
Generation of Recombinant Primary Human B Lymphocytes Using Non-Viral Vectors



Abstract
1. Introduction
2. Results and Discussion
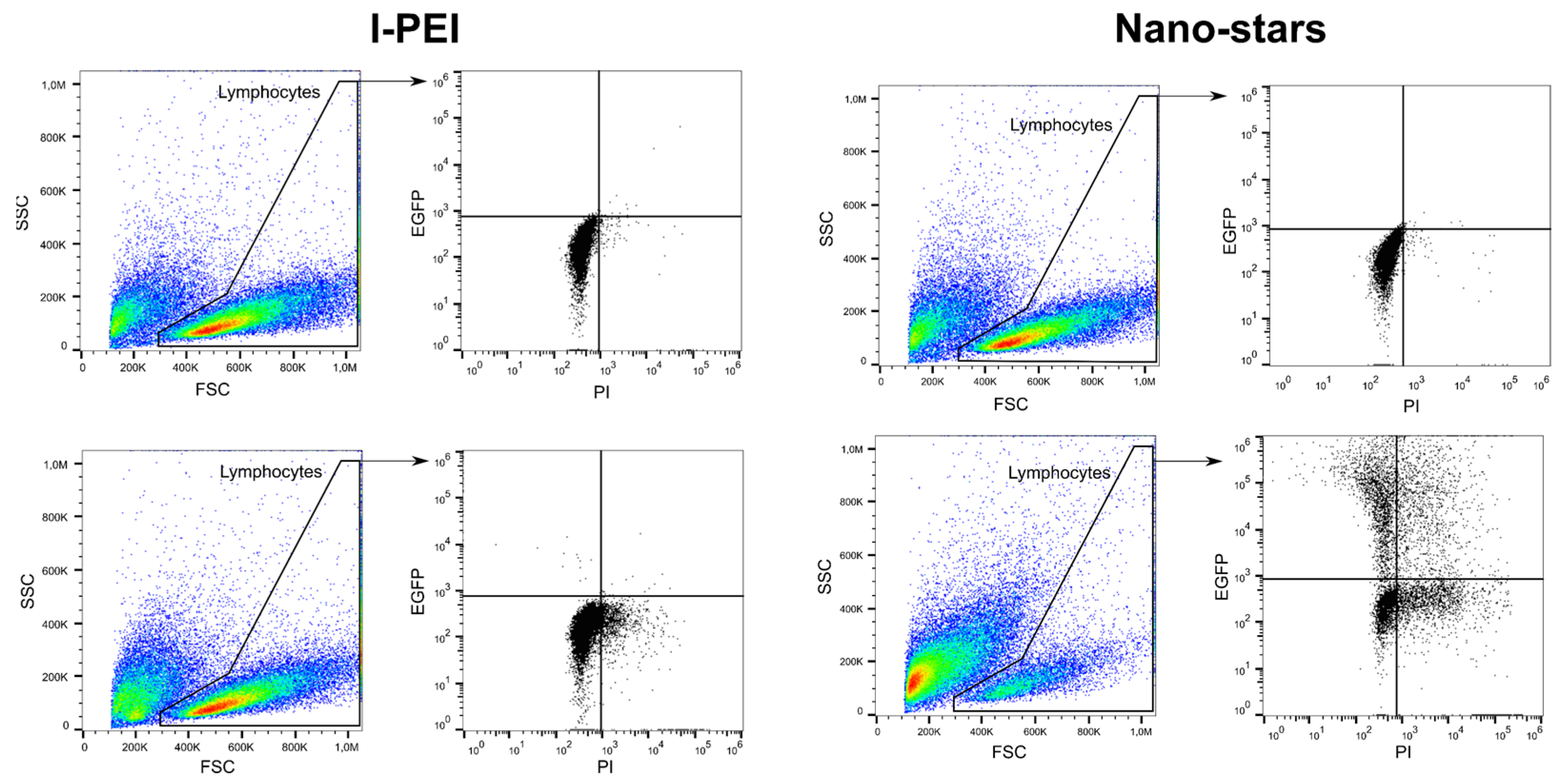
2.1. Comparison of PDMAEMA-Nano-Stars to l-PEI for the Transfection of Primary Human B Lymphocytes
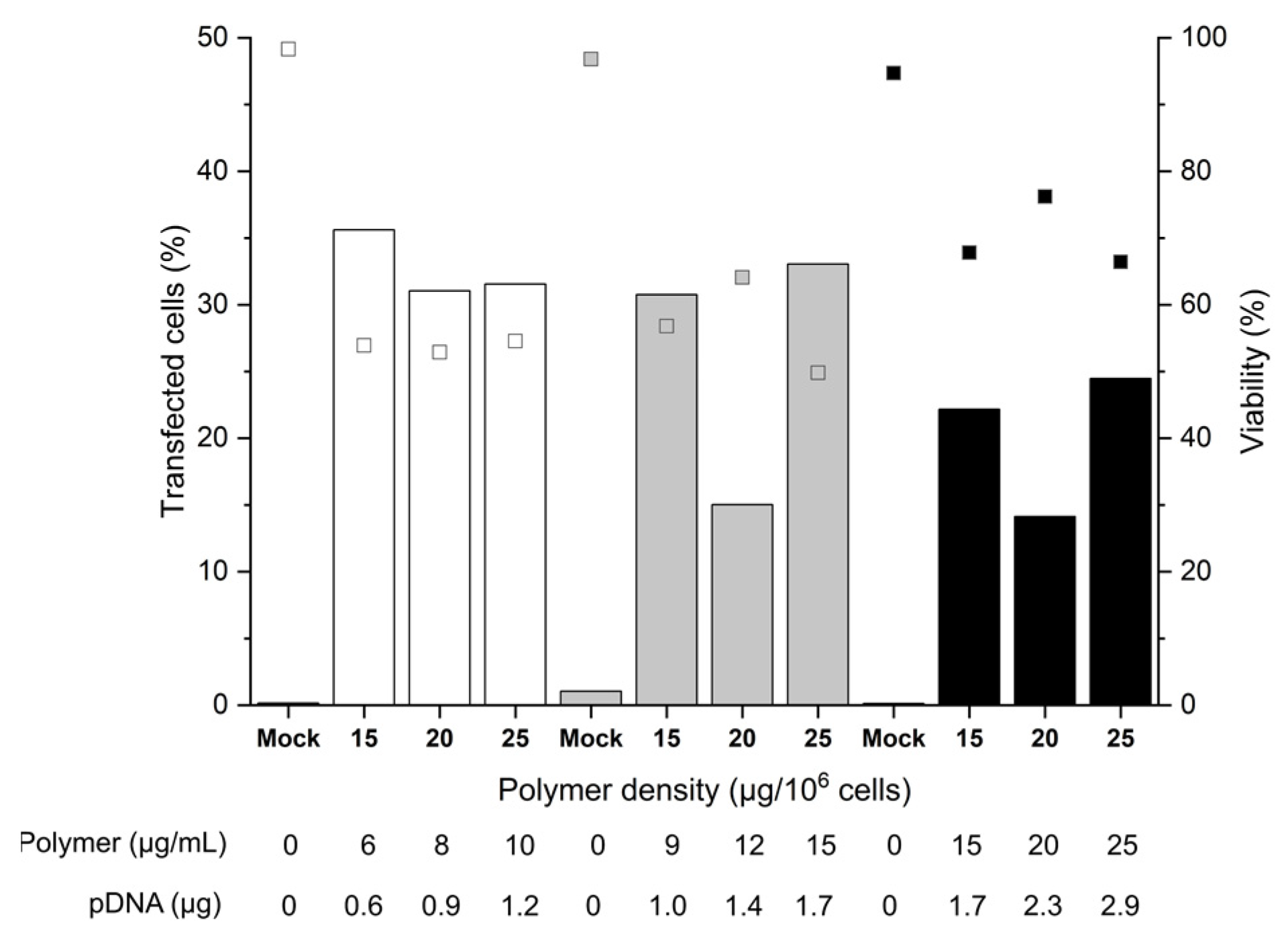
2.2. Influence of the Cell Number and the Polymer Density (Amount per Cell)
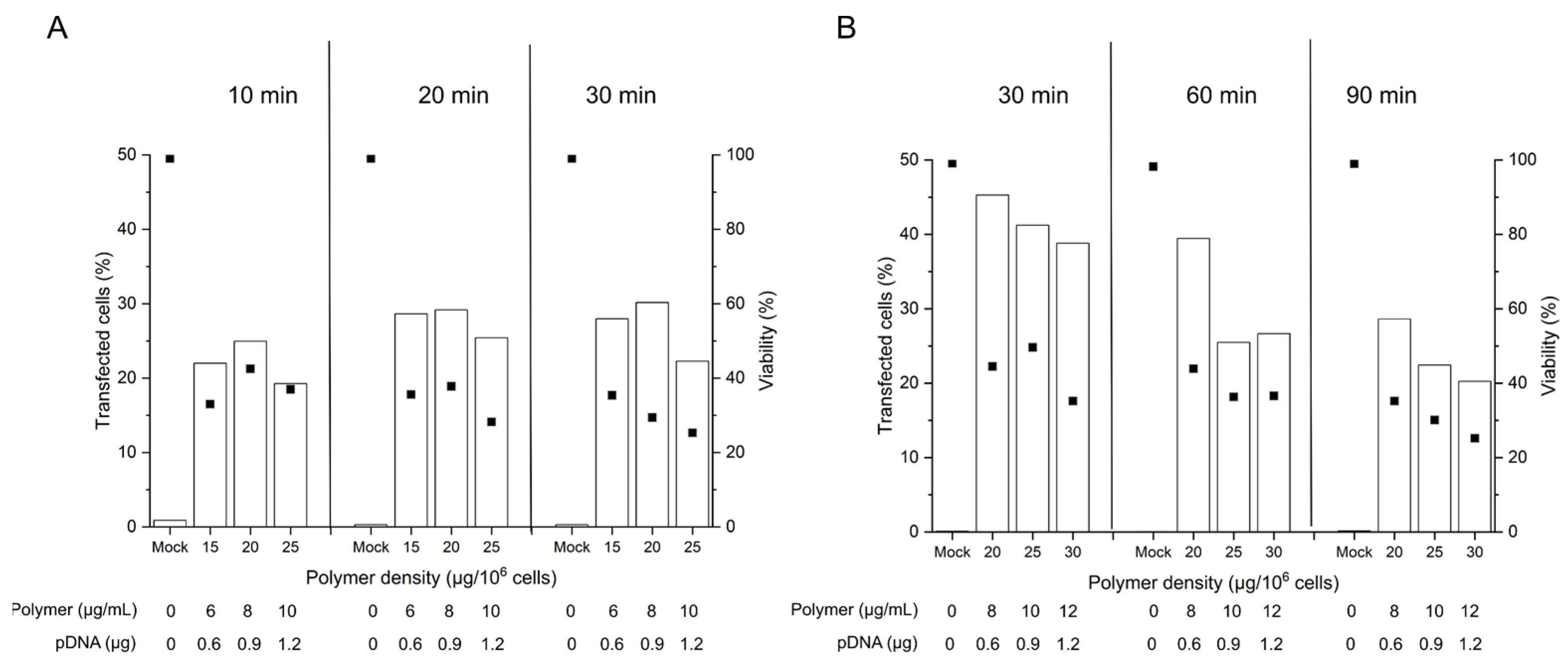
2.3. Influence of the Polyplex Exposure and the Recovery Times in B Cell Transfection
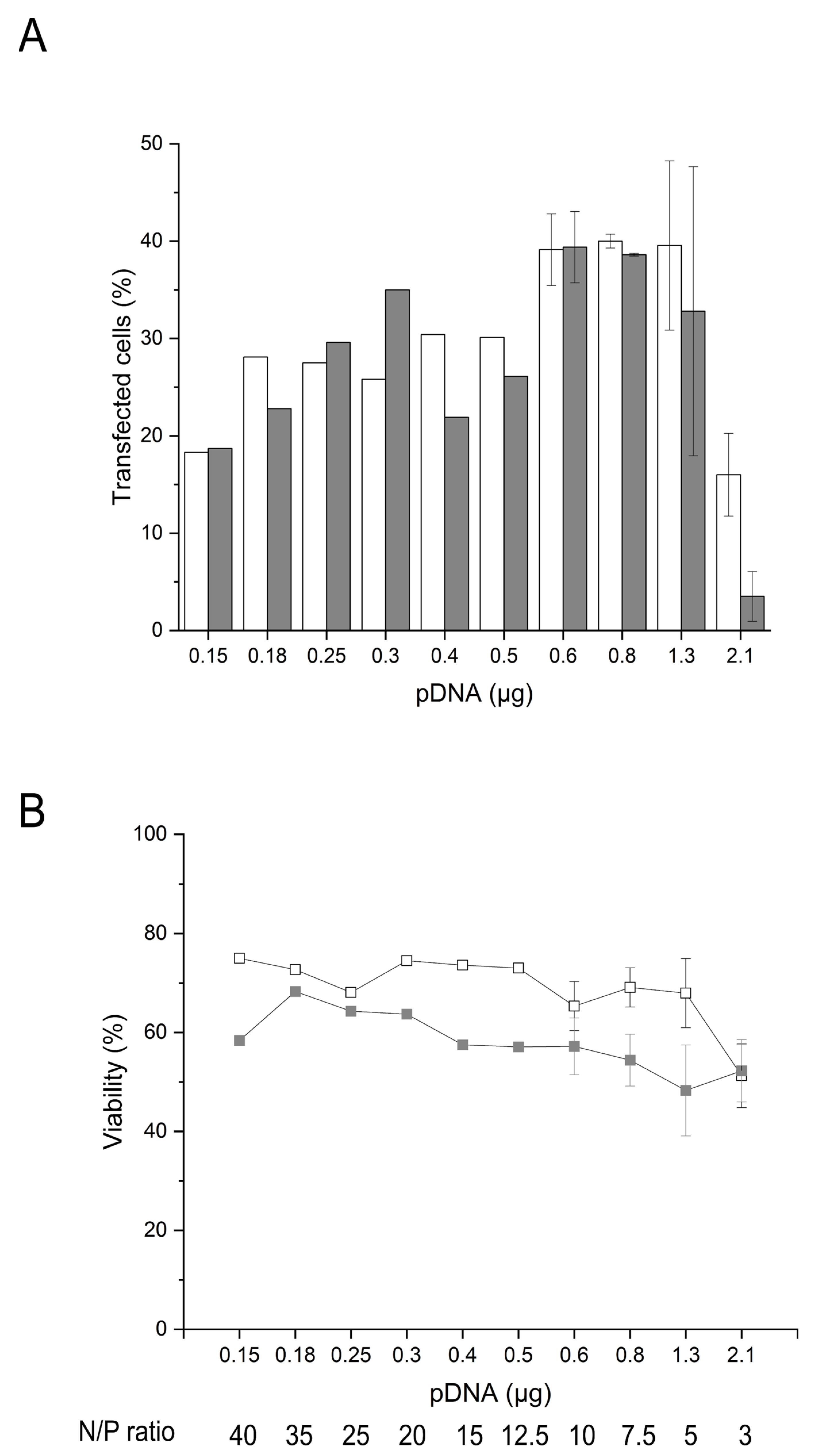
2.4. Influence of the Amount of pDNA and Transgene Expression on the Cell Survival
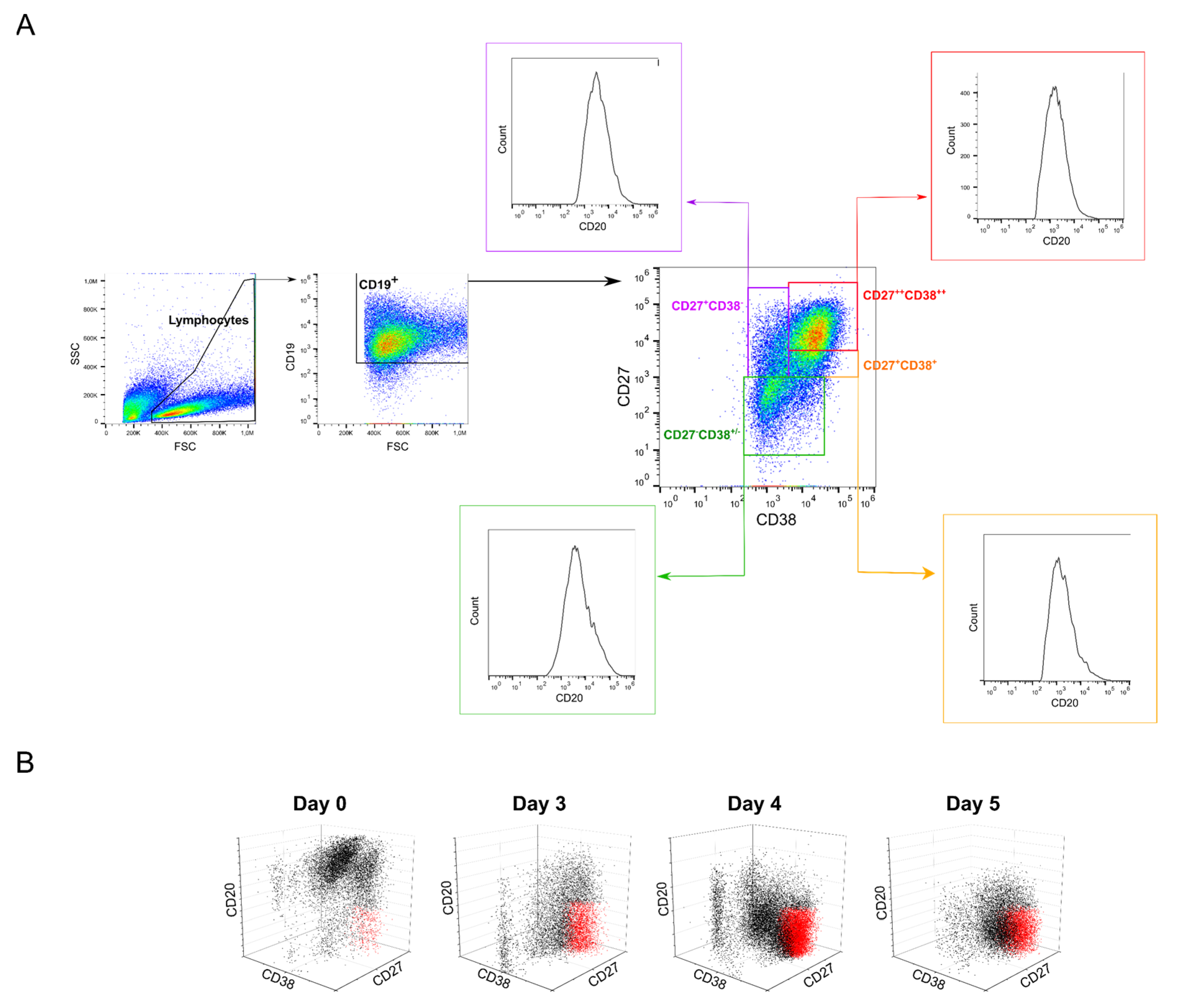
2.5. Possible Effects of B Cell Polyclonality during Transfection
3. Conclusions
4. Materials and Methods
4.1. Materials
4.2. Methods
4.2.1. Cell Culture
4.2.2. Plasmid
4.2.3. Transfection

4.2.4. Analytics
4.2.5. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Liebig, T.M.; Fiedler, A.; Zoghi, S.; Shimabukuro-Vornhagen, A.; von Bergwelt-Baildon, M.S. Generation of human CD40-activated B cells. J. Vis. Exp. 2009, 1373. [Google Scholar] [CrossRef]
- Naito, M.; Hainz, U.; Burkhardt, U.E.; Fu, B.; Ahove, D.; Stevenson, K.E.; Rajasagi, M.; Zhu, B.; Alonso, A.; Witten, E.; et al. CD40L-Tri, a novel formulation of recombinant human CD40L that effectively activates B cells. Cancer Immunol. Immunother. 2013, 62, 347–357. [Google Scholar] [CrossRef]
- Cassese, G.; Arce, S.; Hauser, A.E.; Lehnert, K.; Moewes, B.; Mostarac, M.; Muehlinghaus, G.; Szyska, M.; Radbruch, A.; Manz, R.A. Plasma cell survival is mediated by synergistic effects of cytokines and adhesion-dependent signals. J. Immunol. 2003, 171, 1684–1690. [Google Scholar] [CrossRef] [PubMed]
- Hung, K.L.; Meitlis, I.; Hale, M.; Chen, C.Y.; Singh, S.; Jackson, S.W.; Miao, C.H.; Khan, I.F.; Rawlings, D.J.; James, R.G. Engineering protein-secreting plasma cells by homology-directed repair in primary human B cells. Mol. Ther. 2018, 26, 456–467. [Google Scholar] [CrossRef] [PubMed]
- Kurosaki, T.; Kometani, K.; Ise, W. Memory B cells. Nat. Rev. Immunol. 2015, 15, 149–159. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Lao, X.; Pan, Q.; Ning, N.; Yet, J.; Xu, Y.; Li, S.; Chang, A.E. Adoptive transfer of tumor reactive B cells confers host T cell immunity and tumor regression. Clin. Cancer Res. 2011, 17, 4987–4995. [Google Scholar] [CrossRef] [PubMed]
- Nutt, S.L.; Hodgkin, P.D.; Tarlinton, D.M.; Corcoran, L.M. The generation of antibody-secreting plasma cells. Nat. Rev. Immunol. 2015, 15, 160–171. [Google Scholar] [CrossRef]
- Pesch, T.; Bonati, L.; Kelton, W.; Parola, C.; Ehling, R.A.; Csepregi, L.; Kitamura, D.; Reddy, S.T. Molecular design, optimization, and genomic integration of chimeric B cell receptors in murine B cells. Front. Immunol. 2019, 10, 1–16. [Google Scholar] [CrossRef]
- Tangye, S.G.; Tarlinton, D.M. Memory B cells: Effectors of long-lived immune responses. Eur. J. Immunol. 2009, 39, 2065–2075. [Google Scholar] [CrossRef] [PubMed]
- Brekke, O.H.; Sandlie, I. Therapeutic antibodies for human diseases at the dawn of the twenty-first century. Nat. Rev. Drug Discov. 2003, 2, 52–62. [Google Scholar] [CrossRef] [PubMed]
- Guijarro-Munoz, I.; Compte, M.; Alvarez-Vallina, L.; Sanz, L. Antibody gene therapy: Getting closer to clinical application? Curr. Gene Ther. 2013, 13, 282–290. [Google Scholar] [CrossRef] [PubMed]
- Moffett, H.F.; Harms, C.K.; Fitzpatrick, K.S.; Tooley, M.R.; Boonyaratanakornkit, J.; Taylor, J.J. B cells engineered to express pathogen-specific antibodies protect against infection. Sci. Immunol. 2019, 4, eaax0644. [Google Scholar] [CrossRef] [PubMed]
- Voss, J.E.; Gonzalez-Martin, A.; Andrabi, R.; Fuller, R.P.; Murrell, B.; McCoy, L.E.; Porter, K.; Huang, D.; Li, W.; Sok, D.; et al. Reprogramming the antigen specificity of B cells using genome-editing technologies. Elife 2019, 8, e42995. [Google Scholar] [CrossRef]
- Laoharawee, K.; Johnson, M.J.; Lahr, W.S.; Peterson, J.J.; Webber, B.R.; Moriarity, B.S. Genome engineering of primary human B cells using CRISPR/Cas9. JoVE 2020, 3, e61855. [Google Scholar]
- Greiner, V.; Bou Puerto, R.; Liu, S.; Herbel, C.; Carmona, E.M.; Goldberg, M.S. CRISPR-mediated editing of the B cell receptor in primary human B cells. iScience 2019, 12, 369–378. [Google Scholar] [CrossRef]
- Wu, C.-A.M.; Roth, T.L.; Baglaenko, Y.; Ferri, D.M.; Brauer, P.; Zuniga-Pflucker, J.C.; Rosbe, K.W.; Wither, J.E.; Marson, A.; Allen, C.D.C. Genetic engineering in primary human B cells with CRISPR-Cas9 ribonucleoproteins. J. Immunol. Methods 2018, 457, 33–40. [Google Scholar] [CrossRef]
- Walker, L.M.; Burton, D.R. Passive immunotherapy of viral infections: ’super-antibodies’ enter the fray. Nat. Rev. Immunol. 2018, 18, 297–308. [Google Scholar] [CrossRef]
- Hartweger, H.; McGuire, A.T.; Horning, M.; Taylor, J.J.; Dosenovic, P.; Yost, D.; Gazumyan, A.; Seaman, M.S.; Stamatatos, L.; Jankovic, M.; et al. HIV-specific humoral immune responses by CRISPR/Cas9-edited B cells. J. Exp. Med. 2019, 216, 1301–1310. [Google Scholar] [CrossRef] [PubMed]
- Doench, J.G. Am I ready for CRISPR? A user’s guide to genetic screens. Nat. Rev. Genet. 2018, 19, 67–80. [Google Scholar] [CrossRef] [PubMed]
- Lino, C.A.; Harper, J.C.; Carney, J.P.; Timlin, J.A. Delivering CRISPR: A review of the challenges and approaches. Drug Deliv. 2018, 25, 1234–1257. [Google Scholar] [CrossRef]
- Liu, C.; Zhang, L.; Liu, H.; Cheng, K. Delivery strategies of the CRISPR-Cas9 gene-editing system for therapeutic applications. J. Control. Release 2017, 266, 17–26. [Google Scholar] [CrossRef]
- Li, L.H.; Biagi, E.; Allen, C.; Shivakumar, R.; Weiss, J.M.; Feller, S.; Yvon, E.; Fratantoni, J.C.; Liu, L.N. Rapid and efficient nonviral gene delivery of CD154 to primary chronic lymphocytic leukemia cells. Cancer Gene Ther. 2006, 13, 215–224. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Canoy, R.J.; André, F.; Shmakova, A.; Wiels, J.; Lipinski, M.; Vassetzky, Y.; Germini, D. Easy and robust electrotransfection protocol for efficient ectopic gene expression and genome editing in human B cells. Gene Ther. 2020. [Google Scholar] [CrossRef] [PubMed]
- Moghimi, B.; Zolotukhin, I.; Sack, B.K.; Herzog, R.W.; Cao, O. High efficiency ex vivo gene transfer to primary murine B cells using plasmid or viral vectors. J. Genet. Syndr. Gene Ther. 2011, 2, 1000103–1000105. [Google Scholar] [CrossRef] [PubMed]
- Olden, B.R.; Cheng, E.; Cheng, Y.; Pun, S.H. Identifying key barriers in cationic polymer gene delivery to human T cells. Biomat. Sci. 2019, 7, 789–797. [Google Scholar] [CrossRef] [PubMed]
- Smith, T.T.; Stephan, S.B.; Moffett, H.F.; McKnight, L.E.; Ji, W.; Reiman, D.; Bonagofski, E.; Wohlfahrt, M.E.; Pillai, S.P.S.; Stephan, M.T. In situ programming of leukaemia-specific T cells using synthetic DNA nanocarriers. Nat. Nanotechnol. 2017, 12, 813. [Google Scholar] [CrossRef] [PubMed]
- McMahon, S.B.; Norvell, A.; Levine, K.J.; Monroe, J.G. Transient transfection of murine B lymphocyte blasts as a method for examining gene regulation in primary B cells. J. Immunol. Methods 1995, 179, 251–259. [Google Scholar] [CrossRef]
- Agarwal, S.; Zhang, Y.; Maji, S.; Greiner, A. PDMAEMA based gene delivery materials. Mater. Today 2012, 15, 388–393. [Google Scholar] [CrossRef]
- Li, L.; Wei, Y.; Gong, C. Polymeric nanocarriers for non-viral gene delivery. J. Biomed. Nanotechnol. 2015, 11, 739–770. [Google Scholar] [CrossRef]
- Zakeri, A.; Kouhbanani, M.A.J.; Beheshtkhoo, N.; Beigi, V.; Mousavi, S.M.; Hashemi, S.A.R.; Karimi Zade, A.; Amani, A.M.; Savardashtaki, A.; Mirzaei, E.; et al. Polyethylenimine-based nanocarriers in co-delivery of drug and gene: A developing horizon. Nano Rev. Exp. 2018, 9, 1488497. [Google Scholar] [CrossRef]
- Plamper, F.A.; Schmalz, A.; Penott-Chang, E.; Drechsler, M.; Jusufi, A.; Ballauff, M.; Muller, A.H.E. Synthesis and characterization of star-shaped poly(N,N-dimethylaminoethyl methacrylate) and its quaternized ammonium salts. Macromolecules 2007, 40, 5689–5697. [Google Scholar] [CrossRef]
- Diaz Ariza, I.L.; Jérôme, V.; Pérez Pérez, L.D.; Freitag, R. Amphiphilic graft copolymers capable of mixed-mode interaction as alternative nonviral transfection agents. ACS Appl. Bio Mater. 2021, 4, 1268–1282. [Google Scholar] [CrossRef]
- Raup, A.; Jérôme, V.; Freitag, R.; Synatschke, C.V.; Müller, A.H.E. Promoter, transgene, and cell line effects in the transfection of mammalian cells using PDMAEMA-based nano-stars. Biotechnol. Rep. 2016, 11, 53–61. [Google Scholar] [CrossRef] [PubMed]
- Raup, A.; Stahlschmidt, U.; Jérôme, V.; Synatschke, C.V.; Müller, A.H.E.; Freitag, R. Influence of polyplex formation on the performance of star-shaped polycationic transfection agents for mammalian cells. Polymers 2016, 8, 224. [Google Scholar] [CrossRef] [PubMed]
- Synatschke, C.V.; Schallon, A.; Jérôme, V.; Freitag, R.; Müller, A.H.E. Influence of polymer architecture and molecular weight of poly(2-(dimethylamino)ethyl methacrylate) polycations on transfection efficiency and cell viability in gene delivery. Biomacromolecules 2011, 12, 4247–4255. [Google Scholar] [CrossRef] [PubMed]
- Riedl, S.; Kaiser, P.; Raup, A.; Synatschke, C.; Jérôme, V.; Freitag, R. Non-viral transfection of human T lymphocytes. Processes 2018, 6, 188. [Google Scholar] [CrossRef]
- Schallon, A.; Synatschke, C.V.; Jérôme, V.; Müller, A.H.E.; Freitag, R. Nanoparticulate nonviral agent for the effective delivery of pDNA and siRNA to differentiated cells and primary human T lymphocytes. Biomacromolecules 2012, 13, 3463–3474. [Google Scholar] [CrossRef] [PubMed]
- Jérôme, V.; Synatschke, C.V.; Freitag, R. Transient destabilization of biological membranes contributes to the superior performance of star-shaped PDMAEMA in delivering pDNA. ACS Omega 2020, 5, 26640–26654. [Google Scholar] [CrossRef]
- Helm, M.A.B.; Riedl, S.; Gollner, K.; Gollner, U.; Jérôme, V.; Freitag, R. Isolation of primary human B lymphocytes from tonsils compared to blood as alternative source for ex vivo application. J. Chromatogr. B 2021, 1179, 122853. [Google Scholar] [CrossRef]
- Remaut, K.; Symens, N.; Lucas, B.; Demeester, J.; De Smedt, S.C. Cell division responsive peptides for optimized plasmid DNA delivery: The mitotic window of opportunity? J Control. Release 2014, 179, 1–9. [Google Scholar] [CrossRef]
- Zhang, P.; Wagner, E. History of polymeric gene delivery systems. Top. Curr. Chem. 2017, 375, 26. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, S.; Srienc, F. Quantitative analysis of transient gene expression in mammalian cells using the green fluorescent protein. J. Biotechnol. 1996, 49, 137–151. [Google Scholar] [CrossRef]
- Seiffert, M.; Stilgenbauer, S.; Döhner, H.; Lichter, P. Efficient nucleofection of primary human B cells and B-CLL cells induces apoptosis, which depends on the microenvironment and on the structure of transfected nucleic acids. Leukemia 2007, 21, 1977–1983. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Angelats, M.; Cidlowski, J.A. Cell volume control and signal transduction in apoptosis. Toxicol. Pathol. 2002, 30, 541–551. [Google Scholar] [CrossRef]
- Diaz, I.L.; Sierra, C.A.; Jérôme, V.; Freitag, R.; Perez, L.D. Target grafting of poly(2-(dimethylamino)ethyl methacrylate) to biodegradable block copolymers. J. Polym. Sci. 2020, 58, 2168–2180. [Google Scholar] [CrossRef]
- Dias, C.; Nylandsted, J. Plasma membrane integrity in health and disease: Significance and therapeutic potential. Cell Discov. 2021, 7, 4. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Zhang, H.; Mou, W.; Qi, Z.; Ren, X.; Wang, G.; Jiao, H.; Kong, X.; Gui, J. Flow cytometric analyses of the viability, surface marker expression and function of lymphocytes from children following cryopreservation. Mol. Med. Rep. 2016, 14, 4301–4308. [Google Scholar] [CrossRef]
- Przybylski, S.; Gasch, M.; Marschner, A.; Ebert, M.; Ewe, A.; Helmig, G.; Hilger, N.; Fricke, S.; Rudzok, S.; Aigner, A.; et al. Influence of nanoparticle-mediated transfection on proliferation of primary immune cells in vitro and in vivo. PLoS ONE 2017, 12, e0176517. [Google Scholar] [CrossRef]
- Mullins, C.S.; Wegner, T.; Klar, E.; Classen, C.-F.; Linnebacher, M. Optimizing the process of nucleofection for professional antigen presenting cells. BMC Res. Notes 2015, 8, 472. [Google Scholar] [CrossRef]
- Ansari, A.M.; Ahmed, A.K.; Matsangos, A.E.; Lay, F.; Born, L.J.; Marti, G.; Harmon, J.W.; Sun, Z. Cellular GFP toxicity and immunogenicity: Potential confounders in in vivo cell tracking experiments. Stem Cell Rev. Rep. 2016, 12, 553–559. [Google Scholar] [CrossRef]
- Ganini, D.; Leinisch, F.; Kumar, A.; Jiang, J.; Tokar, E.J.; Malone, C.C.; Petrovich, R.M.; Mason, R.P. Fluorescent proteins such as eGFP lead to catalytic oxidative stress in cells. Redox Biol. 2017, 12, 462–468. [Google Scholar] [CrossRef] [PubMed]
- Lesueur, L.L.; Mir, L.M.; Andre, F.M. Overcoming the specific toxicity of large plasmids electrotransfer in primary cells in vitro. Mol. Ther. Nucleic Acids 2016, 5, e291. [Google Scholar] [CrossRef] [PubMed]
- Kitsera, N.; Khobta, A.; Epe, B. Destabilized green fluorescent protein detects rapid removal of transcription blocks after genotoxic exposure. BioTechniques 2007, 43, 222–227. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zhao, X.; Fang, Y.; Jiang, X.; Duong, T.; Fan, C.; Huang, C.C.; Kain, S.R. Generation of destabilized green fluorescent protein as a transcription reporter. J. Biol. Chem. 1998, 273, 34970–34975. [Google Scholar] [CrossRef] [PubMed]
- Hellweg, C.E.; Baumstark-Khan, C.; Horneck, G. Enhanced green fluorescent protein as reporter protein for biomonitoring of cytotoxic effects in mammalian cells. Anal. Chim. Acta 2001, 427, 191–199. [Google Scholar] [CrossRef]
- Jellusova, J. Metabolic control of B cell immune responses. Curr. Opin. Immunol. 2020, 63, 21–28. [Google Scholar] [CrossRef]
- Jackson, S.M.; Wilson, P.C.; James, J.A.; Capra, J.D. Human B cell subsets. Adv. Immunol. 2008, 98, 151–224. [Google Scholar] [PubMed]
- Rasmussen, S.M.; Bilgrau, A.E.; Schmitz, A.; Falgreen, S.; Bergkvist, K.S.; Tramm, A.M.; Bæch, J.; Jacobsen, C.L.; Gaihede, M.; Kjeldsen, M.K.; et al. Stable phenotype of B-cell subsets following cryopreservation and thawing of normal human lymphocytes stored in a tissue biobank. Cytom. Part B Clin. Cytom. 2015, 88, 40–49. [Google Scholar] [CrossRef]
- Marasco, E.; Farroni, C.; Cascioli, S.; Marcellini, V.; Scarsella, M.; Giorda, E.; Piano Mortari, E.; Leonardi, L.; Scarselli, A.; Valentini, D.; et al. B-cell activation with CD40L or CpG measures the function of B-cell subsets and identifies specific defects in immunodeficient patients. Eur. J. Immunol. 2017, 47, 131–143. [Google Scholar] [CrossRef]
- Robinson, M.J.; Pitt, C.; Brodie, E.J.; Valk, A.M.; O’Donnell, K.; Nitschke, L.; Jones, S.; Tarlinton, D.M. BAFF, IL-4 and IL-21 separably program germinal center-like phenotype acquisition, BCL6 expression, proliferation and survival of CD40L-activated B cells in vitro. Immunol. Cell Biol. 2019, 97, 826–839. [Google Scholar] [CrossRef]
- Brown, A.J.; Sweeney, B.; Mainwaring, D.O.; James, D.C. NF-κB, CRE and YY1 elements are key functional regulators of CMV promoter-driven transient gene expression in CHO cells. Biotechnol. J. 2015, 10, 1019–1028. [Google Scholar] [CrossRef] [PubMed]
- Stinski, M.F.; Isomura, H. Role of the cytomegalovirus major immediate early enhancer in acute infection and reactivation from latency. Med. Microbiol. Immunol. 2008, 197, 223–231. [Google Scholar] [CrossRef] [PubMed]
- Elgueta, R.; Benson, M.J.; de Vries, V.C.; Wasiuk, A.; Guo, Y.; Noelle, R.J. Molecular mechanism and function of CD40/CD40L engagement in the immune system. Immunol. Rev. 2009, 229, 152–172. [Google Scholar] [CrossRef]
- Hostager, B.; Bishop, G. CD40-mediated activation of the NF-κB2 pathway. Front. Immunol. 2013, 4, 376. [Google Scholar] [CrossRef] [PubMed]
- Huse, K.; Wogsland, C.E.; Polikowsky, H.G.; Diggins, K.E.; Smeland, E.B.; Myklebust, J.H.; Irish, J.M. Human germinal center B cells differ from naïve and memory B cells in CD40 expression and CD40L-induced signaling response. Cytom. Part A 2019, 95, 442–449. [Google Scholar] [CrossRef]
- Roy, K.; Mitchell, S.; Liu, Y.; Ohta, S.; Lin, Y.S.; Metzig, M.O.; Nutt, S.L.; Hoffmann, A. A regulatory circuit controlling the dynamics of NFκB cRel transitions B cells from proliferation to plasma cell differentiation. Immunity 2019, 50, 616–628.e6. [Google Scholar] [CrossRef]
- Sonam, C.; Ruzgys, P.; Maciulevičius, M.; Šatkauskas, S. Effect of cell passage time on the electrotransfection efficiency. Biol. Bull. 2020, 47, 441–447. [Google Scholar] [CrossRef]
- ISO 10993−5. Biological Evaluation of Medical Devices—Part 5: Tests for In Vitro Cytotoxicity; Beuth Verlag GmbH: Berlin, Germany, 2009. [Google Scholar]
- Thompson, J.M.; Gralow, J.R.; Levy, R.; Miller, R.A. The optimal application of forward and ninety-degree light scatter in flow cytometry for the gating of mononuclear cells. Cytometry 1985, 6, 401–406. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| TE 1 (%) | MFI 2 (a.u.) | Viability (%) | ||||
|---|---|---|---|---|---|---|
| N/P Ratio | Nano-Stars 24 h/48 h | l-PEI 24 h/48 h | Nano-Stars 24 h/48 h | l-PEI 24 h/48 h | Nano-Stars 24 h/48 h | l-PEI 24 h/48 h |
| 3 | 4.9/2.4 | 3.7 /0.1 | 750/1085 | 495/715 | 66.3/48.8 | 90.9/95.9 |
| 5 | 9.3/6.5 | 1.8 /0.2 | 1289/2393 | 483/732 | 62.1/58.3 | 95.8/88.1 |
| 7.5 | 7.5/7.2 | 5.0 /0.3 | 1719/2099 | 500/732 | 60.9/54.3 | 77.7/83.9 |
| 12.5 | 24.3/8.1 | 1.9 /0.2 | 947/1311 | 496/784 | 46.6/44.9 | 80.5/90.5 |
| 15 | 15.1/6.2 | 2.7 /0.3 | 997/1096 | 503/778 | 52.9/31.2 | 75.1/93.8 |
| 20 | 20.5/4.1 | 4.1 /0.1 | 944/1103 | 500/730 | 55.5/42.3 | 80.7/87.3 |
| Cryovial Nb. | Viability Pre-Transfection (%) | Transfection Efficiency (%) | Viability Post-Transfection (%) |
|---|---|---|---|
| I 1 | 22 | 33 | |
| 79.9 | 28.6 | 35.6 | |
| 28 | 35.4 | ||
| II | 90.4 | 41.3 | 54.1 |
| III | 84.4 | 16 | 46.2 |
| IV | 86.9 | 31.8 | 52.9 |
| V | 82.6 | 40.8 | 63.6 |
| Mean ± SD (%) | 83.4 ± 4.1 | 29.8 ± 9.2 | 45.8 ± 11.6 |
| Cultivation Time | |||||
|---|---|---|---|---|---|
| Pre-Transfection (Days) | Post-Transfection (Hours) | TE (%) | MFI 2 (a.u.) | Viability (%) | |
| Tfd3 | 3 | 24 | 39.2 ± 0.9 | 34,537 ± 1532 | 61.8 ± 0.3 |
| (n = 3) | |||||
| 48 | 28.2 ± 6.1 * | 32,220 ± 2695 | 53.0 ± 1.6 * | ||
| (n = 3) | |||||
| Tfd4 1 | 4 | 24 | 37.4 ± 1.8 | 41,564 ± 11,966 | 69.3 ± 1.7 # |
| (n = 2) | |||||
| 48 | 32.5 ± 3.9 | 35,204 ± 9644 | 54.2 ± 2.4 * | ||
| (n = 4) | |||||
| Tfd5 | 5 | 24 | n.a | n.a | n.a |
| 48 | 21.2 ± 5.8 § | 10,940 ± 759 | 38.3 ± 9.8 # | ||
| (n = 3) | |||||
| Total Cultivation Time (Days) | |||||
|---|---|---|---|---|---|
| Subclasses | Classification 2 | 0 | 3 | 4 | 5 |
| CD20+CD27−CD38−/+ | Naive | 21.0 ± 0.1 | 7.7 | 7.9 ± 3.7 | 2.7 ± 0.7 |
| CD20+CD27+CD38− | Memory | 24.6 ± 5.8 | 7.4 | 4.1 ± 2.6 | 0.6 ± 0.04 |
| CD20−/+CD27+CD38+ | GC 3 | 12.8 ± 1.0 | 3.9 | 6.1 ± 2.5 | 7.0 ± 4.4 |
| CD20−CD27++CD38++ | Plasma | 2.8 ± 0.6 | 43.1 | 55.2 ± 6.1 | 69.0 ± 10.6 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Keim, D.; Gollner, K.; Gollner, U.; Jérôme, V.; Freitag, R. Generation of Recombinant Primary Human B Lymphocytes Using Non-Viral Vectors. Int. J. Mol. Sci. 2021, 22, 8239. https://doi.org/10.3390/ijms22158239
Keim D, Gollner K, Gollner U, Jérôme V, Freitag R. Generation of Recombinant Primary Human B Lymphocytes Using Non-Viral Vectors. International Journal of Molecular Sciences. 2021; 22(15):8239. https://doi.org/10.3390/ijms22158239
Chicago/Turabian StyleKeim, Daniel, Katrin Gollner, Ulrich Gollner, Valérie Jérôme, and Ruth Freitag. 2021. "Generation of Recombinant Primary Human B Lymphocytes Using Non-Viral Vectors" International Journal of Molecular Sciences 22, no. 15: 8239. https://doi.org/10.3390/ijms22158239
APA StyleKeim, D., Gollner, K., Gollner, U., Jérôme, V., & Freitag, R. (2021). Generation of Recombinant Primary Human B Lymphocytes Using Non-Viral Vectors. International Journal of Molecular Sciences, 22(15), 8239. https://doi.org/10.3390/ijms22158239