
Comparative Molecular Dynamics Investigation of the Electromotile Hearing Protein Prestin


 , ,
, , 
Abstract
:1. Introduction
2. Methods
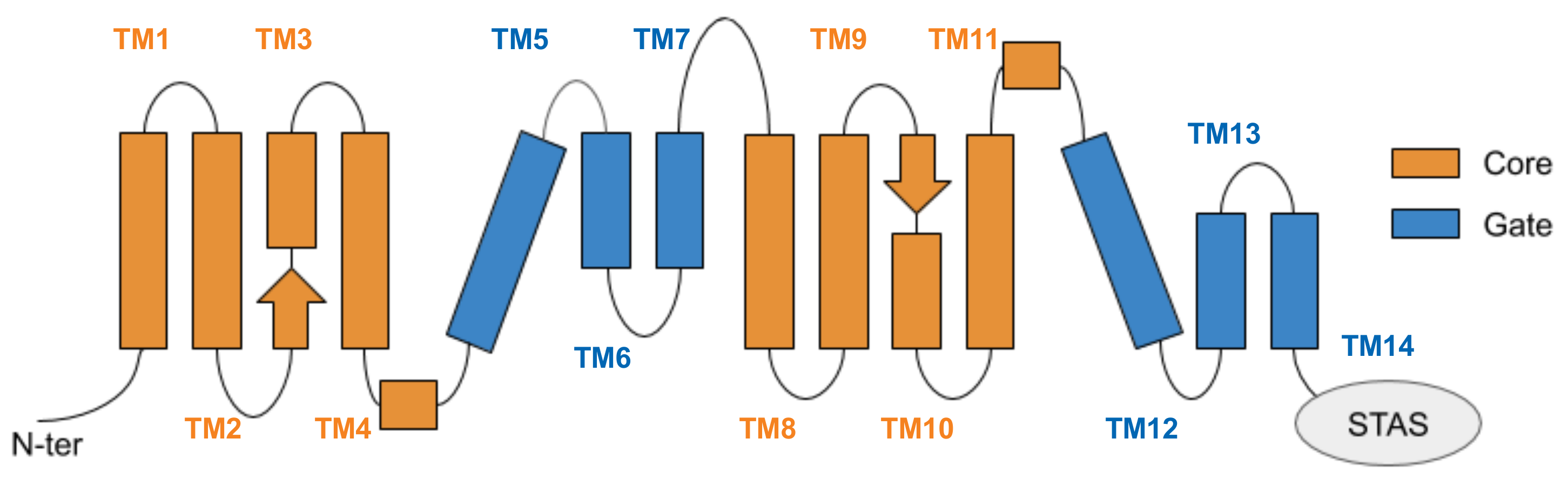
2.1. System Setup
2.2. Simulation Details
2.3. Analysis
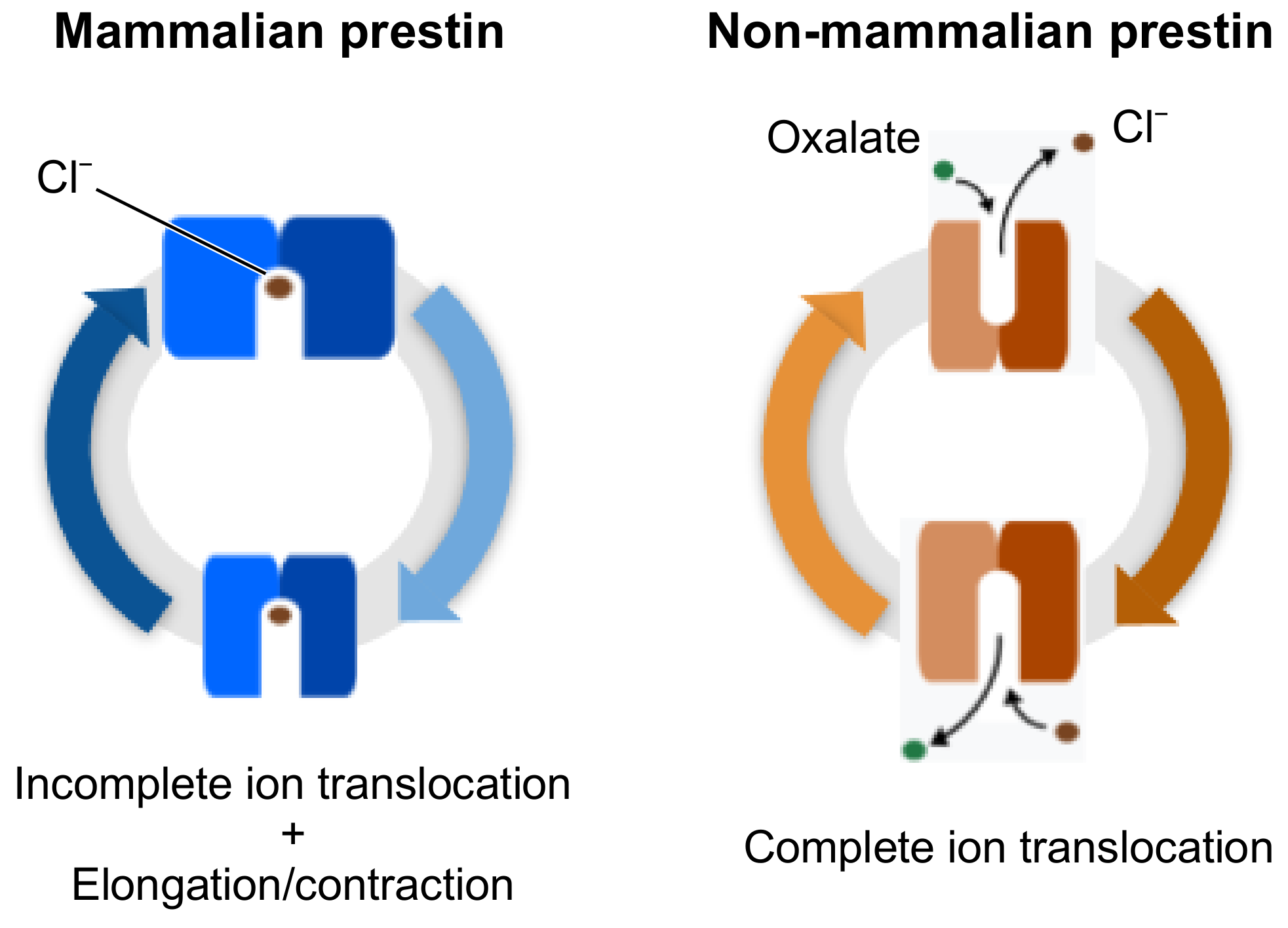
3. Results and Discussion
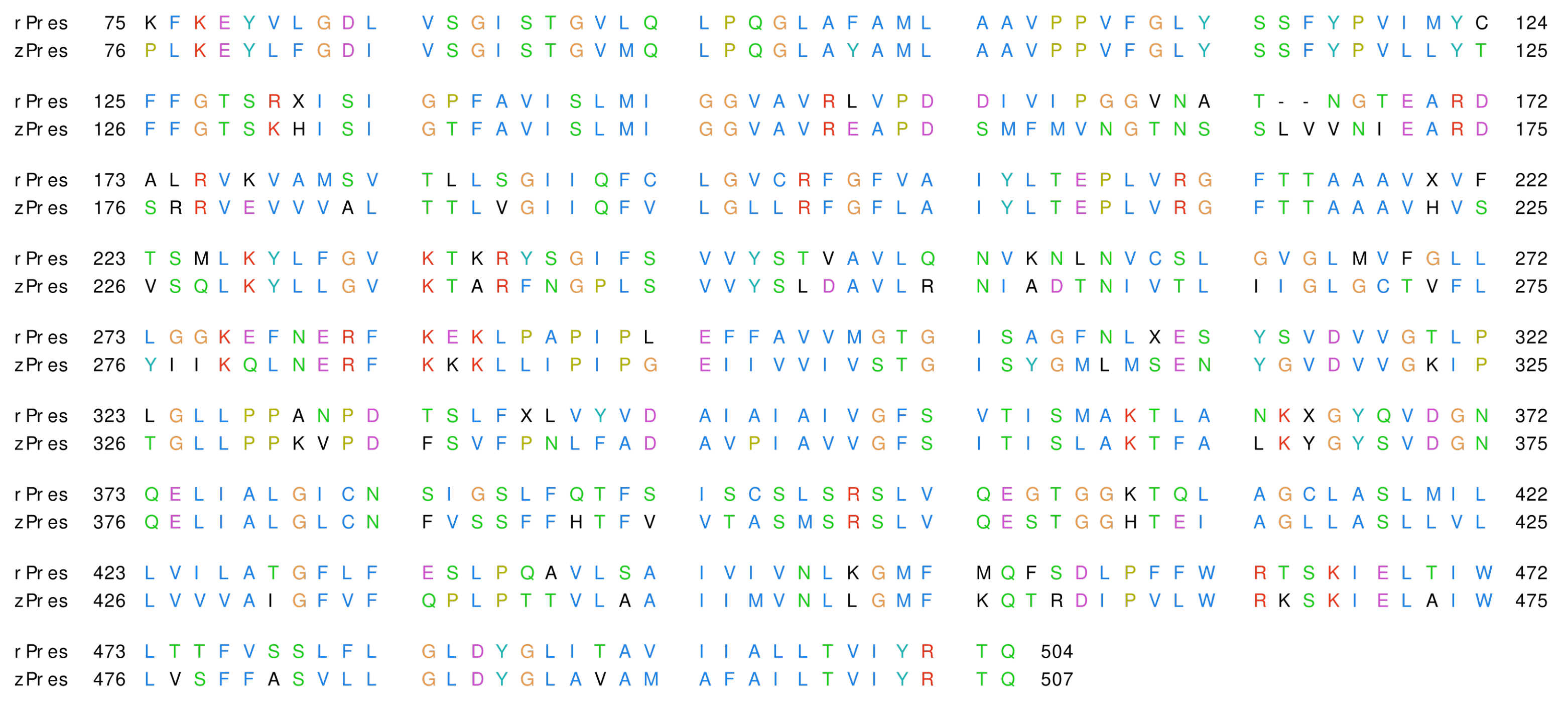
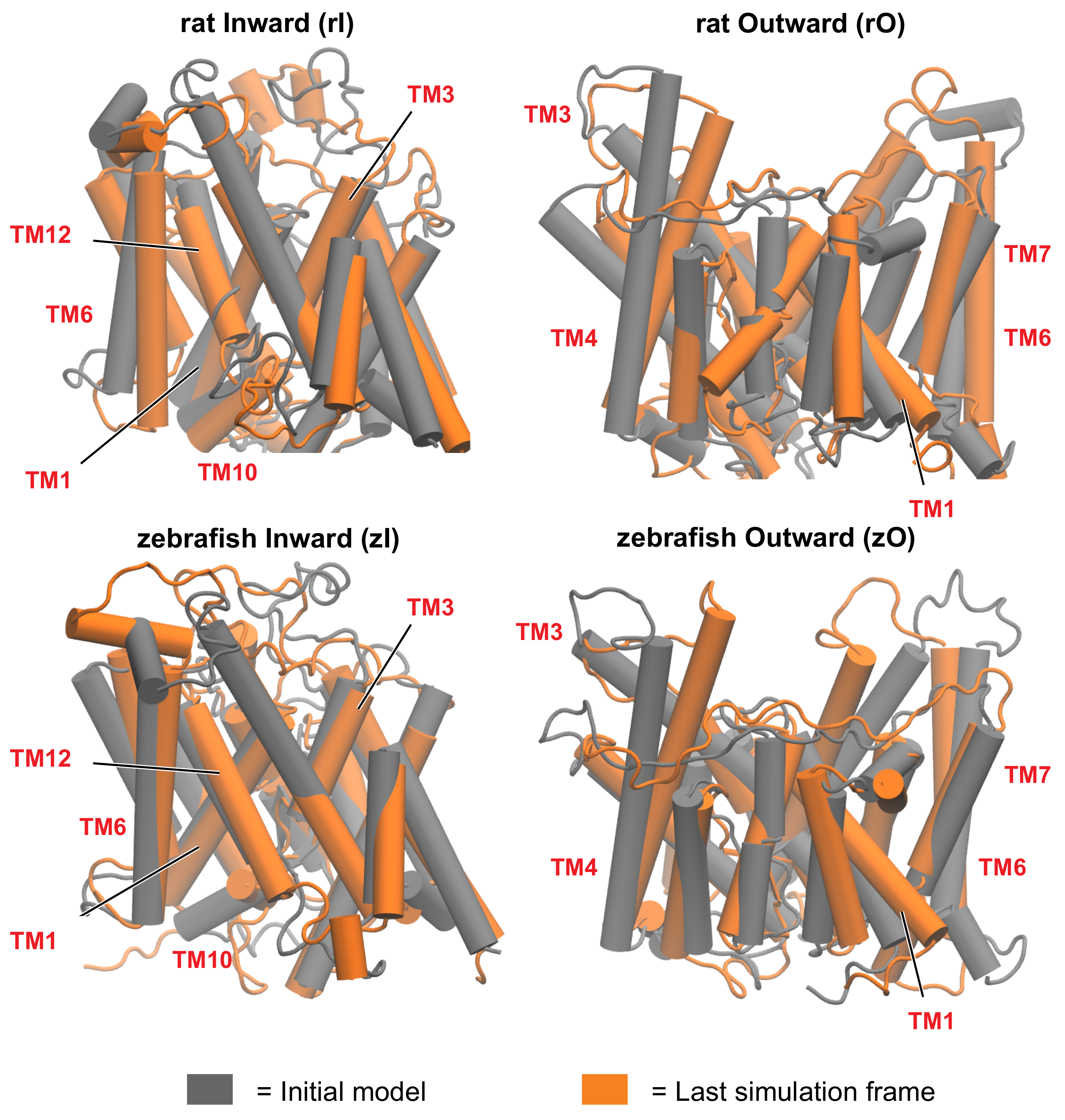
3.1. Structural Models and Validation
3.2. Conformational Variability
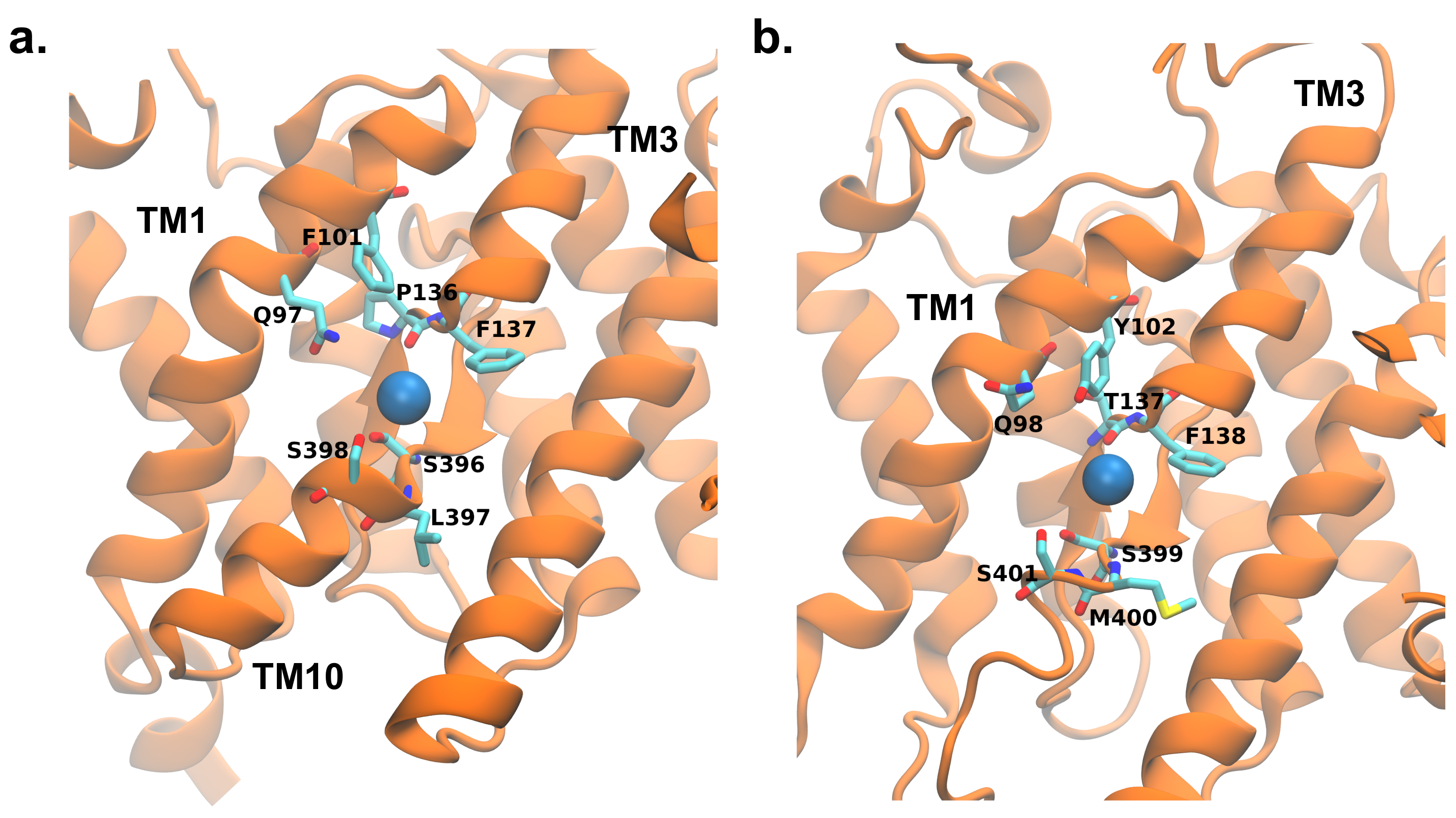
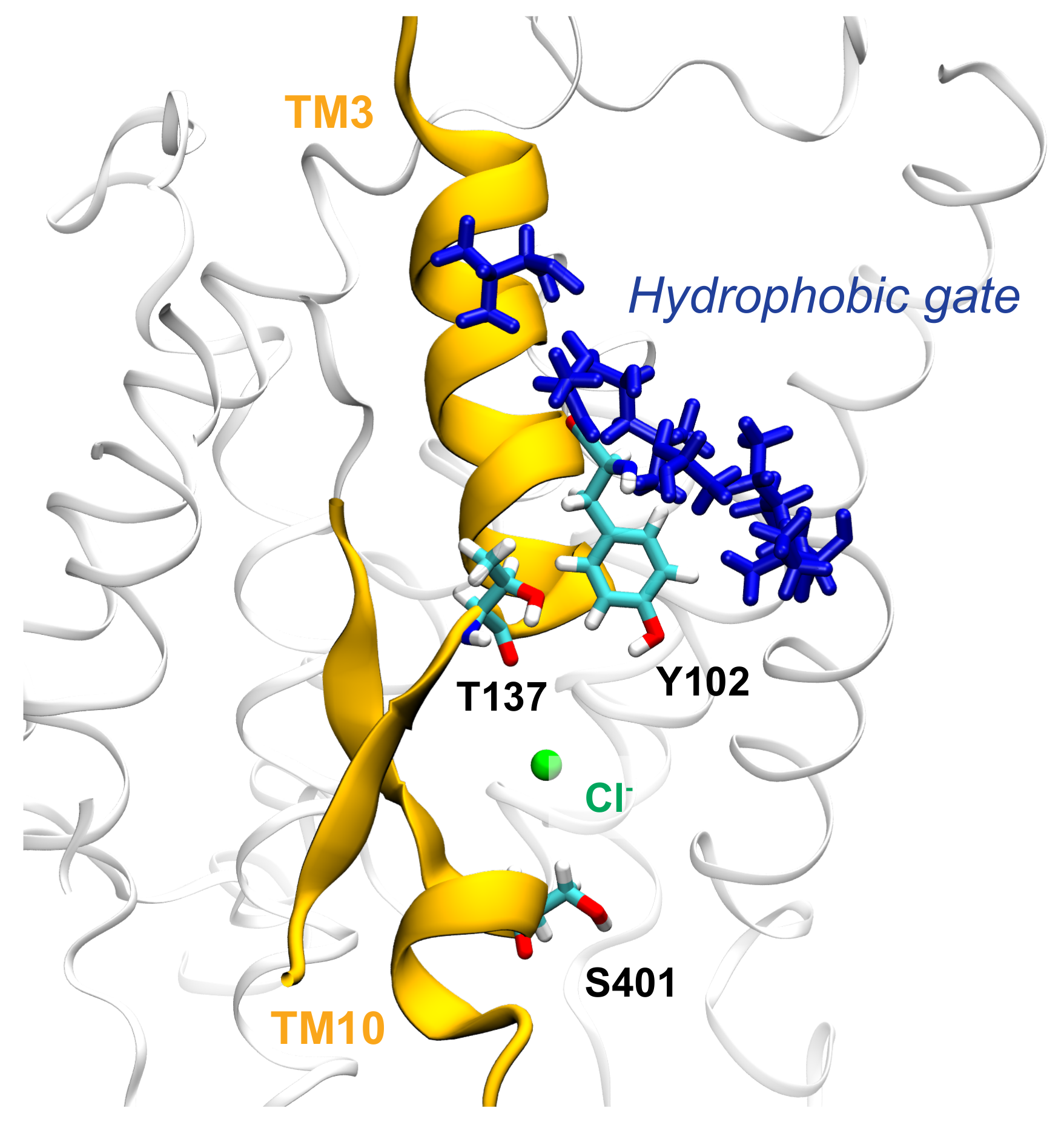
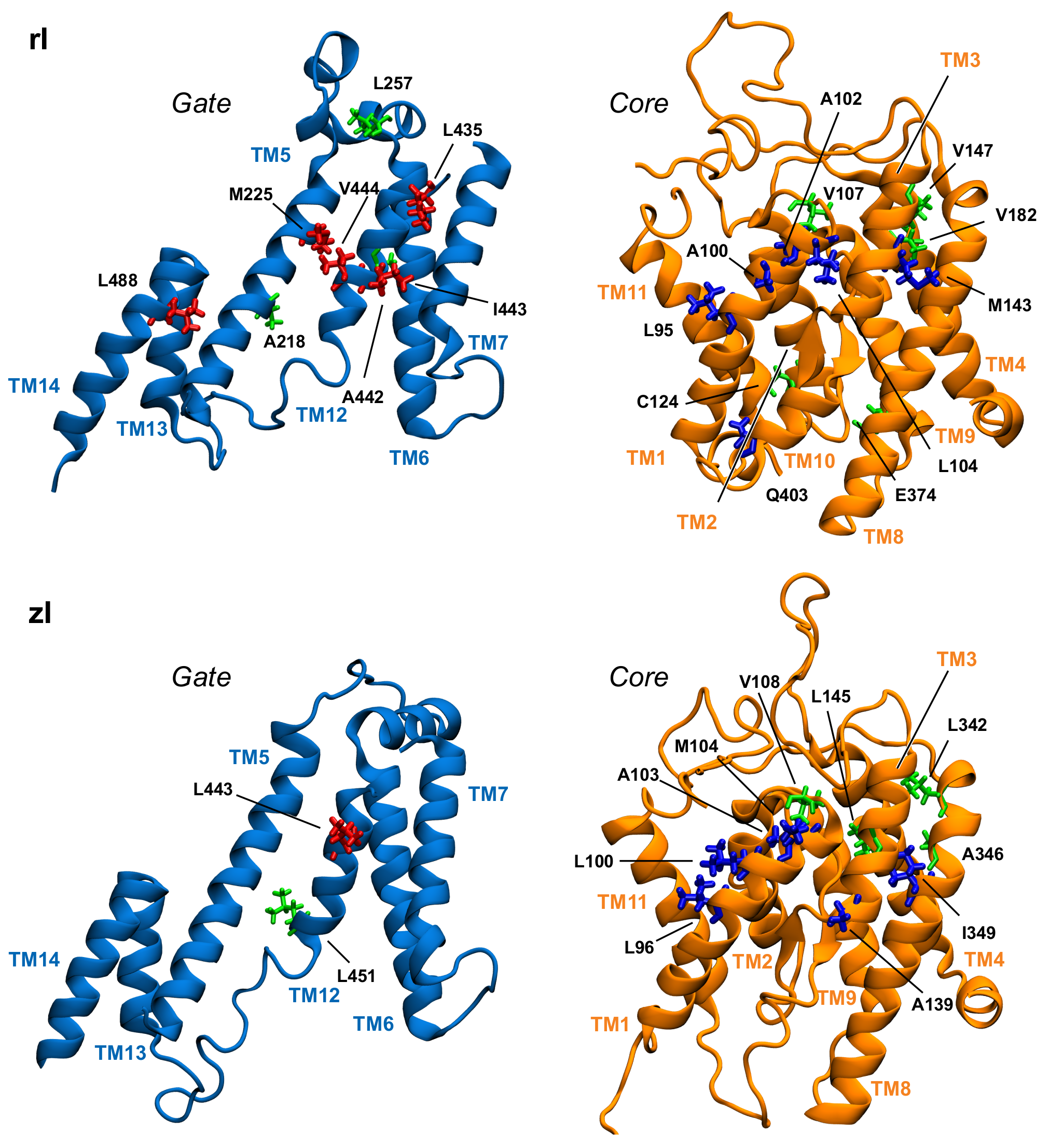
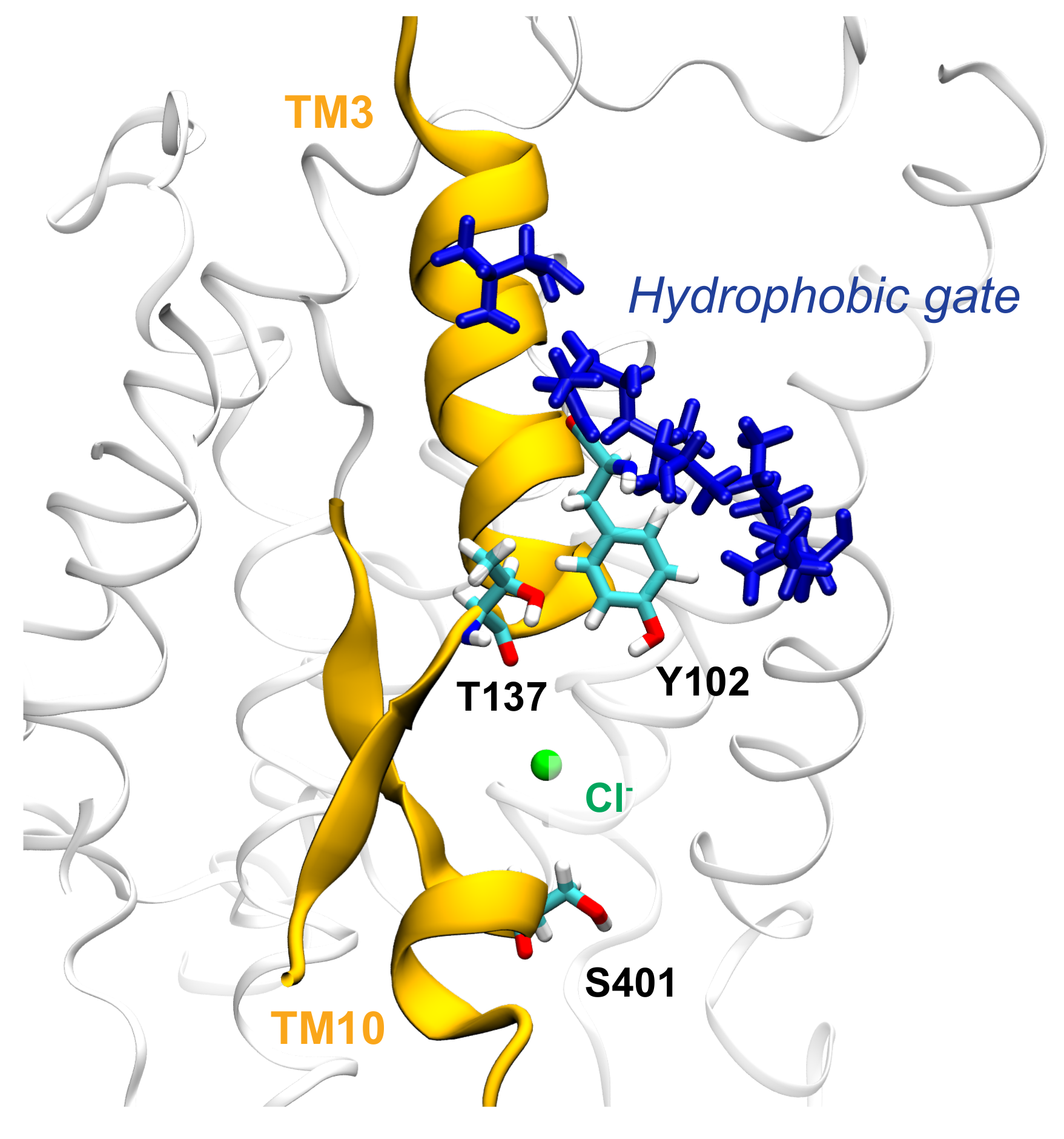
3.3. Key Residues in the Interaction Network
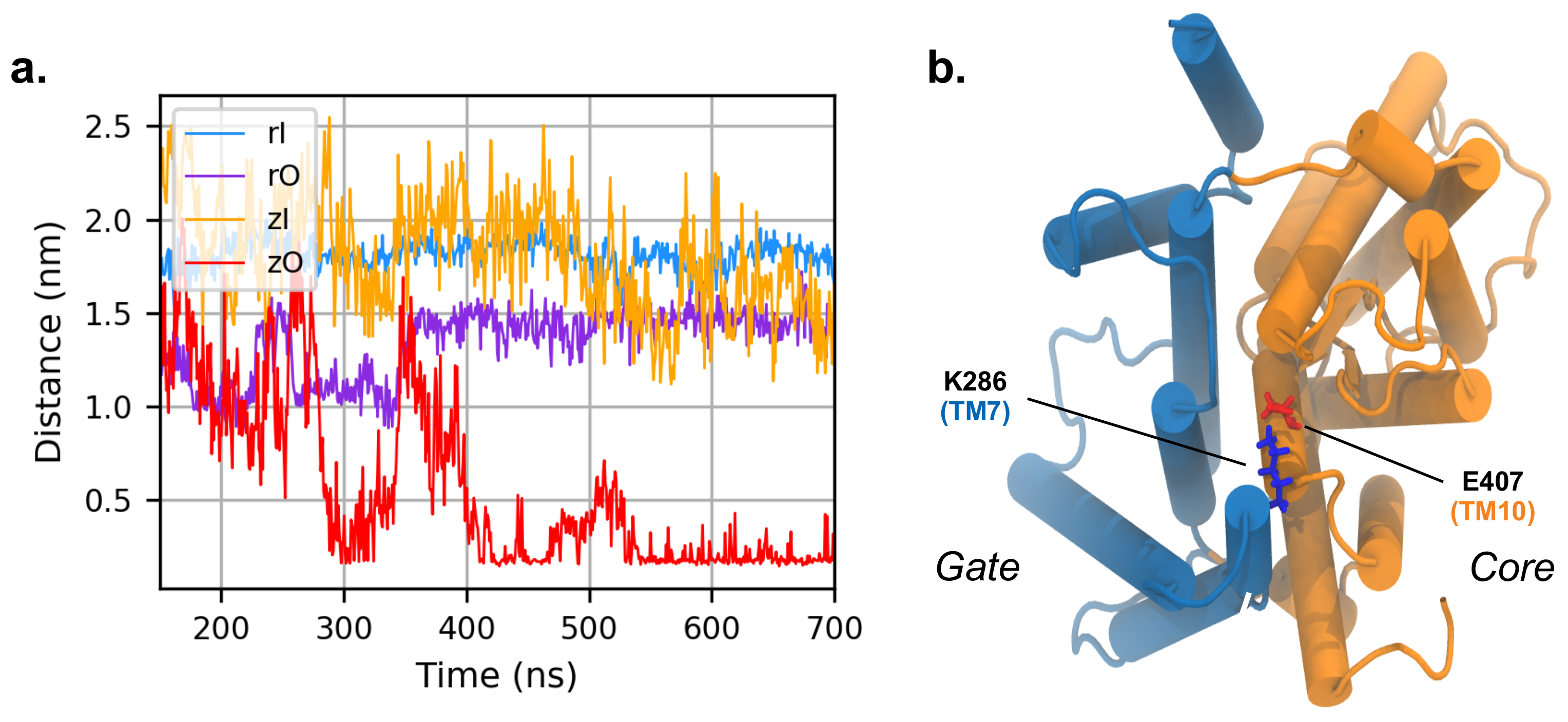
3.4. MD Simulations with an Applied Electric Field
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhang, Y.; Zhang, Y.; Sun, K.; Meng, Z.; Chen, L. The SLC transporter in nutrient and metabolic sensing, regulation, and drug development. J. Mol. Cell Biol. 2019, 11, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Lin, L.; Yee, S.W.; Kim, R.B.; Giacomini, K.M. SLC transporters as therapeutic targets: Emerging opportunities. Nat. Rev. Drug Discov. 2015, 14, 543–560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schumann, T.; König, J.; Henke, C.; Willmes, D.M.; Bornstein, S.R.; Jordan, J.; Fromm, M.F.; Birkenfeld, A.L. Solute Carrier Transporters as Potential Targets for the Treatment of Metabolic Disease. Pharmacol. Rev. 2020, 72, 343–379. [Google Scholar] [CrossRef] [PubMed]
- Alexander, S.P.; Kelly, E.; Mathie, A.; Peters, J.A.; Veale, E.L.; Armstrong, J.F.; Faccenda, E.; Harding, S.D.; Pawson, A.J.; Sharman, J.L.; et al. The concise guide to pharmacology 2019/20: Transporters. Br. J. Pharmacol. 2019, 176, S397–S493. [Google Scholar] [CrossRef] [PubMed]
- Scalise, M.; Pochini, L.; Console, L.; Losso, M.A.; Indiveri, C. The human SLC1A5 (ASCT2) amino acid transporter: From function to structure and role in cell biology. Front. Cell Dev. Biol. 2018, 6, 96. [Google Scholar] [CrossRef]
- Scimemi, A. Structure, function, and plasticity of GABA transporters. Front. Cell. Neurosci. 2014, 8, 161. [Google Scholar] [CrossRef] [Green Version]
- Arakawa, T.; Kobayashi-Yurugi, T.; Alguel, Y.; Iwanari, H.; Hatae, H.; Iwata, M.; Abe, Y.; Hino, T.; Ikeda-Suno, C.; Kuma, H.; et al. Crystal structure of the anion exchanger domain of human erythrocyte band 3. Science 2015, 350, 680–684. [Google Scholar] [CrossRef] [Green Version]
- Rapp, C.; Bai, X.; Reithmeier, R.A. Molecular analysis of human solute carrier SLC26 anion transporter disease-causing mutations using 3-dimensional homology modeling. Biochim. Biophys. Acta (BBA) Biomembr. 2017, 1859, 2420–2434. [Google Scholar] [CrossRef]
- Walter, J.D.; Sawicka, M.; Dutzler, R. Cryo-EM structures and functional characterization of murine Slc26a9 reveal mechanism of uncoupled chloride transport. Elife 2019, 8, e46986. [Google Scholar] [CrossRef]
- Deng, W.; Nies, F.; Feuer, A.; Bočina, I.; Oliver, D.; Jiang, D. Anion translocation through an Slc26 transporter mediates lumen expansion during tubulogenesis. Proc. Natl. Acad. Sci. USA 2013, 110, 14972–14977. [Google Scholar] [CrossRef] [Green Version]
- Alper, S.L.; Sharma, A.K. The SLC26 gene family of anion transporters and channels. Mol. Asp. Med. 2013, 34, 494–515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, Y.N.; Jaumann, E.A.; Reichel, K.; Hartmann, J.; Oliver, D.; Hummer, G.; Joseph, B.; Geertsma, E.R. Structural basis for functional interactions in dimers of SLC26 transporters. Nat. Commun. 2019, 10, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oliver, D.; He, D.Z.; Klöcker, N.; Ludwig, J.; Schulte, U.; Waldegger, S.; Ruppersberg, J.P.; Dallos, P.; Fakler, B. Intracellular anions as the voltage sensor of prestin, the outer hair cell motor protein. Science 2001, 292, 2340–2343. [Google Scholar] [CrossRef] [PubMed]
- Schaechinger, T.J.; Oliver, D. Nonmammalian orthologs of prestin (SLC26A5) are electrogenic divalent/chloride anion exchangers. Proc. Natl. Acad. Sci. USA 2007, 104, 7693–7698. [Google Scholar] [CrossRef] [Green Version]
- Bai, J.P.; Surguchev, A.; Montoya, S.; Aronson, P.S.; Santos-Sacchi, J.; Navaratnam, D. Prestin’s Anion Transport and Voltage-Sensing Capabilities Are Independent. Biophys. J. 2009, 96, 3179–3186. [Google Scholar] [CrossRef] [Green Version]
- Rajagopalan, L.; Patel, N.; Madabushi, S.; Goddard, J.A.; Anjan, V.; Lin, F.; Shope, C.; Farrell, B.; Lichtarge, O.; Davidson, A.L.; et al. Essential Helix Interactions in the Anion Transporter Domain of Prestin Revealed by Evolutionary Trace Analysis. J. Neurosci. 2006, 26, 12727–12734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schaechinger, T.J.; Gorbunov, D.; Halaszovich, C.R.; Moser, T.; Kügler, S.; Fakler, B.; Oliver, D. A synthetic prestin reveals protein domains and molecular operation of outer hair cell piezoelectricity. EMBO J. 2011, 30, 2793–2804. [Google Scholar] [CrossRef] [Green Version]
- Gorbunov, D.; Sturlese, M.; Nies, F.; Kluge, M.; Bellanda, M.; Battistutta, R.; Oliver, D. Molecular architecture and the structural basis for anion interaction in prestin and SLC26 transporters. Nat. Commun. 2014, 5, 3622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santos-Sacchi, J.; Tan, W. The Frequency Response of Outer Hair Cell Voltage-Dependent Motility Is Limited by Kinetics of Prestin. J. Neurosci. Off. J. Soc. Neurosci. 2018, 38, 5495–5506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brownell, W.E.; Bader, C.R.; Bertrand, D.; Ribaupierre, Y.D. Evoked mechanical responses of isolated cochlear outer hair cells. Science 1985, 227, 194–196. [Google Scholar] [CrossRef] [PubMed]
- Dallos, P. Cochlear amplification, outer hair cells and prestin. Curr. Opin. Neurobiol. 2008, 18, 370–376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hudspeth, A. Making an effort to listen: Mechanical amplification in the ear. Neuron 2008, 59, 530–545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ashmore, J. Outer hair cells and electromotility. Cold Spring Harb. Perspect. Med. 2019, 9, a033522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dallos, P.; Fakler, B. Prestin, a new type of motor protein. Nat. Rev. Mol. Cell Biol. 2002, 3, 104–111. [Google Scholar] [CrossRef]
- Ashmore, J.F. A fast motile response in guinea-pig outer hair cells: The cellular basis of the cochlear amplifier. J. Physiol. 1987, 388, 323–347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stelma, F.; Bhutta, M. Non-syndromic hereditary sensorineural hearing loss: Review of the genes involved. J. Laryngol. Otol. 2014, 128, 13. [Google Scholar] [CrossRef]
- Santos-Sacchi, J. Reversible inhibition of voltage-dependent outer hair cell motility and capacitance. J. Neurosci. 1991, 11, 3096–3110. [Google Scholar] [CrossRef]
- Chi, X.; Jin, X.; Chen, Y.; Lu, X.; Tu, X.; Li, X.; Zhang, Y.; Lei, J.; Huang, J.; Huang, Z.; et al. Structural insights into the gating mechanism of human SLC26A9 mediated by its C-terminal sequence. Cell Discov. 2020, 6, 1–10. [Google Scholar] [CrossRef]
- Lovas, S.; He, D.Z.Z.; Liu, H.; Tang, J.; Pecka, J.L.; Hatfield, M.P.D.; Beisel, K.W. Glutamate Transporter Homolog-based Model Predicts That Anion-π Interaction Is the Mechanism for the Voltage-dependent Response of Prestin. J. Biol. Chem. 2015, 290, 24326–24339. [Google Scholar] [CrossRef] [Green Version]
- Geertsma, E.R.; Chang, Y.N.; Shaik, F.R.; Neldner, Y.; Pardon, E.; Steyaert, J.; Dutzler, R. Structure of a prokaryotic fumarate transporter reveals the architecture of the SLC26 family. Nat. Struct. Mol. Biol. 2015, 22, 803–808. [Google Scholar] [CrossRef]
- Kelley, L.A.; Mezulis, S.; Yates, C.M.; Wass, M.N.; Sternberg, M.J. The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 2015, 10, 845–858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molecular Operating Environment (MOE) 2019.01. Chemical Computing Group ULC. Available online: https://www.chemcomp.com/index.htm (accessed on 26 July 2021).
- Labute, P. Protonate3D: Assignment of ionization states and hydrogen coordinates to macromolecular structures. Proteins: Struct. Funct. Bioinform. 2009, 75, 187–205. [Google Scholar] [CrossRef] [Green Version]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Van Meer, G.; Voelker, D.R.; Feigenson, G.W. Membrane lipids: Where they are and how they behave. Nat. Rev. Mol. Cell Biol. 2008, 9, 112–124. [Google Scholar] [CrossRef] [PubMed]
- Enkavi, G.; Javanainen, M.; Kulig, W.; Róg, T.; Vattulainen, I. Multiscale simulations of biological membranes: The challenge to understand biological phenomena in a living substance. Chem. Rev. 2019, 119, 5607–5774. [Google Scholar] [CrossRef] [Green Version]
- Lomize, M.A.; Pogozheva, I.D.; Joo, H.; Mosberg, H.I.; Lomize, A.L. OPM database and PPM web server: Resources for positioning of proteins in membranes. Nucleic Acids Res. 2012, 40, D370–D376. [Google Scholar] [CrossRef]
- Price, D.J.; Brooks, C.L., III. A modified TIP3P water potential for simulation with Ewald summation. J. Chem. Phys. 2004, 121, 10096–10103. [Google Scholar] [CrossRef] [PubMed]
- Phillips, J.C.; Braun, R.; Wang, W.; Gumbart, J.; Tajkhorshid, E.; Villa, E.; Chipot, C.; Skeel, R.D.; Kalé, L.; Schulten, K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005, 26, 1781–1802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, J.; Rauscher, S.; Nawrocki, G.; Ran, T.; Feig, M.; de Groot, B.L.; Grubmüller, H.; MacKerell, A.D., Jr. CHARMM36m: An improved force field for folded and intrinsically disordered proteins. Nat. Methods 2016, 14, 71. [Google Scholar] [CrossRef] [Green Version]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An Nlog (N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef] [Green Version]
- Adelman, S.; Doll, J. Generalized Langevin equation approach for atom/solid-surface scattering: General formulation for classical scattering off harmonic solids. J. Chem. Phys. 1976, 64, 2375–2388. [Google Scholar] [CrossRef]
- Martyna, G.J.; Tobias, D.J.; Klein, M.L. Constant pressure molecular dynamics algorithms. J. Chem. Phys. 1994, 101, 4177–4189. [Google Scholar] [CrossRef]
- Feller, S.E.; Zhang, Y.; Pastor, R.W.; Brooks, B.R. Constant pressure molecular dynamics simulation: The Langevin piston method. J. Chem. Phys. 1995, 103, 4613–4621. [Google Scholar] [CrossRef]
- Ryckaert, J.P.; Ciccotti, G.; Berendsen, H.J.C. Numerical Integration of the cartesian equations of motion of a system with constraints: Molecular dynamics of n-alkanes. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar] [CrossRef] [Green Version]
- Torgerson, W.S. Multidimensional scaling: I. Theory and method. Psychometrika 1952, 17, 401–419. [Google Scholar] [CrossRef]
- Yu, G.; Sapiro, G.; Mallat, S. Solving inverse problems with piecewise linear estimators: From Gaussian mixture models to structured sparsity. IEEE Trans. Image Process. 2011, 21, 2481–2499. [Google Scholar] [PubMed] [Green Version]
- Schwarz, G. Estimating the dimension of a model. Ann. Stat. 1978, 6, 461–464. [Google Scholar] [CrossRef]
- Tiberti, M.; Invernizzi, G.; Lambrughi, M.; Inbar, Y.; Schreiber, G.; Papaleo, E. PyInteraph: A Framework for the Analysis of Interaction Networks in Structural Ensembles of Proteins. J. Chem. Inf. Model. 2014, 54, 1537–1551. [Google Scholar] [CrossRef] [PubMed]
- Vishveshwara, S.; Ghosh, A.; Hansia, P. Intra and inter-molecular communications through protein structure network. Curr. Protein Pept. Sci. 2009, 10, 146–160. [Google Scholar] [CrossRef]
- Michaud-Agrawal, N.; Denning, E.J.; Woolf, T.B.; Beckstein, O. MDAnalysis: A toolkit for the analysis of molecular dynamics simulations. J. Comput. Chem. 2011, 32, 2319–2327. [Google Scholar] [CrossRef] [Green Version]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Li, G.H.; Huang, J.F.; Murphy, R.W.; Shi, P. Hearing aid for vertebrates via multiple episodic adaptive events on prestin genes. Mol. Biol. Evol. 2012, 29, 2187–2198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larkin, M.A.; Blackshields, G.; Brown, N.P.; Chenna, R.; McGettigan, P.A.; McWilliam, H.; Valentin, F.; Wallace, I.M.; Wilm, A.; Lopez, R.; et al. Clustal W and Clustal X version 2.0. bioinformatics 2007, 23, 2947–2948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, F.; Li, S.; Jiang, Y.; Jiang, J.; Fan, H.; Lu, G.; Deng, D.; Dang, S.; Zhang, X.; Wang, J.; et al. Structure and mechanism of the uracil transporter UraA. Nature 2011, 472, 243–246. [Google Scholar] [CrossRef] [PubMed]
- Drew, D.; Boudker, O. Shared molecular mechanisms of membrane transporters. Annu. Rev. Biochem. 2016, 85, 543–572. [Google Scholar] [CrossRef] [PubMed]
- Thurtle-Schmidt, B.H.; Stroud, R.M. Structure of Bor1 supports an elevator transport mechanism for SLC4 anion exchangers. Proc. Natl. Acad. Sci. USA 2016, 113, 10542–10546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ficici, E.; Faraldo-Gómez, J.D.; Jennings, M.L.; Forrest, L.R. Asymmetry of inverted-topology repeats in the AE1 anion exchanger suggests an elevator-like mechanism. J. Gen. Physiol. 2017, 149, 1149–1164. [Google Scholar] [CrossRef] [PubMed]
- Song, L.; Santos-Sacchi, J. Conformational State-Dependent Anion Binding in Prestin: Evidence for Allosteric Modulation. Biophys. J. 2010, 98, 371–376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gumbart, J.; Khalili-Araghi, F.; Sotomayor, M.; Roux, B. Constant electric field simulations of the membrane potential illustrated with simple systems. Biochim. Biophys. Acta (BBA) Biomembr. 2012, 1818, 294–302. [Google Scholar] [CrossRef] [Green Version]
- Le Guilloux, V.; Schmidtke, P.; Tuffery, P. Fpocket: An open source platform for ligand pocket detection. BMC Bioinform. 2009, 10, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Schmidtke, P.; Bidon-Chanal, A.; Luque, F.J.; Barril, X. MDpocket: Open-source cavity detection and characterization on molecular dynamics trajectories. Bioinformatics 2011, 27, 3276–3285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dutzler, R.; Campbell, E.B.; Cadene, M.; Chait, B.T.; MacKinnon, R. X-ray structure of a ClC chloride channel at 3.0 Å reveals the molecular basis of anion selectivity. Nature 2002, 415, 287–294. [Google Scholar] [CrossRef] [PubMed]
- Dani, J.A.; Sanchez, J.A.; Hille, B. Lyotropic anions. Na channel gating and Ca electrode response. J. Gen. Physiol. 1983, 81, 255–281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
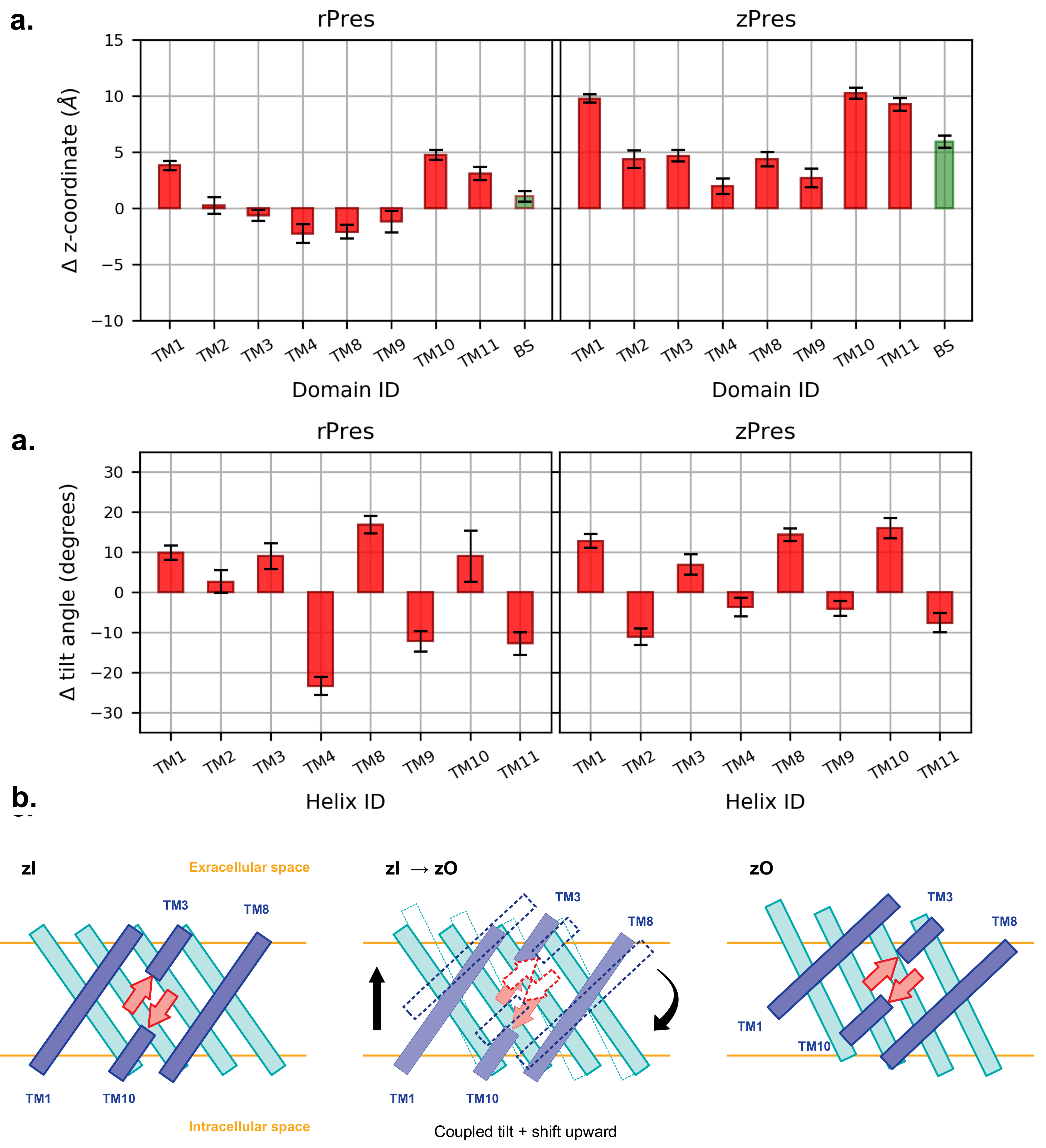
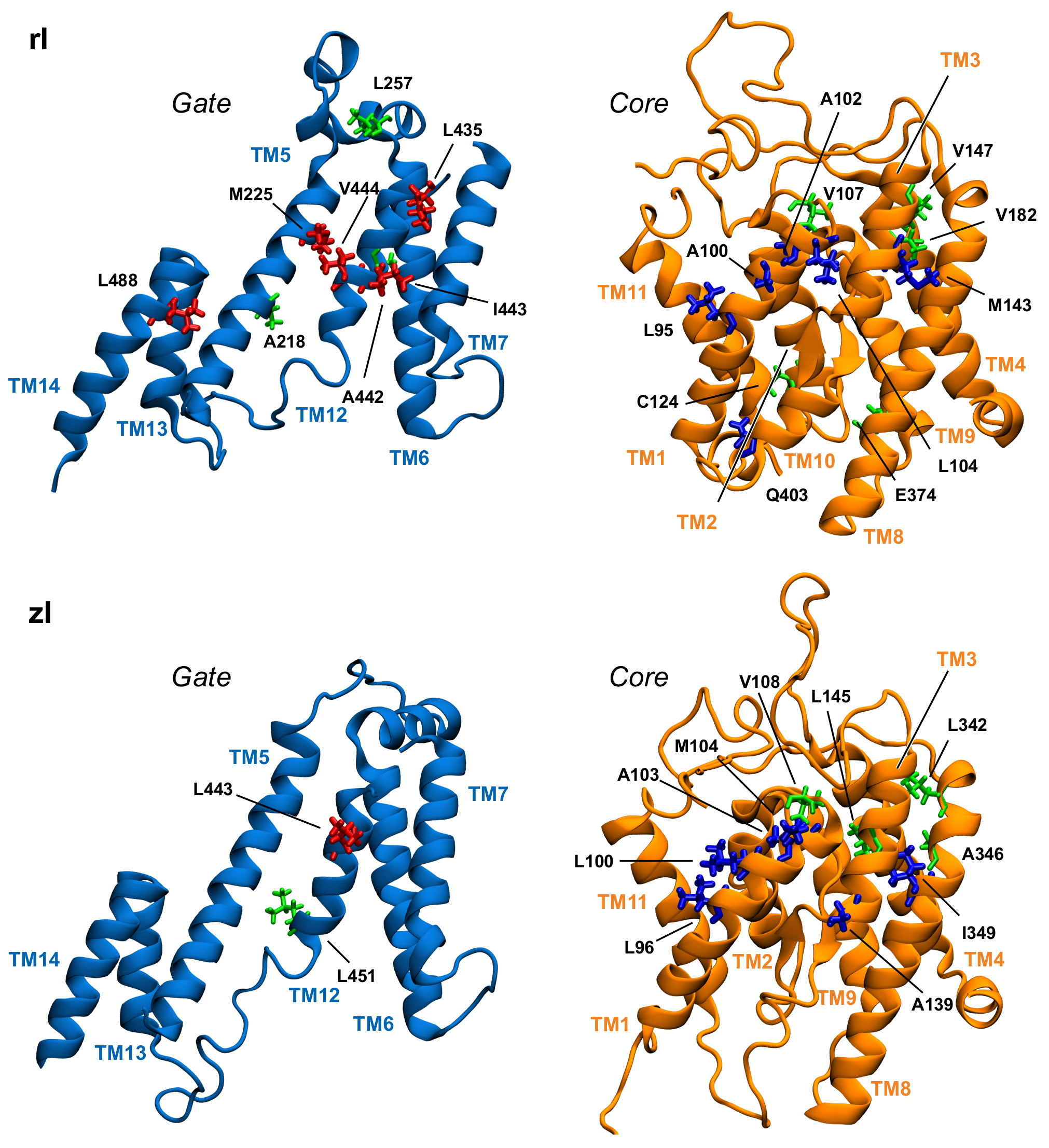
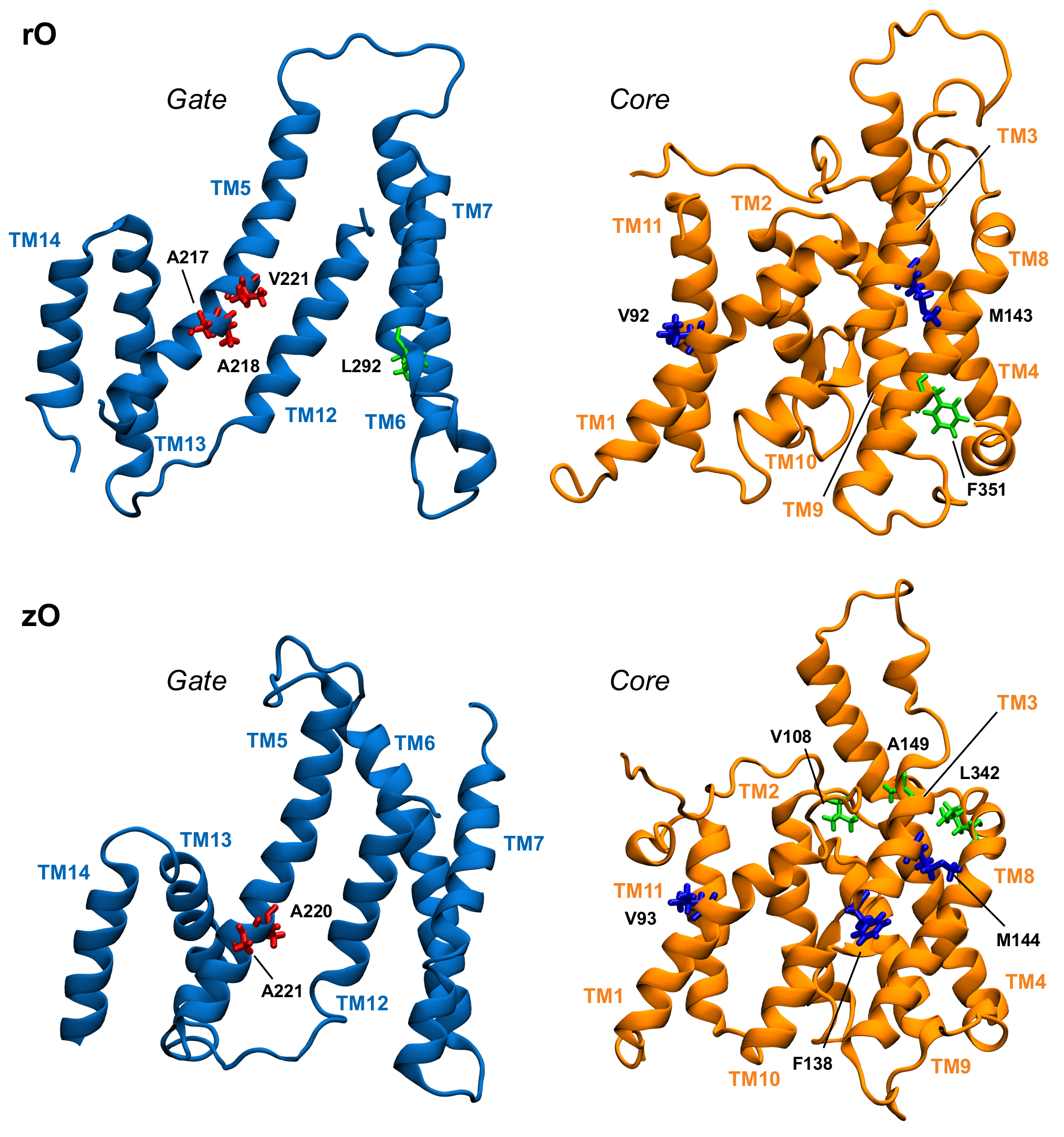
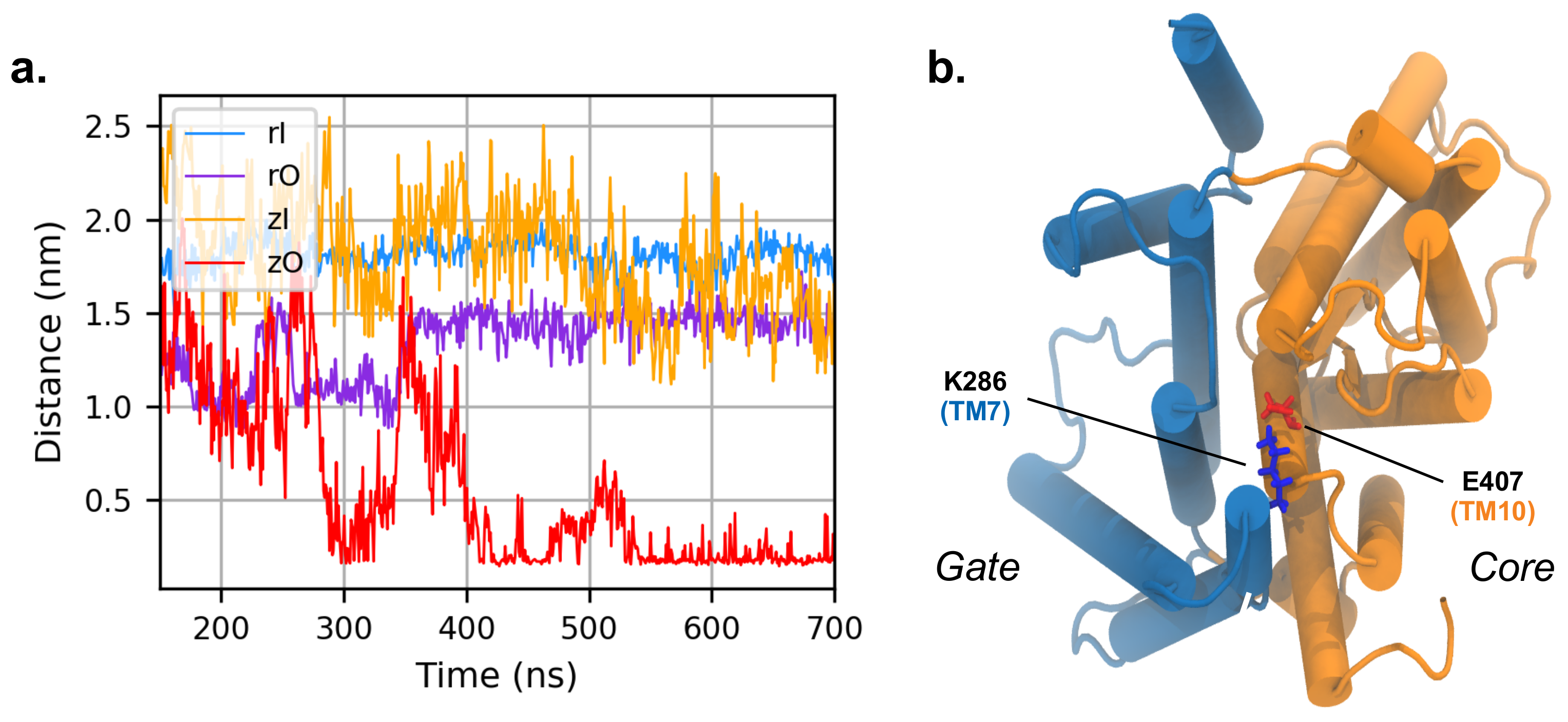

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Domain | rPres In | rPres Out | zPres In | zPres Out |
|---|---|---|---|---|
| Core | L95 (8) | V92 (7) | L100 (8) | V93 (8) |
| A100 (7) | M143 (7) | I145 (8) | V108 (7) | |
| A102 (7) | F351 (7) | A346 (8) | F138 (7) | |
| L104 (7) | L96 (7) | M144 (7) | ||
| V107 (7) | A103 (7) | A149 (7) | ||
| C124 (7) | M104 (7) | L342 (7) | ||
| M143 (7) | V108 (7) | |||
| V147 (7) | A139 (7) | |||
| V182 (7) | L342 (7) | |||
| E374 (7) | I349 (7) | |||
| Q403 (7) | ||||
| Gate | V444 (8) | A217 (8) | L443 (7) | A220 (7) |
| A218 (7) | A218 (8) | L451 (7) | A221 (7) | |
| M225 (7) | V221 (7) | |||
| L257 (7) | L292 (7) | |||
| L435 (7) | ||||
| A442 (7) | ||||
| I443 (7) | ||||
| L488 (7) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abrusci, G.; Tarenzi, T.; Sturlese, M.; Giachin, G.; Battistutta, R.; Lattanzi, G. Comparative Molecular Dynamics Investigation of the Electromotile Hearing Protein Prestin. Int. J. Mol. Sci. 2021, 22, 8318. https://doi.org/10.3390/ijms22158318
Abrusci G, Tarenzi T, Sturlese M, Giachin G, Battistutta R, Lattanzi G. Comparative Molecular Dynamics Investigation of the Electromotile Hearing Protein Prestin. International Journal of Molecular Sciences. 2021; 22(15):8318. https://doi.org/10.3390/ijms22158318
Chicago/Turabian StyleAbrusci, Gianfranco, Thomas Tarenzi, Mattia Sturlese, Gabriele Giachin, Roberto Battistutta, and Gianluca Lattanzi. 2021. "Comparative Molecular Dynamics Investigation of the Electromotile Hearing Protein Prestin" International Journal of Molecular Sciences 22, no. 15: 8318. https://doi.org/10.3390/ijms22158318
APA StyleAbrusci, G., Tarenzi, T., Sturlese, M., Giachin, G., Battistutta, R., & Lattanzi, G. (2021). Comparative Molecular Dynamics Investigation of the Electromotile Hearing Protein Prestin. International Journal of Molecular Sciences, 22(15), 8318. https://doi.org/10.3390/ijms22158318