
Computational Investigation of the pH Dependence of Stability of Melanosome Proteins: Implication for Melanosome formation and Disease

 ,
,  ,
, 
Abstract
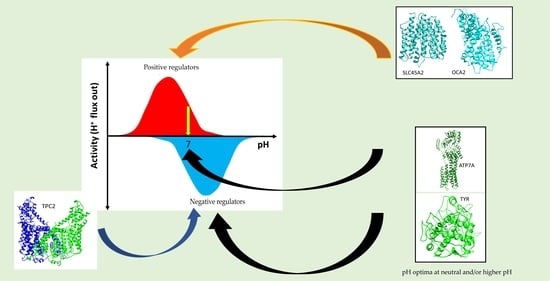
:
1. Introduction
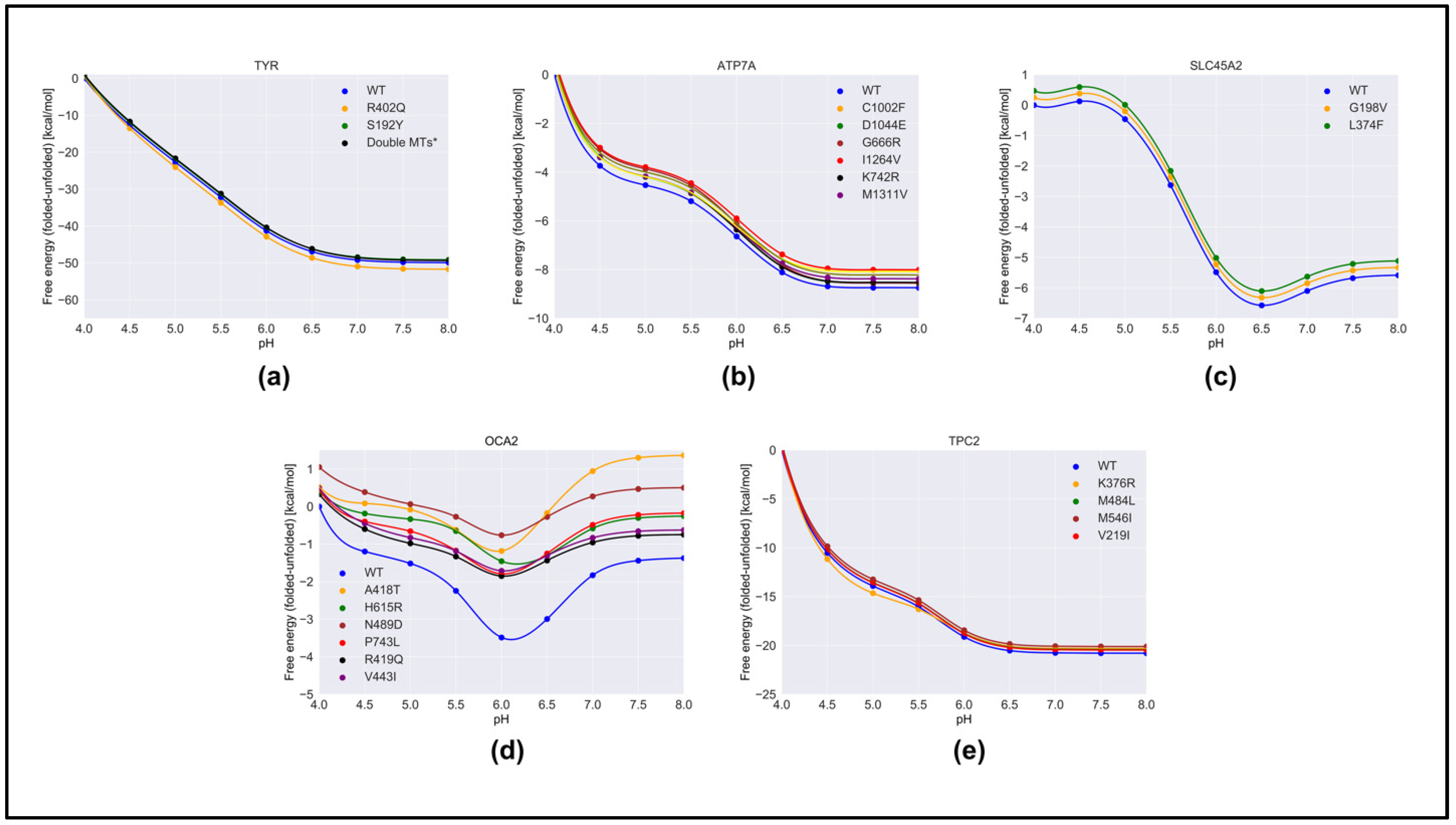
2. Results
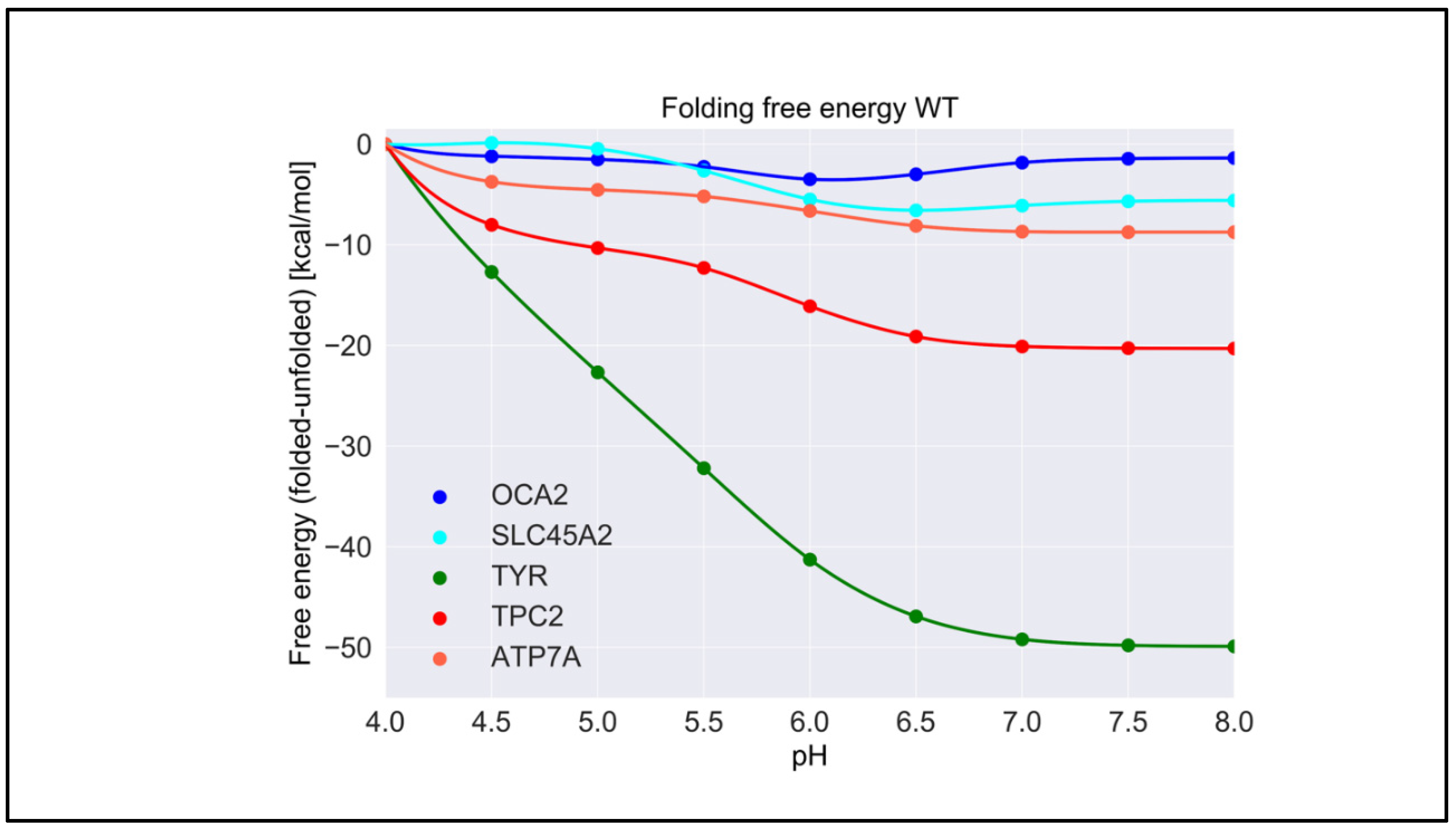
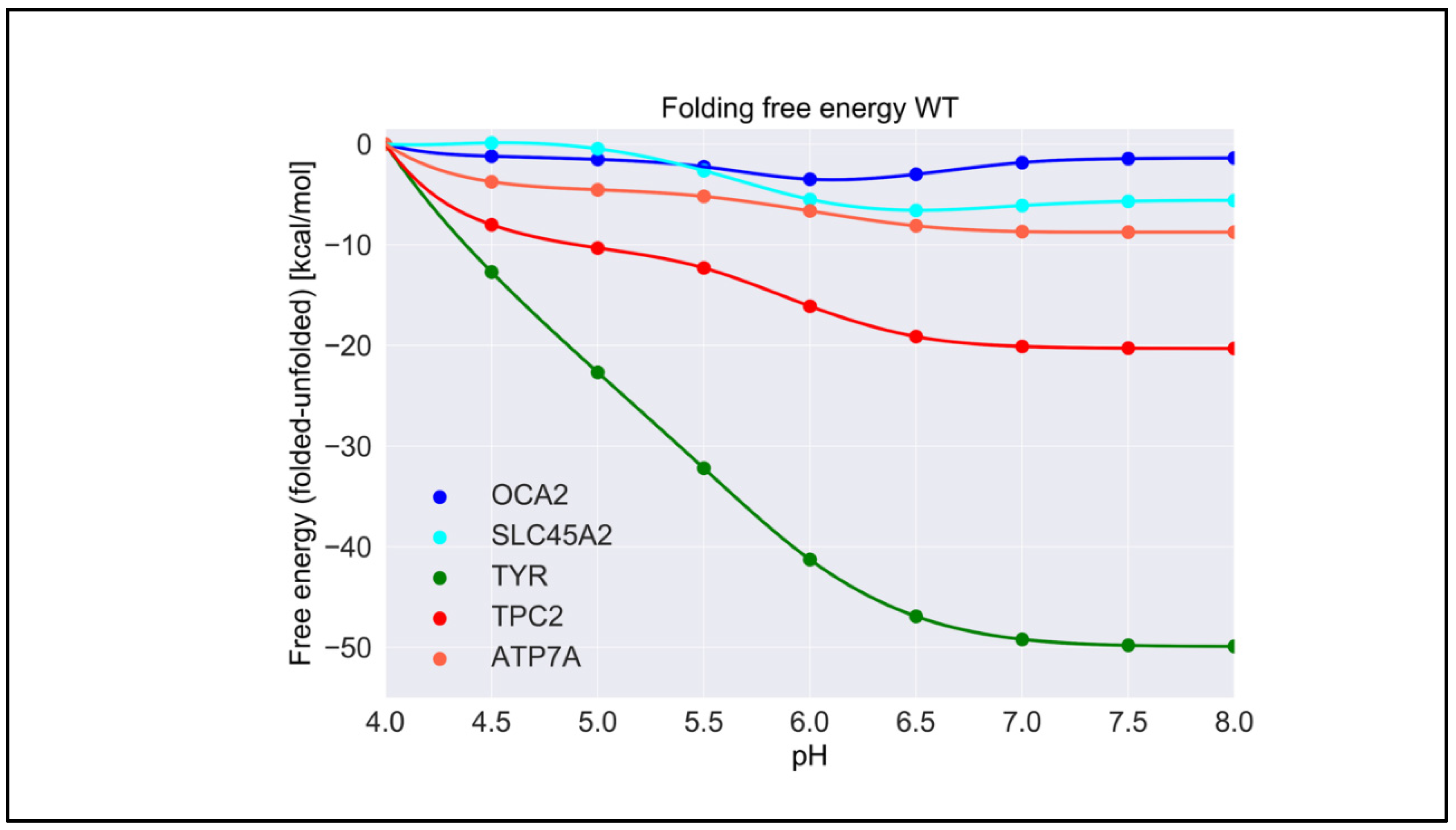
2.1. pH Dependence of Folding Free Energy on Wild-Type Proteins
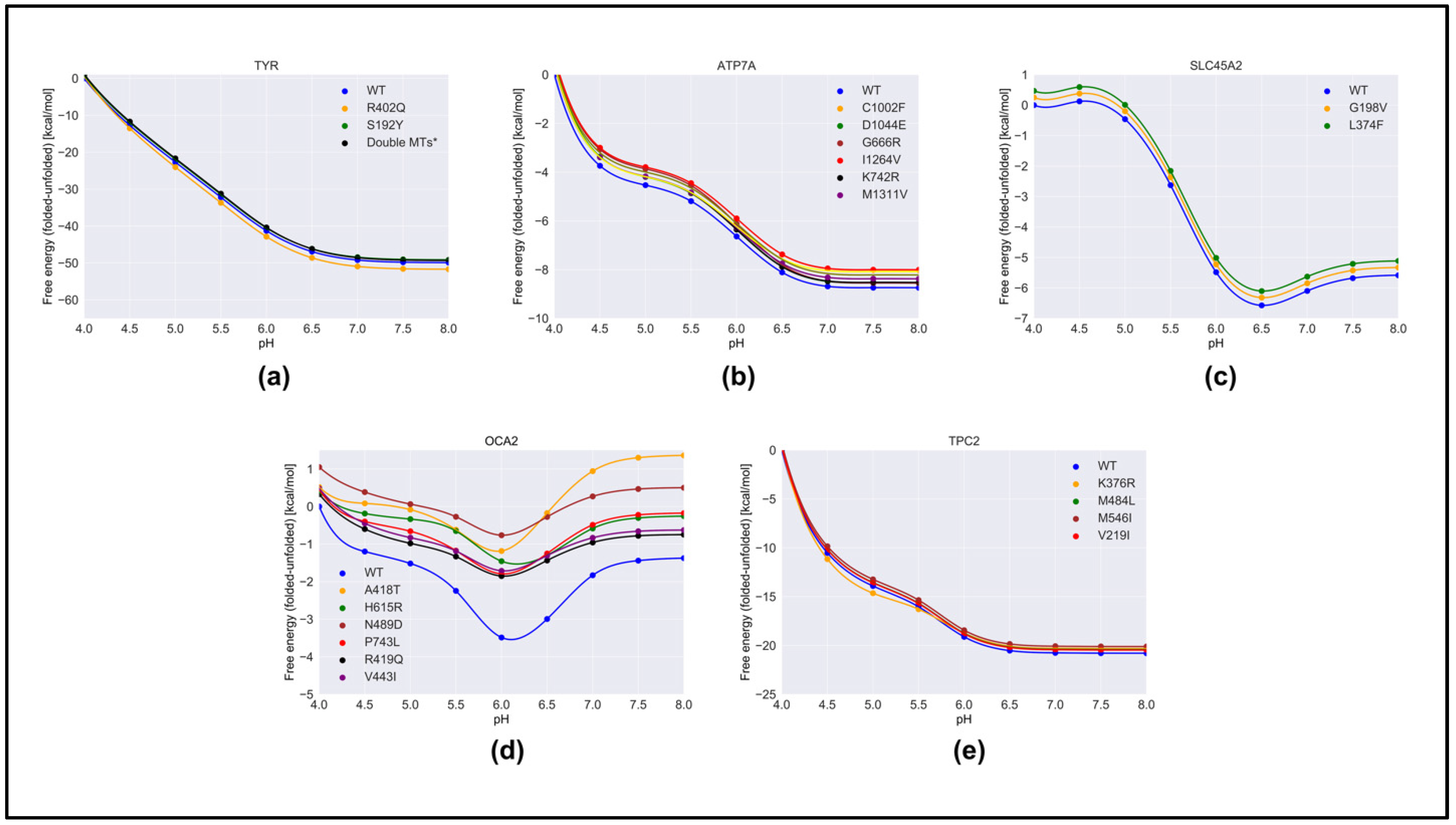
2.2. Effect of Pathogenic Variants on Protein Stability
2.3. pH Dependence of Folding Free Energy on Genetic Variation
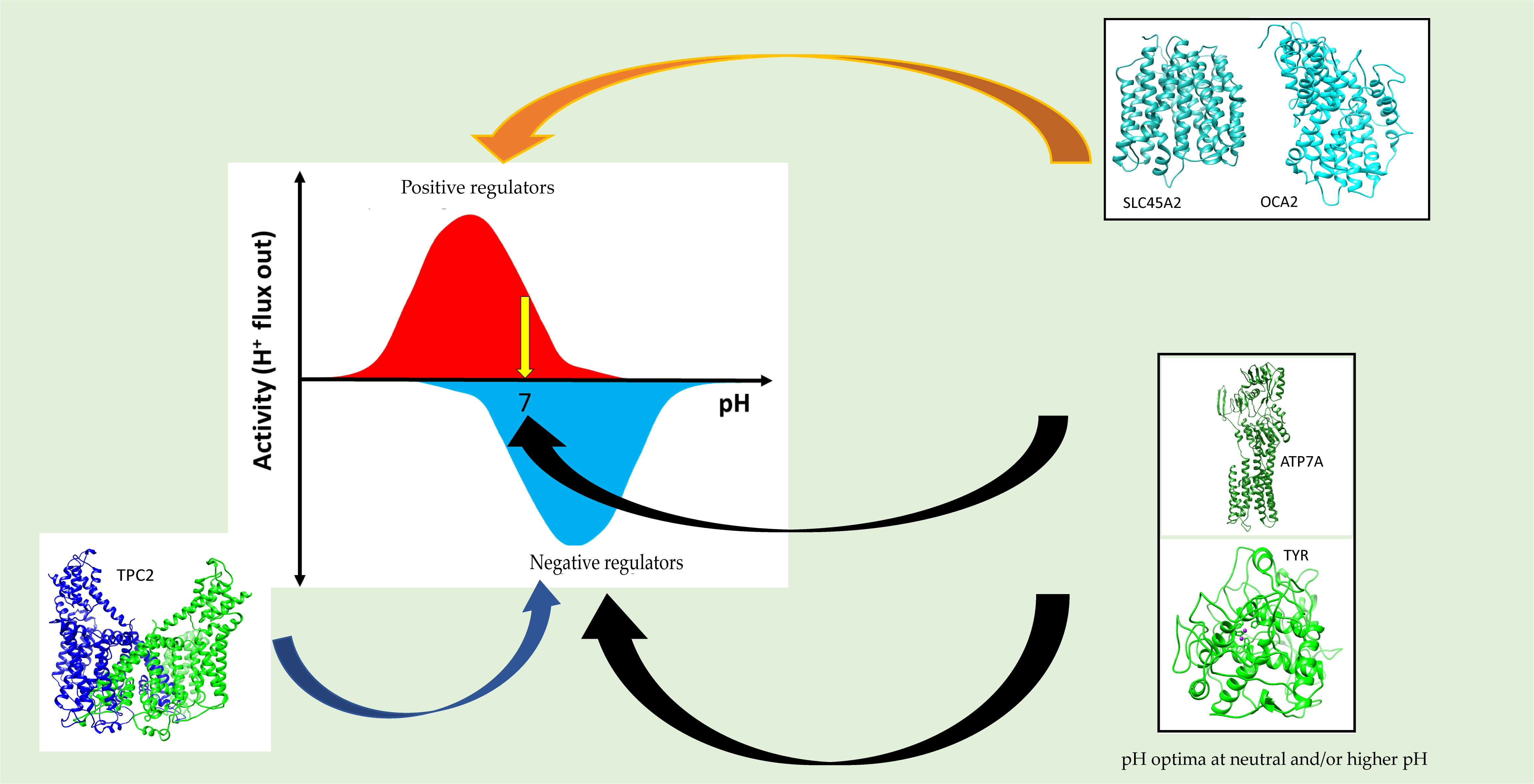
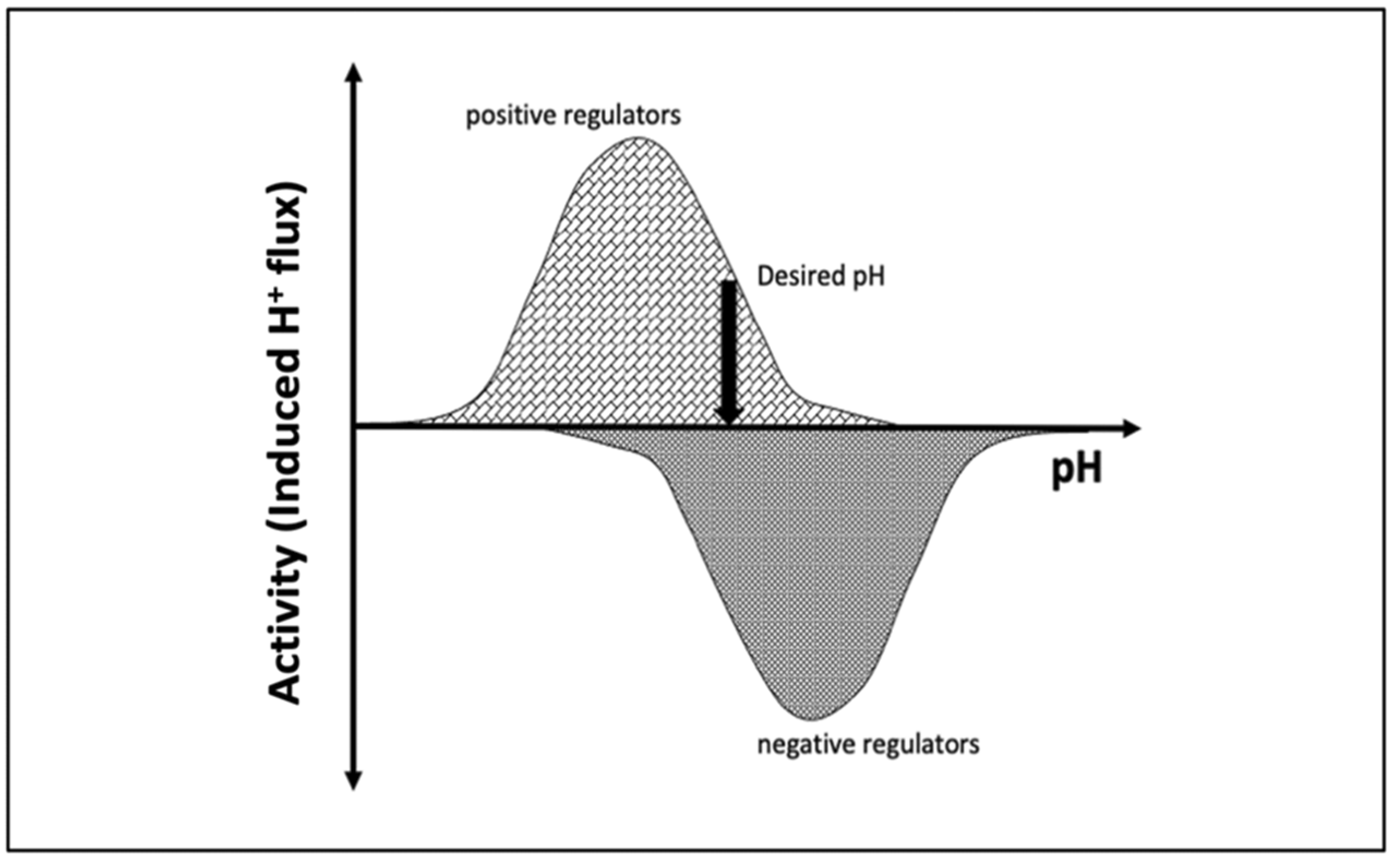
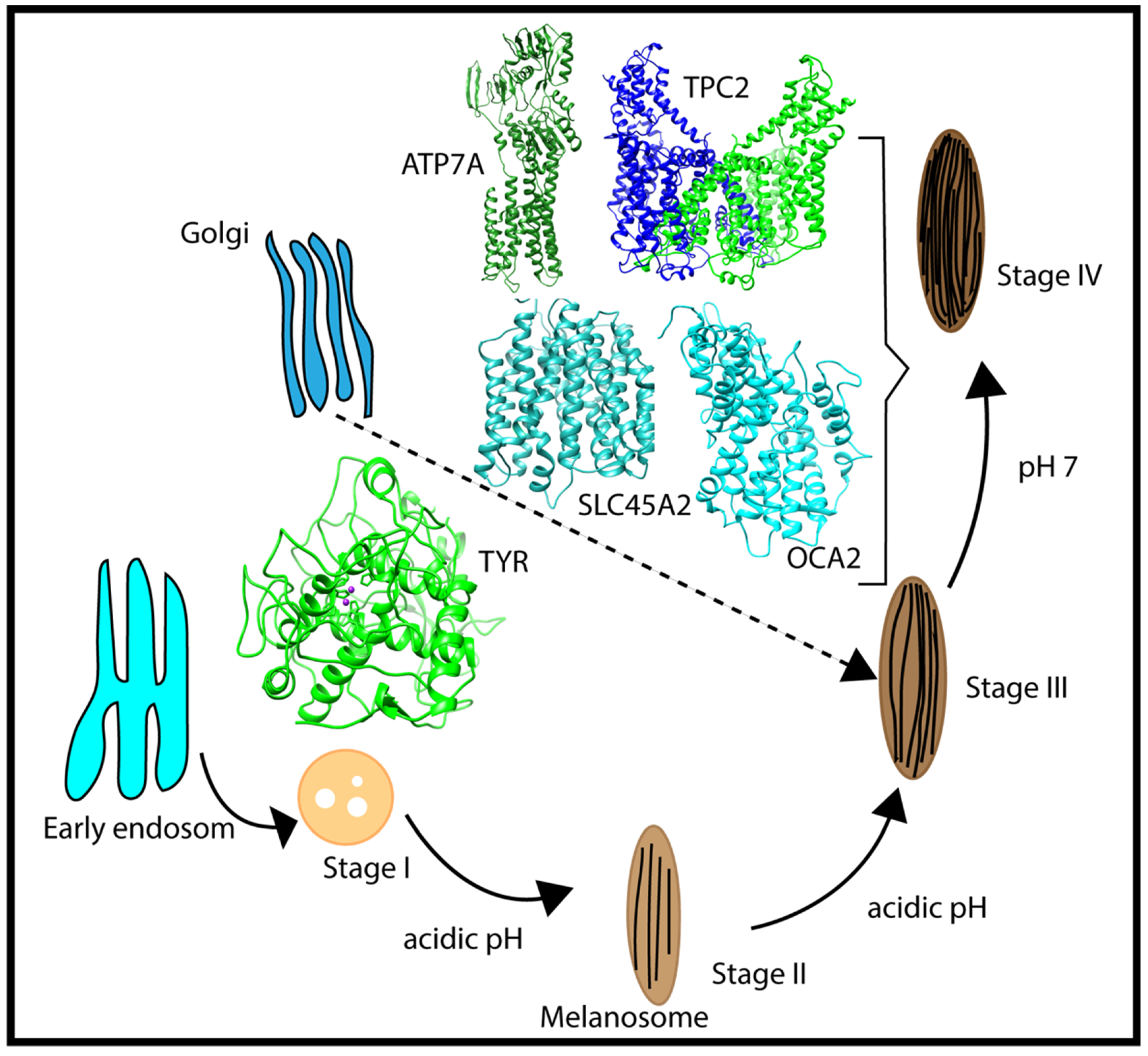
3. Discussions
4. Materials and Methods
4.1. Structures Used in the Modeling
4.2. List of Nonsynonymous GWAS-Identified Pigmentation-Associated Variants
4.3. Generation of Mutants
4.4. Molecular Dynamics (MD) Simulations
4.5. Modeling pH Dependence of Folding Free Energy
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| TYR | tyrosinase |
| OCA2 | oculocutaneous albinism 2 |
| SLC45A2 | solute carrier 45 member 2 |
| TPC2 | two-pore channel 2 |
| ATP7A | copper-transporting ATPase 1 |
References
- Alexov, E. Numerical calculations of the pH of maximal protein stability. The effect of the sequence composition and three-dimensional structure. Eur. J. Biochem. 2004, 271, 173–185. [Google Scholar] [CrossRef]
- Peng, Y.; Alexov, E. Computational investigation of proton transfer, pKa shifts and pH-optimum of protein-DNA and protein-RNA complexes. Proteins 2017, 85, 282–295. [Google Scholar] [CrossRef]
- Garcia-Moreno, B. Adaptations of proteins to cellular and subcellular pH. J. Biol. 2009, 8, 1–4. [Google Scholar] [CrossRef] [Green Version]
- Mitra, R.C.; Zhang, Z.; Alexov, E. In silico modeling of pH-optimum of protein-protein binding. Proteins Struct. Funct. Bioinform. 2011, 79, 925–936. [Google Scholar] [CrossRef] [Green Version]
- Peng, Y.; Kelle, R.; Little, C.; Michonova, E.; Kornev, K.G.; Alexov, E. pH-dependent interactions of Apolipophorin-III with a lipid disk. J. Comput. Biophys. Chem. 2020, 20, 153–164. [Google Scholar] [CrossRef]
- Luo, Z.; Li, Y.; Mousa, J.; Bruner, S.; Zhang, Y.; Pei, Y.; Keyhani, N.O. Bbmsn2 acts as a pH-dependent negative regulator of secondary metabolite production in the entomopathogenic fungus Beauveria bassiana. Environ. Microbiol. 2015, 17, 1189–1202. [Google Scholar] [CrossRef]
- Wiriyasermkul, P.; Moriyama, S.; Nagamori, S. Membrane transport proteins in melanosomes: Regulation of ions for pigmentation. Biochim. Biophys. Acta Biomembr. 2020, 1862, 183318. [Google Scholar] [CrossRef]
- Yamaguchi, Y.; Hearing, V.J. Melanocytes and their diseases. Cold Spring Harb. Perspect. Med. 2014, 4, a017046. [Google Scholar] [CrossRef] [Green Version]
- Pavan, W.J.; Sturm, R.A. The genetics of human skin and hair pigmentation. Annu. Rev. Genom. Hum. Genet. 2019, 20, 41–72. [Google Scholar] [CrossRef]
- Grønskov, K.; Ek, J.; Brondum-Nielsen, K. Oculocutaneous albinism. Orphanet J. Rare Dis. 2007, 2, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Shain, A.H.; Bastian, B.C. From melanocytes to melanomas. Nat. Rev. Cancer 2016, 16, 345–358. [Google Scholar] [CrossRef] [PubMed]
- Talley, K.; Alexov, E. On the pH-optimum of activity and stability of proteins. Proteins Struct. Funct. Bioinform. 2010, 78, 2699–2706. [Google Scholar] [CrossRef] [Green Version]
- Ikehata, K.; Nicell, J.A. Characterization of tyrosinase for the treatment of aqueous phenols. Bioresour. Technol. 2000, 74, 191–199. [Google Scholar] [CrossRef]
- Zaidi, K.U.; Ali, A.S.; Ali, S.A. Purification and characterization of melanogenic enzyme tyrosinase from button mushroom. Enzym. Res. 2014, 2014. [Google Scholar] [CrossRef] [Green Version]
- White, C.; Lee, J.; Kambe, T.; Fritsche, K.; Petris, M.J. A role for the ATP7A copper-transporting ATPase in macrophage bactericidal activity. J. Biol. Chem. 2009, 284, 33949–33956. [Google Scholar] [CrossRef] [Green Version]
- Cobbold, C.; Ponnambalam, S.; Francis, M.J.; Monaco, A.P. Novel membrane traffic steps regulate the exocytosis of the Menkes disease ATPase. Hum. Mol. Genet. 2002, 11, 2855–2866. [Google Scholar] [CrossRef] [PubMed]
- Duffy, D.L.; Montgomery, G.W.; Chen, W.; Zhao, Z.Z.; Le, L.; James, M.R.; Hayward, N.K.; Martin, N.G.; Sturm, R.A. A three-single-nucleotide polymorphism haplotype in intron 1 of OCA2 explains most human eye-color variation. Am. J. Hum. Genet. 2007, 80, 241–252. [Google Scholar] [CrossRef] [Green Version]
- Brilliant, M.H. The mouse p (pink-eyed dilution) and human P genes, oculocutaneous albinism type 2 (OCA2), and melanosomal pH. Pigment. Cell Res. 2001, 14, 86–93. [Google Scholar] [CrossRef]
- Le, L.; Escobar, I.E.; Ho, T.; Lefkovith, A.J.; Latteri, E.; Haltaufderhyde, K.D.; Dennis, M.K.; Plowright, L.; Sviderskaya, E.V.; Bennett, D.C. SLC45A2 protein stability and regulation of melanosome pH determine melanocyte pigmentation. Mol. Biol. Cell 2020, 31, 2687–2702. [Google Scholar] [CrossRef]
- Ancans, J.; Tobin, D.J.; Hoogduijn, M.J.; Smit, N.P.; Wakamatsu, K.; Thody, A.J. Melanosomal pH controls rate of melanogenesis, eumelanin/phaeomelanin ratio and melanosome maturation in melanocytes and melanoma cells. Exp. Cell Res. 2001, 268, 26–35. [Google Scholar] [CrossRef]
- Eiberg, H.; Troelsen, J.; Nielsen, M.; Mikkelsen, A.; Mengel-From, J.; Kjaer, K.; Hansen, L. Blue eye color in humans may be caused by a perfectly associated founder mutation in a regulatory element located within the HERC2 gene inhibiting OCA2 expression. Hum. Genet. 2008, 123, 177. [Google Scholar] [CrossRef]
- Branicki, W.; Brudnik, U.; Draus-Barini, J.; Kupiec, T.; Wojas-Pelc, A. Association of the SLC45A2 gene with physiological human hair colour variation. J. Hum. Genet. 2008, 53, 966–971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernandez, L.; Milne, R.; Pita, G.; Aviles, J.; Lazaro, P.; Benitez, J.; Ribas, G. SLC45A2: A novel malignant melanoma-associated gene. Hum. Mutat. 2008, 29, 1161–1167. [Google Scholar] [CrossRef]
- Ambrosio, A.L.; Boyle, J.A.; Aradi, A.E.; Christian, K.A.; Di Pietro, S.M. TPC2 controls pigmentation by regulating melanosome pH and size. Proc. Natl. Acad. Sci. USA 2016, 113, 5622–5627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chao, Y.-K.; Schludi, V.; Chen, C.-C.; Butz, E.; Nguyen, O.P.; Müller, M.; Krüger, J.; Kammerbauer, C.; Ben-Johny, M.; Vollmar, A.M. TPC2 polymorphisms associated with a hair pigmentation phenotype in humans result in gain of channel function by independent mechanisms. Proc. Natl. Acad. Sci. USA 2017, 114, E8595–E8602. [Google Scholar] [CrossRef] [Green Version]
- Yu, H.; Jacobson, D.R.; Luo, H.; Perkins, T.T. Quantifying the native energetics stabilizing bacteriorhodopsin by single-molecule force spectroscopy. Phys. Rev. Lett. 2020, 125, 068102. [Google Scholar] [CrossRef] [PubMed]
- Hamborg, L.; Horsted, E.W.; Johansson, K.E.; Willemoës, M.; Lindorff-Larsen, K.; Teilum, K. Global analysis of protein stability by temperature and chemical denaturation. Anal. Biochem. 2020, 605, 113863. [Google Scholar] [CrossRef]
- Getov, I.; Petukh, M.; Alexov, E. SAAFEC: Predicting the effect of single point mutations on protein folding free energy using a knowledge-modified MM/PBSA approach. Int. J. Mol. Sci. 2016, 17, 512. [Google Scholar] [CrossRef]
- Tajielyato, N.; Alexov, E. Modeling pKas of unfolded proteins to probe structural models of unfolded state. J. Theor. Comput. Chem. 2019, 18, 1950020. [Google Scholar] [CrossRef]
- Skjørringe, T.; Pedersen, P.A.; Thorborg, S.S.; Nissen, P.; Gourdon, P.; Møller, L.B. Characterization of ATP7A missense mutants suggests a correlation between intracellular trafficking and severity of Menkes disease. Sci. Rep. 2017, 7, 1–18. [Google Scholar] [CrossRef]
- Takano, K.; Liu, D.; Tarpey, P.; Gallant, E.; Lam, A.; Witham, S.; Alexov, E.; Chaubey, A.; Stevenson, R.E.; Schwartz, C.E.; et al. An X-linked channelopathy with cardiomegaly due to a CLIC2 mutation enhancing ryanodine receptor channel activity. Hum. Mol. Genet. 2012, 21, 4497–4507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Witham, S.; Takano, K.; Schwartz, C.; Alexov, E. A missense mutation in CLIC2 associated with intellectual disability is predicted by in silico modeling to affect protein stability and dynamics. Proteins 2011, 79, 2444–2454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grønskov, K.; Jespersgaard, C.; Bruun, G.H.; Harris, P.; Brøndum-Nielsen, K.; Andresen, B.S.; Rosenberg, T. A pathogenic haplotype, common in Europeans, causes autosomal recessive albinism and uncovers missing heritability in OCA1. Sci. Rep. 2019, 9, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sturm, R.A.; Duffy, D.L.; Zhao, Z.Z.; Leite, F.P.; Stark, M.S.; Hayward, N.K.; Martin, N.G.; Montgomery, G.W. A single SNP in an evolutionary conserved region within intron 86 of the HERC2 gene determines human blue-brown eye color. Am. J. Hum. Genet. 2008, 82, 424–431. [Google Scholar] [CrossRef] [Green Version]
- Zhang, T.; Choi, J.; Kovacs, M.A.; Shi, J.; Xu, M.; Goldstein, A.M.; Trower, A.J.; Bishop, D.T.; Iles, M.M.; Duffy, D.L. Cell-type–specific eQTL of primary melanocytes facilitates identification of melanoma susceptibility genes. Genome Res. 2018, 28, 1621–1635. [Google Scholar] [CrossRef] [Green Version]
- Guex, N.; Peitsch, M.C. SWISS-MODEL and the Swiss-Pdb Viewer: An environment for comparative protein modeling. Electrophoresis 1997, 18, 2714–2723. [Google Scholar] [CrossRef]
- Consortium, U. UniProt: A hub for protein information. Nucleic Acids Res. 2015, 43, D204–D212. [Google Scholar] [CrossRef]
- Lai, X.; Wichers, H.J.; Soler-Lopez, M.; Dijkstra, B.W. Structure of human tyrosinase related protein 1 reveals a binuclear zinc active site important for melanogenesis. Angew. Chem. Int. Ed. 2017, 56, 9812–9815. [Google Scholar] [CrossRef]
- Kelley, L.A.; Mezulis, S.; Yates, C.M.; Wass, M.N.; Sternberg, M.J. The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 2015, 10, 845–858. [Google Scholar] [CrossRef] [Green Version]
- Mancusso, R.; Gregorio, G.G.; Liu, Q.; Wang, D.-N. Structure and mechanism of a bacterial sodium-dependent dicarboxylate transporter. Nature 2012, 491, 622–626. [Google Scholar] [CrossRef] [Green Version]
- She, J.; Zeng, W.; Guo, J.; Chen, Q.; Bai, X.-c.; Jiang, Y. Structural mechanisms of phospholipid activation of the human TPC2 channel. eLife 2019, 8, e45222. [Google Scholar] [CrossRef]
- Nomura, N.; Verdon, G.; Kang, H.J.; Shimamura, T.; Nomura, Y.; Sonoda, Y.; Hussien, S.A.; Qureshi, A.A.; Coincon, M.; Sato, Y. Structure and mechanism of the mammalian fructose transporter GLUT5. Nature 2015, 526, 397–401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gourdon, P.; Liu, X.-Y.; Skjørringe, T.; Morth, J.P.; Møller, L.B.; Pedersen, B.P.; Nissen, P. Crystal structure of a copper-transporting PIB-type ATPase. Nature 2011, 475, 59–64. [Google Scholar] [CrossRef]
- Buniello, A.; MacArthur, J.A.L.; Cerezo, M.; Harris, L.W.; Hayhurst, J.; Malangone, C.; McMahon, A.; Morales, J.; Mountjoy, E.; Sollis, E. The NHGRI-EBI GWAS Catalog of published genome-wide association studies, targeted arrays and summary statistics 2019. Nucleic Acids Res. 2019, 47, D1005–D1012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Phillips, J.C.; Braun, R.; Wang, W.; Gumbart, J.; Tajkhorshid, E.; Villa, E.; Chipot, C.; Skeel, R.D.; Kale, L.; Schulten, K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005, 26, 1781–1802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brooks, B.R.; Bruccoleri, R.E.; Olafson, B.D.; States, D.J.; Swaminathan, S.A.; Karplus, M. CHARMM: A program for macromolecular energy, minimization, and dynamics calculations. J. Comput. Chem. 1983, 4, 187–217. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Panday, S.K.; Shashikala, M.H.; Koirala, M.; Pahari, S.; Chakvorty, A.; Peng, Y.; Li, L.; Jia, Z.; Li, C.; Alexov, E. Modeling electrostatics in molecular biology: A tutorial of DelPhi and associated resources [Article v1. 0]. Living J. Comput. Mol. Sci. 2019, 1, 10841. [Google Scholar] [CrossRef]
- Wang, L.; Li, L.; Alexov, E. pKa predictions for proteins, RNA s, and DNA s with the Gaussian dielectric function using DelPhi pKa. Proteins Struct. Funct. Bioinform. 2015, 83, 2186–2197. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Zhang, M.; Alexov, E. DelPhiPKa web server: Predicting p K a of proteins, RNAs and DNAs. Bioinformatics 2016, 32, 614–615. [Google Scholar] [CrossRef]
- Ozenne, V.; Bauer, F.; Salmon, L.; Huang, J.-R.; Jensen, M.R.; Segard, S.; Bernadó, P.; Charavay, C.; Blackledge, M. Flexible-meccano: A tool for the generation of explicit ensemble descriptions of intrinsically disordered proteins and their associated experimental observables. Bioinformatics 2012, 28, 1463–1470. [Google Scholar] [CrossRef] [PubMed]
- Estrada, J.; Bernadó, P.; Blackledge, M.; Sancho, J. ProtSA: A web application for calculating sequence specific protein solvent accessibilities in the unfolded ensemble. BMC Bioinform. 2009, 10, 104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schaefer, M.; Sommer, M.; Karplus, M. pH-dependence of protein stability: Absolute electrostatic free energy differences between conformations. J. Phys. Chem. B 1997, 101, 1663–1683. [Google Scholar] [CrossRef]
- Savojardo, C.; Fariselli, P.; Martelli, P.L.; Casadio, R. INPS-MD: A web server to predict stability of protein variants from sequence and structure. Bioinformatics 2016, 32, 2542–2544. [Google Scholar] [CrossRef] [PubMed]
- Pires, D.E.; Ascher, D.B.; Blundell, T.L. mCSM: Predicting the effects of mutations in proteins using graph-based signatures. Bioinformatics 2014, 30, 335–342. [Google Scholar] [CrossRef] [Green Version]
- Pandurangan, A.P.; Ochoa-Montaño, B.; Ascher, D.B.; Blundell, T.L. SDM: A server for predicting effects of mutations on protein stability. Nucleic Acids Res. 2017, 45, W229–W235. [Google Scholar] [CrossRef] [Green Version]
- Pires, D.E.; Ascher, D.B.; Blundell, T.L. DUET: A server for predicting effects of mutations on protein stability using an integrated computational approach. Nucleic Acids Res. 2014, 42, W314–W319. [Google Scholar] [CrossRef]
- Capriotti, E.; Fariselli, P.; Casadio, R. I-Mutant2.0: Predicting stability changes upon mutation from the protein sequence or structure. Nucleic Acids Res. 2005, 33 (Suppl. 2), W306–W310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, J.; Randall, A.; Baldi, P. Prediction of protein stability changes for single-site mutations using support vector machines. Proteins Struct. Funct. Bioinform. 2006, 62, 1125–1132. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.-W.; Lin, J.; Chu, Y.-W. iStable: off-the-shelf predictor integration for predicting protein stability changes. BMC Bioinform. 2013, 14, S5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, H.; Wang, J.; He, L.; Qi, Y.; Zhang, J.Z. DeepDDG: Predicting the stability change of protein point mutations using neural networks. J. Chem. Inf. Modeling 2019, 59, 1508–1514. [Google Scholar] [CrossRef] [PubMed]

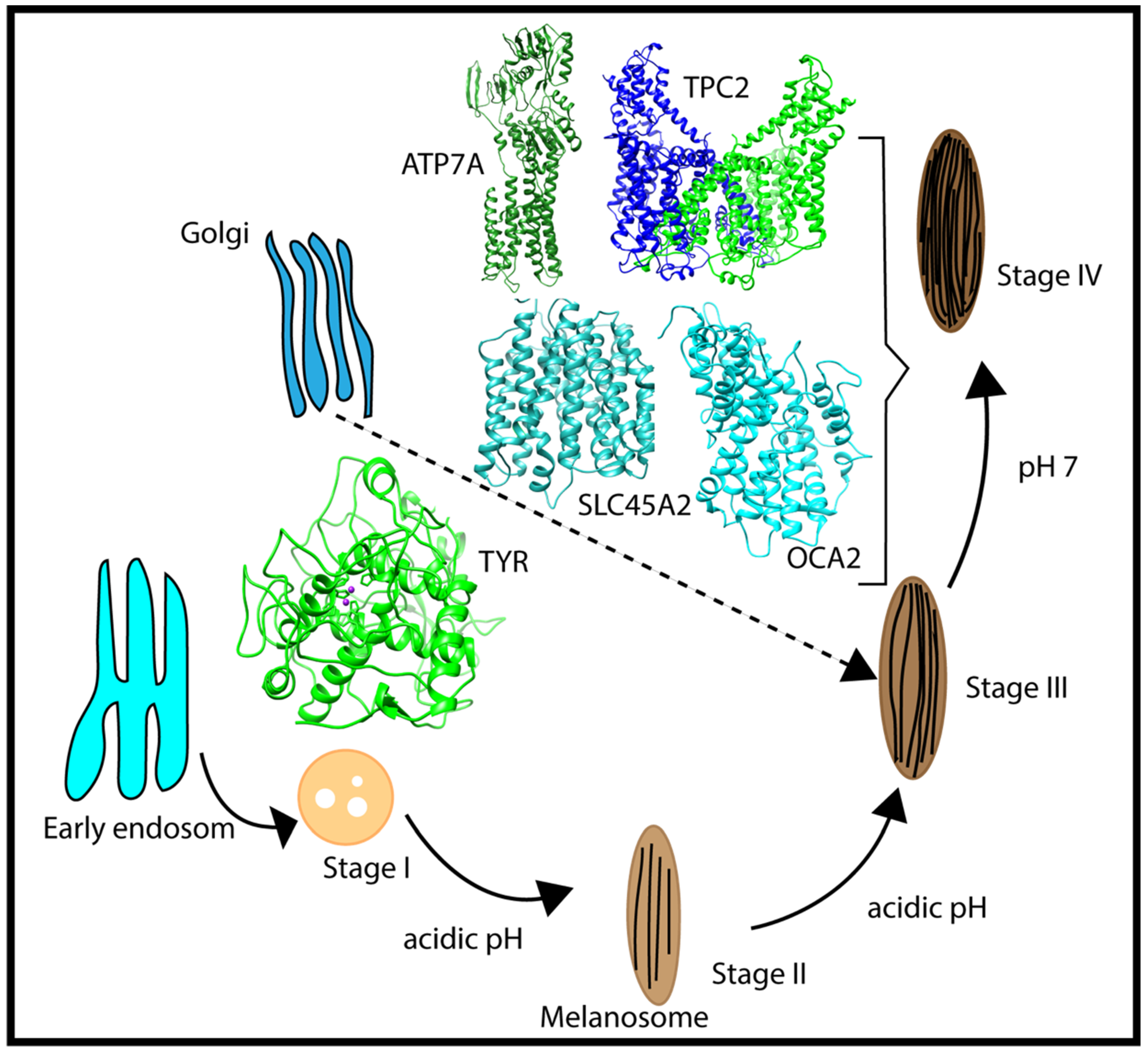

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Change in Folding Free Energy (ΔΔG) Due to Variants(kcal/mol) | |||
|---|---|---|---|
| Protein | Variant | Avg ƊΔG | SD |
| TYR | R402Q | −0.5 | 0.5 |
| S192 YDouble MT * | −0.27 −0.77 | 0.78 1.09 | |
| OCA2 | A481T | −1.01 | 0.52 |
| H615L | 0.17 | 0.39 | |
| N489D | −1.05 | 1.08 | |
| P743L | −0.9 | 0.45 | |
| R419Q | −0.54 | 0.33 | |
| V443I | −0.54 | 0.48 | |
| SLC45A2 | G198V | −0.51 | 0.25 |
| L374F | −0.84 | 0.47 | |
| TPC2 | K376R | −0.49 | 0.3 |
| M484L | −0.86 | 0.33 | |
| M546I | −0.1 | 0.67 | |
| V219I | −0.11 | 0.32 | |
| ATP7A | C1002F | −1.2 | 0.74 |
| G666R | −0.21 | 0.7 | |
| D1044E | −0.8 | 0.53 | |
| I1264V | −1.1 | 0.74 | |
| K742R | 0.01 | 0.35 | |
| M1311V | −0.79 | 0.35 | |
| R844C | −0.48 | 0.39 | |
| S653Y | −0.45 | 0.54 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Koirala, M.; Shashikala, H.B.M.; Jeffries, J.; Wu, B.; Loftus, S.K.; Zippin, J.H.; Alexov, E. Computational Investigation of the pH Dependence of Stability of Melanosome Proteins: Implication for Melanosome formation and Disease. Int. J. Mol. Sci. 2021, 22, 8273. https://doi.org/10.3390/ijms22158273
Koirala M, Shashikala HBM, Jeffries J, Wu B, Loftus SK, Zippin JH, Alexov E. Computational Investigation of the pH Dependence of Stability of Melanosome Proteins: Implication for Melanosome formation and Disease. International Journal of Molecular Sciences. 2021; 22(15):8273. https://doi.org/10.3390/ijms22158273
Chicago/Turabian StyleKoirala, Mahesh, H. B. Mihiri Shashikala, Jacob Jeffries, Bohua Wu, Stacie K. Loftus, Jonathan H. Zippin, and Emil Alexov. 2021. "Computational Investigation of the pH Dependence of Stability of Melanosome Proteins: Implication for Melanosome formation and Disease" International Journal of Molecular Sciences 22, no. 15: 8273. https://doi.org/10.3390/ijms22158273
APA StyleKoirala, M., Shashikala, H. B. M., Jeffries, J., Wu, B., Loftus, S. K., Zippin, J. H., & Alexov, E. (2021). Computational Investigation of the pH Dependence of Stability of Melanosome Proteins: Implication for Melanosome formation and Disease. International Journal of Molecular Sciences, 22(15), 8273. https://doi.org/10.3390/ijms22158273