
A Microfluidic 3D Endothelium-on-a-Chip Model to Study Transendothelial Migration of T Cells in Health and Disease

 ,
,
Abstract
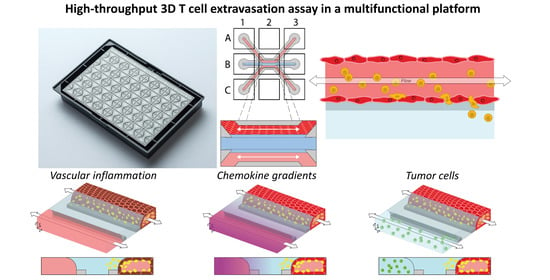
1. Introduction
2. Results
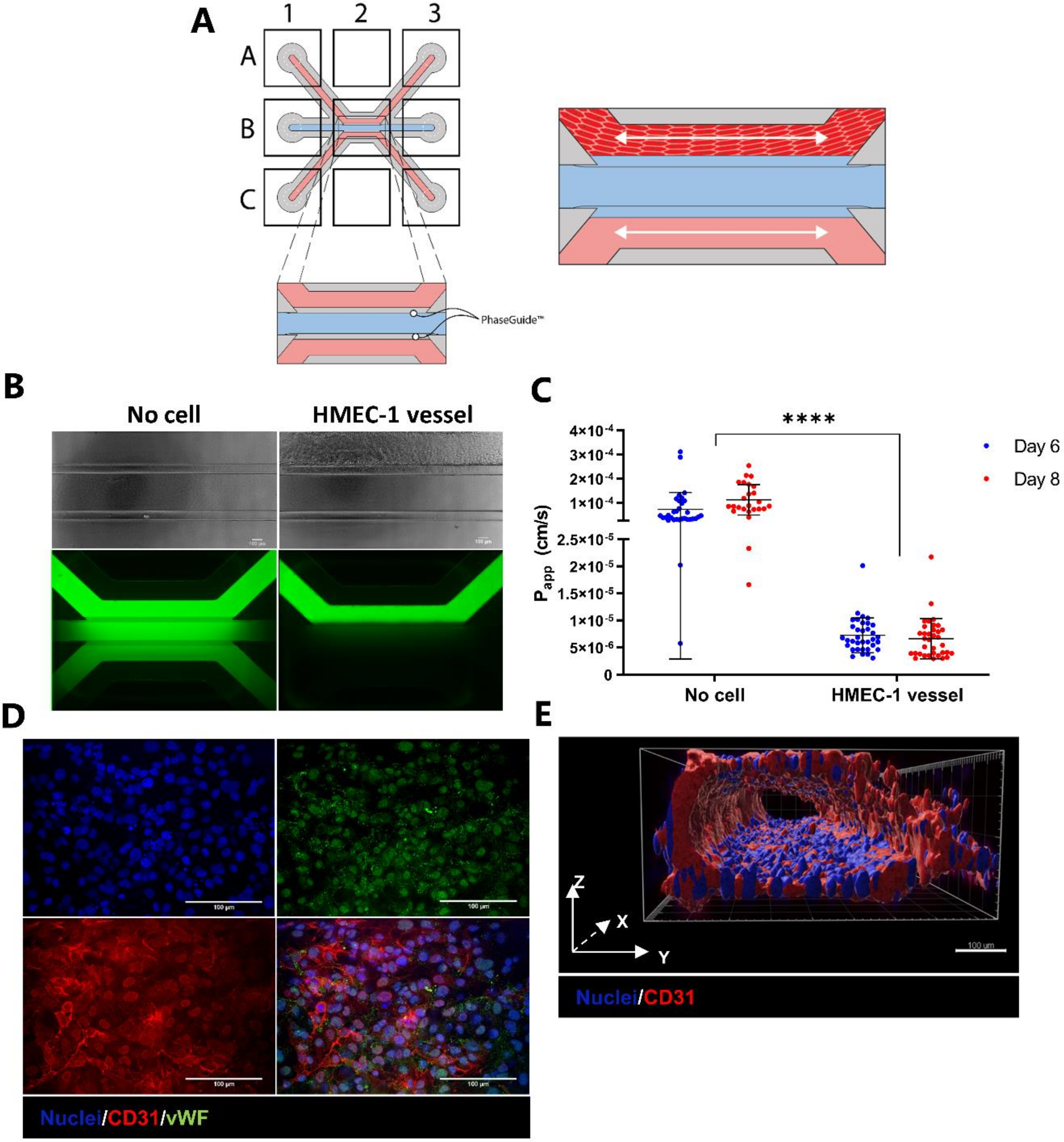
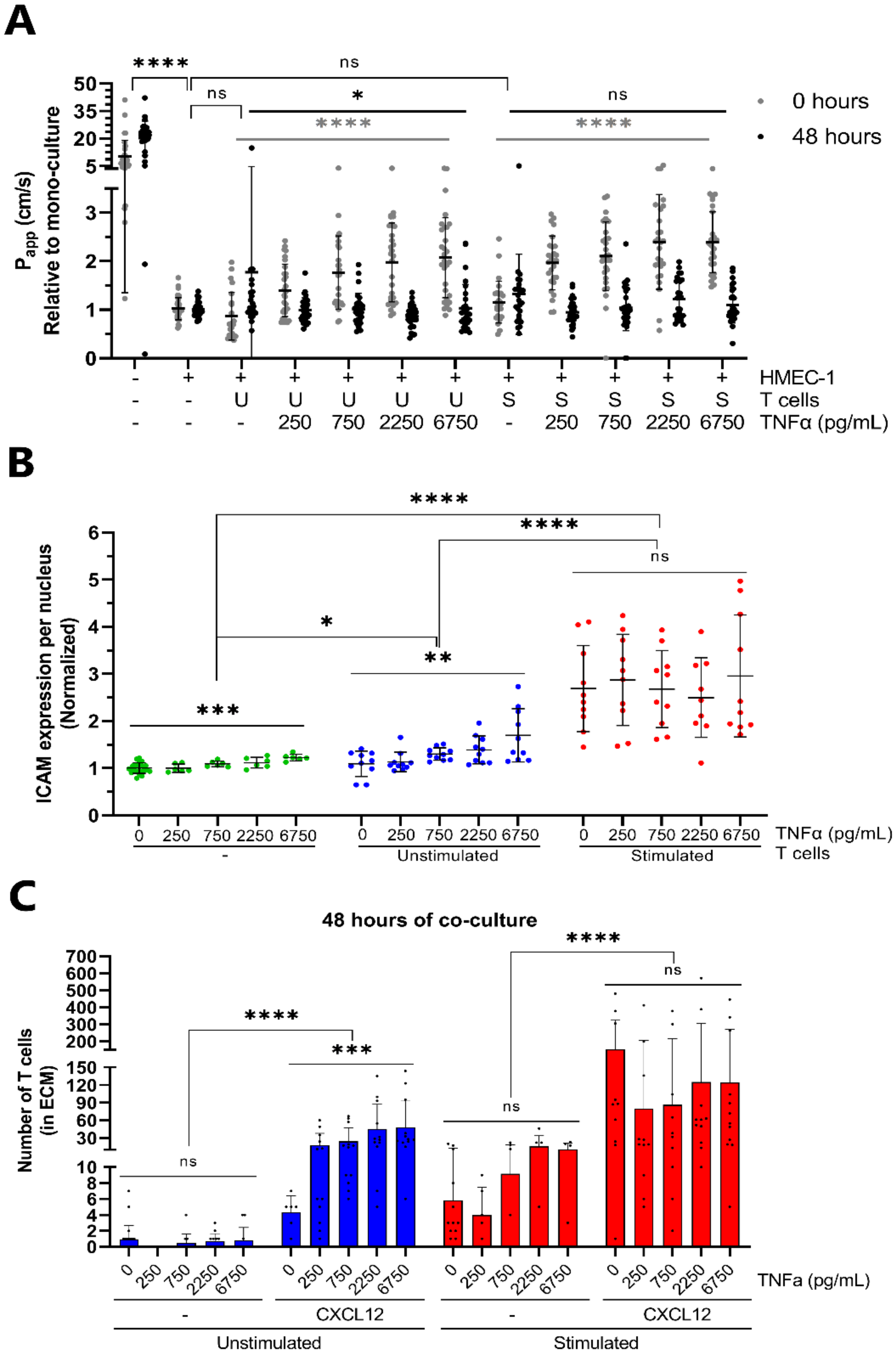
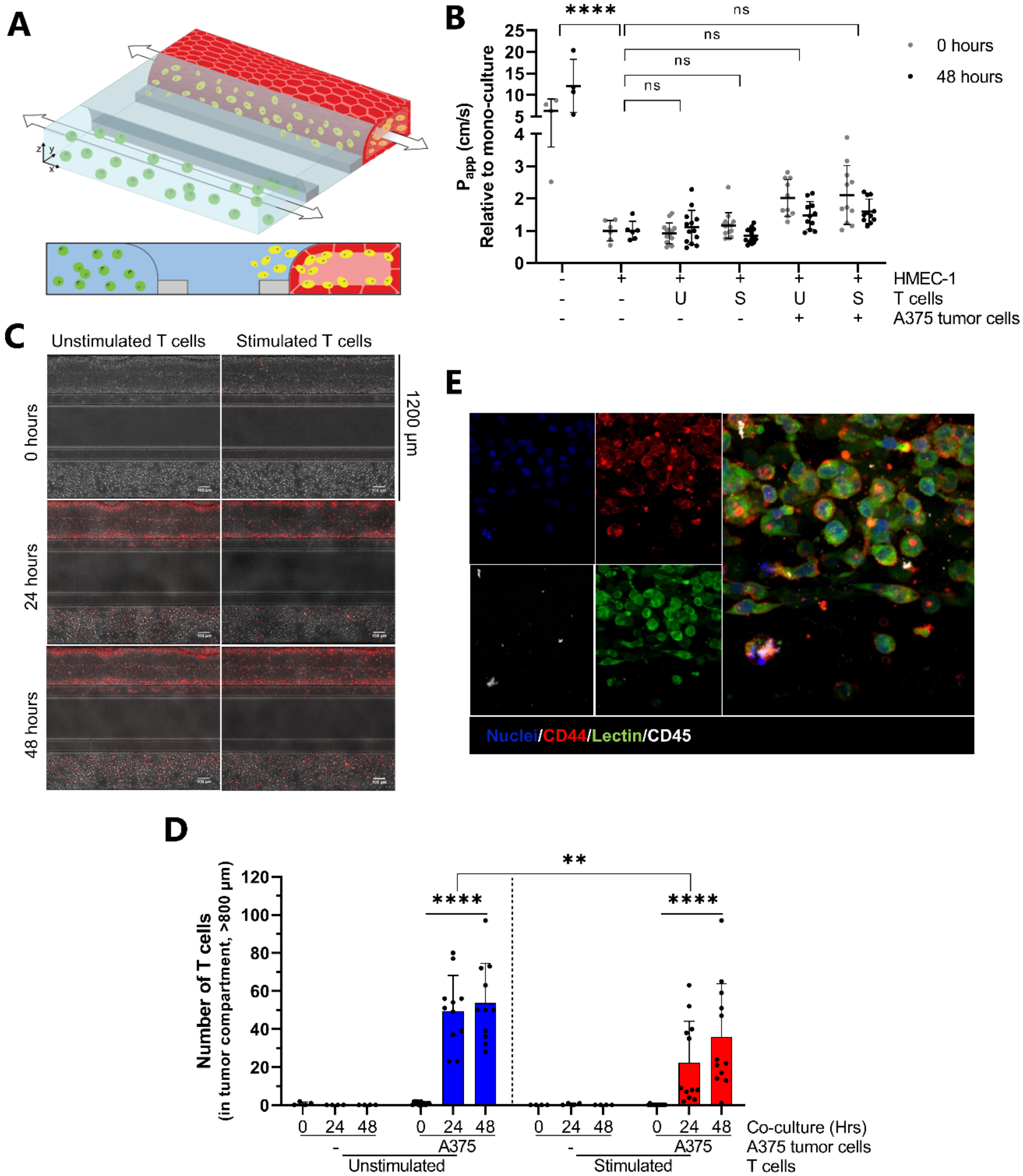
2.1. Development and Characterization of 3D Endothelial Vessels Perfused with T Cells
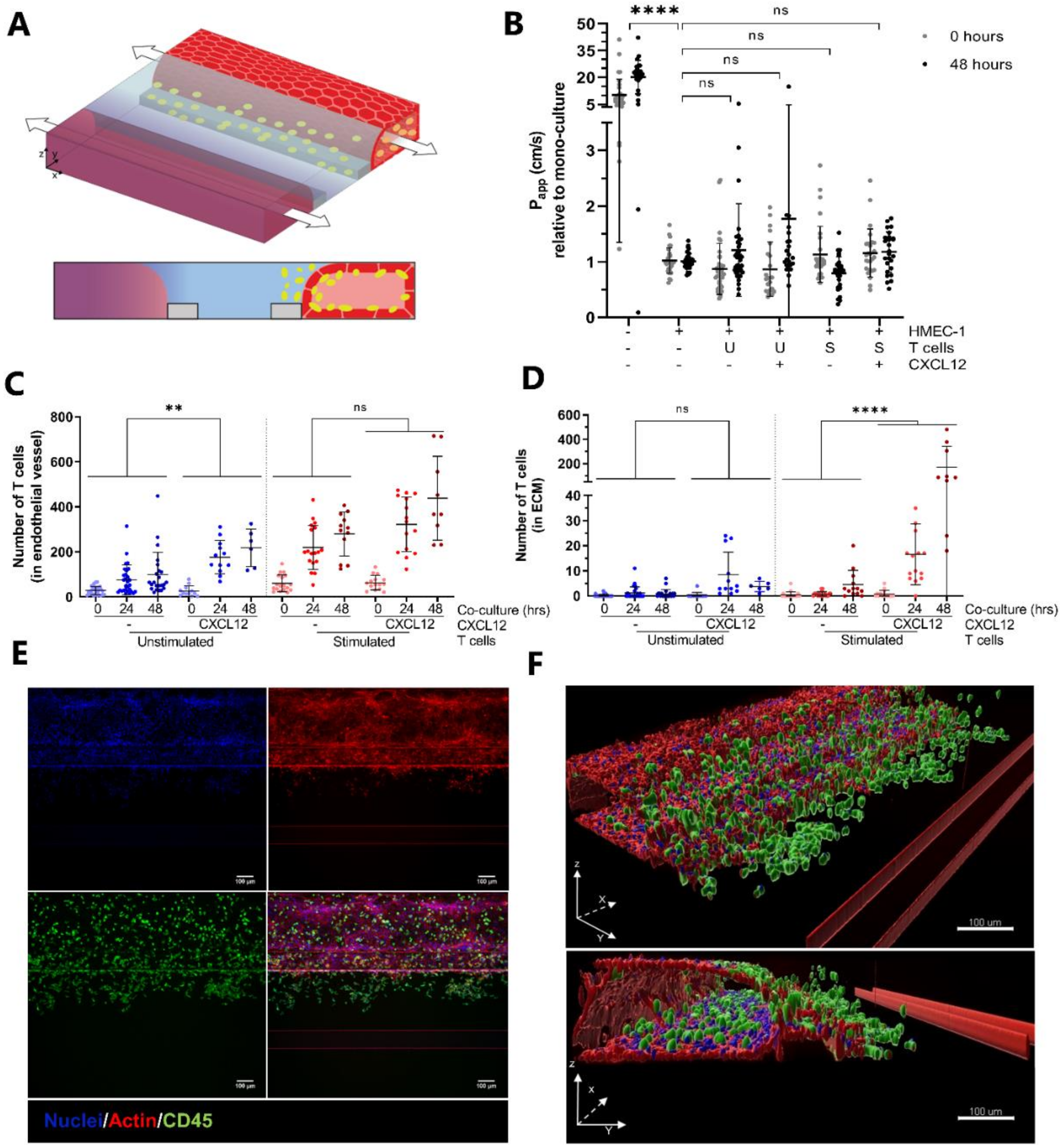
2.2. CXCL12 Stimulates Transendothelial Migration of T Cells into Collagen Hydrogel
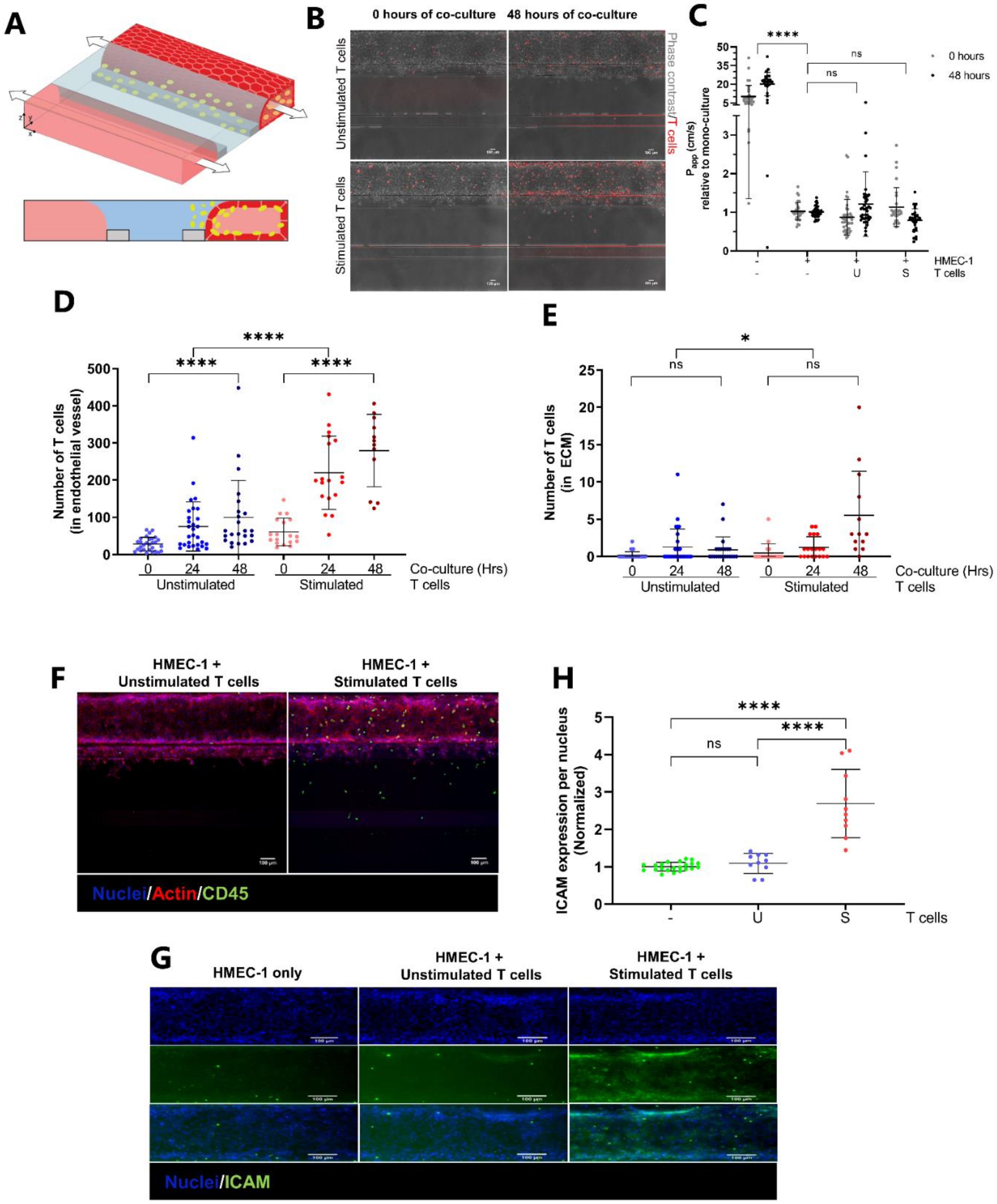
2.3. Transendothelial Migration of T Cells Is Increased by Addition of CXCL12 under Inflammatory Conditions
2.4. Proof-of-Concept; Transendothelial T Cell Migration towards A375 Melanoma Cells
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. T Cell Stimulation and Labelling
4.3. Microfluidic Co-Culture
4.4. Barrier Integrity Assessment
4.5. Quantification of T Cell Dynamics
4.6. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ECM | Extracellular matrix |
| TNFα | Tumor necrosis factor alpha |
| IFNγ | Interferon gamma |
| ICAM-1 | Intercellular adhesion molecule |
| TEM | Transendothelial migration |
| FITC | Fluorescein isothiocyanate |
| TRITC | Tetramethylrhodamine isothiocyanate |
| PECAM-1 | Platelet endothelial cell adhesion molecule-1 |
| vWF | Von Willebrand factor |
| PDMS | Polydimethylsiloxane |
| PBMC | Peripheral blood mononuclear cell |
References
- Parkin, J.; Cohen, B. An Overview of the Immune System. Lancet 2001, 357, 1777–1789. [Google Scholar] [CrossRef]
- Wang, L.; Wang, F.S.; Gershwin, M.E. Human Autoimmune Diseases: A Comprehensive Update. J. Intern. Med. 2015, 278, 369–395. [Google Scholar] [CrossRef]
- Gonzalez, H.; Hagerling, C.; Werb, Z. Roles of the Immune System in Cancer: From Tumor Initiation to Metastatic Progression. Genes Dev. 2018, 32, 1267–1284. [Google Scholar] [CrossRef] [PubMed]
- Medzhitov, R. Inflammation 2010: New Adventures of an Old Flame. Cell 2010, 140, 771–776. [Google Scholar] [CrossRef] [PubMed]
- Moro-García, M.A.; Mayo, J.C.; Sainz, R.M.; Alonso-Arias, R. Influence of Inflammation in the Process of T Lymphocyte Differentiation: Proliferative, Metabolic, and Oxidative Changes. Front. Immunol. 2018, 9, 339. [Google Scholar] [CrossRef] [PubMed]
- Bevilacqua, M.P.; Gimbrone, M.A. Inducible Endothelial Functions in Inflammation and Coagulation. Semin. Thromb. Hemost. 1987, 13, 425–433. [Google Scholar] [CrossRef] [PubMed]
- Esche, C.; Stellato, C.; Beck, L.A. Chemokines: Key Players in Innate and Adaptive Immunity. J. Investig. Dermatol. 2005, 125, 615–628. [Google Scholar] [CrossRef]
- Schimmel, L.; Heemskerk, N.; van Buul, J.D. Leukocyte Transendothelial Migration: A Local Affair. Small GTPases 2017, 8, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Muller, W.A. The Regulation of Transendothelial Migration: New Knowledge and New Questions. Cardiovasc. Res. 2015, 310–320. [Google Scholar] [CrossRef] [PubMed]
- Masopust, D.; Sivula, C.P.; Jameson, S.C. Of Mice, Dirty Mice, and Men: Using Mice To Understand Human Immunology. J. Immunol. 2017, 199, 383–388. [Google Scholar] [CrossRef]
- Mohammad, M.G.; Tsai, V.W.W.; Ruitenberg, M.J.; Hassanpour, M.; Li, H.; Hart, P.H.; Breit, S.N.; Sawchenko, P.E.; Brown, D.A. Immune Cell Trafficking from the Brain Maintains CNS Immune Tolerance. J. Clin. Investig. 2014, 124, 1228–1241. [Google Scholar] [CrossRef] [PubMed]
- Benechet, A.P.; Menon, M.; Khanna, K.M. Visualizing T Cell Migration in Situ. Front. Immunol. 2014, 5, 1–12. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Krzastek, S.C.; Goliadze, E.; Zhou, S.; Petrossian, A.; Youniss, F.; Sundaresan, G.; Wang, L.; Zweit, J.; Guruli, G. Dendritic Cell Trafficking in Tumor-Bearing Mice. Cancer Immunol. Immunother. 2018, 67, 1939–1947. [Google Scholar] [CrossRef] [PubMed]
- Na, I.K.; Markley, J.C.; Tsai, J.J.; Yim, N.L.; Beattie, B.J.; Klose, A.D.; Holland, A.M.; Ghosh, A.; Rao, U.K.; Stephan, M.T.; et al. Concurrent Visualization of Trafficking, Expansion, and Activation of T Lymphocytes and T-Cell Precursors in Vivo. Blood 2010, 116, 18–25. [Google Scholar] [CrossRef]
- Kircher, M.F.; Grimm, J.; Swirski, F.K.; Libby, P.; Gerszten, R.E.; Allport, J.R.; Weissleder, R. Noninvasive in Vivo Imaging of Monocyte Trafficking to Atherosclerotic Lesions. Circulation 2008, 117, 388–395. [Google Scholar] [CrossRef]
- Mestas, J.; Hughes, C.C.W. Of Mice and Not Men: Differences between Mouse and Human Immunology. J. Immunol. 2004, 172, 2731–2738. [Google Scholar] [CrossRef] [PubMed]
- Levy, N. Leading Opinion The Use of Animal as Models: Ethical Considerations. Int. J. Stroke 2012, 7, 440–442. [Google Scholar] [CrossRef]
- Sellick, J. Enhancing the Protection of Animals Used for Scientific Purposes. Environ. Law Manag. 2011, 23, 75–82. [Google Scholar]
- Abbott, A. Cell Culture: Biology’s New Dimension. Nature 2003, 424, 870–872. [Google Scholar] [CrossRef] [PubMed]
- Ledgerwood, L.G.; Lal, G.; Zhang, N.; Garin, A.; Esses, S.J.; Ginhoux, F.; Merad, M.; Peche, H.; Lira, S.A.; Ding, Y.; et al. The Sphingosine 1-Phosphate Receptor 1 Causes Tissue Retention by Inhibiting the Entry of Peripheral Tissue T Lymphocytes into Afferent Lymphatics. Nat. Immunol. 2008, 9, 42–53. [Google Scholar] [CrossRef]
- Irekeola, A.A.; EAR, E.N.S.; Mat Lazim, N.; Mohamud, R.; Yean, C.Y.; Shueb, R.H. Technical Considerations in Ex Vivo Human Regulatory T Cell Migration and Suppression Assays. Cells 2020, 9, 487. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.; Herbert, J.A.; Smith, C.M.; Smyth, R.L. An in Vitro Transepithelial Migration Assay to Evaluate the Role of Neutrophils in Respiratory Syncytial Virus (RSV) Induced Epithelial Damage. Sci. Rep. 2018, 8, 6777. [Google Scholar] [CrossRef] [PubMed]
- Stewart-Hutchinson, P.J.; Szasz, T.P.; Jaeger, E.R.; Onken, M.D.; Cooper, J.A.; Morley, S.C. Technical Advance: New in Vitro Method for Assaying the Migration of Primary B Cells Using an Endothelial Monolayer as Substrate. J. Leukoc. Biol. 2017, 102, 941–948. [Google Scholar] [CrossRef] [PubMed]
- Justus, C.R.; Leffler, N.; Ruiz-echevarria, M.; Yang, L.V. In Vitro Cell Migration and Invasion Assays. Mutat. Res. Rev. Mutat. Res. 2014, 752, 1–8. [Google Scholar] [CrossRef]
- Zhang, B.; Korolj, A.; Lai, B.F.L.; Radisic, M. Advances in Organ-on-a-Chip Engineering. Nat. Rev. Mater. 2018, 3, 257–278. [Google Scholar] [CrossRef]
- Poussin, C.; Kramer, B.; Lanz, H.L.; Van den Heuvel, A.; Laurent, A.; Olivier, T.; Vermeer, M.; Peric, D.; Baumer, K.; Dulize, R.; et al. 3D Human Microvessel-on-a-Chip Model for Studying Monocyte-to-Endothelium Adhesion under Flow—Application in Systems Toxicology. ALTEX 2020, 37, 47–63. [Google Scholar] [CrossRef]
- Mondadori, C.; Crippa, M.; Moretti, M.; Candrian, C.; Lopa, S.; Arrigoni, C. Advanced Microfluidic Models of Cancer and Immune Cell Extravasation: A Systematic Review of the Literature. Front. Bioeng. Biotechnol. 2020, 8, 907. [Google Scholar] [CrossRef]
- Gjorevski, N.; Avignon, B.; Gérard, R.; Cabon, L.; Roth, A.B.; Bscheider, M.; Moisan, A. Neutrophilic Infiltration in Organ-on-a-Chip Model of Tissue Inflammation. Lab Chip 2020, 20, 3365–3374. [Google Scholar] [CrossRef]
- Pavesi, A.; Kamm, R.D.; Bertoletti, A.; Pavesi, A.; Tan, A.T.; Koh, S.; Chia, A.; Colombo, M.; Antonecchia, E.; Miccolis, C.; et al. A 3D Microfluidic Model for Preclinical Evaluation of TCR-Engineered T Cells against Solid Tumors. JCI Insight 2017, 2, e89762. [Google Scholar] [CrossRef]
- Ando, Y.; Siegler, E.L.; Ta, H.P.; Cinay, G.E.; Zhou, H.; Gorrell, K.A.; Au, H.; Jarvis, B.M.; Wang, P.; Shen, K. Evaluating CAR-T Cell Therapy in a Hypoxic 3D Tumor Model. Adv. Healthc. Mater. 2019, 8, 1900001. [Google Scholar] [CrossRef] [PubMed]
- Ayuso, J.M.; Truttschel, R.; Gong, M.M.; Humayun, M.; Vitek, R.; Felder, M.; Gillies, S.D.; Wisinski, K.B.; Patankar, M.; Beebe, D.J.; et al. Evaluating Natural Killer Cell Cytotoxicity against Solid Tumors Using a Microfluidic Model. Oncoimmunology 2018, 8, 1553477. [Google Scholar] [CrossRef]
- Nawroth, J.; Lucchesi, C.; Cheng, D.; Shukla, A.; Ngyuen, J.; Shroff, T.; Karalis, K.; Lee, H.H.; Alves, S.; Hamilton, G.A.; et al. A Micro-Engineered Airway Lung-Chip Models Key Features of Viral-Induced Exacerbation of Asthma. Am. J. Respir. Cell Mol. Biol. 2020, 63, 591–600. [Google Scholar] [CrossRef]
- Trietsch, S.J.; Israels, G.D.; Joore, J.; Hankemeier, T.; Vulto, P. Microfluidic Titer Plate for Stratified 3D Cell Culture. Lab Chip 2013, 13, 3548–3554. [Google Scholar] [CrossRef] [PubMed]
- Van Duinen, V.; Van Den Heuvel, A.; Trietsch, S.J.; Lanz, H.L.; Van Gils, J.M.; Van Zonneveld, A.J.; Vulto, P.; Hankemeier, T. 96 Perfusable Blood Vessels To Study Vascular Permeability in Vitro. Sci. Rep. 2017, 7, 18071. [Google Scholar] [CrossRef] [PubMed]
- Muller, W.A.; Weigl, S.A.; Deng, X.; Phillips, D.M. PECAM-1 Is Required for Transendothelial Migration of Leukocytes. J. Exp. Med. 1993, 178, 449–460. [Google Scholar] [CrossRef]
- Medzhitov, R. Origin and Physiological Roles of Inflammation. Nature 2008, 454, 428–435. [Google Scholar] [CrossRef]
- Haslam, A.; Prasad, V. Estimation of the Percentage of US Patients with Cancer Who Are Eligible for and Respond to Checkpoint Inhibitor Immunotherapy Drugs. JAMA Netw. Open 2019, 2, e192535. [Google Scholar] [CrossRef] [PubMed]
- Sip, C.G.; Bhattacharjee, N.; Folch, A. Microfluidic transwell inserts for generation of tissue culture-friendly gradients in well plates. Lab Chip. 2014, 302–314. [Google Scholar] [CrossRef]
- Zhang, C.; Barrios, M.P.; Alani, R.M.; Cabodi, M.; Wong, J.Y. A Micro Fl Uidic Transwell to Study Chemotaxis. Exp. Cell Res. 2016. [Google Scholar] [CrossRef]
- Lei, K.F.; Tseng, H.P.; Lee, C.Y.; Tsang, N.M. Quantitative Study of Cell Invasion Process under Extracellular Stimulation of Cytokine in a Microfluidic Device. Sci. Rep. 2016, 6, 25557. [Google Scholar] [CrossRef]
- Wong, I.; Ho, C.M. Surface Molecular Property Modifications for Poly(Dimethylsiloxane) (PDMS) Based Microfluidic Devices. Microfluid. Nanofluid. 2009, 7, 291–306. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Chiao, M. Anti-Fouling Coatings of Poly(Dimethylsiloxane) Devices for Biological and Biomedical Applications. J. Med. Biol. Eng. 2015, 35, 143–155. [Google Scholar] [CrossRef] [PubMed]
- Halldorsson, S.; Lucumi, E.; Gómez-Sjöberg, R.; Fleming, R.M.T. Advantages and Challenges of Microfluidic Cell Culture in Polydimethylsiloxane Devices. Biosens. Bioelectron. 2015, 63, 218–231. [Google Scholar] [CrossRef] [PubMed]
- Chistiakov, D.A.; Orekhov, A.N.; Bobryshev, Y.V. Effects of Shear Stress on Endothelial Cells: Go with the Flow. Acta Physiol. 2017, 219, 382–408. [Google Scholar] [CrossRef] [PubMed]
- Wevers, N.R.; Kasi, D.G.; Gray, T.; Wilschut, K.J.; Smith, B.; Vught, R.; Shimizu, F.; Sano, Y.; Kanda, T.; Marsh, G.; et al. A Perfused Human Blood-Brain Barrier on-a-Chip for High-Throughput Assessment of Barrier Function and Antibody Transport. Fluids Barriers CNS 2018, 15, 1–12. [Google Scholar] [CrossRef]
- Schall, T.J.; Bacon, K.B. Chemokines, Leukocyte Trafficking, and Inflammation. Curr. Opin. Immunol. 1994, 6, 865–873. [Google Scholar] [CrossRef]
- Reiss, Y.; Engelhardt, B. T Cell Interaction with ICAM-1-Deficient Endothelium in vitro: Transendothelial Migration of Different T Cell Populations Is Mediated by Endothelial ICAM-1 and ICAM-2. Int. Immunol. 1999, 11, 1527–1539. [Google Scholar] [CrossRef][Green Version]
- Van De Stolpe, A.; Van Der Saag, P.T. Intercellular Adhesion Molecule-1. J. Mol. Med. 1996, 74, 13–33. [Google Scholar] [CrossRef] [PubMed]
- Von Andrian, U.H.; Mackay, C.R. T-Cell Function and Migration. Two Sides of the Same Coin. N. Engl. J. Med. 2000, 343, 1020–1034. [Google Scholar] [CrossRef]
- Clark, P.R.; Manes, T.D.; Pober, J.S.; Kluger, M.S. Increased ICAM-1 Expression Causes Endothelial Cell Leakiness, Cytoskeletal Reorganization and Junctional Alterations. J. Investig. Dermatol. 2007, 127, 762–774. [Google Scholar] [CrossRef]
- Royall, J.A.; Berkow, R.L.; Beckman, J.S.; Cunningham, M.K.; Matalon, S.; Freeman, B.A. Tumor Necrosis Factor and Interleukin 1 Alpha Increase Vascular Endothelial Permeability. Am. J. Physiol. 1989, 257, L399–L410. [Google Scholar] [CrossRef]
- Balkwill, F. Cancer and the Chemokine Network. Nat. Rev. Cancer 2004, 4, 540–550. [Google Scholar] [CrossRef] [PubMed]
- Landskron, G.; De La Fuente, M.; Thuwajit, P.; Thuwajit, C.; Hermoso, M.A. Chronic Inflammation and Cytokines in the Tumor Microenvironment. J. Immunol. Res. 2014, 2014, 149185. [Google Scholar] [CrossRef]
- Salven, P.; Heikkilä, P.; Joensuu, H. Enhanced Expression of Vascular Endothelial Growth Factor in Metastatic Melanoma. Br. J. Cancer 1997, 76, 930–934. [Google Scholar] [CrossRef] [PubMed]
- Ormiston, M.L.; Toshner, M.R.; Kiskin, F.N.; Huang, C.J.Z.; Groves, E.; Morrell, N.W.; Rana, A.A. Generation and Culture of Blood Outgrowth Endothelial Cells from Human Peripheral Blood. JoVE J. Vis. Exp. 2015, 106, e53384. [Google Scholar] [CrossRef]
- Mathur, T.; Singh, K.A.; Pandian, N.K.R.; Tsai, S.; Hein, T.W.; Gaharwar, A.K.; Flanagan, J.M.; Jain, A. Organ-on-chips made of blood: Endothelial progenitor cells from blood reconstitute vascular thromboinflammation in vessel-chips. Lab Chip. 2019, 19, 2500–2511. [Google Scholar] [CrossRef]
- Liston, A.; Carr, E.J.; Linterman, M.A. Shaping Variation in the Human Immune System. Trends Immunol. 2016, 37, 637–646. [Google Scholar] [CrossRef] [PubMed]
- Trietsch, S.J.; Naumovska, E.; Kurek, D.; Setyawati, M.C.; Vormann, M.K.; Wilschut, K.J.; Lanz, H.L.; Nicolas, A.; Ng, C.P.; Joore, J.; et al. Membrane-Free Culture and Real-Time Barrier Integrity Assessment of Perfused Intestinal Epithelium Tubes. Nat. Commun. 2017, 8, 262. [Google Scholar] [CrossRef]
- Sternberg, S.R. Biomedical Image Processing. Computer 1983, 16, 22–34. [Google Scholar] [CrossRef]
- Sbalzarini, I.F.; Koumoutsakos, P. Feature Point Tracking and Trajectory Analysis for Video Imaging in Cell Biology. J. Struct. Biol. 2005, 151, 182–195. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Frequency of Parent | ||
|---|---|---|
| Subset | Unstimulated T cells | Stimulated T cells |
| Single cells | 97.75 ± 0.55% | 88.95 ± 0.75% |
| CD14+, CD3+ | 4.55 ± 0.15% | 6.17 ± 0.08% |
| CD14+, CD3- | 1.69 ± 0.36% | 2.51 ± 0.28% |
| CD14−, CD3+ | 91.20 ± 0.80% | 86.15 ± 1.35% |
| CD8+, CD4+ | 0.11 ± 0.02% | 1.08 ± 0.07% |
| CD8+, CD4- | 20.10 ± 0.70% | 19.80 ± 1.0% |
| CD8−, CD4+ | 78.05 ± 1.45% | 77.60 ± 1.4% |
| CD8−, CD4- | 1.77 ± 0.70% | 1.50 ± 0.45% |
| CD14−, CD3 | 2.60 ± 0.27% | 5.17 ± 1.02% |
| Frequency of Parent | ||
|---|---|---|
| Subset | Unstimulated T cells | Stimulated T cells |
| Single cells | 98.45 ± 0.35% | 94.65 ± 0.35% |
| Live cells | 92.20 ± 0.20% | 94.60 ± 0.10% |
| IFNγ+ | 0.19 ± 0.13% | 38.45 ± 2.85% |
| IFNγ- | 99.82 ± 0.12% | 61.55 ± 2.85% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
de Haan, L.; Suijker, J.; van Roey, R.; Berges, N.; Petrova, E.; Queiroz, K.; Strijker, W.; Olivier, T.; Poeschke, O.; Garg, S.; et al. A Microfluidic 3D Endothelium-on-a-Chip Model to Study Transendothelial Migration of T Cells in Health and Disease. Int. J. Mol. Sci. 2021, 22, 8234. https://doi.org/10.3390/ijms22158234
de Haan L, Suijker J, van Roey R, Berges N, Petrova E, Queiroz K, Strijker W, Olivier T, Poeschke O, Garg S, et al. A Microfluidic 3D Endothelium-on-a-Chip Model to Study Transendothelial Migration of T Cells in Health and Disease. International Journal of Molecular Sciences. 2021; 22(15):8234. https://doi.org/10.3390/ijms22158234
Chicago/Turabian Stylede Haan, Luuk, Johnny Suijker, Ruthger van Roey, Nina Berges, Elissaveta Petrova, Karla Queiroz, Wouter Strijker, Thomas Olivier, Oliver Poeschke, Sakshi Garg, and et al. 2021. "A Microfluidic 3D Endothelium-on-a-Chip Model to Study Transendothelial Migration of T Cells in Health and Disease" International Journal of Molecular Sciences 22, no. 15: 8234. https://doi.org/10.3390/ijms22158234
APA Stylede Haan, L., Suijker, J., van Roey, R., Berges, N., Petrova, E., Queiroz, K., Strijker, W., Olivier, T., Poeschke, O., Garg, S., & van den Broek, L. J. (2021). A Microfluidic 3D Endothelium-on-a-Chip Model to Study Transendothelial Migration of T Cells in Health and Disease. International Journal of Molecular Sciences, 22(15), 8234. https://doi.org/10.3390/ijms22158234