
CYP2E1 in Alcoholic and Non-Alcoholic Liver Injury. Roles of ROS, Reactive Intermediates and Lipid Overload



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. The Discovery of CYP2E1
2. Expression, Functions and Cellular Fate of CYP2E1
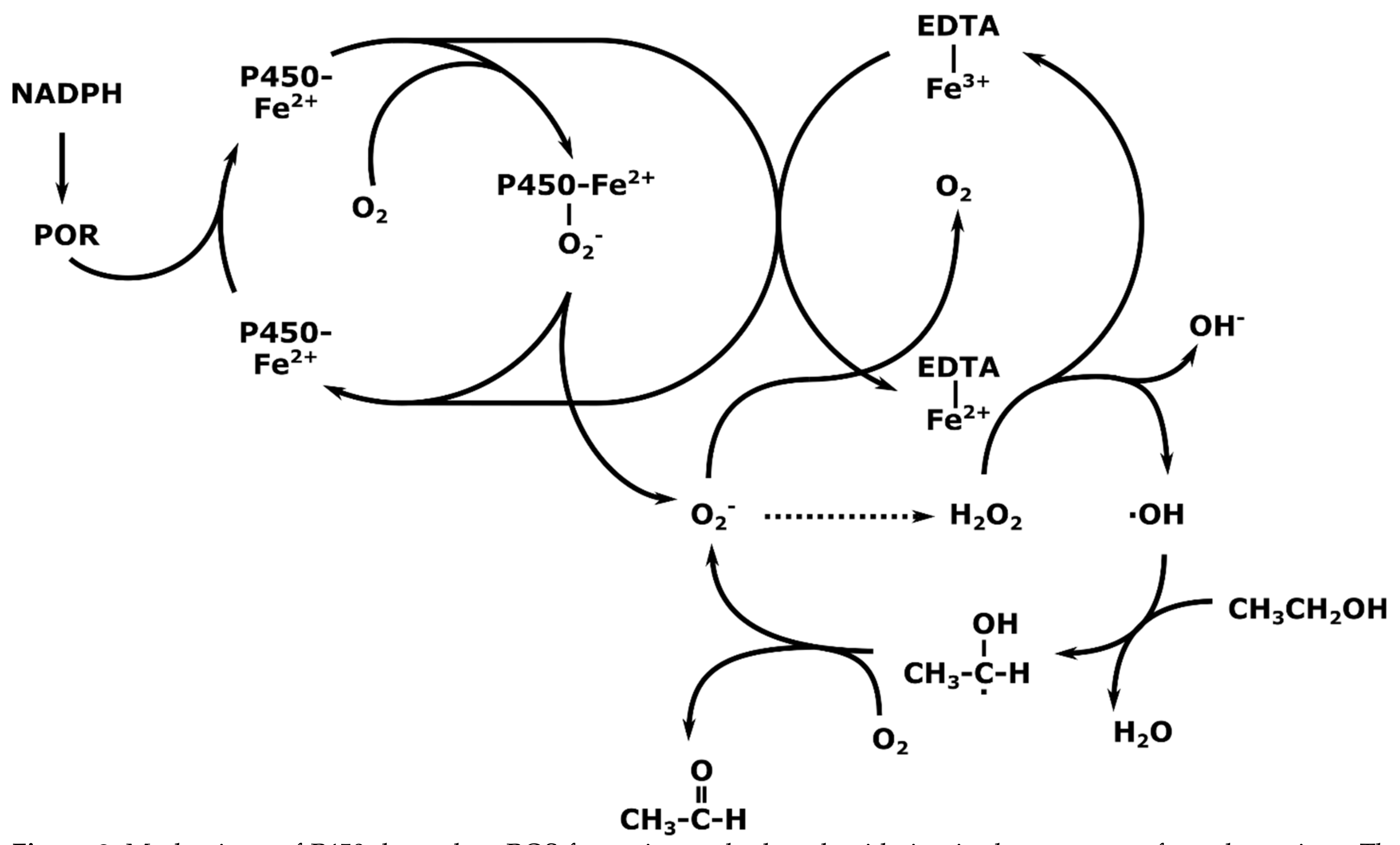
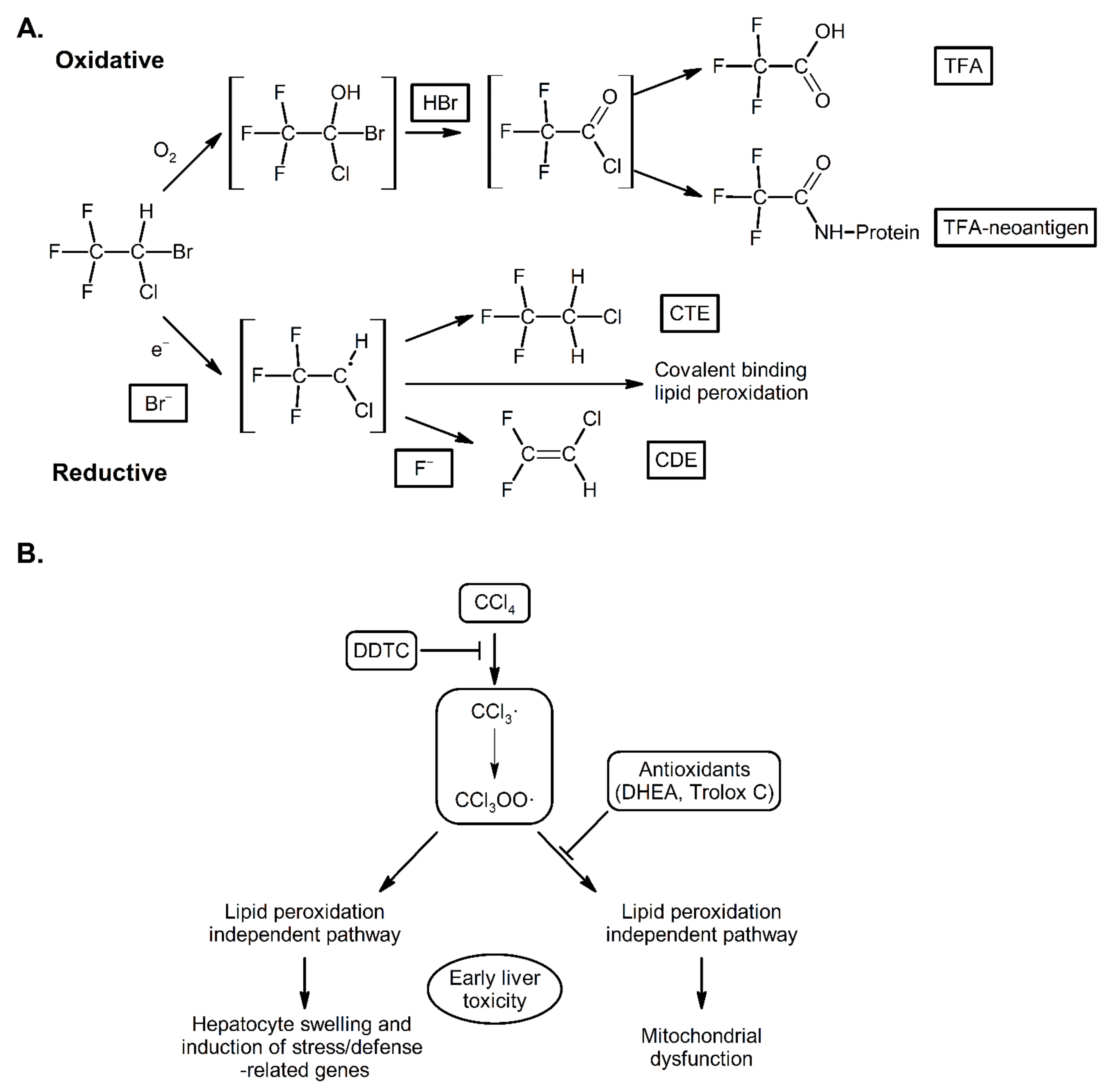
3. Mechanisms of Action of CYP2E1; Radical Mediated Toxicity
4. CYP2E1 in ALD
Intestinal CYP2E1 in ALD
5. CYP2E1 in NAFLD
5.1. CYP2E1 Links Insulin Resistance and NAFLD
5.2. The Role of CYP2E1 in Hepatic Lipid Accumulation
5.3. Complementary Roles of CYP4A and CYP2E1 in Lipid Oxidation through PPARα
5.4. Possible Role of Mitochondrial CYP2E1 in NAFLD
5.5. Kupffer Cells and CYP2E1 in NAFLD
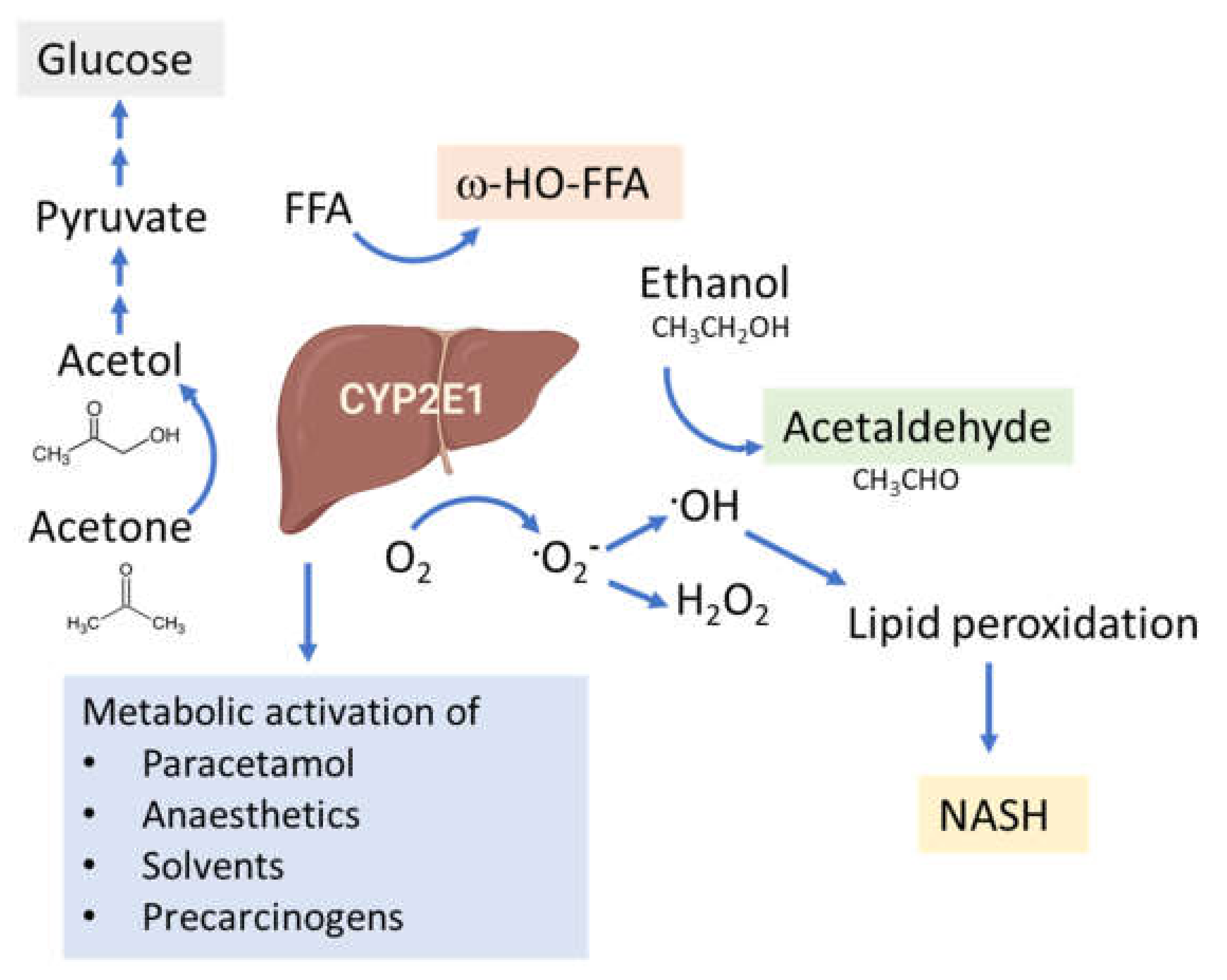
5.6. CYP2E1-Mediated ROS Production and Lipid Peroxidation
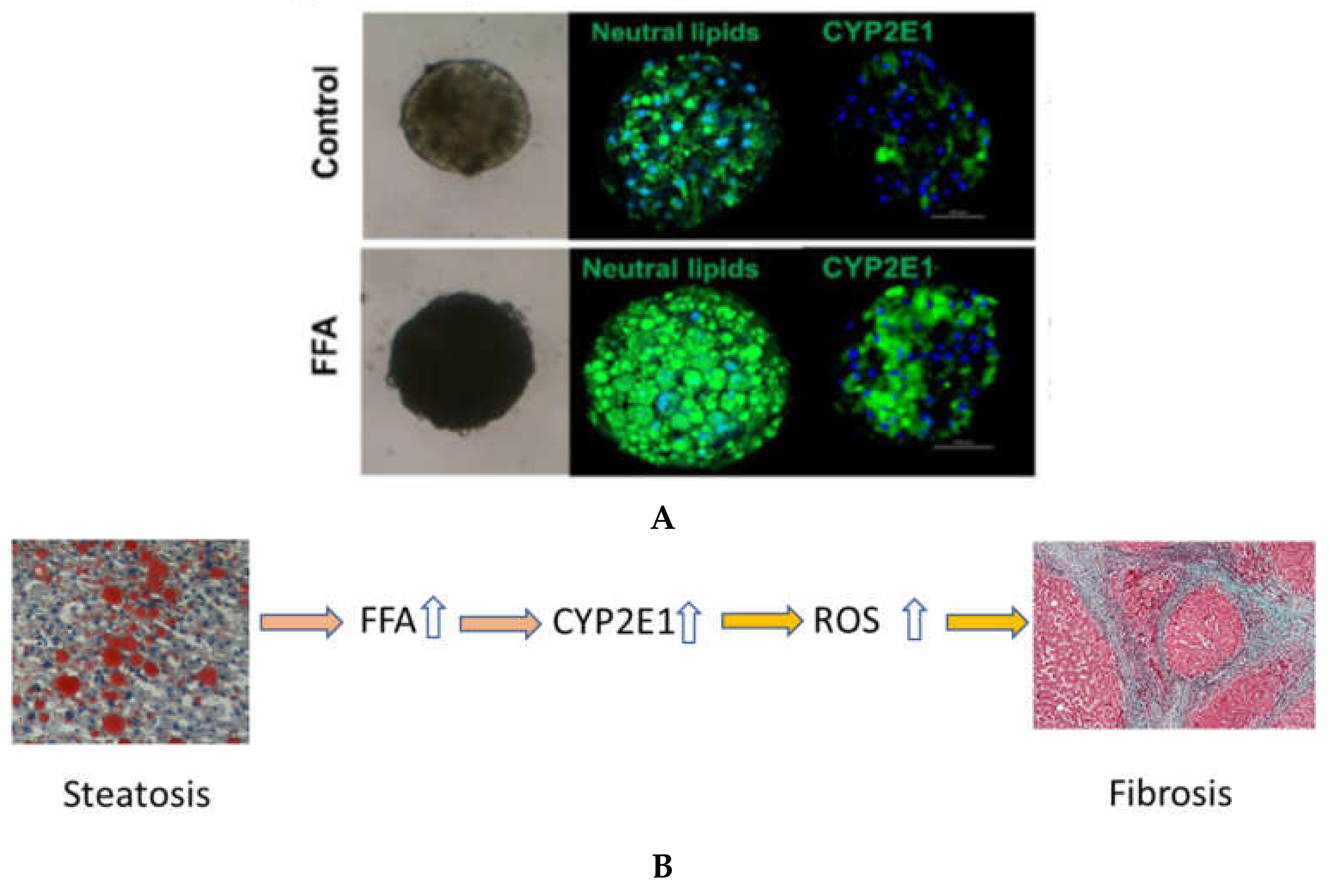
6. In Vitro Models for Examining the Role of CYP2E1 and FFA on Liver Fibrosis and Damage
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| 3D | three dimensional |
| ADH | alcohol dehydrogenase |
| AFLD | alcoholic fatty liver disease |
| ALD | alcoholic liver disease |
| ASH | alcoholic steatohepatitis |
| CDE | chlorodifluoroethylene |
| CTE | chlorotrifluoroethane |
| CYP2E1 | cytochrome P450 2E1 |
| ER | endoplasmic reticulum |
| FFA | free fatty |
| HCC | hepatocellular carcinoma |
| hPSC | human pluripotent stem cell |
| NADPH | nicotinamide adenine dinucleotide phosphate acid |
| NAFL | nonalcoholic fatty liver |
| NAFLD | nonalcoholic fatty liver disease |
| NASH | nonalcoholic steatohepatitis |
| NPC | non-parenchymal cell |
| PHH | human primary hepatocyte |
| POR | P450 reductase |
| ROS | reactive oxygen species |
| TFA | trifluoroacetic acid |
References
- Shigeta, Y.; Nomura, F.; Iida, S.; Leo, M.A.; Felder, M.R.; Lieber, C.S. Ethanol metabolism in vivo by the microsomal ethanol-oxidizing system in deermice lacking alcohol dehydrogenase (ADH). Biochem. Pharmacol. 1984, 33, 807–814. [Google Scholar] [CrossRef]
- Teschke, R.; Hasumura, Y.; Lieber, C.S. Hepatic microsomal alcohol-oxidizing system. Affinity for methanol, ethanol, propanol, and butanol. J. Biol. Chem. 1975, 250, 7397–7404. [Google Scholar] [CrossRef]
- Morgan, E.T.; Devine, M.; Skett, P. Changes in the rat hepatic mixed function oxidase system associated with chronic ethanol vapor inhalation. Biochem. Pharmacol. 1981, 30, 595–600. [Google Scholar] [CrossRef]
- Ingelman-Sundberg, M.; Johansson, I. The mechanism of cytochrome P-450-dependent oxidation of ethanol in reconstituted membrane vesicles. J. Biol. Chem. 1981, 256, 6321–6326. [Google Scholar] [CrossRef]
- I Cederbaum, A.; Dicker, E.; Gutteridge, J.M.; Smith, A.; A Clejan, L.; Groebler, L.K.; Liu, J.; Shanu, A.; Codd, R.; Witting, P.K.; et al. Inhibition of microsomal oxidation of alcohols and of hydroxyl-radical-scavenging agents by the iron-chelating agent desferrioxamine. Biochem. J. 1983, 210, 107–113. [Google Scholar] [CrossRef] [PubMed]
- Teschke, R.; Moreno, F.; Petrides, A.S. Hepatic microsomal ethanol oxidizing system (MEOS): Respective roles of ethanol and carbohydrates for the enhanced activity after chronic alcohol consumption. Biochem. Pharmacol. 1981, 30, 1745–1751. [Google Scholar] [CrossRef]
- Koop, D.R.; Morgan, E.; E Tarr, G.; Coon, M.J. Purification and characterization of a unique isozyme of cytochrome P-450 from liver microsomes of ethanol-treated rabbits. J. Biol. Chem. 1982, 257, 8472–8480. [Google Scholar] [CrossRef]
- Ingelman-Sundberg, M.; Hagbjork, A.L. On the significance of the cytochrome P-450-dependent hydroxyl radical-mediated oxygenation mechanism. Xenobiotica 1982, 12, 673–686. [Google Scholar] [CrossRef]
- Khani, S.C.; Zaphiropoulos, P.; Fujita, V.S.; Porter, T.D.; Koop, D.R.; Coon, M.J. cDNA and derived amino acid sequence of ethanol-inducible rabbit liver cytochrome P-450 isozyme 3a (P-450ALC). Proc. Natl. Acad. Sci. USA 1987, 84, 638–642. [Google Scholar] [CrossRef]
- Nebert, D.W.; Nelson, D.R.; Coon, M.J.; Estabrook, R.W.; Feyereisen, R.; Fujii-Kuriyama, Y.; Gonzalez, F.J.; Guengerich, F.P.; Gunsalus, I.C.; Johnson, E.F.; et al. The P450 Superfamily: Update on New Sequences, Gene Mapping, and Recommended Nomenclature. DNA Cell Biol. 1991, 10, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Umeno, M.; McBride, O.W.; Yang, C.S.; Gelboin, H.V.; Gonzalez, F.J. Human ethanol-inducible P450IIE1: Complete gene sequence, promoter characterization, chromosome mapping, and cDNA-directed expression. Biochemistry 1988, 27, 9006–9013. [Google Scholar] [CrossRef] [PubMed]
- Gaedigk, A.; Sangkuhl, K.; Whirl-Carrillo, M.; Twist, G.P.; Klein, T.E.; Miller, N.A. The PharmVar Steering Committee The Evolution of PharmVar. Clin. Pharmacol. Ther. 2019, 105, 29–32. [Google Scholar] [CrossRef]
- Tanaka, E.; Terada, M.; Misawa, S. Cytochrome P450 2E1: Its clinical and toxicological role. J. Clin. Pharm. Ther. 2000, 25, 165–175. [Google Scholar] [CrossRef] [PubMed]
- Ronis, M.J.J.; Lindros, K.O.; Ingelman-Sundberg, M. The CYP2E subfamily. Cytochromes P 1996, 450, 211–239. [Google Scholar]
- Ma, H.; Chen, S.; I Zheng, K.; Yu, Y.; Wang, X.; Tang, L.; Li, G.; Rios, R.S.; Huang, O.; Zheng, X.; et al. TA allele of rs2070673 in the CYP2E1 gene is associated with lobular inflammation and nonalcoholic steatohepatitis in patients with biopsy-proven nonalcoholic fatty liver disease. J. Gastroenterol. Hepatol. 2021. [Google Scholar] [CrossRef] [PubMed]
- Koop, D.R.; Chernosky, A.; Brass, E.P. Identification and induction of cytochrome P450 2E1 in rat Kupffer cells. J. Pharmacol. Exp. Ther. 1991, 258, 1072–1076. [Google Scholar]
- Koivisto, T.; Mishin, V.M.; Mak, K.M.; Cohen, P.A.; Lieber, C.S. Induction of Cytochrome P-4502E1 by Ethanol in Rat Kupffer Cells. Alcohol. Clin. Exp. Res. 1996, 20, 207–212. [Google Scholar] [CrossRef] [PubMed]
- Ye, Q.; Wang, X.; Wang, Q.; Xia, M.; Zhu, Y.; Lian, F.; Ling, W. Cytochrome P4502E1 inhibitor, chlormethiazole, decreases lipopolysaccharide-induced inflammation in rat Kupffer cells with ethanol treatment. Hepatol. Res. 2013, 43, 1115–1123. [Google Scholar] [CrossRef]
- Neve, E.P.A.; Ingelman-Sundberg, M. Molecular Basis for the Transport of Cytochrome P450 2E1 to the Plasma Membrane. J. Biol. Chem. 2000, 275, 17130–17135. [Google Scholar] [CrossRef] [PubMed]
- Neve, E.P.; Ingelman-Sundberg, M. A soluble NH2-terminally truncated catalytically active form of rat cytochrome P450 2E1 targeted to liver mitochondria. FEBS Lett. 1999, 460, 309–314. [Google Scholar] [CrossRef]
- Lieber, C.S. Cytochrome P-4502E1: Its physiological and pathological role. Physiol. Rev. 1997, 77, 517–544. [Google Scholar] [CrossRef]
- Loeper, J.; Descatoire, V.; Maurice, M.; Beaune, P.; Feldmann, G.; Larrey, D.; Pessayre, D. Presence of functional cytochrome P-450 on isolated rat hepatocyte plasma membrane. Hepatology 1990, 11, 850–858. [Google Scholar] [CrossRef] [PubMed]
- Loeper, J.; Descatoire, V.; Maurice, M.; Beaune, P.; Belghiti, J.; Houssin, D.; Ballet, F.; Feldmann, G.; Guengerich, F.; Pessayre, D. Cytochromes P-450 in human hepatocyte plasma membrane: Recognition by several autoantibodies. Gastroenterology 1993, 104, 203–216. [Google Scholar] [CrossRef]
- Neve, E.P.; Eliasson, E.; Pronzato, M.A.; Albano, E.; Marinari, U.; Ingelman-Sundberg, M. Enzyme-Specific Transport of Rat Liver Cytochrome P450 to the Golgi Apparatus. Arch. Biochem. Biophys. 1996, 333, 459–465. [Google Scholar] [CrossRef]
- Koop, D.R. Oxidative and reductive metabolism by cytochrome P450 2E1. FASEB J. 1992, 6, 724–730. [Google Scholar] [CrossRef]
- Massart, J.; Begriche, K.; Fromenty, B. Cytochrome P450 2E1 should not be neglected for acetaminophen-induced liver injury in metabolic diseases with altered insulin levels or glucose homeostasis. Clin. Res. Hepatol. Gastroenterol. 2021, 45, 101470. [Google Scholar] [CrossRef] [PubMed]
- Seitz, H.K. The role of cytochrome P4502E1 in the pathogenesis of alcoholic liver disease and carcinogenesis. Chem. Interact. 2020, 316, 108918. [Google Scholar] [CrossRef]
- Seitz, H.K.; Mueller, S. Alcohol and Cancer: An Overview with Special Emphasis on the Role of Acetaldehyde and Cytochrome P450 2E1. Adv. Exp. Med. Biol. 2014, 815, 59–70. [Google Scholar] [CrossRef]
- Johansson, I.; Eliasson, E.; Norsten, C.; Ingelman-Sundberg, M. Hydroxylation of acetone by ethanol- and acetone-inducible cytochrome P-450 in liver microsomes and reconstituted membranes. FEBS Lett. 1986, 196, 59–64. [Google Scholar] [CrossRef]
- Song, B.; Cederbaum, A.I. Ethanol-Inducible Cytochrome P450 (CYP2E1): Biochemistry, Molecular Biology and Clinical Relevance: 1996 Update. Alcohol. Clin. Exp. Res. 1996, 20, 138a–146a. [Google Scholar] [CrossRef]
- Gonzalez, F.J. Role of cytochromes P450 in chemical toxicity and oxidative stress: Studies with CYP2E1. Mutat. Res. Mol. Mech. Mutagen. 2005, 569, 101–110. [Google Scholar] [CrossRef]
- Correia, M.A.; Kwon, D. Why Hepatic CYP2E1-Elevation by Itself Is Insufficient for Inciting NAFLD/NASH: Inferences from Two Genetic Knockout Mouse Models. Biology 2020, 9, 419. [Google Scholar] [CrossRef] [PubMed]
- Badger, T.M.; Ronis, M.J.J.; Ingelman-Sundberg, M.; Hakkak, R. Inhibition of CYP2E1 Activity does not Abolish Pulsatile Urine Alcohol Concentrations During Chronic Alcohol Infusions. JBIC J. Biol. Inorg. Chem. 1995, 230, 914–919. [Google Scholar] [CrossRef]
- Porubsky, P.R.; Meneely, K.M.; Scott, E.E. Structures of Human Cytochrome P-450 2E1. J. Biol. Chem. 2008, 283, 33698–33707. [Google Scholar] [CrossRef]
- Porubsky, P.R.; Battaile, K.; Scott, E.E. Human Cytochrome P450 2E1 Structures with Fatty Acid Analogs Reveal a Previously Unobserved Binding Mode. J. Biol. Chem. 2010, 285, 22282–22290. [Google Scholar] [CrossRef]
- Koop, D.R.; Crump, B.L.; Nordblom, G.D.; Coon, M.J. Immunochemical evidence for induction of the alcohol-oxidizing cytochrome P-450 of rabbit liver microsomes by diverse agents: Ethanol, imidazole, trichloroethylene, acetone, pyrazole, and isoniazid. Proc. Natl. Acad. Sci. USA 1985, 82, 4065–4069. [Google Scholar] [CrossRef]
- Koop, D.R.; Tierney, D.J. Multiple mechanisms in the regulation of ethanol-inducible cytochrome P450IIE1. BioEssays 1990, 12, 429–435. [Google Scholar] [CrossRef] [PubMed]
- Song, B.J.; Matsunaga, T.; Hardwick, J.P.; Park, S.S.; Veech, R.L.; Yang, C.S.; Gelboin, H.V.; Gonzalez, F.J. Stabilization of Cytochrome P450j Messenger Ribonucleic Acid in the Diabetic Rat. Mol. Endocrinol. 1987, 1, 542–547. [Google Scholar] [CrossRef]
- Yun, Y.P.; Casazza, J.P.; Sohn, D.H.; Veech, R.L.; Song, B.J. Pretranslational activation of cytochrome P450IIE during ketosis induced by a high fat diet. Mol. Pharmacol. 1992, 41, 474–479. [Google Scholar]
- Novak, R.F.; Woodcroft, K.J. The alcohol-inducible form of cytochrome P450 (CYP2E1): Role in toxicology and regulation of expression. Arch. Pharml. Res. 2000, 23, 267–282. [Google Scholar] [CrossRef] [PubMed]
- Johansson, I.; Lindros, K.O.; Eriksson, H.; Ingelman-Sundberg, M. Transcriptional control of CYP2E1 in the perivenous liver region and during starvation. Biochem. Biophys. Res. Commun. 1990, 173, 331–338. [Google Scholar] [CrossRef]
- Yap, C.G.; Zaini, A.; Othman, I. Targeted CYP2E1 quantification and its correlation to currently acceptable clinical biochemical indices. J. Biol. Res. 2016, 23, 15. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Lee, S.S.; Buters, J.; Pineau, T.; Fernandez-Salguero, P.M.; Gonzalez, F.J. Role of CYP2E1 in the Hepatotoxicity of Acetaminophen. J. Biol. Chem. 1996, 271, 12063–12067. [Google Scholar] [CrossRef]
- Johansson, I.; Ekstroem, G.; Scholte, B.; Puzycki, D.; Joernvall, H.; Ingelman-Sundberg, M. Ethanol-, fasting-, and aceton-inducible cytochromes P-450 in rat liver: Regulation and characteristics of enzymes belonging to the IIB and IIE gene subfamilies. Biochemistry 1988, 27, 1925–1934. [Google Scholar] [CrossRef]
- Koop, D.R.; Coon, M.J. Purification of liver microsomal cytochrome P-450 isozymes 3a and 6 from imidazole-treated rabbits. Evidence for the identity of isozyme 3a with the form obtained by ethanol treatment. Mol. Pharmacol. 1984, 25, 494–501. [Google Scholar] [PubMed]
- Song, B.J.; Gelboin, H.V.; Park, S.S.; Yang, C.S.; Gonzalez, F.J. Complementary DNA and protein sequences of ethanol-inducible rat and human cytochrome P-450s. Transcriptional and post-transcriptional regulation of the rat enzyme. J. Biol. Chem. 1986, 261, 16689–16697. [Google Scholar] [CrossRef]
- Eliasson, E.; Johansson, I.; Ingelman-Sundberg, M. Substrate-, hormone-, and cAMP-regulated cytochrome P450 degradation. Proc. Natl. Acad. Sci. USA 1990, 87, 3225–3229. [Google Scholar] [CrossRef] [PubMed]
- Eliasson, E.; Mkrtchian, S.; Ingelman-Sundberg, M. Hormone- and substrate-regulated intracellular degradation of cytochrome P450 (2E1) involving MgATP-activated rapid proteolysis in the endoplasmic reticulum membranes. J. Biol. Chem. 1992, 267, 15765–15769. [Google Scholar] [CrossRef]
- Lu, Y.; Cederbaum, A.I. CYP2E1 and oxidative liver injury by alcohol. Free Radic. Biol. Med. 2008, 44, 723–738. [Google Scholar] [CrossRef]
- Wang, Y.; Guan, S.; Acharya, P.; Koop, D.R.; Liu, Y.; Liao, M.; Burlingame, A.L.; Correia, M.A. Ubiquitin-dependent Proteasomal Degradation of Human Liver Cytochrome P450 2E1. J. Biol. Chem. 2011, 286, 9443–9456. [Google Scholar] [CrossRef] [PubMed]
- Ronis, M.J.J.; Johansson, I.; Hultenby, K.; Lagercrantz, J.; Glaumann, H.; Ingelman-Sundberg, M. Acetone-regulated synthesis and degradation of cytochrome P4502E2 and cytochrome P4502B1 in rat liver. JBIC J. Biol. Inorg. Chem. 1991, 198, 383–389. [Google Scholar] [CrossRef]
- Bell, C.C.; Hendriks, D.; Moro, S.M.L.; Ellis, E.; Walsh, J.; Renblom, A.; Puigvert, L.F.; Dankers, A.C.A.; Jacobs, F.; Snoeys, J.; et al. Characterization of primary human hepatocyte spheroids as a model system for drug-induced liver injury, liver function and disease. Sci. Rep. 2016, 6, 25187. [Google Scholar] [CrossRef] [PubMed]
- Owen, O.E.; Patel, M.S.; Block, B.S.B.; Kreulen, T.H.; Reichle, F.A.; Mozzoli, M.A. Gluconeogenesis in normal, cirrhotic, and diabetic humans. In Gluconeogenesis: Its Regulation in Mammalian Species; Wiley: New York, NY, USA, 1976; pp. 533–558. [Google Scholar]
- Ingelman-Sundberg, M.; Johansson, I. Mechanisms of hydroxyl radical formation and ethanol oxidation by ethanol-inducible and other forms of rabbit liver microsomal cytochromes P-450. J. Biol. Chem. 1984, 259, 6447–6458. [Google Scholar] [CrossRef]
- Cederbaum, A.I.; Dicker, E.; Cohen, G. Role of hydroxyl radicals in the iron-ethylenediaminetetraacetic acid mediated stimulation of microsomal oxidation of ethanol. Biochemistry 1980, 19, 3698–3704. [Google Scholar] [CrossRef]
- Johansson, I.; Ingelman-Sundberg, M. Carbon tetrachloride-induced lipid peroxidation dependent on an ethanol-inducible form of rabbit liver microsomal cytochrome P-450. FEBS Lett. 1985, 183, 265–269. [Google Scholar] [CrossRef]
- Safari, S.; Motavaf, M.; Siamdoust, S.A.S.; Alavian, S.M. Hepatotoxicity of Halogenated Inhalational Anesthetics. Iran. Red Crescent Med. J. 2014, 16, e20153. [Google Scholar] [CrossRef] [PubMed]
- Spracklin, D.K.; Emery, M.E.; Thummel, K.E.; Kharasch, E.D. Concordance between trifluoroacetic acid and hepatic protein trifluoroacetylation after disulfiram inhibition of halothane metabolism in rats. Acta Anaesthesiol. Scand. 2003, 47, 765–770. [Google Scholar] [CrossRef] [PubMed]
- Clot, P.; Albano, E.; Eliasson, E.; Tabone, M.; Aricò, S.; Israel, Y.; Moncada, C.; Sundberg, M.I.-. Cytochrome P4502E1 hydroxyethyl radical adducts as the major antigen in autoantibody formation among alcoholics. Gastroenterology 1996, 111, 206–216. [Google Scholar] [CrossRef] [PubMed]
- Spracklin, D.K.; Hankins, D.C.; Fisher, J.M.; E Thummel, K.; Kharasch, E.D. Cytochrome P450 2E1 is the principal catalyst of human oxidative halothane metabolism in vitro. J. Pharmacol. Exp. Ther. 1997, 281, 400–411. [Google Scholar] [PubMed]
- Kharasch, E.; Hankins, D.; Mautz, D.; Thummel, K. Identification of the enzyme responsible for oxidative halothane metabolism: Implications for prevention of halothane hepatitis. Lancet 1996, 347, 1367–1371. [Google Scholar] [CrossRef]
- Knockaert, L.; Berson, A.; Ribault, C.; Prost, P.-E.; Fautrel, A.; Pajaud, J.; Lepage, S.; Lucas-Clerc, C.; Bégué, J.-M.; Fromenty, B.; et al. Carbon tetrachloride-mediated lipid peroxidation induces early mitochondrial alterations in mouse liver. Lab. Investig. 2011, 92, 396–410. [Google Scholar] [CrossRef] [PubMed]
- Mann, R.E.; Smart, R.G.; Govoni, R. The epidemiology of alcoholic liver disease. Alcohol Res. Health 2003, 27, 209–219. [Google Scholar]
- Sakaguchi, S.; Takahashi, S.; Sasaki, T.; Kumagai, T.; Nagata, K. Progression of Alcoholic and Non-alcoholic Steatohepatitis: Common Metabolic Aspects of Innate Immune System and Oxidative Stress. Drug Metab. Pharmacokinet. 2011, 26, 30–46. [Google Scholar] [CrossRef]
- Leung, T.-M.; Nieto, N. CYP2E1 and oxidant stress in alcoholic and non-alcoholic fatty liver disease. J. Hepatol. 2013, 58, 395–398. [Google Scholar] [CrossRef] [PubMed]
- Robertson, G.; Leclercq, I.; Farrell, G.C., II. Cytochrome P-450 enzymes and oxidative stress. Am. J. Physiol. Liver Physiol. 2001, 281, G1135–G1139. [Google Scholar] [CrossRef]
- Ekström, G.; Ingelman-Sundberg, M. Rat liver microsomal NADPH-supported oxidase activity and lipid peroxidation dependent on ethanol-inducible cytochrome P-450 (P-450IIE1). Biochem. Pharmacol. 1989, 38, 1313–1319. [Google Scholar] [CrossRef]
- Ingelman-Sundberg, M.; Johansson, I.; Yin, H.; Terelius, Y.; Eliasson, E.; Clot, P.; Albano, E. Ethanol-inducible cytochrome P4502E1: Genetic polymorphism, regulation, and possible role in the etiology of alcohol-induced liver disease. Alcohol 1993, 10, 447–452. [Google Scholar] [CrossRef]
- Zhukov, A.; Ingelman-Sundberg, M. Relationship between cytochrome P450 catalytic cycling and stability: Fast degradation of ethanol-inducible cytochrome P450 2E1 (CYP2E1) in hepatoma cells is abolished by inactivation of its electron donor NADPH–cytochrome P450 reductase. Biochem. J. 1999, 340, 453–458. [Google Scholar] [CrossRef]
- Bell, L.C.; Guengerich, F.P. Oxidation Kinetics of Ethanol by Human Cytochrome P450 2E1. J. Biol. Chem. 1997, 272, 29643–29651. [Google Scholar] [CrossRef] [PubMed]
- Gorsky, L.D.; Koop, D.R.; Coon, M.J. On the stoichiometry of the oxidase and monooxygenase reactions catalyzed by liver microsomal cytochrome P-450. Products of oxygen reduction. J. Biol. Chem. 1984, 259, 6812–6817. [Google Scholar] [CrossRef]
- Morgan, K.; French, S.W.; Morgan, T.R. Production of a cytochrome P450 2E1 transgenic mouse and initial evaluation of alcoholic liver damage. Hepatology 2002, 36, 122–134. [Google Scholar] [CrossRef]
- Butura, A.; Nilsson, K.; Morgan, K.; Morgan, T.R.; French, S.W.; Johansson, I.; Schuppe-Koistinen, I.; Ingelman-Sundberg, M. The impact of CYP2E1 on the development of alcoholic liver disease as studied in a transgenic mouse model. J. Hepatol. 2009, 50, 572–583. [Google Scholar] [CrossRef]
- Abdelmegeed, M.A.; Choi, Y.; Ha, S.-K.; Song, B.-J. Cytochrome P450-2E1 promotes aging-related hepatic steatosis, apoptosis and fibrosis through increased nitroxidative stress. Free Radic. Biol. Med. 2016, 91, 188–202. [Google Scholar] [CrossRef]
- Hu, Y.; Mishin, V.; Johansson, I.; Von Bahr, C.; Cross, A.; Ronis, M.J.; Badger, T.M.; Ingelman-Sundberg, M. Chlormethiazole as an efficient inhibitor of cytochrome P450 2E1 expression in rat liver. J. Pharmacol. Exp. Ther. 1994, 269, 1286–1291. [Google Scholar]
- Gouillon, Z.-Q.; Lucas, D.; Li, J.; Hagbjork, A.L.; French, B.A.; Fu, P.; Fang, C.; Ingelman-Sundberg, M.; Donohue, T.M.; French, S.W. Inhibition of ethanol-induced liver disease in the intragastric feeding rat model by chlormethiazole. Proc. Soc. Exp. Boil. Med. 2000, 224, 302–308. [Google Scholar] [CrossRef] [PubMed]
- Albano, E.; Clot, P.; Morimoto, M.; Tomasi, A.; Ingelman-Sundberg, M.; French, S.W. Role of cytochrome P4502E1-dependent formation of hydroxyethyl free radical in the development of liver damage in rats intragastrically fed with ethanol. Hepatology 1996, 23, 155–163. [Google Scholar] [CrossRef]
- Morimoto, M.; Hagbjork, A.-L.; Wan, Y.-J.Y.; Fu, P.C.; Clot, P.; Albano, E.; Ingelman-Sundberg, M.; French, S.W. Modulation of experimental alcohol-induced liver disease by cytochrome P450 2E1 inhibitors. Hepatology 1995, 21, 1610–1617. [Google Scholar] [CrossRef] [PubMed]
- Seitz, H.; Neuman, M. The History of Alcoholic Liver Disease: From an Unrecognized Disease to One of the Most Frequent Diseases in Hepatology. J. Clin. Med. 2021, 10, 858. [Google Scholar] [CrossRef]
- Ballway, J.; Song, B.-J. Translational Approaches with Antioxidant Phytochemicals against Alcohol-Mediated Oxidative Stress, Gut Dysbiosis, Intestinal Barrier Dysfunction, and Fatty Liver Disease. Antioxidants 2021, 10, 384. [Google Scholar] [CrossRef] [PubMed]
- Abdelmegeed, M.A.; Banerjee, A.; Jang, S.; Yoo, S.-H.; Yun, J.-W.; Gonzalez, F.J.; Keshavarzian, A.; Song, B.-J. CYP2E1 potentiates binge alcohol-induced gut leakiness, steatohepatitis, and apoptosis. Free Radic. Biol. Med. 2013, 65, 1238–1245. [Google Scholar] [CrossRef] [PubMed]
- Abdelmegeed, M.A.; Ha, S.-K.; Choi, Y.; Akbar, M.; Song, B.-J. Role of CYP2E1 in Mitochondrial Dysfunction and Hepatic Injury by Alcohol and Non-Alcoholic Substances. Curr. Mol. Pharmacol. 2017, 10, 207–225. [Google Scholar] [CrossRef]
- Tang, Y.; Forsyth, C.B.; Farhadi, A.; Rangan, J.; Jakate, S.; Shaikh, M.; Banan, A.; Fields, J.Z.; Keshavarzian, A. Nitric Oxide-Mediated Intestinal Injury Is Required for Alcohol-Induced Gut Leakiness and Liver Damage. Alcohol. Clin. Exp. Res. 2009, 33, 1220–1230. [Google Scholar] [CrossRef] [PubMed]
- Forsyth, C.B.; Voigt, R.M.; Shaikh, M.; Tang, Y.; Cederbaum, A.I.; Turek, F.W.; Keshavarzian, A. Role for intestinal CYP2E1 in alcohol-induced circadian gene-mediated intestinal hyperpermeability. Am. J. Physiol. Liver Physiol. 2013, 305, G185–G195. [Google Scholar] [CrossRef]
- Keshavarzian, A.; Fields, J. Alcoholic liver disease: Is it an “extraintestinal” complication of alcohol-induced intestinal injury? J. Lab. Clin. Med. 2003, 142, 285–287. [Google Scholar] [CrossRef]
- Tamai, H.; Kato, S.; Horie, Y.; Ohki, E.; Yokoyama, H.; Ishii, H. Effect of Acute Ethanol Administration on the Intestinal Absorption of Endotoxin in Rats. Alcohol. Clin. Exp. Res. 2000, 24, 390–394. [Google Scholar] [CrossRef] [PubMed]
- Bode, C.; Kugler, V.; Bode, J. Endotoxemia in patients with alcoholic and non-alcoholic cirrhosis and in subjects with no evidence of chronic liver disease following acute alcohol excess. J. Hepatol. 1987, 4, 8–14. [Google Scholar] [CrossRef]
- Mathurin, P.; Deng, Q.; Keshavarzian, A.; Choudhary, S.; Holmes, E.; Tsukamoto, H. Exacerbation of Alcoholic Liver Injury by Enteral Endotoxin in Rats. Hepatology 2000, 32, 1008–1017. [Google Scholar] [CrossRef]
- Enomoto, N.; Ikejima, K.; Bradford, B.; Rivera, C.; Kono, H.; Brenner, D.A.; Thurman, R.G. Alcohol causes both tolerance and sensitization of rat Kupffer cells via mechanisms dependent on endotoxin. Gastroenterology 1998, 115, 443–451. [Google Scholar] [CrossRef]
- Luther, J.; Garber, J.J.; Khalili, H.; Dave, M.; Bale, S.S.; Jindal, R.; Motola, D.L.; Luther, S.; Bohr, S.; Jeoung, S.W.; et al. Hepatic Injury in Nonalcoholic Steatohepatitis Contributes to Altered Intestinal Permeability. Cell. Mol. Gastroenterol. Hepatol. 2015, 1, 222–232.e2. [Google Scholar] [CrossRef]
- Jennison, E.; Byrne, C.D. The role of the gut microbiome and diet in the pathogenesis of non-alcoholic fatty liver disease. Clin. Mol. Hepatol. 2021, 27, 22–43. [Google Scholar] [CrossRef] [PubMed]
- Gäbele, E.; Dostert, K.; Hofmann, C.; Wiest, R.; Schölmerich, J.; Hellerbrand, C.; Obermeier, F. DSS induced colitis increases portal LPS levels and enhances hepatic inflammation and fibrogenesis in experimental NASH. J. Hepatol. 2011, 55, 1391–1399. [Google Scholar] [CrossRef] [PubMed]
- Henao-Mejia, J.; Elinav, E.; Jin, C.; Hao, L.; Mehal, W.Z.; Strowig, T.; Thaiss, C.A.; Kau, A.; Eisenbarth, S.; Jurczak, M.; et al. Inflammasome-mediated dysbiosis regulates progression of NAFLD and obesity. Nat. Cell Biol. 2012, 482, 179–185. [Google Scholar] [CrossRef]
- Ludwig, J.; Viggiano, T.R.; McGill, D.B.; Ott, B.J. Nonalcoholic steatohepatitis. Mayo Clinic experiences with a hitherto un-named disease. Mayo Clin. Proc. 1980, 55, 434–438. [Google Scholar] [PubMed]
- Bugianesi, E.; Gastaldelli, A.; Vanni, E.; Gambino, R.; Cassader, M.; Baldi, S.; Ponti, V.; Pagano, G.; Ferrannini, E.; Rizzetto, M. Insulin resistance in non-diabetic patients with non-alcoholic fatty liver disease: Sites and mechanisms. Diabetologia 2005, 48, 634–642. [Google Scholar] [CrossRef]
- Fabbrini, E.; Sullivan, S.; Klein, S. Obesity and nonalcoholic fatty liver disease: Biochemical, metabolic, and clinical implications. Hepatology 2010, 51, 679–689. [Google Scholar] [CrossRef] [PubMed]
- Vega, G.L.; Chandalia, M.; Szczepaniak, L.S.; Grundy, S.M. Metabolic correlates of nonalcoholic fatty liver in women and men. Hepatology 2007, 46, 716–722. [Google Scholar] [CrossRef]
- Sanyal, A.J.; Campbell–Sargent, C.; Mirshahi, F.; Rizzo, W.B.; Contos, M.J.; Sterling, R.K.; Luketic, V.A.; Shiffman, M.L.; Clore, J.N. Nonalcoholic steatohepatitis: Association of insulin resistance and mitochondrial abnormalities. Gastroenterology 2001, 120, 1183–1192. [Google Scholar] [CrossRef]
- Abenavoli, L.; Milic, N.; Di Renzo, L.; Preveden, T.; Medić-Stojanoska, M.; De Lorenzo, A. Metabolic aspects of adult patients with nonalcoholic fatty liver disease. World J. Gastroenterol. 2016, 22, 7006–7016. [Google Scholar] [CrossRef]
- Almeda-Valdes, P.; Aguilar-Olivos, N.; Uribe, M.; Méndez-Sánchez, N. Common features of the metabolic syndrome and nonalcoholic fatty liver disease. Rev. Recent Clin. Trials 2015, 9, 148–158. [Google Scholar] [CrossRef]
- Buzzetti, E.; Pinzani, M.; Tsochatzis, E.A. The multiple-hit pathogenesis of non-alcoholic fatty liver disease (NAFLD). Metabolism 2016, 65, 1038–1048. [Google Scholar] [CrossRef]
- Eslam, M.; Sanyal, A.J.; George, J. International Consensus Panel. MAFLD: A Consensus-Driven Proposed Nomenclature for Metabolic Associated Fatty Liver Disease. Gastroenterology 2020, 158, 1999–2014.e1. [Google Scholar] [CrossRef] [PubMed]
- El-Serag, H.B.; Kanwal, F.; Feng, Z.; Marrero, J.A.; Khaderi, S.; Singal, A.G. Risk Factors for Cirrhosis in Contemporary Hepatology Practices—Findings From the Texas Hepatocellular Carcinoma Consortium Cohort. Gastroenterology 2020, 159, 376–377. [Google Scholar] [CrossRef]
- Bianco, C.; Casirati, E.; Malvestiti, F.; Valenti, L. Genetic predisposition similarities between NASH and ASH: Identification of new therapeutic targets. JHEP Rep. 2021, 3, 100284. [Google Scholar] [CrossRef]
- Tilg, H.; Moschen, A. Evolution of inflammation in nonalcoholic fatty liver disease: The multiple parallel hits hypothesis. Hepatology 2010, 52, 1836–1846. [Google Scholar] [CrossRef]
- Bessone, F.; Razori, M.V.; Roma, M.G. Molecular pathways of nonalcoholic fatty liver disease development and progression. Cell. Mol. Life Sci. 2019, 76, 99–128. [Google Scholar] [CrossRef]
- Masarone, M.; Rosato, V.; Dallio, M.; Gravina, A.G.; Aglitti, A.; Loguercio, C.; Federico, A.; Persico, M. Role of Oxidative Stress in Pathophysiology of Nonalcoholic Fatty Liver Disease. Oxid. Med. Cell. Longev. 2018, 2018, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Pan, M.; Cederbaum, A.I.; Zhang, Y.-L.; Ginsberg, H.N.; Williams, K.J.; Fisher, E.A. Lipid peroxidation and oxidant stress regulate hepatic apolipoprotein B degradation and VLDL production. J. Clin. Investig. 2004, 113, 1277–1287. [Google Scholar] [CrossRef]
- Weltman, M.D.; Farrell, G.C.; Liddle, C. Increased hepatocyte CYP2E1 expression in a rat nutritional model of hepatic steatosis with inflammation. Gastroenterology 1996, 111, 1645–1653. [Google Scholar] [CrossRef]
- Brunt, E.M.; Janney, C.G.; Di Bisceglie, A.M.; Neuschwander-Tetri, B.A.; Bacon, B.R. Nonalcoholic Steatohepatitis: A Proposal for Grading and Staging The Histological Lesions. Am. J. Gastroenterol. 1999, 94, 2467–2474. [Google Scholar] [CrossRef]
- Weltman, M.D.; Farrell, G.C.; Hall, P.; Ingelman-Sundberg, M.; Liddle, C. Hepatic cytochrome P450 2E1 is increased in patients with nonalcoholic steatohepatitis. Hepatology 1998, 27, 128–133. [Google Scholar] [CrossRef] [PubMed]
- Aubert, J.; Begriche, K.; Knockaert, L.; Robin, M.; Fromenty, B. Increased expression of cytochrome P450 2E1 in nonalcoholic fatty liver disease: Mechanisms and pathophysiological role. Clin. Res. Hepatol. Gastroenterol. 2011, 35, 630–637. [Google Scholar] [CrossRef]
- Videla, L.A.; Rodrigo, R.; Orellana, M.; Fernandez, V.; Tapia, G.; Quiñones, L.; Varela, N.; Contreras, J.; Lazarte, R.; Csendes, A.; et al. Oxidative stress-related parameters in the liver of non-alcoholic fatty liver disease patients. Clin. Sci. 2004, 106, 261–268. [Google Scholar] [CrossRef] [PubMed]
- Zangar, R.C.; Novak, R.F. Effects of Fatty Acids and Ketone Bodies on Cytochromes P450 2B, 4A, and 2E1 Expression in Primary Cultured Rat Hepatocytes. Arch. Biochem. Biophys. 1997, 337, 217–224. [Google Scholar] [CrossRef]
- Woodcroft, K.J.; Novak, R.F. Insulin effects on CYP2E1, 2B, 3A, and 4A expression in primary cultured rat hepatocytes. Chem. Interact. 1997, 107, 75–91. [Google Scholar] [CrossRef]
- Zong, H.; Armoni, M.; Harel, C.; Karnieli, E.; Pessin, J.E. Cytochrome P-450 CYP2E1 knockout mice are protected against high-fat diet-induced obesity and insulin resistance. Am. J. Physiol. Metab. 2012, 302, E532–E539. [Google Scholar] [CrossRef]
- Lee, H.Y.; Lee, G.-H.; Bhattarai, K.R.; Park, B.-H.; Koo, S.-H.; Kim, H.-R.; Chae, H.J. Bax Inhibitor-1 regulates hepatic lipid accumulation via ApoB secretion. Sci. Rep. 2016, 6, 27799. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.-R.; Lee, G.-H.; Cho, E.Y.; Chae, S.-W.; Ahn, T.; Chae, H.-J. Bax inhibitor 1 regulates ER-stress-induced ROS accumulation through the regulation of cytochrome P450 2E1. J. Cell Sci. 2009, 122, 1126–1133. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.-M.; Grenert, J.P.; Patterson, C.; Correia, M.A. CHIP−/−-Mouse Liver: Adiponectin-AMPK-FOXO-Activation Overrides CYP2E1-Elicited JNK1-Activation, Delaying Onset of NASH: Therapeutic Implications. Sci. Rep. 2016, 6, 29423. [Google Scholar] [CrossRef] [PubMed]
- Leclercq, I.A.; Farrell, G.C.; Field, J.; Bell, D.R.; Gonzalez, F.J.; Robertson, G.R. CYP2E1 and CYP4A as microsomal catalysts of lipid peroxides in murine nonalcoholic steatohepatitis. J. Clin. Investig. 2000, 105, 1067–1075. [Google Scholar] [CrossRef]
- Adas, F.; Berthou, F.; Salaün, J.; Dréano, Y.; Amet, Y. Interspecies variations in fatty acid hydroxylations involving cytochromes P450 2E1 and 4A. Toxicol. Lett. 1999, 110, 43–55. [Google Scholar] [CrossRef]
- Adas, F.; Salaün, J.; Berthou, F.; Picart, D.; Simon, B.; Amet, Y. Requirement for ω and (ω–1)-hydroxylations of fatty acids by human cytochromes P450 2E1 and 4A11. J. Lipid Res. 1999, 40, 1990–1997. [Google Scholar] [CrossRef]
- Gao, H.; Cao, Y.; Xia, H.; Zhu, X.; Jin, Y. CYP4A11 is involved in the development of nonalcoholic fatty liver disease via ROS-induced lipid peroxidation and inflammation. Int. J. Mol. Med. 2020, 45, 1121–1129. [Google Scholar] [CrossRef]
- Diesinger, T.; Buko, V.; Lautwein, A.; Dvorsky, R.; Belonovskaya, E.; Lukivskaya, O.; Naruta, E.; Kirko, S.; Andreev, V.; Buckert, D.; et al. Drug targeting CYP2E1 for the treatment of early-stage alcoholic steatohepatitis. PLoS ONE 2020, 15, e0235990. [Google Scholar] [CrossRef]
- Diesinger, T.; Lautwein, A.; Buko, V.; Belonovskaya, E.; Lukivskaya, O.; Naruta, E.; Kirko, S.; Andreev, V.; Dvorsky, R.; Buckert, D.; et al. ω-Imidazolyl-alkyl derivatives as new preclinical drug candidates for treating non-alcoholic steatohepatitis. Physiol. Rep. 2021, 9, e14795. [Google Scholar] [CrossRef]
- Lambert, J.E.; Ramos–Roman, M.A.; Browning, J.D.; Parks, E. Increased De Novo Lipogenesis Is a Distinct Characteristic of Individuals With Nonalcoholic Fatty Liver Disease. Gastroenterology 2014, 146, 726–735. [Google Scholar] [CrossRef]
- Buyco, D.G.; Martin, J.; Jeon, S.; Hooks, R.; Lin, C.; Carr, R. Experimental models of metabolic and alcoholic fatty liver disease. World J. Gastroenterol. 2021, 27, 1–18. [Google Scholar] [CrossRef]
- Houten, S.M.; Wanders, R.J.A. A general introduction to the biochemistry of mitochondrial fatty acid β-oxidation. J. Inherit. Metab. Dis. 2010, 33, 469–477. [Google Scholar] [CrossRef]
- Berardo, C.; Di Pasqua, L.G.; Cagna, M.; Richelmi, P.; Vairetti, M.; Ferrigno, A. Nonalcoholic Fatty Liver Disease and Non-Alcoholic Steatohepatitis: Current Issues and Future Perspectives in Preclinical and Clinical Research. Int. J. Mol. Sci. 2020, 21, 9646. [Google Scholar] [CrossRef]
- Yoon, M. The role of PPARα in lipid metabolism and obesity: Focusing on the effects of estrogen on PPARα actions. Pharmacol. Res. 2009, 60, 151–159. [Google Scholar] [CrossRef]
- Kroetz, D.L.; Yook, P.; Costet, P.; Bianchi, P.; Pineau, T. Peroxisome Proliferator-activated Receptor α Controls the Hepatic CYP4A Induction Adaptive Response to Starvation and Diabetes. J. Biol. Chem. 1998, 273, 31581–31589. [Google Scholar] [CrossRef]
- Wan, Y. Regulation of peroxisome proliferator activated receptor α-mediated pathways in alcohol fed cytochrome P450 2E1 deficient mice. Hepatol. Res. 2001, 19, 117–130. [Google Scholar] [CrossRef]
- Abdelmegeed, M.A.; Yoo, S.-H.; Henderson, L.E.; Gonzalez, F.J.; Woodcroft, K.J.; Song, B.-J. PPARα Expression Protects Male Mice from High Fat–Induced Nonalcoholic Fatty Liver. J. Nutr. 2011, 141, 603–610. [Google Scholar] [CrossRef]
- Anandatheerthavarada, H.K.; Addya, S.; Dwivedi, R.S.; Biswas, G.; Mullick, J.; Avadhani, N.G. Localization of Multiple Forms of Inducible Cytochromes P450 in Rat Liver Mitochondria: Immunological Characteristics and Patterns of Xenobiotic Substrate Metabolism. Arch. Biochem. Biophys. 1997, 339, 136–150. [Google Scholar] [CrossRef]
- Robin, M.-A.; Anandatheerthavarada, H.K.; Fang, J.-K.; Cudic, M.; Otvos, L.; Avadhani, N.G. Mitochondrial Targeted Cytochrome P450 2E1 (P450 MT5) Contains an Intact N Terminus and Requires Mitochondrial Specific Electron Transfer Proteins for Activity. J. Biol. Chem. 2001, 276, 24680–24689. [Google Scholar] [CrossRef] [PubMed]
- Hartman, J.H.; Miller, G.; Meyer, J.N. Toxicological implications of mitochondrial localization of CYP2E1. Toxicol. Res. 2017, 6, 273–289. [Google Scholar] [CrossRef]
- Bansal, S.; Liu, C.-P.; Sepuri, N.B.; Anandatheerthavarada, H.K.; Selvaraj, V.; Hoek, J.; Milne, G.; Guengerich, F.P.; Avadhani, N.G. Mitochondria-targeted Cytochrome P450 2E1 Induces Oxidative Damage and Augments Alcohol-mediated Oxidative Stress. J. Biol. Chem. 2010, 285, 24609–24619. [Google Scholar] [CrossRef]
- Robin, M.-A.; Sauvage, I.; Grandperret, T.; Descatoire, V.; Pessayre, D.; Fromenty, B. Ethanol increases mitochondrial cytochrome P450 2E1 in mouse liver and rat hepatocytes. FEBS Lett. 2005, 579, 6895–6902. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Zhou, Y.; Wang, H.; Zhang, M.; Qiu, P.; Zhang, R.; Zhao, Q.; Liu, J. Crosstalk Between Liver Macrophages and Surrounding Cells in Nonalcoholic Steatohepatitis. Front. Immunol. 2020, 11, 1169. [Google Scholar] [CrossRef]
- Mihm, S. Danger-Associated Molecular Patterns (DAMPs): Molecular Triggers for Sterile Inflammation in the Liver. Int. J. Mol. Sci. 2018, 19, 3104. [Google Scholar] [CrossRef]
- Pessayre, D.; Berson, A.; Fromenty, B.; Mansouri, A. Mitochondria in Steatohepatitis. Semin. Liver Dis. 2001, 21, 057–070. [Google Scholar] [CrossRef]
- Cao, Q.; Mak, K.M.; Lieber, C.S. Cytochrome P4502E1 primes macrophages to increase TNF-α production in response to lipopolysaccharide. Am. J. Physiol. Liver Physiol. 2005, 289, G95–G107. [Google Scholar] [CrossRef]
- Kim, S.Y.; Jeong, J.-M.; Kim, S.J.; Seo, W.; Kim, M.-H.; Choi, W.-M.; Yoo, W.; Lee, J.-H.; Shim, Y.-R.; Yi, H.-S.; et al. Pro-inflammatory hepatic macrophages generate ROS through NADPH oxidase 2 via endocytosis of monomeric TLR4–MD2 complex. Nat. Commun. 2017, 8, 1–15. [Google Scholar] [CrossRef]
- Chance, B.; Sies, H.; Boveris, A. Hydroperoxide metabolism in mammalian organs. Physiol. Rev. 1979, 59, 527–605. [Google Scholar] [CrossRef]
- Ingelman-Sundberg, M.; Johansson, I. Cytochrome b5 as electron donor to rabbit liver cytochrome P-450LM2 in reconstituted phospholipid vesicles. Biochem. Biophys. Res. Commun. 1980, 97, 582–589. [Google Scholar] [CrossRef]
- White, R.E. The involvement of free radicals in the mechanisms of monooxygenases. Pharmacol. Ther. 1991, 49, 21–42. [Google Scholar] [CrossRef]
- Linhart, K.; Bartsch, H.; Seitz, H.K. The role of reactive oxygen species (ROS) and cytochrome P-450 2E1 in the generation of carcinogenic etheno-DNA adducts. Redox Biol. 2014, 3, 56–62. [Google Scholar] [CrossRef] [PubMed]
- Schattenberg, J.M.; Czaja, M.J. Regulation of the effects of CYP2E1-induced oxidative stress by JNK signaling. Redox Biol. 2014, 3, 7–15. [Google Scholar] [CrossRef]
- Liu, R.-M.; Desai, L.P. Reciprocal regulation of TGF-β and reactive oxygen species: A perverse cycle for fibrosis. Redox Biol. 2015, 6, 565–577. [Google Scholar] [CrossRef] [PubMed]
- Miñana, J.B.; Gómez-Cambronero, L.; Lloret, A.; Pallardó, F.V.; Del Olmo, J.; Escudero, A.; Rodrigo, J.M.; Pellíin, A.; Viña, J.R.; Viña, J.; et al. Mitochondrial oxidative stress and CD95 ligand: A dual mechanism for hepatocyte apoptosis in chronic alcoholism. Hepatology 2002, 35, 1205–1214. [Google Scholar] [CrossRef]
- Ganji, S.H.; Kashyap, M.; Kamanna, V.S. Niacin inhibits fat accumulation, oxidative stress, and inflammatory cytokine IL-8 in cultured hepatocytes: Impact on non-alcoholic fatty liver disease. Metabolism 2015, 64, 982–990. [Google Scholar] [CrossRef] [PubMed]
- Yin, M.; Gäbele, E.; Wheeler, M.D.; Connor, H.; Bradford, B.U.; Dikalova, A.; Rusyn, I.; Mason, R.; Thurman, R.G. Alcohol-induced free radicals in mice: Direct toxicants or signaling molecules? Hepatology 2001, 34, 935–942. [Google Scholar] [CrossRef]
- Pessayre, D.; Fromenty, B. NASH: A mitochondrial disease. J. Hepatol. 2005, 42, 928–940. [Google Scholar] [CrossRef]
- Loguercio, C.; De Girolamo, V.; de Sio, I.; Tuccillo, C.; Ascione, A.; Baldi, F.; Budillon, G.; Cimino, L.; Di Carlo, A.; Di Marino, M.P.; et al. Non-alcoholic fatty liver disease in an area of southern Italy: Main clinical, histological, and pathophysiological aspects. J. Hepatol. 2001, 35, 568–574. [Google Scholar] [CrossRef]
- Begriche, K.; Igoudjil, A.; Pessayre, D.; Fromenty, B. Mitochondrial dysfunction in NASH: Causes, consequences and possible means to prevent it. Mitochondrion 2006, 6, 1–28. [Google Scholar] [CrossRef] [PubMed]
- Begriche, K.; Knockaert, L.; Massart, J.; Robin, M.-A.; Fromenty, B. Mitochondrial dysfunction in nonalcoholic steatohepatitis (NASH): Are there drugs able to improve it? Drug Discov. Today Dis. Mech. 2009, 6, e11–e23. [Google Scholar] [CrossRef]
- Abdel-Maboud, M.; Menshawy, A.; Menshawy, E.; Emara, A.; Alshandidy, M.; Eid, M. The efficacy of vitamin E in reducing non-alcoholic fatty liver disease: A systematic review, meta-analysis, and meta-regression. Ther. Adv. Gastroenterol. 2020, 13, 13. [Google Scholar] [CrossRef] [PubMed]
- Amanullah, I.; Khan, Y.H.; Anwar, I.; Gulzar, A.; Mallhi, T.H.; Raja, A.A. Effect of vitamin E in non-alcoholic fatty liver disease: A systematic review and meta-analysis of randomised controlled trials. Postgrad. Med. J. 2019, 95, 601–611. [Google Scholar] [CrossRef]
- Schürks, M.; Glynn, R.J.; Rist, P.M.; Tzourio, C.; Kurth, T. Effects of vitamin E on stroke subtypes: Meta-analysis of randomised controlled trials. BMJ 2010, 341, c5702. [Google Scholar] [CrossRef]
- Wang, T.; Xu, L. Circulating Vitamin E Levels and Risk of Coronary Artery Disease and Myocardial Infarction: A Mendelian Randomization Study. Nutrients 2019, 11, 2153. [Google Scholar] [CrossRef]
- Klein, E.A.; Thompson, I.M.; Tangen, C.M.; Crowley, J.J.; Lucia, M.S.; Goodman, P.J.; Minasian, L.M.; Ford, L.G.; Parnes, H.L.; Gaziano, J.M.; et al. Vitamin E and the Risk of Prostate Cancer: The selenium and vitamin e cancer prevention trial (SELECT). JAMA 2011, 306, 1549–1556. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Tan, W.; Liu, X.; Deng, L.; Huang, L.; Wang, X.; Gao, X. New insight and potential therapy for NAFLD: CYP2E1 and flavonoids. Biomed. Pharmacother. 2021, 137, 111326. [Google Scholar] [CrossRef]
- Yao, H.; Qiao, Y.-J.; Zhao, Y.-L.; Tao, X.-F.; Xu, L.-N.; Yin, L.-H.; Qi, Y.; Peng, J.-Y. Herbal medicines and nonalcoholic fatty liver disease. World J. Gastroenterol. 2016, 22, 6890–6905. [Google Scholar] [CrossRef] [PubMed]
- Hurrell, T.; Kastrinou-Lampou, V.; Fardellas, A.; Hendriks, D.F.G.; Nordling, Å.; Johansson, I.; Baze, A.; Parmentier, C.; Richert, L.; Ingelman-Sundberg, M. Human Liver Spheroids as a Model to Study Aetiology and Treatment of Hepatic Fibrosis. Cells 2020, 9, 964. [Google Scholar] [CrossRef] [PubMed]
- Soret, P.-A.; Magusto, J.; Housset, C.; Gautheron, J. In Vitro and In Vivo Models of Non-Alcoholic Fatty Liver Disease: A Critical Appraisal. J. Clin. Med. 2020, 10, 36. [Google Scholar] [CrossRef] [PubMed]
- Kozyra, M.; Johansson, I.; Nordling, Å.; Ullah, S.; Lauschke, V.M.; Ingelman-Sundberg, M. Human hepatic 3D spheroids as a model for steatosis and insulin resistance. Sci. Rep. 2018, 8, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Vorrink, S.U.; Ullah, S.; Schmidt, S.; Nandania, J.; Velagapudi, V.; Beck, O.; Ingelman-Sundberg, M.; Lauschke, V.M. Endogenous and xenobiotic metabolic stability of primary human hepatocytes in long-term 3D spheroid cultures revealed by a combination of targeted and untargeted metabolomics. FASEB J. 2017, 31, 2696–2708. [Google Scholar] [CrossRef]
- Prill, S.; Caddeo, A.; Baselli, G.A.; Jamialahmadi, O.; Dongiovanni, P.; Rametta, R.; Kanebratt, K.P.; Pujia, A.; Pingitore, P.; Mancina, R.M.; et al. The TM6SF2 E167K genetic variant induces lipid biosynthesis and reduces apolipoprotein B secretion in human hepatic 3D spheroids. Sci. Rep. 2019, 9, 1–12. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Harjumäki, R.; Pridgeon, C.S.; Ingelman-Sundberg, M. CYP2E1 in Alcoholic and Non-Alcoholic Liver Injury. Roles of ROS, Reactive Intermediates and Lipid Overload. Int. J. Mol. Sci. 2021, 22, 8221. https://doi.org/10.3390/ijms22158221
Harjumäki R, Pridgeon CS, Ingelman-Sundberg M. CYP2E1 in Alcoholic and Non-Alcoholic Liver Injury. Roles of ROS, Reactive Intermediates and Lipid Overload. International Journal of Molecular Sciences. 2021; 22(15):8221. https://doi.org/10.3390/ijms22158221
Chicago/Turabian StyleHarjumäki, Riina, Chris S. Pridgeon, and Magnus Ingelman-Sundberg. 2021. "CYP2E1 in Alcoholic and Non-Alcoholic Liver Injury. Roles of ROS, Reactive Intermediates and Lipid Overload" International Journal of Molecular Sciences 22, no. 15: 8221. https://doi.org/10.3390/ijms22158221
APA StyleHarjumäki, R., Pridgeon, C. S., & Ingelman-Sundberg, M. (2021). CYP2E1 in Alcoholic and Non-Alcoholic Liver Injury. Roles of ROS, Reactive Intermediates and Lipid Overload. International Journal of Molecular Sciences, 22(15), 8221. https://doi.org/10.3390/ijms22158221