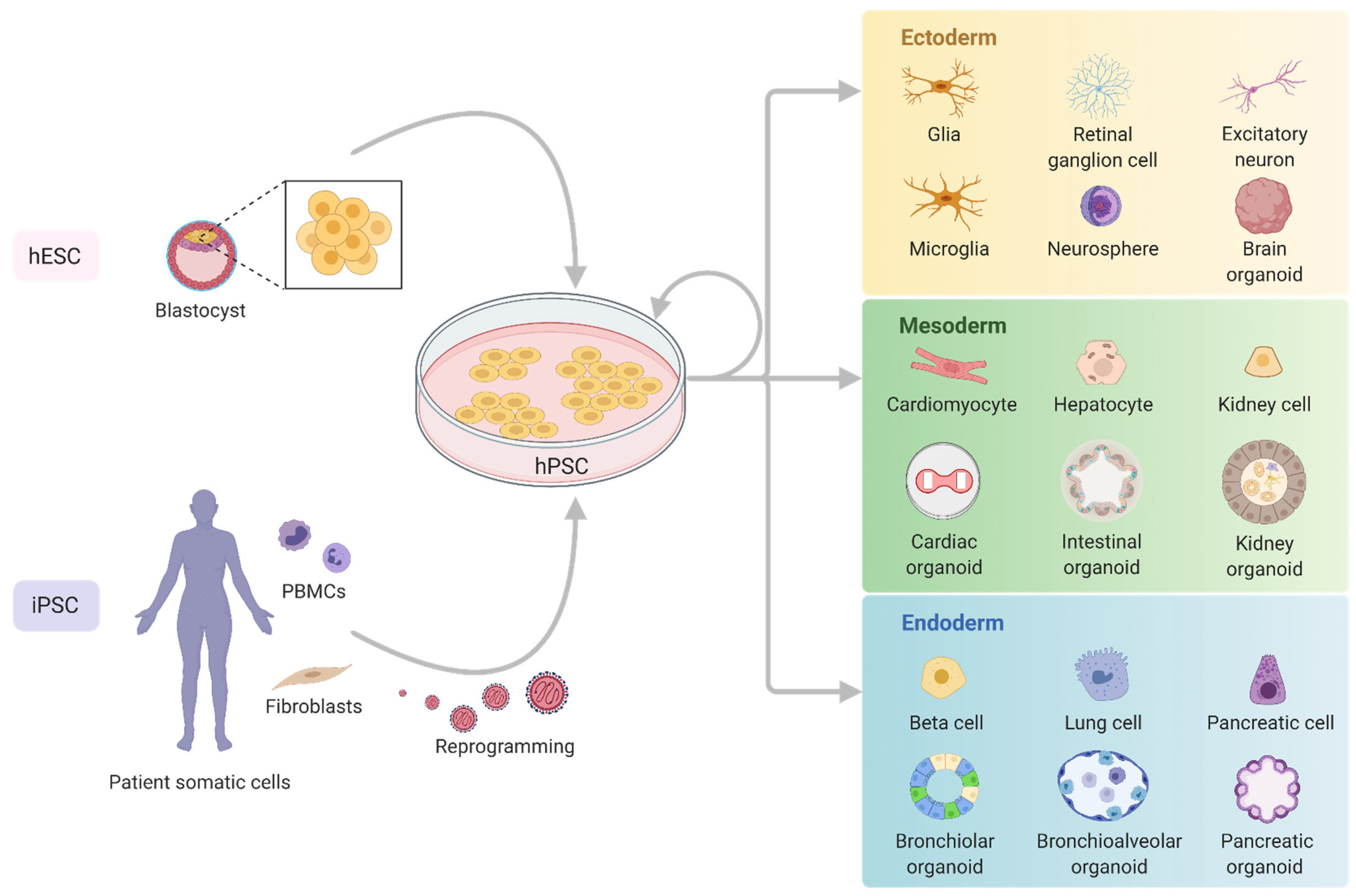
2. Pluripotent Stem Cells in Mitochondrial Disease Modelling
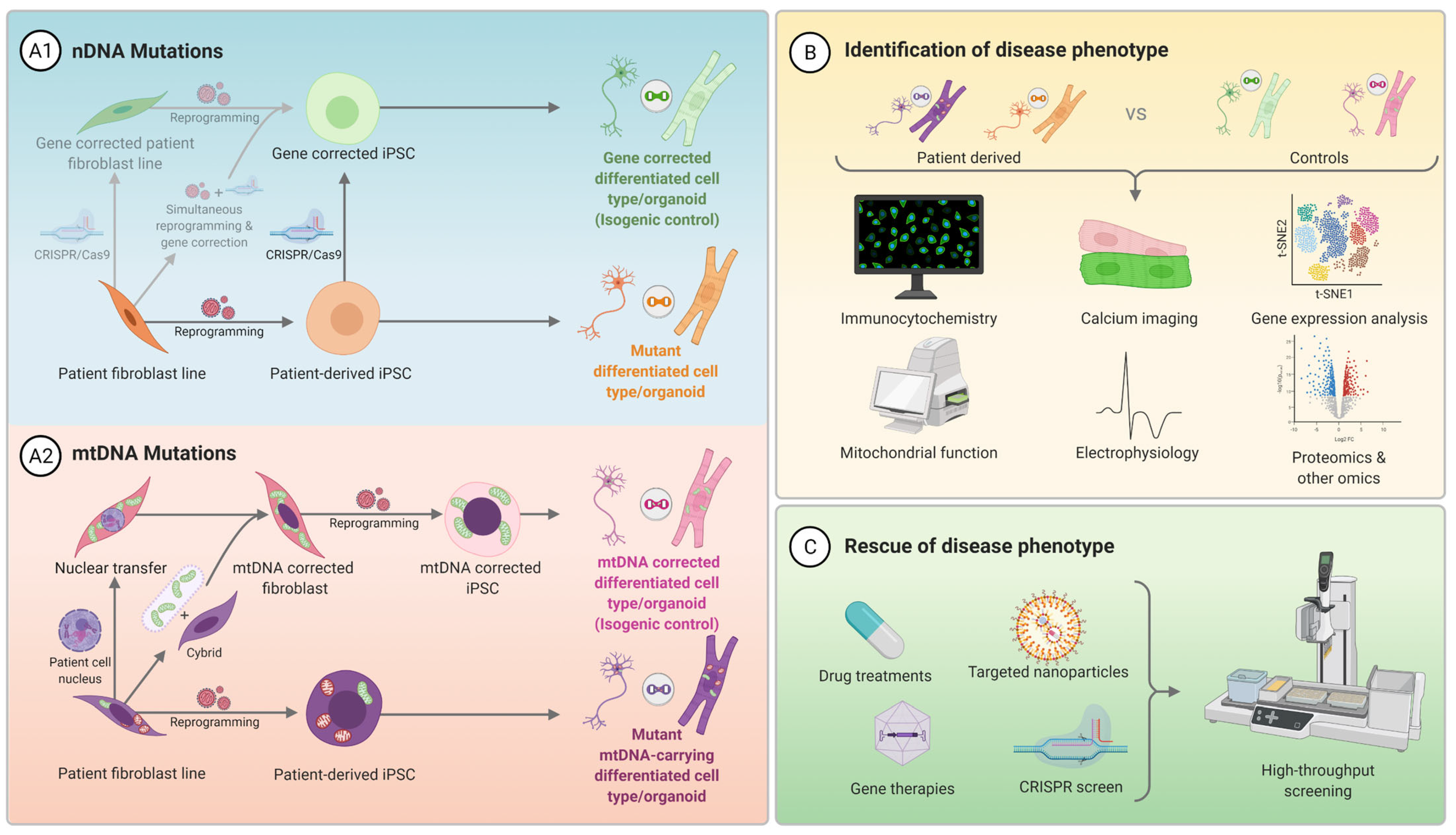
3. Generation of Human Pluripotent Stem Cell Mitochondrial Disease Models
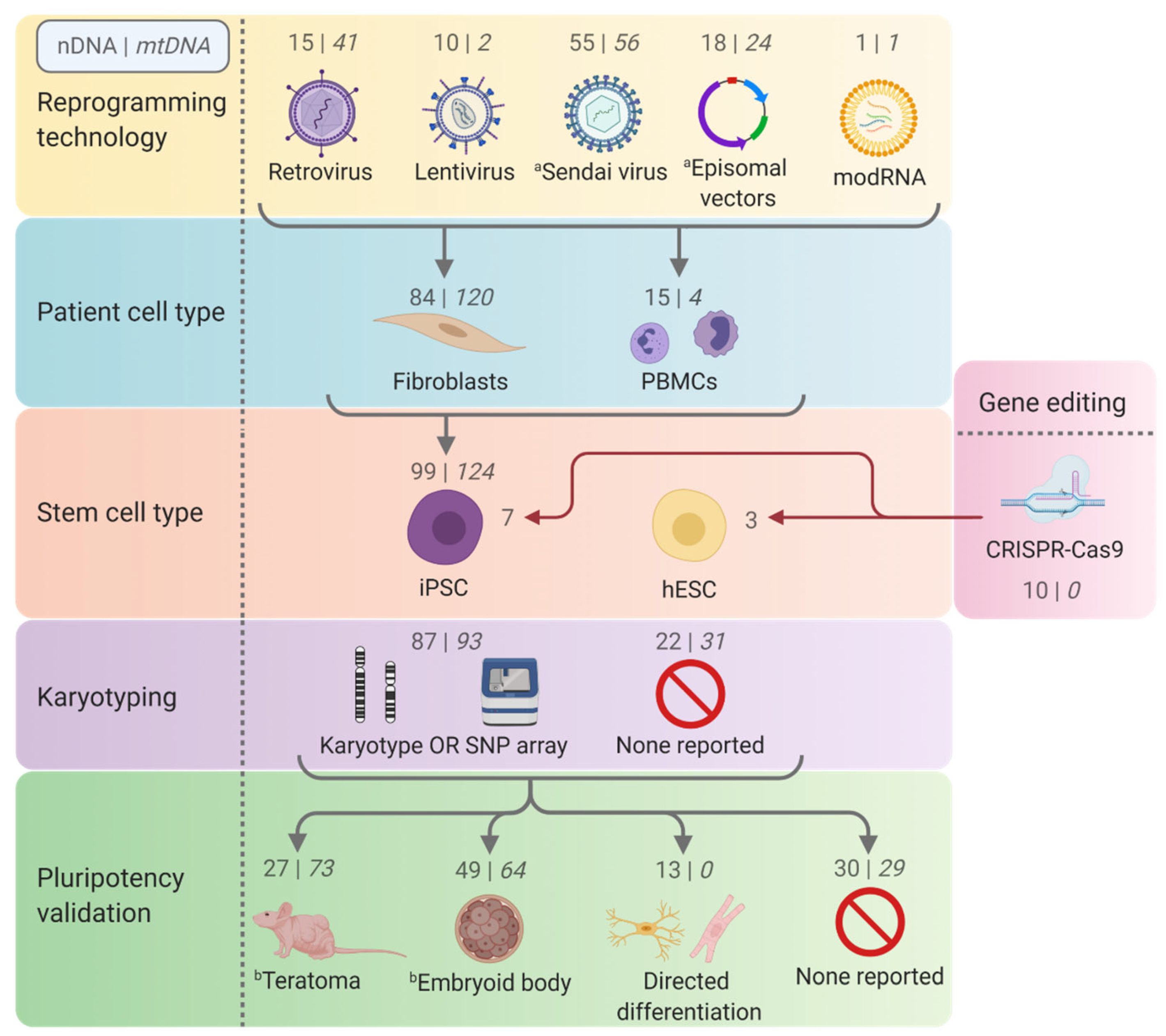
3.1. Technologies and Considerations for Generating hPSC Disease Models
3.1.1. Reprogramming of Somatic Cells into iPSCs

3.1.2. Gene Editing
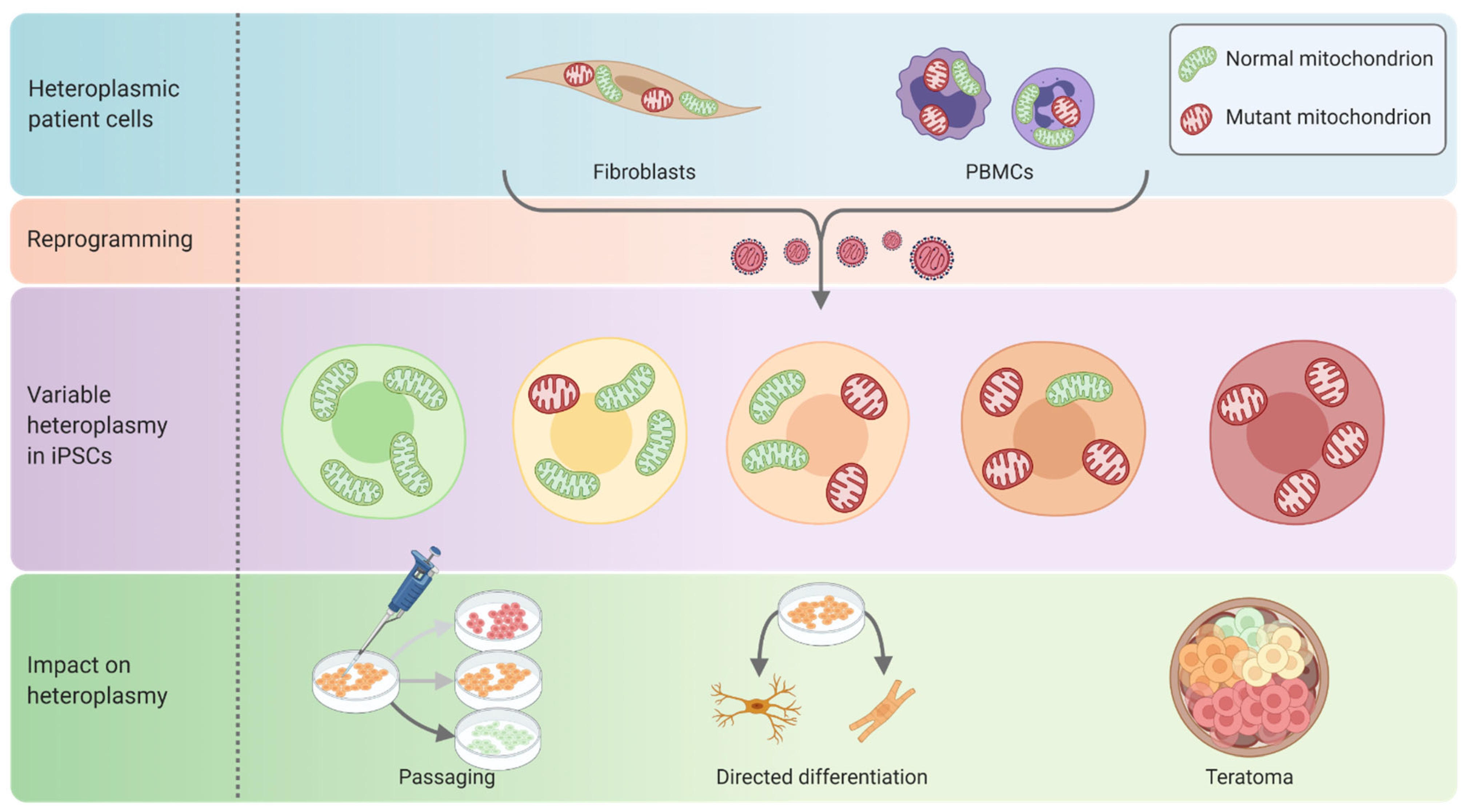
3.2. Quality Control and Characterisation of Pluripotent Stem Cell Disease Models
4. Disease Modelling
5. Functional Studies
5.1. Barth Syndrome—TAFAZZIN
5.2. DOA and Parkinson’s Disease—OPA1
5.3. PEO and Alpers Syndrome—POLG
5.4. mtDNA Depletion Syndromes—DGUOK and RRM2B
5.5. Leigh Syndrome
5.5.1. Complex IV Assembly Factors—SURF1 and SCO2
5.5.2. MT-ATP6
5.6. LHON
5.7. MELAS—MT-TL1
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Disease | Cell Line ID | Mutation a | Cell Line Origin b | Gene Editing /Reprogramming | Pluripotency | Trilineage Potential | Karyotype; Lineage Validation c | Mycoplasma Check | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|
| AARS2 | COXPD8 | LUMCi024-A | p.[(R958*)]; [(R592W);(V730M)] | M; 4 days; Fib | Sendai virus | Morphology; IF; FACS | EB differentiation | G-banding; Yes | Yes | [193] |
| AARS2 | COXPD8 | LUMCi025-A | p.[(R958*)]; [(R592W);(V730M)] | M; 1 day; Fib | Sendai virus | Morphology; IF; FACS | EB differentiation | G-banding; Yes | Yes | [193] |
| ACO2 | DOA | IISHDOi006-A | p.[(E667K)];[=] | M; 30 yr; Fib | Sendai virus | Morphology; AP; qPCR; IF | EB differentiation | G-banding; Yes | Yes | [194] |
| AIFM1 | AN | CPGHi003-A | p.[(R422Q)];[0] | M; 49 yr; PBMC | Episomal vectors | Morphology; RT-qPCR; IF | EB differentiation | G-banding; Yes | Yes | [195] |
| ATAD3A | HSP | HEL142 (2 clones) | p.[(G355D)];[=] | F; 35 yr; Fib | Episomal vectors | qPCR; IF | N/D | N/D | N/D | [196] |
| C1QBP | COXPD | XACHi010-A | p.[(L275F)]; [(L275F)] | M; 14 yr; PBMC | Sendai virus | Morphology; IF; FACS | Trilineage differentiation | G-banding; Yes | Yes | [197] |
| COQ2 | MSA-C | MSA_A (3 clones) | p.[(R387*)]; [(V393A)] | M; 61 yr; PBMC | Episomal vectors | Morphology; IF | Teratoma | G-banding; N/D | N/D | [198] |
| COQ4 | CoQ10 deficiency | CQ4-Ipsc (4 clones) | p.[(E161D)];[=] | F; 4 yr; Fib | Sendai virus | Morphology; AP; RT-qPCR; IF; FACS; hypomethylation | Teratoma | G-banding; Yes | Yes | [199,200] |
| COX6A2 | CIV deficiency | WAe009-A-47 | p.[A16Lfs*18]; [A16Lfs*18] | F; hESC-WA09 | CRISPR-Cas9 induced mutation | Morphology; RT-qPCR; IF; FACS | Teratoma | G-banding; Yes | Yes | [105] |
| DGUOK | MTDPS3 | DGUOKΔ14/Δ5 iPSC | p.[W166*]; [H167Lfs*213] | M; iPS-SV20 | CRISPR-Cas9 induced mutation | N/D | N/D | N/D | N/D | [131] |
| DGUOK | MTDPS | Patient 1 | p.[F256*];[F256*] | F; 2 mo; Fib | Retrovirus | Morphology | N/D | G-banding; N/D | N/D | [132] |
| DGUOK | MTDPS | Patient 2 | [p.A2S; c.591G > A]; [c.142 + 1G > A], | M; 2 mo; Fib | Retrovirus | Morphology | N/D | G-banding; N/D | N/D | [132] |
| DNAJC19 | DCMA | Patient 1 | c.[130-1G > C]; [130-1G > C] | F; 1.5 yr; PBMC | Sendai virus | IF | N/D | SNP microarray; N/D | N/D | [201] |
| DNAJC19 | DCMA | Patient 2 | c.[130-1G > C]; [130-1G > C] | M; 11 yr; PBMC | Sendai virus | IF | N/D | SNP microarray; N/D | N/D | [201] |
| DNAJC19 | DCMA | LIBUCi001-A | c.[130-1G > C]; [130-1G > C] | M; 8 yr; Fib | Sendai virus | Morphology; IF; FACS | EB differentiation | G-banding; Yes | Yes | [202] |
| DNAJC19 | DCMA | LIBUCi002-A | c.[130-1G > C]; [130-1G > C] | F; 10 yr; Fib | Sendai virus | Morphology; IF; FACS | EB differentiation | G-banding; Yes | Yes | [202] |
| DNAJC19 | DCMA | JMUi001-A | p.[(A44Vfs*12)];[(S46Rfs*3)] | M; iPS- JMUi001-A. | CRISPR-Cas9 induced mutation | Morphology; IF; FACS | EB differentiation | G-banding; Yes | Yes | [202] |
| ECHS1 | Leigh-like syndrome | UOMi001-A | p.[(A172V)]; [(K284Pfs*7)] | M; 13 yr; PBMC | Sendai virus | Morphology; qPCR; IF | EB differentiation | KaryoStat analysis; Yes | Yes | [203] |
| FBXL4 | MTDPS13 | SHCDNi001-A | p.[(L332Tfs*3)];[(L332Tfs*3)] | F; 1 yr; PBMC; | Sendai virus | Morphology; IF; FACS | Trilineage differentiation | G-banding; Yes | Yes | [204] |
| GDAP1 | CMT2K | CMT2-FiPS4F1 | p.[(Q163*)]; [(T288Nfs*3)] | M; 45 yr; Fib | Sendai virus | Morphology; qPCR; IF | EB differentiation | G-banding; Yes | N/D | [205] |
| GFM1 | Mitochondrial encephalopathy | GFM1SV.25 | p.[G469Vfs*84];[R671C] | F; 5.5 yr; Fib | Sendai virus | Morphology; AP; qPCR; IF; hypomethylation | EB differentiation | G-banding; Yes | N/D | [206] |
| MFN2 | CMT2A | Patient 1 (3 clones) | p.[(R364W)];[=] | M; 42 yr; Fib | Retrovirus | Morphology; qPCR; IF | EB differentiation | G-banding; Yes | N/D | [207] |
| MFN2 | CMT2A | CMT2A-1 (3 clones) | p.[(A383V)];[=] | F; 7 yr; Fib | Episomal vectors | Morphology; RT-PCR; IF | Yes (data not shown) | N/D | N/D | [191] |
| MFN2 | CMT2A | CMT2A-2 (3 clones) | p.[(A383V)];[=] | F; 12 yr; Fib | Episomal vectors | Morphology; RT-qPCR; IF | Yes (data not shown) | N/D | N/D | [191] |
| MFN2 | CMT2A | ZJUCHi002-A | p.[(P251L)];[=] | M; 8 yr; Urine cells | Retrovirus | Morphology; AP; qPCR; IF | EB differentiation | G-banding; Yes | Yes | [208] |
| MFN2 | MSL | JUCTCi012-A | p.[(R707W)]; [(R707W)] | F; 39 yr; Fib | Sendai virus | Morphology; IF; FACS | EB differentiation | G-banding; Yes | Yes | [209] |
| NDUFS4 | Leigh syndrome | NDU_1 | p.[(K154fs)];[(K154fs)] | M; 5 mo; Fib | Sendai virus | N/D | N/D | SNP microarray; Yes | Yes | [99] |
| NDUFS4 | Leigh syndrome | NDU_2 | p.[(R106*)]; [(R106*)] | F; 4 mo; Fib | Sendai virus | N/D | N/D | SNP microarray; Yes | Yes | [99] |
| NDUFV1 | Leigh syndrome | UOMi002-A | p.[(Y177Lfs*2)];[(E214K)] | F; 2.5 yr; PBMC | Sendai virus | Morphology; IF | EB differentiation | hPSC Genetic Analysis Kit; Yes | Yes | [210] |
| OPA1 | DOA | VO-iPSC | c.[(2496+1G > T)];[=] | Fib | Retrovirus/ Sendai virus | IF | Teratoma | N/D | N/D | [211] |
| OPA1 | DOA | OL-iPSC | c.[(2496+1G > T)];[=] | Fib | Retrovirus/ Sendai virus | IF | Teratoma | N/D | N/D | [211] |
| OPA1 | DOA ‘plus’ | Oex2054SV.4 | p.[(Q621*)];[=] | M; Fib | Sendai virus | Morphology; AP; qPCR; IF; hypomethylation | EB differentiation | G-banding; Yes | N/D | [212] |
| OPA1 | Behr syndrome | iPS-OPA1-BEHR | c.[610+364G>A];p.[I437M] | F; 48 yr; Fib | Episomal vectors | AP; RT-qPCR; IF | EB differentiation | SNP microarray; Yes | N/D | [213] |
| OPA1 | DOA ‘plus’ | IISHDOi003-A | p.[(S545R)];[=] | M; 43 yr; Fib | Sendai virus | Morphology; AP; qPCR; IF | EB differentiation | G-banding; Yes | Yes | [214] |
| OPA1 | Parkinson’s disease | PD-OPA1 G488R (2 clones) | p.[G488R];[=] | M; 74 yr; Fib | Sendai virus | Morphology; qPCR; IF | Trilineage differentiation | G-banding; N/D | N/D | [119] |
| OPA1 | Parkinson’s disease | PD-OPA1 A495V #72 | p.[A495V[;[=] | M; 70 yr; Fib | Sendai virus | Morphology; qPCR; IF | Trilineage differentiation | G-banding; N/D | N/D | [119] |
| OPA1 | Parkinson’s disease | Opa1P (2 clones) | c.[(33-34ins9)];[=] | M; 62 yr; Fib | Sendai virus | Morphology; Pluritest; FACS | N/D | SNP microarray; Yes | Yes | [120] |
| OPA1 | DOA | Opa1 (2 clones) | c.[(33-34ins9)];[=] | F; 84 yr; Fib | Sendai virus | Morphology; Pluritest; FACS | N/D | SNP microarray; Yes | Yes | [120] |
| OPA1 | DOA | OPA1+/− hESCs (2 clones) | N/D (haploinsufficiency) | F; hESC-WA22 | CRISPR-Cas9 induced mutation | IF | N/D | SNP microarray; N/D | N/D | [104] |
| OPA1 | DOA | OPA1+/- iPSC 1 | p.[(V958Gfs*3)];[=] | M; PBMC | Sendai virus | N/D | N/D | N/D | N/D | [104] |
| OPA1 | DOA | OPA1+/- iPSC2 | p.[(V958Gfs*3)];[=] | M; PBMC | Sendai virus | N/D | N/D | N/D | N/D | [104] |
| OPA1 | DOA | BIOi002-A | c.[2708_ 2711delTTAG];[=] | M; 27 yr; PBMC | Episomal vectors | Morphology; RT-PCR; FACS | Teratoma | G-banding; Yes | Yes | [215] |
| PDK3 | CMTX6 | iPSCCMTX6 | p.[(R158H)];[0] | M; Fib | Episomal vectors | RT-qPCR; IF | N/D | G-banding; Yes | N/D | [216] |
| PMPCB | Leigh-like syndrome | DII-2 iPSC (3 clones) | p.[I422T];[I422T] | M; Fib | Episomal vectors | IF; PluriTest | N/D | G-banding; N/D | Yes | [217] |
| POLG | Alpers syndrome | AHS iPS 1 | p.[A467T];c.[1251-2A > T] | M; 3.5 yr; Fib | Retrovirus | Morphology; AP; RT-qPCR; IF | Teratoma; EB differentiation | G-banding; N/D | N/D | [129] |
| POLG | Alpers syndrome | AHS iPS 2 | p.[A467T];c.[3626_3629dup] | F; 2 yr; Fib | Retrovirus | AP; Morphology; RT-qPCR; IF | Teratoma; EB differentiation | G-banding; N/D | N/D | [129] |
| POLG | N/D | PG64SV.2 | p.[(W748S)]; [(W748S)] | F; 36 yr; Fib | Sendai virus | Morphology; AP; qPCR; IF; hypomethylation | EB differentiation | G-banding; Yes | N/D | [218] |
| POLG | PEO and Parkinson’s disease | CSC-35 (3 clones) | p.[(Q811R)];[=] | F; 24 yr; Fib | Sendai virus | Morphology; AP; IF | EB differentiation | G-banding; Yes | N/D | [190] |
| POLG | PEO | WS5A (3 clones) | p.[W748S];[W748S] | F; Fib | Retrovirus | Morphology; RT-qPCR; IF; FACS; | Hep, CM, and neuronal differentiation | G-banding; N/D | Yes | [125,126] |
| POLG | PEO | CP2A (2 clones) | p.[A467T]; [W748S] | M; Fib | Retrovirus | Morphology; RT-qPCR; IF; FACS | Hep, CM, and neuronal differentiation | G-banding; N/D | Yes | [125,126] |
| RRM2B | MTDPS8A/B | RRM2B−/− iPSC | p.[R36Sfs*55]; [R36Sfs*55] | M; iPS-SV20 | CRISPR-Cas9 induced mutation | N/D | N/D | N/D | N/D | [131] |
| SAMHD1 | AGS | PEIi002 (3 clones) | homozygous exon 14 and 15 deletion | M; PBMC | Sendai virus | Morphology; RT-qPCR; IF | Trilineage differentiation | CGH-array; Yes | Yes | [219] |
| SAMHD1 | AGS | hSAMHD1-R290H+Q548X | p.[(R290H)];[(Q548*)] | M; PBMC | Sendai virus | FACS | EB differentiation | G-banding; Yes | N/D | [220] |
| SCO2 | CIV deficiency | SCO2G193S | p.[(G193S)]; [(G193S)] | M; 4 mo; Fib | Lentivirus | IF | Teratoma | G-banding; N/D | N/D | [137] |
| SCO2 | CIV deficiency | SCO2E140K | p.[(E140K)]; c.[(17ins(19))] d | M; 13 wk; Fib | Lentivirus | IF | Teratoma | G-banding; N/D | N/D | [137] |
| SURF1 | Leigh syndrome | SURF1_Mut: S1 | p.[(V177G)]; [(V177G)] | M; 9 yr; Fib | Sendai virus | RT-PCR; IF | EB differentiation | SNP microarray and G-banding; Yes | Yes | [99] |
| SURF1 | Leigh syndrome | SURF1_Mut: S2 | p.[(G257R)]; [(G257R)] | M; 20 mo; Fib | Sendai virus | RT-PCR; IF | EB differentiation | SNP microarray, G-banding and WGS; Yes | Yes | [99] |
| SURF1 | Leigh syndrome | C1_Mut (2 clones) | p.[(G257R)]; [(G257R)] | F; iPS-XM001 | CRISPR-Cas9 induced mutation | N/D | N/D | SNP microarray; Yes | Yes | [99] |
| TAZ | Barth syndrome | TAZ10 (2 clones) | p.[G197V];[0] | M; Fib | Lentivirus | Morphology; AP; RT-PCR; IF; hypomethylation | Teratoma; EB differentiation | N/D | N/D | [221] |
| TAZ | Barth syndrome | TAZ13 (3 clones) | c.[110-1G > C];[0] | M; Fib | Lentivirus | Morphology; AP; RT-PCR; IF; hypomethylation | Teratoma; EB differentiation | N/D | N/D | [221] |
| TAZ | Barth syndrome | TAZ15 (3 clones) | p.[R57L];[0] | M; Fib | Lentivirus | Morphology; AP; RT-PCR; IF; hypomethylation | Teratoma; EB differentiation | N/D | N/D | [221] |
| TAZ | Barth syndrome | BTH-H | p.[(D173Tfs*12)];[0] | M; Fib | Retrovirus | Morphology; RT-qPCR; IF | Teratoma | G-banding; N/D | N/D | [109] |
| TAZ | Barth syndrome | BTH-C | p.[(S110P)];[0] | M; Fib | Modified RNA | Morphology; RT-qPCR; IF | Teratoma | G-banding; N/D | N/D | [109] |
| TAZ | Barth syndrome | PGP1-TAZc.517delG | p.[(D173Tfs*12)];[0] | M; iPS-PGP1 | CRISPR-Cas9 induced mutation | Morphology; RT-qPCR; IF | Teratoma | G-banding; N/D | N/D | [109] |
| TAZ | Barth syndrome | PGP1-TAZc.517ins | p.[(D173Efs)];[0] | M; iPS-PGP1 | CRISPR-Cas9 induced mutation | Morphology; RT-qPCR; IF | Teratoma | G-banding; N/D | N/D | [109] |
| TAZ | Barth syndrome | WMUi002-A | p.[(D173Efs)];[0] | M; 6yr; urine cells | Sendai virus | qPCR; IF | EB differentiation | G-banding; Yes | Yes | [222] |
| TRNT1 | RP | P1 (4 clones) | p.[(E43del)]; [(S418Vfs)] | M; 19 yr; Fib | Sendai virus | Morphology; RT-PCR; IF | Taqman mRNA scorecard | G-banding; N/D | N/D | [223] |
| TRNT1 | RP | P2 (4 clones) | p.[(S418Kfs)]; c.[609-26T > C] | M; 21 yr; Fib | Sendai virus | Morphology; RT-PCR; IF | Taqman mRNA scorecard | G-banding; N/D | N/D | [223] |
| TRNT1 | RP | P3 (4 clones) | p.[(S418Kfs)]; c.[609-26T > C] | M; 18 yr; Fib | Sendai virus | Morphology; RT-PCR; IF | Taqman mRNA scorecard | G-banding; N/D | N/D | [223] |
| Gene(s) | Disease | Cell Line ID | Mutation | Cell Line Origin a | Reprogramming | Pluripotency | Trilineage Potential | Karyotype; Lineage Validation b | Heteroplasmy Before (After) Reprogramming c | Mycoplasma Check | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|---|
| MT-ATP6 | Leigh syndrome | iPSC (8993T/G) (8 clones) | m.8993T > G p.(L156R) | Fib | Sendai virus | Morphology; IF | Teratoma | G-banding; Yes | 52% (32–87%) | Yes | [65] |
| MT-ATP6 | Leigh syndrome | Leigh-iPSC (5 clones) | m.8993T > G p.(L156R) | Fib | Sendai virus | Morphology; IF | Teratoma | G-banding; Yes | 100% (100%) | Yes | [65] |
| MT-ATP6 | Leigh syndrome | LS1-hiPSC | m.8993T > G p.(L156R) | F; 3 yr; Fib | mRNA-miRNA combination | Morphology; RT-PCR; IF | Taqman mRNA scorecard; EB differentiation | N/D | 85% (≤80%) | N/D | [171] |
| MT-ATP6 | Leigh syndrome | L749.1 | m.8993T > G p.(L156R) | M; 8 mo; Fib | Retrovirus | Morphology; AP; RT-PCR; IF; Hypomethylation | EB differentiation | G-banding; Yes | 90% (N/D) | N/D | [224] |
| MT-ATP6 | Leigh syndrome | GM13411 (3 clones) | m.8993T > G p.(L156R) | M; 8 mo; Fib | Retrovirus | RT-PCR; IF | N/D | G-banding; N/D | 100% (100%) | N/D | [136] |
| MT-ATP6 | Leigh syndrome | TFA1 (3 clones) | m.9185T > C p.(L220P) | F; 80 yr; Fib | Episomal vectors | N/D | Teratoma; EB differentiation | G-banding; Yes | 100% (100%) | Yes | [98] |
| MT-ATP6 | Leigh syndrome | TDA2.3 | m.9185T > C p.(L220P) | F; 47 yr; Fib | Retrovirus | N/D | Teratoma; EB differentiation | G-banding; Yes | 100% (100%) | Yes | [98] |
| MT-ATP6 | Leigh syndrome | TDA3.1 | m.9185T > C p.(L220P) | F; 20 yr; Fib | Retrovirus | N/D | Teratoma; EB differentiation | G-banding; Yes | 100% (100%) | Yes | [98] |
| MT-ND1 | LHON | FINCBi001-A | m.3460G > A p.(A52T) | F; 21 yr; Fib | Sendai virus | Morphology; AP; RT-PCR; IF | EB differentiation | CGH-array; Yes | 100% (100%) | Yes | [225] |
| MT-ND4 | LHON | LHON V31-1 (3 clones) | m.11778G > C p.(R340P) d | M; 18 yr; Fib | Episomal vector | Morphology; IF | Teratoma; EB differentiation | N/D | 100% (N/D) | N/D | [226] |
| MT-ND4 | LHON | LHON T1-20 (3 clones) | m.11778G > C p.(R340P) d | M; 33 yr; Fib | Episomal vector | Morphology; IF | Teratoma; EB differentiation | N/D | 100% (N/D) | N/D | [226] |
| MT-ND4 | LHON | TVGH-iPSC-010 | m.11778G > A p.(R340H) | F; 61 yr; PBMC | Sendai virus | Morphology; RT-PCR; FACS | Teratoma; EB differentiation | G-banding; Yes | 100% (N/D) | Yes | [227] |
| MT-ND4 | LHON | LHON-affected | m.11778G > A p.(R340H) | PBMC | Sendai virus | AP; RT-PCR; IF | N/D | N/D | 100% (>98%) | N/D | [165] |
| MT-ND4 | LHON (unaffected) | LHON-carrier | m.11778G > A p.(R340H) | PBMC | Sendai virus | AP; RT-PCR; IF | N/D | N/D | 100% (>98%) | N/D | [165] |
| MT-ND4 | LHON | LHON-iPSC | m.11778G > A p.(R340H) | M; 18 yr; PBMC | Sendai virus | Morphology; RT-qPCR | N/D | N/D | N/D | N/D | [101] |
| MT-ND6 and MT-ND1 | ‘LHON plus’ | LHON Q1-4 (3 clones) | m.14484T > C p.(M64V) and m.4160T > C p.(L285P) | F; 30 yr; Fib | Episomal vector | Morphology; IF | Teratoma; EB differentiation | N/D | 100% (N/D) | N/D | [226] |
| MT-ND5 | MELAS | M-iPS (2 clones) | m.13513G > A p.(D393N) | Fib | Lentivirus | Morphology; RT-qPCR; IF | EB differentiation | N/D; Yes | 47% (50%) | N/D | [69] |
| MT-ND5 | Leigh syndrome | iPSC (13513G/A) (5 clones) | m.13513G > Ap.(D393N) | Fib | Sendai virus | Morphology; IF | Teratoma | G-banding; Yes | 84% (32–100%) | Yes | [65] |
| MT-ND5 | Leigh syndrome | LND554SV.3 | m.13513G > A p.(D393N) | M; Fib | Sendai virus | Morphology; AP; qPCR; IF; hypomethylation | EB differentiation | G-banding; Yes | 55% (32%) | N/D | [228] |
| MT-ND5 | MELAS | MELAS-iPSC A01 (2 clones) | m.13513G > A p.(D393N) | M; 16 yr; Fib | Episomal vectors | Morphology; RT-PCR; IF | EB differentiation | G-banding; N/D | 20% (>60%) | Yes | [81] |
| MT-RNR1 | SNHL | IBMSi004-A | m.1555A > G | F; 39 yr; PBMC | Sendai virus | RT-PCR; IF; FACS; | Teratoma; EB differentiation | G-banding; Yes | N/D (84.7%) | Yes | [229] |
| MT-RNR2e | HCM | HCM-iPSC | m.2336T > C e | Urine cells | Retrovirus | AP; qPCR; IF; hypomethylation | Teratoma; EB differentiation | G-banding; N/D | 100% (N/D) | Yes | [230] |
| MT-TK | MERRF | M1-iPSC | m.8344A > G | F; 15 yr; Fib | Retrovirus | Morphology; AP; RT-PCR; IF | Teratoma; EB differentiation | N/D | 90% (70%) | N/D | [231] |
| MT-TK | MERRF | M2-iPSC | m.8344A > G | F; 13 yr; Fib | Retrovirus | Morphology; AP; RT-PCR; IF | Teratoma; EB differentiation | N/D | 60% (60%) | N/D | [231] |
| MT-TK | MERRF | TVGH-iPSC-MRF-MHigh | m.8344A > G | F; 15 yr; Fib | Retrovirus | RT-PCR; IF | Teratoma; EB differentiation | G-banding; Yes | N/D (76%) | Yes | [232] |
| MT-TL1 | MIDD | Mt1 (2 clones) | m.3243A > G | M; 38 yr; Fib | Retrovirus | Morphology; AP; IF; hypomethylation | Teratoma; EB differentiation | G-banding; Yes | 18% (51% and 87%) | N/D | [233] |
| MT-TL1 | MIDD and MELAS | Mt2 (4 clones) | m.3243A > G | F; 46 yr; Fib | Retrovirus | Morphology; AP; IF; hypomethylation | Teratoma; EB differentiation | G-banding; Yes | 24% (69–83%) | N/D | [233] |
| MT-TL1 | MIDD | MH1, MH2 and MH3 (3 clones) | m.3243A > G | M; 39 yr; Fib | Retrovirus | Morphology; IF; RT-PCR | Teratoma | G-banding; Yes | 22% (>80%) | N/D | [21] |
| MT-TL1 | CM | P3-iPSC | m.3243A > G | F; 55 yr; Fib | Retrovirus | N/D | N/D | N/D; Yes | 35% (>80%) | N/D | [21] |
| MT-TL1 | MELAS | MELAS-iPSC (5 clones) | m.3243A > G | Fib | Sendai virus | Morphology; IF | Teratoma | G-banding; Yes | 29% (33–100%) | Yes | [65] |
| MT-TL1 | MELAS | Patient #1- iPSCs | m.3243A > G | Fib | Episomal plasmid | Morphology; IF | EB differentiation | N/D | ~99% (100%) | N/D | [90] |
| MT-TL1 | MELAS | Patient #2- iPSCs (3 clones) | m.3243A > G | Fib | Episomal plasmid | Morphology; IF | EB differentiation | N/D | ~69% (>70–100%) | N/D | [90] |
| MT-TL1 | MELAS | Patient #3- iPSCs (3 clones) | m.3243A > G | Fib | Episomal plasmid | Morphology; IF | EB differentiation | N/D | ~55% (60–100%) | N/D | [90] |
| MT-TL1 | MELAS | MELAS-iPSC (16 clones) | m.3243A > G | Fib | Retrovirus | Morphology; IF | Teratoma; EB differentiation | G-banding; N/D | 78% (40–99%) | N/D | [234] |
| MT-TL1 | MELAS | MiPSC5 | m.3243A > G | M; Fib | Sendai virus | Morphology; qPCR; IF | Teratoma | G-banding; Yes | 90% (>80%) | N/D | [80] |
| MT-TL1 | MELAS | MitoA hiPSCs (7 clones) | m.3243A > G | F; 17 yr; Fib | Sendai virus | N/D | N/D | G-banding; Yes | 17% (47–82%) | N/D | [87] |
| MT-TL1 | MELAS | MitoB hiPSCs (5 clones) | m.3243A > G | F; 79 yr; Fib | Sendai virus | N/D | N/D | G-banding; Yes | 45% (38–81%) | N/D | [87] |
| MT-TL1 | MELAS | MitoC hiPSCs (9 clones) | m.3243A > G | M; 31 yr; Fib | Sendai virus | N/D | N/D | G-banding; Yes | 47% (38–83%) | N/D | [87] |
| MT-TL1 | MELAS | HH1 | m.3243A > G | M; 42 yr; Fib | Retrovirus | qPCR; IF | N/D | G-banding; N/D | 85% (71%) | N/D | [97] |
| MT-TL1 | MELAS | P1 iPSCs (2 clones) | m.3243A > G | F; 73 yr; Fib | Sendai virus | IF; FACS | Spontaneous monolayer differentiation | SNP-array; Yes | ~70% (50% and 70% f) | N/D | [235] |
| MT-TW | MELAS | Patient (3 clones) | m.5541C > T g | M; <15 yr; Myoblast | Episomal plasmid | Morphology; IF | EB differentiation | N/D | ~100% (~100%) | Yes | [46] |
| MT-ND4/5; MT-TL2/TS2/TH | Pearson syndrome | PS-iPS (3 clones) | m.10949_13449del (2501bp deletion) | F; 3 yr; Fib | Retrovirus | Morphology; AP; RT-PCR;IF | Teratoma | N/D | ~70% (55–70%) | N/D | [45] |
| MT-ATP6; MT-CO3; MT-ND3/4/4L/5/6; MT-CYB; MT-TG/TR/TL2/ TS2/TH/TE | Pearson syndrome | GM04516PS-iPSC | m.8824_15854del (7031bp deletion) | F; 5 yr; Fib | Retrovirus | AP; IF | Teratoma | N/D | 6% (20%) | N/D | [45] |
| MT-CO3; MT-ND3/4/4L/5/6 | Pearson syndrome | FINCBi002-A | m.9449_14550del (5102bp deletion) | M; 5 mo; Fib | Sendai virus | Morphology; AP; RT-PCR; IF | EB differentiation | Microarray; Yes | 50% (80%) | Yes | [236] |
| MT-ATP6; MT-CO3; MT-ND3/4/4L/5 | Pearson syndrome | FINCBi003-A | m.8469_13460del (4992bp deletion) | M; 8 yr; Fib | Sendai virus | Morphology; AP; RT-PCR; IF | EB differentiation | Microarray; Yes | 10% (30%) | Yes | [236] |
| Disease (Gene) | Cell Line ID | Genetic Mutation | Isogenic Controls a | Cell Type | OXPHOS Defects b | Other Mitochondrial Defects b | Cellular and Physiological Defects b | In Vitro Therapeutic Studies | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| HSP (ATAD3A) | HEL142 | p.[(G355D)];[=] | − | Neurons | N/D | Mitochondrial network abnormalities | ↑ lysosome accumulation | N/D | [196] |
| MTDPS3 (DGUOK) | DGUOKΔ14/Δ5 iPSC | p.[W166*]; [H167Lfs*213] | + + | Hep, Hep- org. | ↓ maximal and basal OCR (3/3) ↓ ATP c ↓ MMP (3/3) ↑ lactate levels (3/3) | Abnormal mitochondrial ultrastructure (1/3) ↑ ROS levels (3/3) ↓ mtDNA depletion (3/3) | ↑ susceptibility to iron overload- induced ferroptosis (2/2)NCOA4-dependent ferritin degradation in lysosomes (2/2) | Drug screen on library of 2400 drugs; NAD treatment restored mitochondrial bioenergetics and improved expression of ETC genes. DFO, Fer-1 (ferroptosis inhibitors) and NAC rescued iron-overload induced ferroptosis. | [131,132] |
| Patient 1 | p.[F256*];[F256 *] | + − | |||||||
| Patient 2 | [p.A2S; c.591G>A]; [c.142+1G>A], | + − | |||||||
| DCMA (DNAJC19) | Patient 1 | c.[130-1G>C]; [130-1G>C] | − | CM | N/D | Fragmented mitochondrial network (2/2) | ↓ L-OPA1 isoform expression (2/2) | Elamipretide improved mitochondrial fragmentation and restored OPA1 isoform balance | [201] |
| Patient 2 | c.[130-1G>C]; [130-1G>C] | − | |||||||
| CMT2A (MFN2) | Patient 1 (2 clones) | p.[(R364W)];[=] | − − | SpMN | N/D | Impaired mitochondrial motility (2/2) Clustering of mitochondria around nucleus (1/1) ↑ mitophagy; ↓ mtDNA content (1/1) | Aberrant electrophysiological activity (hyperpolarised action potential, increased sodium channel density) (1/1) Altered gene expression (↑ OXPHOS; ↓ neurogenesis) (1/1) ↓ sensitivity to apoptosis (1/1) | N/D | [191,207] |
| CMT2A-1/2 | p.[(A383V)];[=] | − − | |||||||
| MSA-C (COQ2) | MSA_A (3 clones) | p.[(R387*)]; [(V393A)] | + + + − − | Neurons | ↓ maximal and basal OCR ↓ ATP d | ↓ CoQ10 (and vitamin E) levels ↑ ROS levels | ↑ apoptosis (in galactose-based media) | N/D | [198] |
| CoQ10 deficiency (COQ4) | CQ4-iPSC | p.[(E161D)];[=] | + − | SkMC | ↓ Complex I + III activity | ↑ ROS levels Mitochondrial morphology defects | Differentiation defect e | N/D | [200] |
| DOA and Parkinson’s disease (OPA1) | VO-iPSC | c.[(2496+1G>T)];[=] | − | NSC, NPC, RGC, and/or Neurons | ↓ maximal and basal OCR (4/5) ↓ ATP c,d (4/5) ↑ glycolysis (2/2) ↓ MMP (2/2) CI defect (↓ CI subunit expression, activity and/or CI related ATP synthesis) (4/4) | Fragmented mitochondrial network (4/5) ↑ ROS levels (3/3) | Differentiation defects e and/or increased neurodegeneration over prolonged culture (7/7) Reduced mitochondrial mass and loss of active synaptic terminals throughout prolonged culture (microfluidic neuronal Nigro-striatal pathway model) (1/1) | NAC, Z-VAD (apoptosis inhibitor), and Nec-1 (necrosis inhibitor) improved survival of neurons derived from NPCs. | [104,119,120,123,211] |
| OL-iPSC | c.[(2496+1G>T)];[=] | − | |||||||
| PD-OPA1 G488R (2 clones) | p.[G488R];[=] | + − | |||||||
| PD-OPA1 A495V #72 | p.[A495V[;[=] | + − | |||||||
| Opa1P (2 clones) | c.[(33-34ins9)];[=] | − − | |||||||
| Opa1 (2 clones) | c.[(33-34ins9)];[=] | − − | |||||||
| OPA1+/− hESC | N/D (haploinsufficiency) | + | |||||||
| CMTX6 (PDK3) | iPSCCMTX6 | p.[(R158H)];[0] | + | SpMN | ↓ ATP c,d | Fragmented mitochondrial network Mitochondrial trafficking defect | PDC E1 hyperphosphorylation | Treatment with the pan PDK inhibitor DCA reduced PDC E1 hyperphosphorylation, and improved mitochondrial fragmentation and motility. | [216] |
| Leigh-like syndrome (PMPCB) | DII-2 iPSC (3 clones) | p.[I422T];[I422T] | − − − | NESC | N/D | N/D | Inefficient processing and accumulation of intermediate Frataxin; increased ISD complex formation; increased Fe-S cluster biogenesis | N/D | [217] |
| Autosomal dominant POE and parkinsonism (POLG) | CSC-35 (3 clones) | p.[(Q811R)];[=] | − | MDNS | ↑ glycolysis | N/D | Accumulation of oligomeric αSYN Altered gene expression ↑ pigmentation in dopaminergic neurons | N/D | [190] |
| Autosomal recessive POE (POLG) | WS5A (3 clones) | p.[W748S]; [W748S] | − − − | NSC, and/or Neurons | ↓ ATP c (2/2) CI defect (↓ CI expression) (2/2) | ↑ ROS levels (2/2) mtDNA depletion (2/2) | N/D | NAC treatment on neurons reduced ROS levels and improved MMP. | [125,126] |
| CP2A (2 clones) | p.[A467T]; [W748S] | − − − | |||||||
| Alpers syndrome (POLG) | AHS iPS 1 | p.[A467T];c.[1251-2A>T] | − − | Hep | ↓ ATP c (2/2) | ↑ frequency of mPTP opening and superoxide release (2/2) Abnormal mitochondrial ultrastructure (2/2) ↑ numbers of mitochondria without mtDNA (2/2) | ↑ VA induced apoptosis (2/2) | CsA (mPTP specific inhibitor) reduced VA induced apoptosis. MnTMPyP and TEMPO (antioxidants) no effect. NAC and carnitine reduced VA induced apoptosis. | [129] |
| AHS iPS 2 | p.[A467T];c.[3626_3629dup] | − − | |||||||
| MTDPS8A/B (RRM2B) | RRM2B−/− iPSC | p.[(R36Sfs*55)]; [(R36Sfs*55)] | + | Hep | ↓ ATP c | mtDNA depletion | N/D | NAD treatment improved ATP levels. | [131] |
| CIV deficiency (SCO2) | SCO2G193S | p.[(G193S)]; [(G193S)] | − − − | CM | N/D | Abnormal mitochondrial ultrastructure (1/2) | Calcium homeostasis defects (2/2) Contractile defects (2/2) | Poor/no response to ionotropic agents. | [137] |
| SCO2E140K | p.[(E140K)]; c.[(17ins(19))] f | − − − | |||||||
| Leigh syndrome (SURF1) | SURF1_Mut: S1 | p.[(V177G)]; [(V177G)] | − − | NPC, Neurons, and/or Neural-org. | ↓ CIV activity (3/3) ↓ maximal and basal OCR (2/2) ↑ lactate levels (2/2) ↑ glycolysis (1/1) | N/D | Differentiation defects e (3/3) ↓ neurite length and branching (3/3) Aberrant action potentials kinetics (3/3) ↑ proliferation and pluripotency markers (1/1) Disrupted WNT, TGFß, and SHH signalling (1/1) | Antioxidants (NAC and AT3), glucose and pyruvate supplementation showed minimal improvement in NPC and neurons. Hypoxia negatively impacted neurite length and branching defects. 400uM bezafibrate treatment alleviated OXPHOS and maturation defects in iPSC-NPCs. | [99] |
| SURF1_Mut: S2 | p.[(G257R)]; [(G257R)] | + + | |||||||
| C1_Mut (2 clones) | p.[(G257R)]; [(G257R)] | + + | |||||||
| Barth syndrome (TAFAZZIN) | TAZ10 | p.[G197V];[0] | - | CM, Cardiac-org. | ↑ basal OCR (compensatory mechanism) (5/5) ↓ maximal OCR (5/5) ↓ ATP c in galactose culture (4/4) ↓ CII expression (1/1) | ↑ ROS levels (2/2) Cardiolipin remodelling defects (4/4) Mitochondrial network fragmentation (1/1) Disrupted OXPHOS supercomplex assembly (1/1) | Disrupted sarcomeric organization at single cell level (4/5) Contractile defects (4/4) Metabolic alterations and substrate utilization (1/1) Abnormal calcium handling (2/2); Increased diastolic calcium leak through RYR2 (1/1) ↓ HIF1α signalling under hypoxic conditions (1/1) | BL corrected cardiolipin ratio. Arg + Cys supplementation improved ATP levels. LA improved MLCL:CL ratio, increased ATP levels, restored basal OCR. LA and MitoTEMPO reduced ROS, and improved sarcomere organisation and contractile defects. | [103,109,110,113,237] |
| BTH-H | p.[(D173Tfs*12)];[0] | + − − − | |||||||
| BTH-C | p.[(S110P)];[0] | − − − | |||||||
| PGP1-TAZ c.517delG | p.[(D173Tfs*12)];[0] | + | |||||||
| PGP1-TAZ c.517ins | p.[(D173Efs)];[0] | + | |||||||
| RP (TRNT1) | P1 | p.[(E43del)]; [(S418Vfs)] | − − − − − − − | Retinal-org. | N/D | ↑ oxidative stress (3/3) | ↑ autophagy (accumulation of LC3-II; decreased LAMP1 expression) (3/3) | N/D | [223] |
| P2 | p.[(S418Kfs)]; c.[609-26T>C] | − − − − − − − | |||||||
| P3 | p.[(S418Kfs)]; c.[609-26T>C] | − − − − − − − |
| Disease (Gene) | Cell Line ID | Genetic Mutation a | Isogenic Controls b | Cell Type | OXPHOS Defects c | Other Mitochondrial Defects c | Cellular and Physiological Defects c | In Vitro Therapeutic Studies | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| Leigh syndrome (MT-ATP6) | TFA1 | m.9185T > C (100%; p.L220P) | − − − − − | NPC, Neurons | ↓ ATP d,f (4/4) ↑ MMP (4/4) ↓ basal OCR (1/1) | ↑ ROS (1/1) | Calcium homeostasis defect (↓ calcium-induced calcium release; (↓ mitochondrial calcium release) (3/3) ↑ neurodegeneration (1/1) | Tested 130 FDA approved drugs; Avanafil (PDE5 inhibitor) partially rescued calcium homeostasis defect in NPCs and neuronsRapamycin treatment improved ATP production, decreased aberrant AMPK activation, and decreased glutamate induced toxicity | [98,136] |
| TDA2.3 | m.9185T > C (100%; p.L220P) | − − − − − | |||||||
| TDA3.1 | m.9185T > C (100%; p.L220P) | − − − − − | |||||||
| GM13411(3 clones) | m.8993T > G (100%; p.L156R) | − − | |||||||
| Leigh syndrome (MT-ND5) | LND554SV.3 | m.13513G > A (19%; p.D393N) | − | NSC, Neurons | ↓ basal and maximal OCR | N/D | ↑ cell death Calcium homeostasis defect (↓ calcium buffering capacity) | Treatment with the succinate prodrug NV241 improved mitochondrial respiration; however, similar observations were detected from DMSO control | [102] |
| Leigh syndrome (MT-ATP6) | Leigh iPSC | m.8993T > G (100% i; p.L156R) | + | SkMC | ↓ ATP e | N/D | N/D | N/D | [65] |
| LHON (MT-ND4) | LHON-affected | m.11778G > A (98.25%; p.R340H) | − − | RGC | ↓ basal OCR (2/2) | ↑ mtDNA copy number ↑ ROS (2/2) | Differentiation defect g (2/3) Aberrant electrophysiological activity (↓ action potential peaks) (1/1) Defect in glutamate uptake (1/1) ↑ apoptosis (1/2) Changes in mitochondrial transport; reduced KIF5A expression (1/2) | NAC reduced ROS levels and apoptosis, and restored KIF5A expression and mitochondrial motility | [101,165,166] |
| LHON-unaffected | m.11778G > A (98.42%; p.R340H) | − | |||||||
| LHON-iPSC | m.11778G > A (N/D; p.R340H) | − | |||||||
| LHON (MT-ND1 and MT-ND6) | LHON Q1-4 (3 clones) | m.4160T > C (p.L285P)/ m.14484T > C (p.M64V) (100% i) | + − | RGC | ↑ ROS | ↑ apoptosis | N/D | [64] | |
| HCM (MT-RNR2) h | HCM-iPSC | m.2336T > C (N/D) | − − − | CM | ↓ MMP | Mitochondrial ultrastructural defects ↑ mtDNA copy number ↓ stability of 16s rRNA | Calcium homeostasis defects Aberrant electrophysiological activity | N/D | [230] |
| MERRF (MT-TK) | M1-iPSC | m.8344A > G (≥40%) | + | CM, NPC | ↓ basal and maximal OCR (2/2) ↓ ATP e (2/2) | ↑ ROS (2/2) Mitochondrial fragmentation/morphology defects (2/2) | ↑ expression of antioxidant genes catalase and CuZnSOD (2/2) | N/D | [231] |
| M2-iPSC | m.8344A > G (>40%) | − | |||||||
| MELAS (MT-TL1) | P1 iPSCs (2 clones j) | m.3243A > G (>55%) | − − − | RPE | N/D | Features of mitochondrial fragmentation/morphology defects | Atypical spatial distribution of RPE Underdeveloped microvilliAberrant melanosome morphology ↓ phagocytosis of POS | N/D | [235] |
| MELAS (MT-TL1) | MiPSC5 | m.3243A > G (~80%) | + − | EC | N/D | ↑ mitochondrial biogenesis ↑ ROS | Differentiation defects g ↓ cell migration and tube formation ↑ apoptosis ↑ uptake and oxidation of LDL ↑ expression of VCAM-1 isoform b ↑ monocyte adhesion to EC | Edaravone (anti-oxidant), CoQ10 and Vit C improved endothelial tube formation and reduced ROS levels. Edaravone also reduced basal inflammation | [48] |
| MIDD and MELAS (MT-TL1) | HH1 | m.3243A > G (>60%) | + − | Neurons, NPC, Spinal- organoid | ↓ basal and maximal OCR (3/3) ↓ ATP e (3/3) ↑ glycolysis (1/1) CI deficiency (1/1) | ↓ mitochondrial content along axons (1/1) Changes in mitochondrial translation (1/1) | Differentiation defectsg (1/1) Structural/morphological defects (2/2) ↓ synaptic density (1/1) ↓ frequency of spontaneous excitatory activity at single cell level (1/1) ↓ spontaneous MFR and NBR at a network level (3/3) ↑ PRS outside network bursts (3/3) Selective accumulation of CI in autophagosomes and clearance by mitophagy upon neuronal differentiation (1/1) Hyperactive Notch signalling (1/1) | DAPT (Notch signalling inhibitor) improved neurodevelopmental defects (improved motor neuron differentiation efficiency and spinal organoid neurite outgrowth defects) | [21,65,80,97,168] |
| MitoA hiPSCs (HH2) | m.3243A > G (>60% i) | + − | |||||||
| MitoB hiPSCs (HH3) | m.3243A > G (65%) | + − | |||||||
| MELAS-iPSC (2 clones) | m.3243A > G (100% h) | + | |||||||
| MH1, MH2 and MH3 (3 clones) | m.3243A > G (>80%) | + − − | |||||||
| MiPSC5 | m.3243A > G (80%) | + − | |||||||
| Pearson syndrome (2501bp macro-deletion) | PS-iPS | m.10949_13449del (50% i) | + + | HPC | N/D | N/D | Accumulation of iron granules in erythroid precursors (i.e., increased formation of sideroblasts) | N/D | [45] |
References
- McBride, H.M.; Neuspiel, M.; Wasiak, S. Mitochondria: More than just a powerhouse. Curr. Biol. 2006, 16, R551–R560. [Google Scholar] [CrossRef]
- Krabbendam, I.E.; Honrath, B.; Culmsee, C.; Dolga, A.M. Mitochondrial Ca2+-activated K+ channels and their role in cell life and death pathways. Cell Calcium 2018, 69, 101–111. [Google Scholar] [CrossRef] [PubMed]
- Russell, O.M.; Gorman, G.S.; Lightowlers, R.N.; Turnbull, D.M. Mitochondrial diseases: Hope for the future. Cell 2020, 181, 168–188. [Google Scholar] [CrossRef]
- Calvo, S.E.; Clauser, K.R.; Mootha, V.K. MitoCarta2.0: An updated inventory of mammalian mitochondrial proteins. Nucleic Acids Res. 2016, 44, D1251–D1257. [Google Scholar] [CrossRef] [PubMed]
- Chinnery, P.F.; Hudson, G. Mitochondrial genetics. Br. Med. Bull. 2013, 106, 135–159. [Google Scholar] [CrossRef] [PubMed]
- Rius, R.; Cowley, M.J.; Riley, L.; Puttick, C.; Thorburn, D.R.; Christodoulou, J. Biparental inheritance of mitochondrial DNA in humans is not a common phenomenon. Genet. Med. 2019, 21, 2823–2826. [Google Scholar] [CrossRef] [PubMed]
- Shmookler Reis, R.J.; Goldstein, S. Mitochondrial DNA in mortal and immortal human cells. Genome number, integrity, and methylation. J. Biol. Chem. 1983, 258, 9078–9085. [Google Scholar] [CrossRef]
- Wai, T.; Ao, A.; Zhang, X.; Cyr, D.; Dufort, D.; Shoubridge, E.A. The role of mitochondrial DNA copy number in mammalian fertility. Biol. Reprod. 2010, 83, 52–62. [Google Scholar] [CrossRef] [PubMed]
- D’Erchia, A.M.; Atlante, A.; Gadaleta, G.; Pavesi, G.; Chiara, M.; De Virgilio, C.; Manzari, C.; Mastropasqua, F.; Prazzoli, G.M.; Picardi, E.; et al. Tissue-specific mtDNA abundance from exome data and its correlation with mitochondrial transcription, mass and respiratory activity. Mitochondrion 2015, 20, 13–21. [Google Scholar] [CrossRef]
- Wachsmuth, M.; Hubner, A.; Li, M.; Madea, B.; Stoneking, M. Age-related and heteroplasmy-related variation in human mtDNA copy number. PLoS Genet. 2016, 12, e1005939. [Google Scholar] [CrossRef]
- Wallace, D.C.; Chalkia, D. Mitochondrial DNA genetics and the heteroplasmy conundrum in evolution and disease. Cold Spring Harb. Perspect. Biol. 2013, 5, a021220. [Google Scholar] [CrossRef] [PubMed]
- Ryzhkova, A.I.; Sazonova, M.A.; Sinyov, V.V.; Galitsyna, E.V.; Chicheva, M.M.; Melnichenko, A.A.; Grechko, A.V.; Postnov, A.Y.; Orekhov, A.N.; Shkurat, T.P. Mitochondrial diseases caused by mtDNA mutations: A mini-review. Ther. Clin. Risk Manag. 2018, 14, 1933–1942. [Google Scholar] [CrossRef]
- Floros, V.I.; Pyle, A.; Dietmann, S.; Wei, W.; Tang, W.C.W.; Irie, N.; Payne, B.; Capalbo, A.; Noli, L.; Coxhead, J.; et al. Segregation of mitochondrial DNA heteroplasmy through a developmental genetic bottleneck in human embryos. Nat. Cell Biol. 2018, 20, 144–151. [Google Scholar] [CrossRef] [PubMed]
- Wonnapinij, P.; Chinnery, P.F.; Samuels, D.C. The distribution of mitochondrial DNA heteroplasmy due to random genetic drift. Am. J. Hum. Genet. 2008, 83, 582–593. [Google Scholar] [CrossRef] [PubMed]
- Papa, S.; Martino, P.L.; Capitanio, G.; Gaballo, A.; De Rasmo, D.; Signorile, A.; Petruzzella, V. The Oxidative Phosphorylation System in Mammalian Mitochondria; Springer: Amsterdam, The Netherlands, 2012; pp. 3–37. [Google Scholar]
- Hock, D.H.; Robinson, D.R.L.; Stroud, D.A. Blackout in the powerhouse: Clinical phenotypes associated with defects in the assembly of OXPHOS complexes and the mitoribosome. Biochem. J. 2020, 477, 4085–4132. [Google Scholar] [CrossRef]
- Jackson, T.D.; Palmer, C.S.; Stojanovski, D. Mitochondrial diseases caused by dysfunctional mitochondrial protein import. Biochem. Soc. Trans. 2018, 46, 1225–1238. [Google Scholar] [CrossRef]
- Pfanner, N.; Warscheid, B.; Wiedemann, N. Mitochondrial proteins: From biogenesis to functional networks. Nat. Rev. Mol. Cell Biol. 2019, 20, 267–284. [Google Scholar] [CrossRef]
- Frazier, A.E.; Thorburn, D.R.; Compton, A.G. Mitochondrial energy generation disorders: Genes, mechanisms, and clues to pathology. J. Biol. Chem. 2019, 294, 5386–5395. [Google Scholar] [CrossRef]
- Stenton, S.L.; Prokisch, H. Genetics of mitochondrial diseases: Identifying mutations to help diagnosis. EBioMedicine 2020, 56, 102784. [Google Scholar] [CrossRef]
- Hamalainen, R.H.; Manninen, T.; Koivumaki, H.; Kislin, M.; Otonkoski, T.; Suomalainen, A. Tissue- and cell-type-specific manifestations of heteroplasmic mtDNA 3243A>G mutation in human induced pluripotent stem cell-derived disease model. Proc. Natl. Acad. Sci. USA 2013, 110, E3622–E3630. [Google Scholar] [CrossRef]
- Komen, J.C.; Thorburn, D.R. Turn up the power—Pharmacological activation of mitochondrial biogenesis in mouse models. Br. J. Pharmacol. 2014, 171, 1818–1836. [Google Scholar] [CrossRef]
- Quadalti, C.; Brunetti, D.; Lagutina, I.; Duchi, R.; Perota, A.; Lazzari, G.; Cerutti, R.; Di Meo, I.; Johnson, M.; Bottani, E.; et al. SURF1 knockout cloned pigs: Early onset of a severe lethal phenotype. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 2131–2142. [Google Scholar] [CrossRef]
- Deepa, S.S.; Pharaoh, G.; Kinter, M.; Diaz, V.; Fok, W.C.; Riddle, K.; Pulliam, D.; Hill, S.; Fischer, K.E.; Soto, V.; et al. Lifelong reduction in complex IV induces tissue-specific metabolic effects but does not reduce lifespan or healthspan in mice. Aging Cell 2018, 17, e12769. [Google Scholar] [CrossRef]
- Dell’agnello, C.; Leo, S.; Agostino, A.; Szabadkai, G.; Tiveron, C.; Zulian, A.; Prelle, A.; Roubertoux, P.; Rizzuto, R.; Zeviani, M. Increased longevity and refractoriness to Ca2+-dependent neurodegeneration in Surf1 knockout mice. Hum. Mol. Genet. 2007, 16, 431–444. [Google Scholar] [CrossRef]
- Ruzzenente, B.; Rotig, A.; Metodiev, M.D. Mouse models for mitochondrial diseases. Hum. Mol. Genet. 2016, 25, R115–R122. [Google Scholar] [CrossRef] [PubMed]
- Pfeffer, G.; Majamaa, K.; Turnbull, D.M.; Thorburn, D.; Chinnery, P.F. Treatment for mitochondrial disorders. Cochrane Database Syst. Rev. 2012, CD004426. [Google Scholar] [CrossRef]
- Liufu, T.; Wang, Z. Treatment for mitochondrial diseases. Rev. Neurosci. 2020, 32, 35–47. [Google Scholar] [CrossRef] [PubMed]
- Parikh, S.; Goldstein, A.; Karaa, A.; Koenig, M.K.; Anselm, I.; Brunel-Guitton, C.; Christodoulou, J.; Cohen, B.H.; Dimmock, D.; Enns, G.M.; et al. Patient care standards for primary mitochondrial disease: A consensus statement from the Mitochondrial Medicine Society. Genet. Med. 2017, 19, 1380. [Google Scholar] [CrossRef] [PubMed]
- Zakrzewski, W.; Dobrzynski, M.; Szymonowicz, M.; Rybak, Z. Stem cells: Past, present, and future. Stem Cell Res. Ther. 2019, 10, 68. [Google Scholar] [CrossRef]
- Thomson, J.A.; Itskovitz-Eldor, J.; Shapiro, S.S.; Waknitz, M.A.; Swiergiel, J.J.; Marshall, V.S.; Jones, J.M. Embryonic stem cell lines derived from human blastocysts. Science 1998, 282, 1145–1147. [Google Scholar] [CrossRef]
- Takahashi, K.; Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [PubMed]
- Narsinh, K.H.; Plews, J.; Wu, J.C. Comparison of human induced pluripotent and embryonic stem cells: Fraternal or identical twins? Mol. Ther. 2011, 19, 635–638. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, E.F.; Tzanakakis, E.S. Who will win: Induced pluripotent stem cells versus embryonic stem cells for beta cell replacement and diabetes disease modeling? Curr. Diab. Rep. 2018, 18, 133. [Google Scholar] [CrossRef] [PubMed]
- KalantarMotamedi, Y.; Peymani, M.; Baharvand, H.; Nasr-Esfahani, M.H.; Bender, A. Systematic selection of small molecules to promote differentiation of embryonic stem cells and experimental validation for generating cardiomyocytes. Cell Death Discov. 2016, 2, 16007. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Pak, C.; Han, Y.; Ahlenius, H.; Zhang, Z.; Chanda, S.; Marro, S.; Patzke, C.; Acuna, C.; Covy, J.; et al. Rapid single-step induction of functional neurons from human pluripotent stem cells. Neuron 2013, 78, 785–798. [Google Scholar] [CrossRef]
- Hofer, M.; Lutolf, M.P. Engineering organoids. Nat. Rev. Mater. 2021, 6, 402–420. [Google Scholar] [CrossRef]
- Worsdorfer, P.; Takashi, I.; Asahina, I.; Sumita, Y.; Ergun, S. Do not keep it simple: Recent advances in the generation of complex organoids. J. Neural Transm. 2020, 127, 1569–1577. [Google Scholar] [CrossRef]
- Lees, J.G.; Gardner, D.K.; Harvey, A.J. Pluripotent stem cell metabolism and mitochondria: Beyond ATP. Stem Cells Int. 2017, 2017, 2874283. [Google Scholar] [CrossRef]
- O’Brien, L.C.; Keeney, P.M.; Bennett, J.P., Jr. Differentiation of human neural stem cells into motor neurons stimulates mitochondrial biogenesis and decreases glycolytic flux. Stem Cells Dev. 2015, 24, 1984–1994. [Google Scholar] [CrossRef]
- Wanet, A.; Remacle, N.; Najar, M.; Sokal, E.; Arnould, T.; Najimi, M.; Renard, P. Mitochondrial remodeling in hepatic differentiation and dedifferentiation. Int. J. Biochem. Cell Biol. 2014, 54, 174–185. [Google Scholar] [CrossRef]
- Theeuwes, W.F.; Gosker, H.R.; Langen, R.C.J.; Pansters, N.A.M.; Schols, A.; Remels, A.H.V. Inactivation of glycogen synthase kinase 3beta (GSK-3beta) enhances mitochondrial biogenesis during myogenesis. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 2913–2926. [Google Scholar] [CrossRef] [PubMed]
- Venkatesh, S.; Baljinnyam, E.; Tong, M.; Kashihara, T.; Yan, L.; Liu, T.; Li, H.; Xie, L.H.; Nakamura, M.; Oka, S.I.; et al. Proteomic analysis of mitochondrial biogenesis in cardiomyocytes differentiated from human induced pluripotent stem cells. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2021, 320, R547–R562. [Google Scholar] [CrossRef] [PubMed]
- Keller, A.; Dziedzicka, D.; Zambelli, F.; Markouli, C.; Sermon, K.; Spits, C.; Geens, M. Genetic and epigenetic factors which modulate differentiation propensity in human pluripotent stem cells. Hum. Reprod. Update 2018, 24, 162–175. [Google Scholar] [CrossRef]
- Cherry, A.B.; Gagne, K.E.; McLoughlin, E.M.; Baccei, A.; Gorman, B.; Hartung, O.; Miller, J.D.; Zhang, J.; Zon, R.L.; Ince, T.A.; et al. Induced pluripotent stem cells with a mitochondrial DNA deletion. Stem Cells 2013, 31, 1287–1297. [Google Scholar] [CrossRef] [PubMed]
- Hatakeyama, H.; Katayama, A.; Komaki, H.; Nishino, I.; Goto, Y. Molecular pathomechanisms and cell-type-specific disease phenotypes of MELAS caused by mutant mitochondrial tRNA(Trp). Acta Neuropathol. Commun. 2015, 3, 52. [Google Scholar] [CrossRef]
- Wanet, A.; Arnould, T.; Najimi, M.; Renard, P. Connecting mitochondria, metabolism, and stem cell fate. Stem Cells Dev. 2015, 24, 1957–1971. [Google Scholar] [CrossRef]
- Pek, N.M.Q.; Phua, Q.H.; Ho, B.X.; Pang, J.K.S.; Hor, J.H.; An, O.; Yang, H.H.; Yu, Y.; Fan, Y.; Ng, S.Y.; et al. Mitochondrial 3243A > G mutation confers pro-atherogenic and pro-inflammatory properties in MELAS iPS derived endothelial cells. Cell Death Dis. 2019, 10, 802. [Google Scholar] [CrossRef]
- Adak, S.; Magdalene, D.; Deshmukh, S.; Das, D.; Jaganathan, B.G. A review on mesenchymal stem cells for treatment of retinal diseases. Stem Cell Rev. Rep. 2021. [Google Scholar] [CrossRef]
- Lester Sequiera, G.; Srivastava, A.; Alagarsamy, K.N.; Rockman-Greenberg, C.; Dhingra, S. Generation and evaluation of isogenic iPSC as a source of cell replacement therapies in patients with Kearns Sayre syndrome. Cells 2021, 10, 568. [Google Scholar] [CrossRef]
- Abdelalim, E.M. Modeling different types of diabetes using human pluripotent stem cells. Cell. Mol. Life Sci. 2021, 78, 2459–2483. [Google Scholar] [CrossRef]
- Zhang, H.; Menzies, K.J.; Auwerx, J. The role of mitochondria in stem cell fate and aging. Development 2018, 145, dev143420. [Google Scholar] [CrossRef] [PubMed]
- Weykopf, B.; Haupt, S.; Jungverdorben, J.; Flitsch, L.J.; Hebisch, M.; Liu, G.H.; Suzuki, K.; Belmonte, J.C.I.; Peitz, M.; Blaess, S.; et al. Induced pluripotent stem cell-based modeling of mutant LRRK2-associated Parkinson’s disease. Eur. J. Neurosci. 2019, 49, 561–589. [Google Scholar] [CrossRef] [PubMed]
- Shaltouki, A.; Sivapatham, R.; Pei, Y.; Gerencser, A.A.; Momcilovic, O.; Rao, M.S.; Zeng, X. Mitochondrial alterations by PARKIN in dopaminergic neurons using PARK2 patient-specific and PARK2 knockout isogenic iPSC lines. Stem Cell Rep. 2015, 4, 847–859. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Marquez, A.; Alonso-Barroso, E.; Cerro-Tello, G.; Bravo-Alonso, I.; Arribas-Carreira, L.; Briso-Montiano, A.; Navarrete, R.; Perez-Cerda, C.; Ugarte, M.; Perez, B.; et al. Generation and characterization of a human iPSC line (UAMi004-A) from a patient with propionic acidemia due to defects in the PCCB gene. Stem Cell Res. 2019, 38, 101469. [Google Scholar] [CrossRef] [PubMed]
- Alonso-Barroso, E.; Brasil, S.; Briso-Montiano, A.; Navarrete, R.; Perez-Cerda, C.; Ugarte, M.; Perez, B.; Desviat, L.R.; Richard, E. Generation and characterization of a human iPSC line from a patient with propionic acidemia due to defects in the PCCA gene. Stem Cell Res. 2017, 23, 173–177. [Google Scholar] [CrossRef]
- Knottnerus, S.J.G.; Mengarelli, I.; Wust, R.C.I.; Baartscheer, A.; Bleeker, J.C.; Coronel, R.; Ferdinandusse, S.; Guan, K.; IJlst, L.; Li, W.; et al. Electrophysiological abnormalities in VLCAD deficient hiPSC-cardiomyocytes can be improved by lowering accumulation of fatty acid oxidation intermediates. Int. J. Mol. Sci. 2020, 21, 2589. [Google Scholar] [CrossRef]
- Berry, B.J.; Smith, A.S.T.; Young, J.E.; Mack, D.L. Advances and current challenges associated with the use of human induced pluripotent stem cells in modeling neurodegenerative disease. Cells Tissues Organs 2018, 205, 331–349. [Google Scholar] [CrossRef]
- Shi, Y.; Inoue, H.; Wu, J.C.; Yamanaka, S. Induced pluripotent stem cell technology: A decade of progress. Nat. Rev. Drug Discov. 2017, 16, 115–130. [Google Scholar] [CrossRef]
- Martufi, M.; Good, R.B.; Rapiteanu, R.; Schmidt, T.; Patili, E.; Tvermosegaard, K.; New, M.; Nanthakumar, C.B.; Betts, J.; Blanchard, A.D.; et al. Single-step, high-efficiency CRISPR-Cas9 genome editing in primary human disease-derived fibroblasts. CRISPR J. 2019, 2, 31–40. [Google Scholar] [CrossRef]
- Grobarczyk, B.; Franco, B.; Hanon, K.; Malgrange, B. Generation of isogenic human iPS cell line precisely corrected by genome editing using the CRISPR/Cas9 system. Stem Cell Rev. Rep. 2015, 11, 774–787. [Google Scholar] [CrossRef]
- Howden, S.E.; Maufort, J.P.; Duffin, B.M.; Elefanty, A.G.; Stanley, E.G.; Thomson, J.A. Simultaneous reprogramming and gene correction of patient fibroblasts. Stem Cell Rep. 2015, 5, 1109–1118. [Google Scholar] [CrossRef] [PubMed]
- Wilkins, H.M.; Carl, S.M.; Swerdlow, R.H. Cytoplasmic hybrid (cybrid) cell lines as a practical model for mitochondriopathies. Redox Biol. 2014, 2, 619–631. [Google Scholar] [CrossRef] [PubMed]
- Wong, R.C.B.; Lim, S.Y.; Hung, S.S.C.; Jackson, S.; Khan, S.; Van Bergen, N.J.; De Smit, E.; Liang, H.H.; Kearns, L.S.; Clarke, L.; et al. Mitochondrial replacement in an iPSC model of Leber’s hereditary optic neuropathy. Aging 2017, 9, 1341–1350. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.; Folmes, C.D.; Wu, J.; Morey, R.; Mora-Castilla, S.; Ocampo, A.; Ma, L.; Poulton, J.; Wang, X.; Ahmed, R.; et al. Metabolic rescue in pluripotent cells from patients with mtDNA disease. Nature 2015, 524, 234–238. [Google Scholar] [CrossRef]
- Prigione, A.; Lichtner, B.; Kuhl, H.; Struys, E.A.; Wamelink, M.; Lehrach, H.; Ralser, M.; Timmermann, B.; Adjaye, J. Human induced pluripotent stem cells harbor homoplasmic and heteroplasmic mitochondrial DNA mutations while maintaining human embryonic stem cell-like metabolic reprogramming. Stem Cells 2011, 29, 1338–1348. [Google Scholar] [CrossRef]
- Takahashi, K.; Okita, K.; Nakagawa, M.; Yamanaka, S. Induction of pluripotent stem cells from fibroblast cultures. Nat. Protoc. 2007, 2, 3081–3089. [Google Scholar] [CrossRef] [PubMed]
- Al Abbar, A.; Ngai, S.C.; Nograles, N.; Alhaji, S.Y.; Abdullah, S. Induced pluripotent stem cells: Reprogramming platforms and applications in cell replacement therapy. Biores. Open Access 2020, 9, 121–136. [Google Scholar] [CrossRef]
- Folmes, C.D.; Martinez-Fernandez, A.; Perales-Clemente, E.; Li, X.; McDonald, A.; Oglesbee, D.; Hrstka, S.C.; Perez-Terzic, C.; Terzic, A.; Nelson, T.J. Disease-causing mitochondrial heteroplasmy segregated within induced pluripotent stem cell clones derived from a patient with MELAS. Stem Cells 2013, 31, 1298–1308. [Google Scholar] [CrossRef]
- Mikkelsen, T.S.; Hanna, J.; Zhang, X.; Ku, M.; Wernig, M.; Schorderet, P.; Bernstein, B.E.; Jaenisch, R.; Lander, E.S.; Meissner, A. Dissecting direct reprogramming through integrative genomic analysis. Nature 2008, 454, 49–55. [Google Scholar] [CrossRef]
- Fusaki, N.; Ban, H.; Nishiyama, A.; Saeki, K.; Hasegawa, M. Efficient induction of transgene-free human pluripotent stem cells using a vector based on Sendai virus, an RNA virus that does not integrate into the host genome. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2009, 85, 348–362. [Google Scholar] [CrossRef]
- Yu, J.; Hu, K.; Smuga-Otto, K.; Tian, S.; Stewart, R.; Slukvin, I.I.; Thomson, J.A. Human induced pluripotent stem cells free of vector and transgene sequences. Science 2009, 324, 797–801. [Google Scholar] [CrossRef]
- Kogut, I.; McCarthy, S.M.; Pavlova, M.; Astling, D.P.; Chen, X.; Jakimenko, A.; Jones, K.L.; Getahun, A.; Cambier, J.C.; Pasmooij, A.M.G.; et al. High-efficiency RNA-based reprogramming of human primary fibroblasts. Nat. Commun. 2018, 9, 745. [Google Scholar] [CrossRef]
- Wang, A.Y.L.; Loh, C.Y.Y. Episomal induced pluripotent stem cells: Functional and potential therapeutic applications. Cell Transplant. 2019, 28, 112S–131S. [Google Scholar] [CrossRef]
- Yaffe, M.P.; Noggle, S.A.; Solomon, S.L. Raising the standards of stem cell line quality. Nat. Cell Biol. 2016, 18, 236–237. [Google Scholar] [CrossRef]
- Valenti, M.T.; Serena, M.; Carbonare, L.D.; Zipeto, D. CRISPR/Cas system: An emerging technology in stem cell research. World J. Stem Cells 2019, 11, 937–956. [Google Scholar] [CrossRef] [PubMed]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef]
- Gammage, P.A.; Moraes, C.T.; Minczuk, M. Mitochondrial genome engineering: The revolution may not be CRISPR-Ized. Trends Genet. 2018, 34, 101–110. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, M.; Bacman, S.R.; Peralta, S.; Falk, M.J.; Chomyn, A.; Chan, D.C.; Williams, S.L.; Moraes, C.T. MitoTALEN: A general approach to reduce mutant mtDNA loads and restore oxidative phosphorylation function in mitochondrial diseases. Mol. Ther. 2015, 23, 1592–1599. [Google Scholar] [CrossRef]
- Yang, Y.; Wu, H.; Kang, X.; Liang, Y.; Lan, T.; Li, T.; Tan, T.; Peng, J.; Zhang, Q.; An, G.; et al. Targeted elimination of mutant mitochondrial DNA in MELAS-iPSCs by mitoTALENs. Protein Cell 2018, 9, 283–297. [Google Scholar] [CrossRef] [PubMed]
- Yahata, N.; Matsumoto, Y.; Omi, M.; Yamamoto, N.; Hata, R. TALEN-mediated shift of mitochondrial DNA heteroplasmy in MELAS-iPSCs with m.13513G>A mutation. Sci. Rep. 2017, 7, 15557. [Google Scholar] [CrossRef] [PubMed]
- Bacman, S.R.; Williams, S.L.; Pinto, M.; Moraes, C.T. The use of mitochondria-targeted endonucleases to manipulate mtDNA. Methods Enzymol. 2014, 547, 373–397. [Google Scholar] [CrossRef] [PubMed]
- Mok, B.Y.; de Moraes, M.H.; Zeng, J.; Bosch, D.E.; Kotrys, A.V.; Raguram, A.; Hsu, F.; Radey, M.C.; Peterson, S.B.; Mootha, V.K.; et al. A bacterial cytidine deaminase toxin enables CRISPR-free mitochondrial base editing. Nature 2020, 583, 631–637. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Li, C.; Zheng, K.; Zhao, X.; Xu, X.; Yang, A.; Yi, M.; Tao, H.; Xie, B.; Qiu, M.; et al. Chromosomal aberration arises during somatic reprogramming to pluripotent stem cells. Cell Div. 2020, 15, 12. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, S.; Stacey, G.N.; Akazawa, C.; Aoyama, N.; Baptista, R.; Bedford, P.; Bennaceur Griscelli, A.; Chandra, A.; Elwood, N.; Girard, M.; et al. Quality control guidelines for clinical-grade human induced pluripotent stem cell lines. Regen. Med. 2018, 13, 859–866. [Google Scholar] [CrossRef] [PubMed]
- Popp, B.; Krumbiegel, M.; Grosch, J.; Sommer, A.; Uebe, S.; Kohl, Z.; Plotz, S.; Farrell, M.; Trautmann, U.; Kraus, C.; et al. Need for high-resolution genetic analysis in iPSC: Results and lessons from the ForIPS consortium. Sci. Rep. 2018, 8, 17201. [Google Scholar] [CrossRef]
- Perales-Clemente, E.; Cook, A.N.; Evans, J.M.; Roellinger, S.; Secreto, F.; Emmanuele, V.; Oglesbee, D.; Mootha, V.K.; Hirano, M.; Schon, E.A.; et al. Natural underlying mtDNA heteroplasmy as a potential source of intra-person hiPSC variability. EMBO J. 2016, 35, 1979–1990. [Google Scholar] [CrossRef] [PubMed]
- Yokota, M.; Hatakeyama, H.; Ono, Y.; Kanazawa, M.; Goto, Y.I. Mitochondrial respiratory dysfunction disturbs neuronal and cardiac lineage commitment of human iPSCs. Cell Death Dis. 2017, 8, e2551. [Google Scholar] [CrossRef]
- Sercel, A.J.; Carlson, N.M.; Patananan, A.N.; Teitell, M.A. Mitochondrial DNA dynamics in reprogramming to pluripotency. Trends Cell Biol. 2021, 31, 311–323. [Google Scholar] [CrossRef]
- Yokota, M.; Hatakeyama, H.; Okabe, S.; Ono, Y.; Goto, Y. Mitochondrial respiratory dysfunction caused by a heteroplasmic mitochondrial DNA mutation blocks cellular reprogramming. Hum. Mol. Genet. 2015, 24, 4698–4709. [Google Scholar] [CrossRef]
- Kosanke, M.; Davenport, C.; Szepes, M.; Wiehlmann, L.; Kohrn, T.; Dorda, M.; Gruber, J.; Menge, K.; Sievert, M.; Melchert, A.; et al. iPSC culture expansion selects against putatively actionable mutations in the mitochondrial genome. bioRxiv 2020. [Google Scholar] [CrossRef]
- Zambelli, F.; Mertens, J.; Dziedzicka, D.; Sterckx, J.; Markouli, C.; Keller, A.; Tropel, P.; Jung, L.; Viville, S.; Van de Velde, H.; et al. Random mutagenesis, clonal events, and embryonic or somatic origin determine the mtDNA variant type and load in human pluripotent stem cells. Stem Cell Rep. 2018, 11, 102–114. [Google Scholar] [CrossRef] [PubMed]
- Russell, O.M.; Fruh, I.; Rai, P.K.; Marcellin, D.; Doll, T.; Reeve, A.; Germain, M.; Bastien, J.; Rygiel, K.A.; Cerino, R.; et al. Preferential amplification of a human mitochondrial DNA deletion in vitro and in vivo. Sci. Rep. 2018, 8, 1799. [Google Scholar] [CrossRef] [PubMed]
- Zambelli, F.; Spits, C. A step forward in disease modelling for mitochondrial diseases. Stem Cell Investig. 2017, 4, 89. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kargaran, P.K.; Mosqueira, D.; Kozicz, T. Mitochondrial medicine: Genetic underpinnings and disease modeling using induced pluripotent stem cell technology. Front. Cardiovasc. Med. 2020, 7, 604581. [Google Scholar] [CrossRef] [PubMed]
- Inak, G.; Lorenz, C.; Lisowski, P.; Zink, A.; Mlody, B.; Prigione, A. Concise review: Induced pluripotent stem cell-based drug discovery for mitochondrial disease. Stem Cells 2017, 35, 1655–1662. [Google Scholar] [CrossRef]
- Klein Gunnewiek, T.M.; Van Hugte, E.J.H.; Frega, M.; Guardia, G.S.; Foreman, K.; Panneman, D.; Mossink, B.; Linda, K.; Keller, J.M.; Schubert, D.; et al. m.3243A > G-induced mitochondrial dysfunction impairs human neuronal development and reduces neuronal network activity and synchronicity. Cell Rep. 2020, 31, 107538. [Google Scholar] [CrossRef]
- Lorenz, C.; Lesimple, P.; Bukowiecki, R.; Zink, A.; Inak, G.; Mlody, B.; Singh, M.; Semtner, M.; Mah, N.; Aure, K.; et al. Human iPSC-derived neural progenitors are an effective drug discovery model for neurological mtDNA disorders. Cell Stem Cell 2017, 20, 659–674.e9. [Google Scholar] [CrossRef]
- Inak, G.; Rybak-Wolf, A.; Lisowski, P.; Pentimalli, T.M.; Juttner, R.; Glazar, P.; Uppal, K.; Bottani, E.; Brunetti, D.; Secker, C.; et al. Defective metabolic programming impairs early neuronal morphogenesis in neural cultures and an organoid model of Leigh syndrome. Nat. Commun. 2021, 12, 1929. [Google Scholar] [CrossRef]
- Bayzigitov, D.R.; Medvedev, S.P.; Dementyeva, E.V.; Bayramova, S.A.; Pokushalov, E.A.; Karaskov, A.M.; Zakian, S.M. Human induced pluripotent stem cell-derived cardiomyocytes afford new opportunities in inherited cardiovascular disease modeling. Cardiol. Res. Pract. 2016, 2016, 3582380. [Google Scholar] [CrossRef]
- Yang, Y.P.; Nguyen, P.N.N.; Lin, T.C.; Yarmishyn, A.A.; Chen, W.S.; Hwang, D.K.; Chiou, G.Y.; Lin, T.W.; Chien, C.S.; Tsai, C.Y.; et al. Glutamate stimulation dysregulates AMPA receptors-induced signal transduction pathway in Leber’s inherited optic neuropathy patient-specific hiPSC-derived retinal ganglion cells. Cells 2019, 8, 625. [Google Scholar] [CrossRef]
- Galera-Monge, T.; Zurita-Diaz, F.; Canals, I.; Hansen, M.G.; Rufian-Vazquez, L.; Ehinger, J.K.; Elmer, E.; Martin, M.A.; Garesse, R.; Ahlenius, H.; et al. Mitochondrial dysfunction and calcium dysregulation in leigh syndrome induced pluripotent stem cell derived neurons. Int. J. Mol. Sci. 2020, 21, 3191. [Google Scholar] [CrossRef]
- Liu, X.; Wang, S.; Guo, X.; Li, Y.; Ogurlu, R.; Lu, F.; Prondzynski, M.; de la Serna Buzon, S.; Ma, Q.; Zhang, D.; et al. Increased reactive oxygen species-mediated Ca2+/calmodulin-dependent protein kinase II activation contributes to calcium handling abnormalities and impaired contraction in Barth syndrome. Circulation 2021, 143, 1894–1911. [Google Scholar] [CrossRef] [PubMed]
- Caglayan, S.; Hashim, A.; Cieslar-Pobuda, A.; Jensen, V.; Behringer, S.; Talug, B.; Chu, D.T.; Pecquet, C.; Rogne, M.; Brech, A.; et al. Optic atrophy 1 controls human neuronal development by preventing aberrant nuclear DNA methylation. iScience 2020, 23, 101154. [Google Scholar] [CrossRef]
- Hang, C.; Song, Y.; Wu, F.; Dong, T.; Jiang, M.; Saleem, A.; Zhang, S.; Chang, Y.; Lu, W.; Cui, M. Generation of a homozygous COX6A2 knockout human embryonic stem cell line (WAe009-A-47) via an epiCRISPR/Cas9 system. Stem Cell Res. 2021, 50, 102152. [Google Scholar] [CrossRef]
- Schreiber, A.M.; Misiorek, J.O.; Napierala, J.S.; Napierala, M. Progress in understanding Friedreich’s ataxia using human induced pluripotent stem cells. Expert Opin. Orphan Drugs 2019, 7, 81–90. [Google Scholar] [CrossRef]
- Paradies, G.; Paradies, V.; Ruggiero, F.M.; Petrosillo, G. Role of cardiolipin in mitochondrial function and dynamics in health and disease: Molecular and pharmacological aspects. Cells 2019, 8, 728. [Google Scholar] [CrossRef]
- Musatov, A.; Sedlak, E. Role of cardiolipin in stability of integral membrane proteins. Biochimie 2017, 142, 102–111. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; McCain, M.L.; Yang, L.; He, A.; Pasqualini, F.S.; Agarwal, A.; Yuan, H.; Jiang, D.; Zhang, D.; Zangi, L.; et al. Modeling the mitochondrial cardiomyopathy of Barth syndrome with induced pluripotent stem cell and heart-on-chip technologies. Nat. Med. 2014, 20, 616–623. [Google Scholar] [CrossRef] [PubMed]
- Dudek, J.; Cheng, I.F.; Chowdhury, A.; Wozny, K.; Balleininger, M.; Reinhold, R.; Grunau, S.; Callegari, S.; Toischer, K.; Wanders, R.J.; et al. Cardiac-specific succinate dehydrogenase deficiency in Barth syndrome. EMBO Mol. Med. 2016, 8, 139–154. [Google Scholar] [CrossRef]
- Van Werkhoven, M.A.; Thorburn, D.R.; Gedeon, A.K.; Pitt, J.J. Monolysocardiolipin in cultured fibroblasts is a sensitive and specific marker for Barth Syndrome. J. Lipid Res. 2006, 47, 2346–2351. [Google Scholar] [CrossRef] [PubMed]
- Kolwicz, S.C., Jr.; Purohit, S.; Tian, R. Cardiac metabolism and its interactions with contraction, growth, and survival of cardiomyocytes. Circ. Res. 2013, 113, 603–616. [Google Scholar] [CrossRef]
- Fatica, E.M.; DeLeonibus, G.A.; House, A.; Kodger, J.V.; Pearce, R.W.; Shah, R.R.; Levi, L.; Sandlers, Y. Barth syndrome: Exploring cardiac metabolism with induced pluripotent stem cell-derived cardiomyocytes. Metabolites 2019, 9, 306. [Google Scholar] [CrossRef]
- Saric, A.; Andreau, K.; Armand, A.S.; Moller, I.M.; Petit, P.X. Barth syndrome: From mitochondrial dysfunctions associated with aberrant production of reactive oxygen species to pluripotent stem cell studies. Front. Genet. 2015, 6, 359. [Google Scholar] [CrossRef]
- Reid Thompson, W.; Hornby, B.; Manuel, R.; Bradley, E.; Laux, J.; Carr, J.; Vernon, H.J. A phase 2/3 randomized clinical trial followed by an open-label extension to evaluate the effectiveness of elamipretide in Barth syndrome, a genetic disorder of mitochondrial cardiolipin metabolism. Genet. Med. 2021, 23, 471–478. [Google Scholar] [CrossRef] [PubMed]
- Alexander, C.; Votruba, M.; Pesch, U.E.; Thiselton, D.L.; Mayer, S.; Moore, A.; Rodriguez, M.; Kellner, U.; Leo-Kottler, B.; Auburger, G.; et al. OPA1, encoding a dynamin-related GTPase, is mutated in autosomal dominant optic atrophy linked to chromosome 3q28. Nat. Genet. 2000, 26, 211–215. [Google Scholar] [CrossRef]
- Delettre, C.; Lenaers, G.; Griffoin, J.M.; Gigarel, N.; Lorenzo, C.; Belenguer, P.; Pelloquin, L.; Grosgeorge, J.; Turc-Carel, C.; Perret, E.; et al. Nuclear gene OPA1, encoding a mitochondrial dynamin-related protein, is mutated in dominant optic atrophy. Nat. Genet. 2000, 26, 207–210. [Google Scholar] [CrossRef]
- Kasahara, A.; Cipolat, S.; Chen, Y.; Dorn, G.W., 2nd; Scorrano, L. Mitochondrial fusion directs cardiomyocyte differentiation via calcineurin and Notch signaling. Science 2013, 342, 734–737. [Google Scholar] [CrossRef]
- Iannielli, A.; Bido, S.; Folladori, L.; Segnali, A.; Cancellieri, C.; Maresca, A.; Massimino, L.; Rubio, A.; Morabito, G.; Caporali, L.; et al. Pharmacological inhibition of necroptosis protects from dopaminergic neuronal cell death in Parkinson’s disease models. Cell Rep. 2018, 22, 2066–2079. [Google Scholar] [CrossRef]
- Jonikas, M.; Madill, M.; Mathy, A.; Zekoll, T.; Zois, C.E.; Wigfield, S.; Kurzawa-Akanbi, M.; Browne, C.; Sims, D.; Chinnery, P.F.; et al. Stem cell modeling of mitochondrial parkinsonism reveals key functions of OPA1. Ann. Neurol. 2018, 83, 915–925. [Google Scholar] [CrossRef]
- Lee, Y.J.; Jeong, S.Y.; Karbowski, M.; Smith, C.L.; Youle, R.J. Roles of the mammalian mitochondrial fission and fusion mediators Fis1, Drp1, and Opa1 in apoptosis. Mol. Biol. Cell 2004, 15, 5001–5011. [Google Scholar] [CrossRef]
- Lynch, D.S.; Loh, S.H.Y.; Harley, J.; Noyce, A.J.; Martins, L.M.; Wood, N.W.; Houlden, H.; Plun-Favreau, H. Nonsyndromic Parkinson disease in a family with autosomal dominant optic atrophy due to OPA1 mutations. Neurol. Genet. 2017, 3, e188. [Google Scholar] [CrossRef]
- Iannielli, A.; Ugolini, G.S.; Cordiglieri, C.; Bido, S.; Rubio, A.; Colasante, G.; Valtorta, M.; Cabassi, T.; Rasponi, M.; Broccoli, V. Reconstitution of the human nigro-striatal pathway on-a-chip reveals OPA1-dependent mitochondrial defects and loss of dopaminergic synapses. Cell Rep. 2019, 29, 4646–4656. [Google Scholar] [CrossRef] [PubMed]
- Rahman, S.; Copeland, W.C. POLG-related disorders and their neurological manifestations. Nat. Rev. Neurol. 2019, 15, 40–52. [Google Scholar] [CrossRef] [PubMed]
- Liang, K.X.; Kristiansen, C.K.; Mostafavi, S.; Vatne, G.H.; Zantingh, G.A.; Kianian, A.; Tzoulis, C.; Hoyland, L.E.; Ziegler, M.; Perez, R.M.; et al. Disease-specific phenotypes in iPSC-derived neural stem cells with POLG mutations. EMBO Mol. Med. 2020, 12, e12146. [Google Scholar] [CrossRef]
- Liang, K.X.; Vatne, G.H.; Kristiansen, C.K.; Ievglevskyi, O.; Kondratskaya, E.; Glover, J.C.; Chen, A.; Sullivan, G.J.; Bindoff, L.A. N-acetylcysteine amide ameliorates mitochondrial dysfunction and reduces oxidative stress in hiPSC-derived dopaminergic neurons with POLG mutation. Exp. Neurol. 2021, 337, 113536. [Google Scholar] [CrossRef]
- Hernansanz-Agustin, P.; Enriquez, J.A. Generation of reactive oxygen species by mitochondria. Antioxidants 2021, 10, 415. [Google Scholar] [CrossRef] [PubMed]
- Harding, B.N. Progressive neuronal degeneration of childhood with liver disease (Alpers-Huttenlocher syndrome): A personal review. J. Child Neurol. 1990, 5, 273–287. [Google Scholar] [CrossRef]
- Li, S.; Guo, J.; Ying, Z.; Chen, S.; Yang, L.; Chen, K.; Long, Q.; Qin, D.; Pei, D.; Liu, X. Valproic acid-induced hepatotoxicity in Alpers syndrome is associated with mitochondrial permeability transition pore opening-dependent apoptotic sensitivity in an induced pluripotent stem cell model. Hepatology 2015, 61, 1730–1739. [Google Scholar] [CrossRef]
- Mandel, H.; Szargel, R.; Labay, V.; Elpeleg, O.; Saada, A.; Shalata, A.; Anbinder, Y.; Berkowitz, D.; Hartman, C.; Barak, M. The deoxyguanosine kinase gene is mutated in individuals with depleted hepatocerebral mitochondrial DNA. Nat. Genet. 2001, 29, 337–341. [Google Scholar] [CrossRef]
- Jing, R.; Corbett, J.L.; Cai, J.; Beeson, G.C.; Beeson, C.C.; Chan, S.S.; Dimmock, D.P.; Lazcares, L.; Geurts, A.M.; Lemasters, J.J.; et al. A screen using iPSC-derived hepatocytes reveals NAD+ as a potential treatment for mtDNA depletion syndrome. Cell Rep. 2018, 25, 1469–1484. [Google Scholar] [CrossRef]
- Guo, J.; Duan, L.; He, X.; Li, S.; Wu, Y.; Xiang, G.; Bao, F.; Yang, L.; Shi, H.; Gao, M.; et al. A combined model of human iPSC-derived liver organoids and hepatocytes reveals ferroptosis in DGUOK mutant mtDNA depletion syndrome. Adv. Sci. 2021, 8, 2004680. [Google Scholar] [CrossRef] [PubMed]
- Nemoto, S.; Fergusson, M.M.; Finkel, T. SIRT1 functionally interacts with the metabolic regulator and transcriptional coactivator PGC-1{alpha}. J. Biol. Chem. 2005, 280, 16456–16460. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.D.; Puigserver, P.; Andersson, U.; Zhang, C.Y.; Adelmant, G.; Mootha, V.; Troy, A.; Cinti, S.; Lowell, B.; Scarpulla, R.C.; et al. Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC-1. Cell 1999, 98, 115–124. [Google Scholar] [CrossRef]
- Lake, N.J.; Compton, A.G.; Rahman, S.; Thorburn, D.R. Leigh syndrome: One disorder, more than 75 monogenic causes. Ann. Neurol. 2016, 79, 190–203. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Boyer, L.; Jin, M.; Kim, Y.; Fan, W.; Bardy, C.; Berggren, T.; Evans, R.M.; Gage, F.H.; Hunter, T. Alleviation of neuronal energy deficiency by mTOR inhibition as a treatment for mitochondria-related neurodegeneration. eLife 2016, 5, e13378. [Google Scholar] [CrossRef] [PubMed]
- Hallas, T.; Eisen, B.; Shemer, Y.; Ben Jehuda, R.; Mekies, L.N.; Naor, S.; Schick, R.; Eliyahu, S.; Reiter, I.; Vlodavsky, E.; et al. Investigating the cardiac pathology of SCO2-mediated hypertrophic cardiomyopathy using patients induced pluripotent stem cell-derived cardiomyocytes. J. Cell. Mol. Med. 2018, 22, 913–925. [Google Scholar] [CrossRef]
- Rahman, S.; Blok, R.B.; Dahl, H.H.; Danks, D.M.; Kirby, D.M.; Chow, C.W.; Christodoulou, J.; Thorburn, D.R. Leigh syndrome: Clinical features and biochemical and DNA abnormalities. Ann. Neurol. 1996, 39, 343–351. [Google Scholar] [CrossRef]
- Sofou, K.; De Coo, I.F.; Isohanni, P.; Ostergaard, E.; Naess, K.; De Meirleir, L.; Tzoulis, C.; Uusimaa, J.; De Angst, I.B.; Lonnqvist, T.; et al. A multicenter study on Leigh syndrome: Disease course and predictors of survival. Orphanet J. Rare Dis. 2014, 9, 52. [Google Scholar] [CrossRef]
- Ma, Y.Y.; Wu, T.F.; Liu, Y.P.; Wang, Q.; Song, J.Q.; Li, X.Y.; Shi, X.Y.; Zhang, W.N.; Zhao, M.; Hu, L.Y.; et al. Genetic and biochemical findings in Chinese children with Leigh syndrome. J. Clin. Neurosci. 2013, 20, 1591–1594. [Google Scholar] [CrossRef]
- Mick, D.U.; Dennerlein, S.; Wiese, H.; Reinhold, R.; Pacheu-Grau, D.; Lorenzi, I.; Sasarman, F.; Weraarpachai, W.; Shoubridge, E.A.; Warscheid, B.; et al. MITRAC links mitochondrial protein translocation to respiratory-chain assembly and translational regulation. Cell 2012, 151, 1528–1541. [Google Scholar] [CrossRef]
- Kovarova, N.; Pecina, P.; Nuskova, H.; Vrbacky, M.; Zeviani, M.; Mracek, T.; Viscomi, C.; Houstek, J. Tissue- and species-specific differences in cytochrome c oxidase assembly induced by SURF1 defects. Biochim. Biophys. Acta 2016, 1862, 705–715. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.; Yao, J.; Johns, T.; Fu, K.; De Bie, I.; Macmillan, C.; Cuthbert, A.P.; Newbold, R.F.; Wang, J.; Chevrette, M.; et al. SURF1, encoding a factor involved in the biogenesis of cytochrome c oxidase, is mutated in Leigh syndrome. Nat. Genet. 1998, 20, 337–343. [Google Scholar] [CrossRef] [PubMed]
- Sue, C.M.; Karadimas, C.; Checcarelli, N.; Tanji, K.; Papadopoulou, L.C.; Pallotti, F.; Guo, F.L.; Shanske, S.; Hirano, M.; De Vivo, D.C.; et al. Differential features of patients with mutations in two COX assembly genes, SURF-1 and SCO2. Ann. Neurol. 2000, 47, 589–595. [Google Scholar] [CrossRef]
- Leary, S.C.; Kaufman, B.A.; Pellecchia, G.; Guercin, G.H.; Mattman, A.; Jaksch, M.; Shoubridge, E.A. Human SCO1 and SCO2 have independent, cooperative functions in copper delivery to cytochrome c oxidase. Hum. Mol. Genet. 2004, 13, 1839–1848. [Google Scholar] [CrossRef]
- Joost, K.; Rodenburg, R.; Piirsoo, A.; van den Heuvel, B.; Zordania, R.; Ounap, K. A novel mutation in the SCO2 gene in a neonate with early-onset cardioencephalomyopathy. Pediatr. Neurol. 2010, 42, 227–230. [Google Scholar] [CrossRef] [PubMed]
- Szymanska-Debinska, T.; Karkucinska-Wieckowska, A.; Piekutowska-Abramczuk, D.; Jurkiewicz, E.; Iwanicka-Pronicka, K.; Rokicki, D.; Pronicki, M. Leigh disease due to SCO2 mutations revealed at extended autopsy. J. Clin. Pathol. 2015, 68, 397–399. [Google Scholar] [CrossRef]
- Papadopoulou, L.C.; Sue, C.M.; Davidson, M.M.; Tanji, K.; Nishino, I.; Sadlock, J.E.; Krishna, S.; Walker, W.; Selby, J.; Glerum, D.M.; et al. Fatal infantile cardioencephalomyopathy with COX deficiency and mutations in SCO2, a COX assembly gene. Nat. Genet. 1999, 23, 333–337. [Google Scholar] [CrossRef]
- Jaksch, M.; Ogilvie, I.; Yao, J.; Kortenhaus, G.; Bresser, H.G.; Gerbitz, K.D.; Shoubridge, E.A. Mutations in SCO2 are associated with a distinct form of hypertrophic cardiomyopathy and cytochrome c oxidase deficiency. Hum. Mol. Genet. 2000, 9, 795–801. [Google Scholar] [CrossRef]
- Wedatilake, Y.; Brown, R.M.; McFarland, R.; Yaplito-Lee, J.; Morris, A.A.; Champion, M.; Jardine, P.E.; Clarke, A.; Thorburn, D.R.; Taylor, R.W.; et al. SURF1 deficiency: A multi-centre natural history study. Orphanet J. Rare Dis. 2013, 8, 96. [Google Scholar] [CrossRef]
- Benit, P.; El-Khoury, R.; Schiff, M.; Sainsard-Chanet, A.; Rustin, P. Genetic background influences mitochondrial function: Modeling mitochondrial disease for therapeutic development. Trends Mol. Med. 2010, 16, 210–217. [Google Scholar] [CrossRef]
- Jain, I.H.; Zazzeron, L.; Goli, R.; Alexa, K.; Schatzman-Bone, S.; Dhillon, H.; Goldberger, O.; Peng, J.; Shalem, O.; Sanjana, N.E.; et al. Hypoxia as a therapy for mitochondrial disease. Science 2016, 352, 54–61. [Google Scholar] [CrossRef]
- Ferrari, M.; Jain, I.H.; Goldberger, O.; Rezoagli, E.; Thoonen, R.; Cheng, K.H.; Sosnovik, D.E.; Scherrer-Crosbie, M.; Mootha, V.K.; Zapol, W.M. Hypoxia treatment reverses neurodegenerative disease in a mouse model of Leigh syndrome. Proc. Natl. Acad. Sci. USA 2017, 114, E4241–E4250. [Google Scholar] [CrossRef]
- Jain, I.H.; Zazzeron, L.; Goldberger, O.; Marutani, E.; Wojtkiewicz, G.R.; Ast, T.; Wang, H.; Schleifer, G.; Stepanova, A.; Brepoels, K.; et al. Leigh syndrome mouse model can be rescued by interventions that normalize brain hyperoxia, but not HIF activation. Cell Metab. 2019, 30, 824–832.e3. [Google Scholar] [CrossRef]
- Augustyniak, J.; Lenart, J.; Gaj, P.; Kolanowska, M.; Jazdzewski, K.; Stepien, P.P.; Buzanska, L. Bezafibrate upregulates mitochondrial biogenesis and influence neural differentiation of human-induced pluripotent stem cells. Mol. Neurobiol. 2019, 56, 4346–4363. [Google Scholar] [CrossRef]
- Steele, H.; Gomez-Duran, A.; Pyle, A.; Hopton, S.; Newman, J.; Stefanetti, R.J.; Charman, S.J.; Parikh, J.D.; He, L.; Viscomi, C.; et al. Metabolic effects of bezafibrate in mitochondrial disease. EMBO Mol. Med. 2020, 12, e11589. [Google Scholar] [CrossRef]
- Montero, R.; Yubero, D.; Villarroya, J.; Henares, D.; Jou, C.; Rodriguez, M.A.; Ramos, F.; Nascimento, A.; Ortez, C.I.; Campistol, J.; et al. GDF-15 is elevated in children with mitochondrial diseases and is induced by mitochondrial dysfunction. PLoS ONE 2016, 11, e0148709. [Google Scholar] [CrossRef] [PubMed]
- Lehtonen, J.M.; Forsstrom, S.; Bottani, E.; Viscomi, C.; Baris, O.R.; Isoniemi, H.; Hockerstedt, K.; Osterlund, P.; Hurme, M.; Jylhava, J.; et al. FGF21 is a biomarker for mitochondrial translation and mtDNA maintenance disorders. Neurology 2016, 87, 2290–2299. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, R.; Censi, S.; Mastrorilli, F.; Boraso, A. Prognostic benefits of heart rate reduction in cardiovascular disease. Eur. Heart J. Suppl. 2003, 5, G10–G14. [Google Scholar] [CrossRef]
- Garbern, J.C.; Lee, R.T. Mitochondria and metabolic transitions in cardiomyocytes: Lessons from development for stem cell-derived cardiomyocytes. Stem Cell Res. Ther. 2021, 12, 177. [Google Scholar] [CrossRef] [PubMed]
- Cortes-Hernandez, P.; Vazquez-Memije, M.E.; Garcia, J.J. ATP6 homoplasmic mutations inhibit and destabilize the human F1F0-ATP synthase without preventing enzyme assembly and oligomerization. J. Biol. Chem. 2007, 282, 1051–1058. [Google Scholar] [CrossRef] [PubMed]
- Manfredi, G.; Gupta, N.; Vazquez-Memije, M.E.; Sadlock, J.E.; Spinazzola, A.; De Vivo, D.C.; Schon, E.A. Oligomycin induces a decrease in the cellular content of a pathogenic mutation in the human mitochondrial ATPase 6 gene. J. Biol. Chem. 1999, 274, 9386–9391. [Google Scholar] [CrossRef]
- Mattiazzi, M.; Vijayvergiya, C.; Gajewski, C.D.; DeVivo, D.C.; Lenaz, G.; Wiedmann, M.; Manfredi, G. The mtDNA T8993G (NARP) mutation results in an impairment of oxidative phosphorylation that can be improved by antioxidants. Hum. Mol. Genet. 2004, 13, 869–879. [Google Scholar] [CrossRef]
- Trounce, I.; Neill, S.; Wallace, D.C. Cytoplasmic transfer of the mtDNA nt 8993 T-->G (ATP6) point mutation associated with Leigh syndrome into mtDNA-less cells demonstrates cosegregation with a decrease in state III respiration and ADP/O ratio. Proc. Natl. Acad. Sci. USA 1994, 91, 8334–8338. [Google Scholar] [CrossRef]
- Wu, Y.R.; Wang, A.G.; Chen, Y.T.; Yarmishyn, A.A.; Buddhakosai, W.; Yang, T.C.; Hwang, D.K.; Yang, Y.P.; Shen, C.N.; Lee, H.C.; et al. Bioactivity and gene expression profiles of hiPSC-generated retinal ganglion cells in MT-ND4 mutated Leber’s hereditary optic neuropathy. Exp. Cell Res. 2018, 363, 299–309. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.C.; Yarmishyn, A.A.; Yang, Y.P.; Lu, P.C.; Chou, S.J.; Wang, M.L.; Lin, T.C.; Hwang, D.K.; Chou, Y.B.; Chen, S.J.; et al. Mitochondrial transport mediates survival of retinal ganglion cells in affected LHON patients. Hum. Mol. Genet. 2020, 29, 1454–1464. [Google Scholar] [CrossRef] [PubMed]
- El-Hattab, A.W.; Adesina, A.M.; Jones, J.; Scaglia, F. MELAS syndrome: Clinical manifestations, pathogenesis, and treatment options. Mol. Genet. Metab. 2015, 116, 4–12. [Google Scholar] [CrossRef] [PubMed]
- Winanto; Khong, Z.J.; Soh, B.S.; Fan, Y.; Ng, S.Y. Organoid cultures of MELAS neural cells reveal hyperactive Notch signaling that impacts neurodevelopment. Cell Death Dis. 2020, 11, 182. [Google Scholar] [CrossRef] [PubMed]
- Koga, Y.; Povalko, N.; Nishioka, J.; Katayama, K.; Kakimoto, N.; Matsuishi, T. MELAS and L-arginine therapy: Pathophysiology of stroke-like episodes. Ann. N. Y. Acad. Sci. 2010, 1201, 104–110. [Google Scholar] [CrossRef] [PubMed]
- Rothstein, J.D. Edaravone: A new drug approved for ALS. Cell 2017, 171, 725. [Google Scholar] [CrossRef] [PubMed]
- Grace, H.E.; Galdun, P., 3rd; Lesnefsky, E.J.; West, F.D.; Iyer, S. mRNA Reprogramming of T8993G Leigh’s syndrome fibroblast cells to create induced pluripotent stem cell models for mitochondrial disorders. Stem Cells Dev. 2019, 28, 846–859. [Google Scholar] [CrossRef] [PubMed]
- Grady, J.P.; Pickett, S.J.; Ng, Y.S.; Alston, C.L.; Blakely, E.L.; Hardy, S.A.; Feeney, C.L.; Bright, A.A.; Schaefer, A.M.; Gorman, G.S.; et al. mtDNA heteroplasmy level and copy number indicate disease burden in m.3243A>G mitochondrial disease. EMBO Mol. Med. 2018, 10, e8262. [Google Scholar] [CrossRef]
- Karbassi, E.; Fenix, A.; Marchiano, S.; Muraoka, N.; Nakamura, K.; Yang, X.; Murry, C.E. Cardiomyocyte maturation: Advances in knowledge and implications for regenerative medicine. Nat. Rev. Cardiol. 2020, 17, 341–359. [Google Scholar] [CrossRef]
- Terryn, J.; Tricot, T.; Gajjar, M.; Verfaillie, C. Recent advances in lineage differentiation from stem cells: Hurdles and opportunities? F1000Research 2018, 7, 220. [Google Scholar] [CrossRef]
- Vieira, M.S.; Santos, A.K.; Vasconcellos, R.; Goulart, V.A.M.; Parreira, R.C.; Kihara, A.H.; Ulrich, H.; Resende, R.R. Neural stem cell differentiation into mature neurons: Mechanisms of regulation and biotechnological applications. Biotechnol. Adv. 2018, 36, 1946–1970. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Koo, B.K.; Knoblich, J.A. Human organoids: Model systems for human biology and medicine. Nat. Rev. Mol. Cell Biol. 2020, 21, 571–584. [Google Scholar] [CrossRef] [PubMed]
- Patterson, M.; Chan, D.N.; Ha, I.; Case, D.; Cui, Y.; Van Handel, B.; Mikkola, H.K.; Lowry, W.E. Defining the nature of human pluripotent stem cell progeny. Cell Res. 2012, 22, 178–193. [Google Scholar] [CrossRef] [PubMed]
- Ban, K.; Bae, S.; Yoon, Y.S. Current strategies and challenges for purification of cardiomyocytes derived from human pluripotent stem cells. Theranostics 2017, 7, 2067–2077. [Google Scholar] [CrossRef] [PubMed]
- Velmurugan, B.K.; Bharathi Priya, L.; Poornima, P.; Lee, L.J.; Baskaran, R. Biomaterial aided differentiation and maturation of induced pluripotent stem cells. J. Cell. Physiol. 2019, 234, 8443–8454. [Google Scholar] [CrossRef]
- Ahmed, R.E.; Anzai, T.; Chanthra, N.; Uosaki, H. A brief review of current maturation methods for human induced pluripotent stem cells-derived cardiomyocytes. Front. Cell Dev. Biol. 2020, 8, 178. [Google Scholar] [CrossRef] [PubMed]
- Lam, D.; Enright, H.A.; Cadena, J.; Peters, S.K.G.; Sales, A.P.; Osburn, J.J.; Soscia, D.A.; Kulp, K.S.; Wheeler, E.K.; Fischer, N.O. Tissue-specific extracellular matrix accelerates the formation of neural networks and communities in a neuron-glia co-culture on a multi-electrode array. Sci. Rep. 2019, 9, 4159. [Google Scholar] [CrossRef] [PubMed]
- Hedegaard, A.; Monzon-Sandoval, J.; Newey, S.E.; Whiteley, E.S.; Webber, C.; Akerman, C.J. Pro-maturational effects of human iPSC-derived cortical astrocytes upon iPSC-derived cortical neurons. Stem Cell Rep. 2020, 15, 38–51. [Google Scholar] [CrossRef]
- Son, G.; Han, J. Roles of mitochondria in neuronal development. BMB Rep. 2018, 51, 549–556. [Google Scholar] [CrossRef]
- Jensen, C.; Teng, Y. Is It Time to Start Transitioning From 2D to 3D Cell Culture? Front. Mol. Biosci. 2020, 7, 33. [Google Scholar] [CrossRef] [PubMed]
- Kathuria, A.; Lopez-Lengowski, K.; Watmuff, B.; Karmacharya, R. Comparative transcriptomic analysis of cerebral organoids and cortical neuron cultures derived from human induced pluripotent stem cells. Stem Cells Dev. 2020, 29, 1370–1381. [Google Scholar] [CrossRef] [PubMed]
- Bhaduri, A.; Andrews, M.G.; Kriegstein, A.R.; Nowakowski, T.J. Are organoids ready for prime time? Cell Stem Cell 2020, 27, 361–365. [Google Scholar] [CrossRef] [PubMed]
- Doss, M.X.; Sachinidis, A. Current challenges of iPSC-based disease modeling and therapeutic implications. Cells 2019, 8, 403. [Google Scholar] [CrossRef] [PubMed]
- Elitt, M.S.; Barbar, L.; Tesar, P.J. Drug screening for human genetic diseases using iPSC models. Hum. Mol. Genet. 2018, 27, R89–R98. [Google Scholar] [CrossRef]
- Khan, S.; Ince-Dunn, G.; Suomalainen, A.; Elo, L.L. Integrative omics approaches provide biological and clinical insights: Examples from mitochondrial diseases. J. Clin. Investig. 2020, 130, 20–28. [Google Scholar] [CrossRef]
- Chumarina, M.; Russ, K.; Azevedo, C.; Heuer, A.; Pihl, M.; Collin, A.; Frostner, E.A.; Elmer, E.; Hyttel, P.; Cappelletti, G.; et al. Cellular alterations identified in pluripotent stem cell-derived midbrain spheroids generated from a female patient with progressive external ophthalmoplegia and parkinsonism who carries a novel variation (p.Q811R) in the POLG1 gene. Acta Neuropathol. Commun. 2019, 7, 208. [Google Scholar] [CrossRef]
- Rizzo, F.; Ronchi, D.; Salani, S.; Nizzardo, M.; Fortunato, F.; Bordoni, A.; Stuppia, G.; Del Bo, R.; Piga, D.; Fato, R.; et al. Selective mitochondrial depletion, apoptosis resistance, and increased mitophagy in human charcot-marie-tooth 2A motor neurons. Hum. Mol. Genet. 2016, 25, 4266–4281. [Google Scholar] [CrossRef] [PubMed]
- Son, D.; Zheng, J.; Kim, I.Y.; You, S. Self-replicative mRNA-mediated generation of induced pluripotent stem cell line from a 1-year-old Leigh syndrome patient with mitochondrial DNA cytochrome b mutation. Stem Cell Res. 2021, 54, 102392. [Google Scholar] [CrossRef]
- Van Helden, R.W.J.; Birket, M.J.; Freund, C.; Arendzen, C.H.; Mikkers, H.M.; Orlova, V.; de Coo, R.I.; Mummery, C.L.; Bellin, M. Generation of three human induced pluripotent stem cell lines, LUMCi024-A, LUMCi025-A, and LUMCi026-A, from two patients with combined oxidative phosphorylation deficiency 8 and a related control. Stem Cell Res. 2021, 53, 102374. [Google Scholar] [CrossRef] [PubMed]
- Cerrada, V.; Garcia-Lopez, M.; Moreno-Izquierdo, A.; Villaverde, C.; Zurita, O.; Martin-Merida, M.I.; Arenas, J.; Ayuso, C.; Gallardo, M.E. Derivation of a human DOA iPSC line, IISHDOi006-A, with a mutation in the ACO2 gene: C.1999G>A; p.Glu667Lys. Stem Cell Res. 2019, 40, 101566. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Wu, K.; Guan, J.; Wang, Q.; Wang, H. Generation of a human induced pluripotent stem cell line (CPGHi003-A) from an auditory neuropathy patient with AIFM1 p.R422Q mutation. Stem Cell Res. 2021, 53, 102376. [Google Scholar] [CrossRef] [PubMed]
- Cooper, H.M.; Yang, Y.; Ylikallio, E.; Khairullin, R.; Woldegebriel, R.; Lin, K.L.; Euro, L.; Palin, E.; Wolf, A.; Trokovic, R.; et al. ATPase-deficient mitochondrial inner membrane protein ATAD3A disturbs mitochondrial dynamics in dominant hereditary spastic paraplegia. Hum. Mol. Genet. 2017, 26, 1432–1443. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, J.; Zhou, Y.; Li, H.; Li, A.; Tan, X.; Wang, G.; Lei, M. Generation of two induced pluripotent stem cell lines (XACHi0010-A, XACHi0011-A) from a Chinese family with combined oxidative phosphorylation deficiency carrying homozygous and heterozygous C1QBP-L275F mutation. Stem Cell Res. 2020, 47, 101912. [Google Scholar] [CrossRef] [PubMed]
- Nakamoto, F.K.; Okamoto, S.; Mitsui, J.; Sone, T.; Ishikawa, M.; Yamamoto, Y.; Kanegae, Y.; Nakatake, Y.; Imaizumi, K.; Ishiura, H.; et al. The pathogenesis linked to coenzyme Q10 insufficiency in iPSC-derived neurons from patients with multiple-system atrophy. Sci. Rep. 2018, 8, 14215. [Google Scholar] [CrossRef] [PubMed]
- Romero-Moya, D.; Castano, J.; Santos-Ocana, C.; Navas, P.; Menendez, P. Generation, genome edition and characterization of iPSC lines from a patient with coenzyme Q10 deficiency harboring a heterozygous mutation in COQ4 gene. Stem Cell Res. 2017, 24, 144–147. [Google Scholar] [CrossRef]
- Romero-Moya, D.; Santos-Ocana, C.; Castano, J.; Garrabou, G.; Rodriguez-Gomez, J.A.; Ruiz-Bonilla, V.; Bueno, C.; Gonzalez-Rodriguez, P.; Giorgetti, A.; Perdiguero, E.; et al. Genetic rescue of mitochondrial and skeletal muscle impairment in an induced pluripotent stem cells model of coenzyme Q10 deficiency. Stem Cells 2017, 35, 1687–1703. [Google Scholar] [CrossRef]
- Rohani, L.; Machiraju, P.; Sabouny, R.; Meng, G.; Liu, S.; Zhao, T.; Iqbal, F.; Wang, X.; Ravandi, A.; Wu, J.C.; et al. Reversible mitochondrial fragmentation in iPSC-derived cardiomyocytes from children with DCMA, a mitochondrial cardiomyopathy. Can. J. Cardiol. 2020, 36, 554–563. [Google Scholar] [CrossRef]
- Janz, A.; Chen, R.; Regensburger, M.; Ueda, Y.; Rost, S.; Klopocki, E.; Gunther, K.; Edenhofer, F.; Duff, H.J.; Ergun, S.; et al. Generation of two patient-derived iPSC lines from siblings (LIBUCi001-A and LIBUCi002-A) and a genetically modified iPSC line (JMUi001-A-1) to mimic dilated cardiomyopathy with ataxia (DCMA) caused by a homozygous DNAJC19 mutation. Stem Cell Res. 2020, 46, 101856. [Google Scholar] [CrossRef] [PubMed]
- Sequiera, G.L.; Rockman-Greenberg, C.; Dhingra, S. Generation of human induced pluripotent stem cell (hiPSC) line UOMi001-A from a patient with Leigh-like syndrome harbouring compound heterozygous variants in ECHS1 gene. Stem Cell Res. 2020, 48, 101934. [Google Scholar] [CrossRef] [PubMed]
- Yuan, F.; Wang, C.; Xi, J.; Wang, S.; Lin, L.; Wang, Y.; Wang, A.; Wang, C.; Luo, X.; Xu, Q.; et al. Generation of an induced pluripotent stem cell line SHCDNi001-A from a one-year-old Chinese girl with mitochondrial DNA depletion syndrome 13. Stem Cell Res. 2020, 45, 101832. [Google Scholar] [CrossRef]
- Marti, S.; Leon, M.; Orellana, C.; Prieto, J.; Ponsoda, X.; Lopez-Garcia, C.; Vilchez, J.J.; Sevilla, T.; Torres, J. Generation of a disease-specific iPS cell line derived from a patient with Charcot-Marie-Tooth type 2K lacking functional GDAP1 gene. Stem Cell Res. 2017, 18, 1–4. [Google Scholar] [CrossRef]
- Zurita-Diaz, F.; Galera-Monge, T.; Moreno-Izquierdo, A.; Fraga, M.F.; Ayuso, C.; Fernandez, A.F.; Garesse, R.; Gallardo, M.E. Generation of a human iPSC line from a patient with a mitochondrial encephalopathy due to mutations in the GFM1 gene. Stem Cell Res. 2016, 16, 124–127. [Google Scholar] [CrossRef][Green Version]
- Saporta, M.A.; Dang, V.; Volfson, D.; Zou, B.; Xie, X.S.; Adebola, A.; Liem, R.K.; Shy, M.; Dimos, J.T. Axonal Charcot-Marie-Tooth disease patient-derived motor neurons demonstrate disease-specific phenotypes including abnormal electrophysiological properties. Exp. Neurol. 2015, 263, 190–199. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Fu, Y.; Xia, W.; He, J.; Zou, Y.; Ruan, W.; Lou, Q.; Li, Y.; Pan, J.; Li, H.; et al. Generation of induced pluripotent stem cell line, ZJUCHi002-A, from Charcot-Marie-Tooth disease type 2A (CMT2A) patient with a mutation of c.752C>T in MFN2. Stem Cell Res. 2019, 36, 101411. [Google Scholar] [CrossRef] [PubMed]
- Ababneh, N.A.; Ali, D.; Barham, R.; Al-Kurdi, B.; Sharar, N.; Al Hadidi, S.; Qanno, O.; Ryalat, A.T.; Salah, B.; Awidi, A. Establishment of a human induced pluripotent stem cell line, JUCTCi012-A, from multiple symmetric lipomatosis (MSL) patient carrying a homozygous Arg707Trp (c.2119C > T) mutation in the MFN2 gene. Stem Cell Res. 2020, 48, 101967. [Google Scholar] [CrossRef]
- Sequiera, G.L.; Rockman-Greenberg, C.; Dhingra, S. Induced pluripotent stem cell line UOMi002-A from a patient with Leigh syndrome with compound heterozygous mutations in the NDUFV1 gene. Stem Cell Res. 2020, 48, 101964. [Google Scholar] [CrossRef]
- Chen, J.; Riazifar, H.; Guan, M.X.; Huang, T. Modeling autosomal dominant optic atrophy using induced pluripotent stem cells and identifying potential therapeutic targets. Stem Cell Res. Ther. 2016, 7, 2. [Google Scholar] [CrossRef]
- Galera-Monge, T.; Zurita-Diaz, F.; Moreno-Izquierdo, A.; Fraga, M.F.; Fernandez, A.F.; Ayuso, C.; Garesse, R.; Gallardo, M.E. Generation of a human iPSC line from a patient with an optic atrophy ‘plus’ phenotype due to a mutation in the OPA1 gene. Stem Cell Res. 2016, 16, 673–676. [Google Scholar] [CrossRef][Green Version]
- Hauser, S.; Schuster, S.; Theurer, Y.; Synofzik, M.; Schols, L. Generation of optic atrophy 1 patient-derived induced pluripotent stem cells (iPS-OPA1-BEHR) for disease modeling of complex optic atrophy syndromes (Behr syndrome). Stem Cell Res. 2016, 17, 426–429. [Google Scholar] [CrossRef] [PubMed]
- Zurita-Diaz, F.; Galera-Monge, T.; Moreno-Izquierdo, A.; Corton, M.; Ayuso, C.; Garesse, R.; Gallardo, M.E. Establishment of a human DOA ‘plus’ iPSC line, IISHDOi003-A, with the mutation in the OPA1 gene: C.1635C>A; p.Ser545Arg. Stem Cell Res. 2017, 24, 81–84. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.H.; Xie, Y.; Xu, K.; Li, Y. Generation of an induced pluripotent stem cell line BIOi002-A from a patient with autosomal dominant optic atrophy. Stem Cell Res. 2021, 53, 102278. [Google Scholar] [CrossRef]
- Perez-Siles, G.; Cutrupi, A.; Ellis, M.; Screnci, R.; Mao, D.; Uesugi, M.; Yiu, E.M.; Ryan, M.M.; Choi, B.O.; Nicholson, G.; et al. Energy metabolism and mitochondrial defects in X-linked Charcot-Marie-Tooth (CMTX6) iPSC-derived motor neurons with the p.R158H PDK3 mutation. Sci. Rep. 2020, 10, 9262. [Google Scholar] [CrossRef]
- Vogtle, F.N.; Brandl, B.; Larson, A.; Pendziwiat, M.; Friederich, M.W.; White, S.M.; Basinger, A.; Kucukkose, C.; Muhle, H.; Jahn, J.A.; et al. Mutations in PMPCB encoding the catalytic subunit of the mitochondrial presequence protease cause neurodegeneration in early childhood. Am. J. Hum. Genet. 2018, 102, 557–573. [Google Scholar] [CrossRef] [PubMed]
- Zurita, F.; Galera, T.; Gonzalez-Paramos, C.; Moreno-Izquierdo, A.; Schneiderat, P.; Fraga, M.F.; Fernandez, A.F.; Garesse, R.; Gallardo, M.E. Generation of a human iPSC line from a patient with a defect of intergenomic communication. Stem Cell Res. 2016, 16, 120–123. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, N.V.; Schieck, M.; Neuenkirch, M.; Tondera, C.; Schmitz, H.; Wendeburg, L.; Steinemann, D.; Elpers, C.; Rutsch, F.; Konig, R. Generation of three induced pluripotent cell lines (iPSCs) from an Aicardi-Goutieres syndrome (AGS) patient harboring a deletion in the genomic locus of the sterile alpha motif and HD domain containing protein 1 (SAMHD1). Stem Cell Res. 2020, 43, 101697. [Google Scholar] [CrossRef]
- Surun, D.; Schneider, A.; Mircetic, J.; Neumann, K.; Lansing, F.; Paszkowski-Rogacz, M.; Hanchen, V.; Lee-Kirsch, M.A.; Buchholz, F. Efficient generation and correction of mutations in human iPS cells utilizing mRNAs of CRISPR base editors and prime editors. Genes 2020, 11, 511. [Google Scholar] [CrossRef]
- Dudek, J.; Cheng, I.F.; Balleininger, M.; Vaz, F.M.; Streckfuss-Bomeke, K.; Hubscher, D.; Vukotic, M.; Wanders, R.J.; Rehling, P.; Guan, K. Cardiolipin deficiency affects respiratory chain function and organization in an induced pluripotent stem cell model of Barth syndrome. Stem Cell Res. 2013, 11, 806–819. [Google Scholar] [CrossRef]
- Guo, X.; Wang, L.; Chen, K.; Song, S.; Wang, X.; Gu, X.; Niu, C.; Chu, M. Generation of urine-derived iPS cell line via a non-integrative method from a Barth syndrome patient with TAZ gene mutation. Stem Cell Res. 2020, 47, 101886. [Google Scholar] [CrossRef]
- Sharma, T.P.; Wiley, L.A.; Whitmore, S.S.; Anfinson, K.R.; Cranston, C.M.; Oppedal, D.J.; Daggett, H.T.; Mullins, R.F.; Tucker, B.A.; Stone, E.M. Patient-specific induced pluripotent stem cells to evaluate the pathophysiology of TRNT1-associated Retinitis pigmentosa. Stem Cell Res. 2017, 21, 58–70. [Google Scholar] [CrossRef] [PubMed]
- Galera-Monge, T.; Zurita-Diaz, F.; Gonzalez-Paramos, C.; Moreno-Izquierdo, A.; Fraga, M.F.; Fernandez, A.F.; Garesse, R.; Gallardo, M.E. Generation of a human iPSC line from a patient with Leigh syndrome caused by a mutation in the MT-ATP6 gene. Stem Cell Res. 2016, 16, 766–769. [Google Scholar] [CrossRef]
- Peron, C.; Mauceri, R.; Cabassi, T.; Segnali, A.; Maresca, A.; Iannielli, A.; Rizzo, A.; Sciacca, F.L.; Broccoli, V.; Carelli, V.; et al. Generation of a human iPSC line, FINCBi001-A, carrying a homoplasmic m.G3460A mutation in MT-ND1 associated with Leber’s Hereditary optic Neuropathy (LHON). Stem Cell Res. 2020, 48, 101939. [Google Scholar] [CrossRef] [PubMed]
- Hung, S.S.; Van Bergen, N.J.; Jackson, S.; Liang, H.; Mackey, D.A.; Hernandez, D.; Lim, S.Y.; Hewitt, A.W.; Trounce, I.; Pebay, A.; et al. Study of mitochondrial respiratory defects on reprogramming to human induced pluripotent stem cells. Aging 2016, 8, 945–957. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.E.; Yang, Y.P.; Chen, Y.T.; Wu, Y.R.; Wang, C.L.; Tsai, F.T.; Hwang, D.K.; Lin, T.C.; Chen, S.J.; Wang, A.G.; et al. Generation of patient-specific induced pluripotent stem cells from Leber’s hereditary optic neuropathy. Stem Cell Res. 2018, 28, 56–60. [Google Scholar] [CrossRef] [PubMed]
- Galera, T.; Zurita, F.; Gonzalez-Paramos, C.; Moreno-Izquierdo, A.; Fraga, M.F.; Fernandez, A.F.; Garesse, R.; Gallardo, M.E. Generation of a human iPSC line from a patient with Leigh syndrome. Stem Cell Res. 2016, 16, 63–66. [Google Scholar] [CrossRef] [PubMed]
- Hsu, Y.H.; Wu, Y.T.; Huang, C.Y.; Ho, M.C.; Cheng, Y.C.; Syu, S.H.; Lu, H.E.; Chen, Y.C.; Tsai, C.L.; Lin, H.C.; et al. Generation of an induced pluripotent stem cell line from a 39-year-old female patient with severe-to-profound non-syndromic sensorineural hearing loss and a A1555G mutation in the mitochondrial MTRNR1 gene. Stem Cell Res. 2017, 25, 245–249. [Google Scholar] [CrossRef]
- Li, S.; Pan, H.; Tan, C.; Sun, Y.; Song, Y.; Zhang, X.; Yang, W.; Wang, X.; Li, D.; Dai, Y.; et al. Mitochondrial dysfunctions contribute to hypertrophic cardiomyopathy in patient iPSC-derived cardiomyocytes with MT-RNR2 mutation. Stem Cell Rep. 2018, 10, 808–821. [Google Scholar] [CrossRef]
- Chou, S.J.; Tseng, W.L.; Chen, C.T.; Lai, Y.F.; Chien, C.S.; Chang, Y.L.; Lee, H.C.; Wei, Y.H.; Chiou, S.H. Impaired ROS scavenging system in human induced pluripotent stem cells generated from patients with MERRF syndrome. Sci. Rep. 2016, 6, 23661. [Google Scholar] [CrossRef]
- Chou, S.J.; Ko, Y.L.; Yang, Y.H.; Yarmishyn, A.A.; Wu, Y.T.; Chen, C.T.; Lee, H.C.; Wei, Y.H.; Chiou, S.H. Generation of two isogenic human induced pluripotent stem cell lines from a 15year-old female patient with MERRF syndrome and A8344G mutation of mitochondrial DNA. Stem Cell Res. 2018, 30, 201–205. [Google Scholar] [CrossRef] [PubMed]
- Fujikura, J.; Nakao, K.; Sone, M.; Noguchi, M.; Mori, E.; Naito, M.; Taura, D.; Harada-Shiba, M.; Kishimoto, I.; Watanabe, A.; et al. Induced pluripotent stem cells generated from diabetic patients with mitochondrial DNA A3243G mutation. Diabetologia 2012, 55, 1689–1698. [Google Scholar] [CrossRef] [PubMed]
- Kodaira, M.; Hatakeyama, H.; Yuasa, S.; Seki, T.; Egashira, T.; Tohyama, S.; Kuroda, Y.; Tanaka, A.; Okata, S.; Hashimoto, H.; et al. Impaired respiratory function in MELAS-induced pluripotent stem cells with high heteroplasmy levels. FEBS Open Bio 2015, 5, 219–225. [Google Scholar] [CrossRef]
- Chichagova, V.; Hallam, D.; Collin, J.; Buskin, A.; Saretzki, G.; Armstrong, L.; Yu-Wai-Man, P.; Lako, M.; Steel, D.H. Human iPSC disease modelling reveals functional and structural defects in retinal pigment epithelial cells harbouring the m.3243A > G mitochondrial DNA mutation. Sci. Rep. 2017, 7, 12320. [Google Scholar] [CrossRef]
- Peron, C.; Mauceri, R.; Iannielli, A.; Cavaliere, A.; Legati, A.; Rizzo, A.; Sciacca, F.L.; Broccoli, V.; Tiranti, V. Generation of two human iPSC lines, FINCBi002-A and FINCBi003-A, carrying heteroplasmic macrodeletion of mitochondrial DNA causing Pearson’s syndrome. Stem Cell Res. 2021, 50, 102151. [Google Scholar] [CrossRef]
- Chowdhury, A.; Aich, A.; Jain, G.; Wozny, K.; Luchtenborg, C.; Hartmann, M.; Bernhard, O.; Balleiniger, M.; Alfar, E.A.; Zieseniss, A.; et al. Defective mitochondrial cardiolipin remodeling dampens HIF-1alpha expression in hypoxia. Cell Rep. 2018, 25, 561–570.e6. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
McKnight, C.L.; Low, Y.C.; Elliott, D.A.; Thorburn, D.R.; Frazier, A.E. Modelling Mitochondrial Disease in Human Pluripotent Stem Cells: What Have We Learned? Int. J. Mol. Sci. 2021, 22, 7730. https://doi.org/10.3390/ijms22147730
McKnight CL, Low YC, Elliott DA, Thorburn DR, Frazier AE. Modelling Mitochondrial Disease in Human Pluripotent Stem Cells: What Have We Learned? International Journal of Molecular Sciences. 2021; 22(14):7730. https://doi.org/10.3390/ijms22147730
Chicago/Turabian StyleMcKnight, Cameron L., Yau Chung Low, David A. Elliott, David R. Thorburn, and Ann E. Frazier. 2021. "Modelling Mitochondrial Disease in Human Pluripotent Stem Cells: What Have We Learned?" International Journal of Molecular Sciences 22, no. 14: 7730. https://doi.org/10.3390/ijms22147730
APA StyleMcKnight, C. L., Low, Y. C., Elliott, D. A., Thorburn, D. R., & Frazier, A. E. (2021). Modelling Mitochondrial Disease in Human Pluripotent Stem Cells: What Have We Learned? International Journal of Molecular Sciences, 22(14), 7730. https://doi.org/10.3390/ijms22147730