
Heme Burden and Ensuing Mechanisms That Protect the Kidney: Insights from Bench and Bedside


Abstract
:1. Introduction
2. Clinical Significance
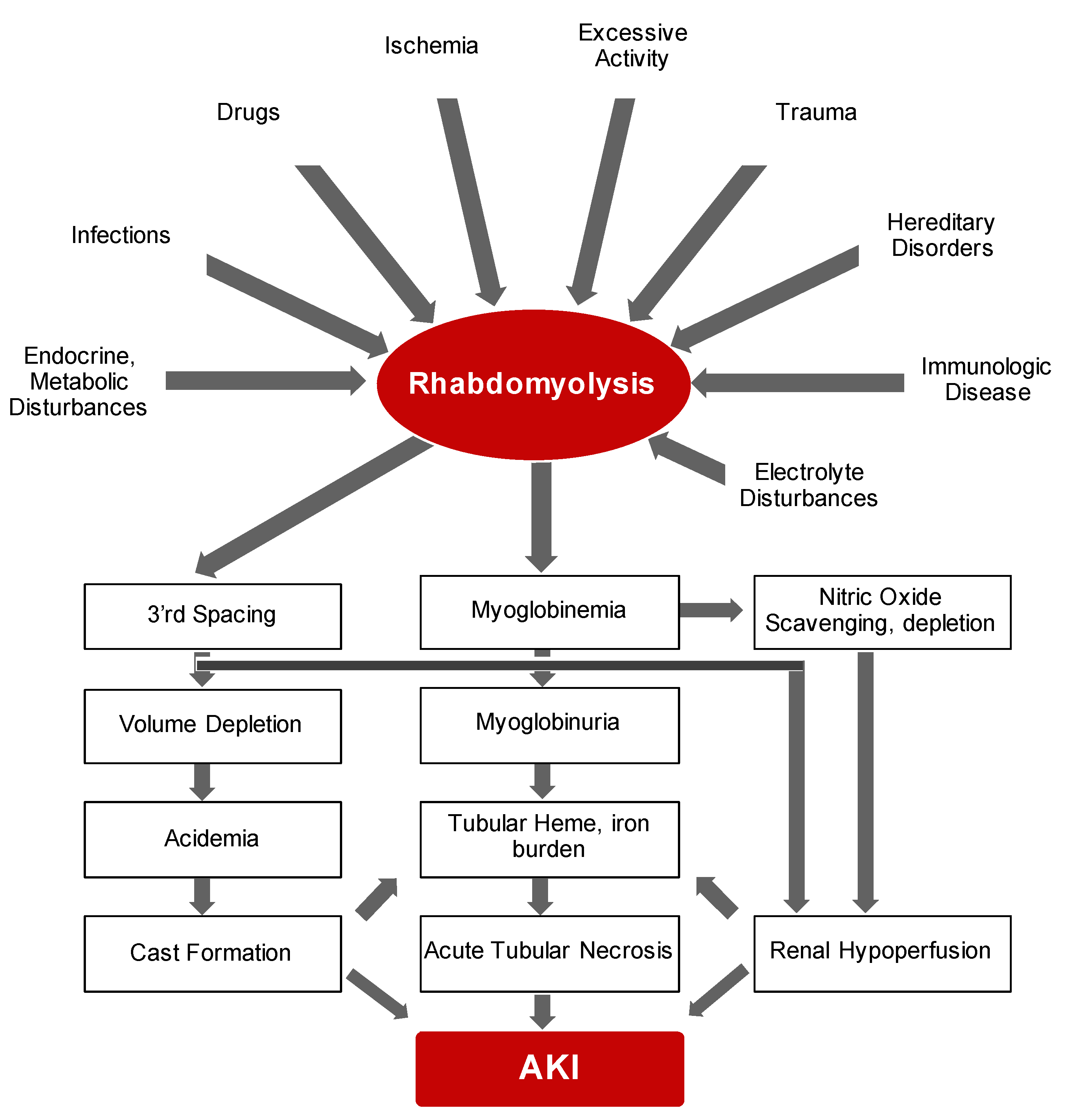
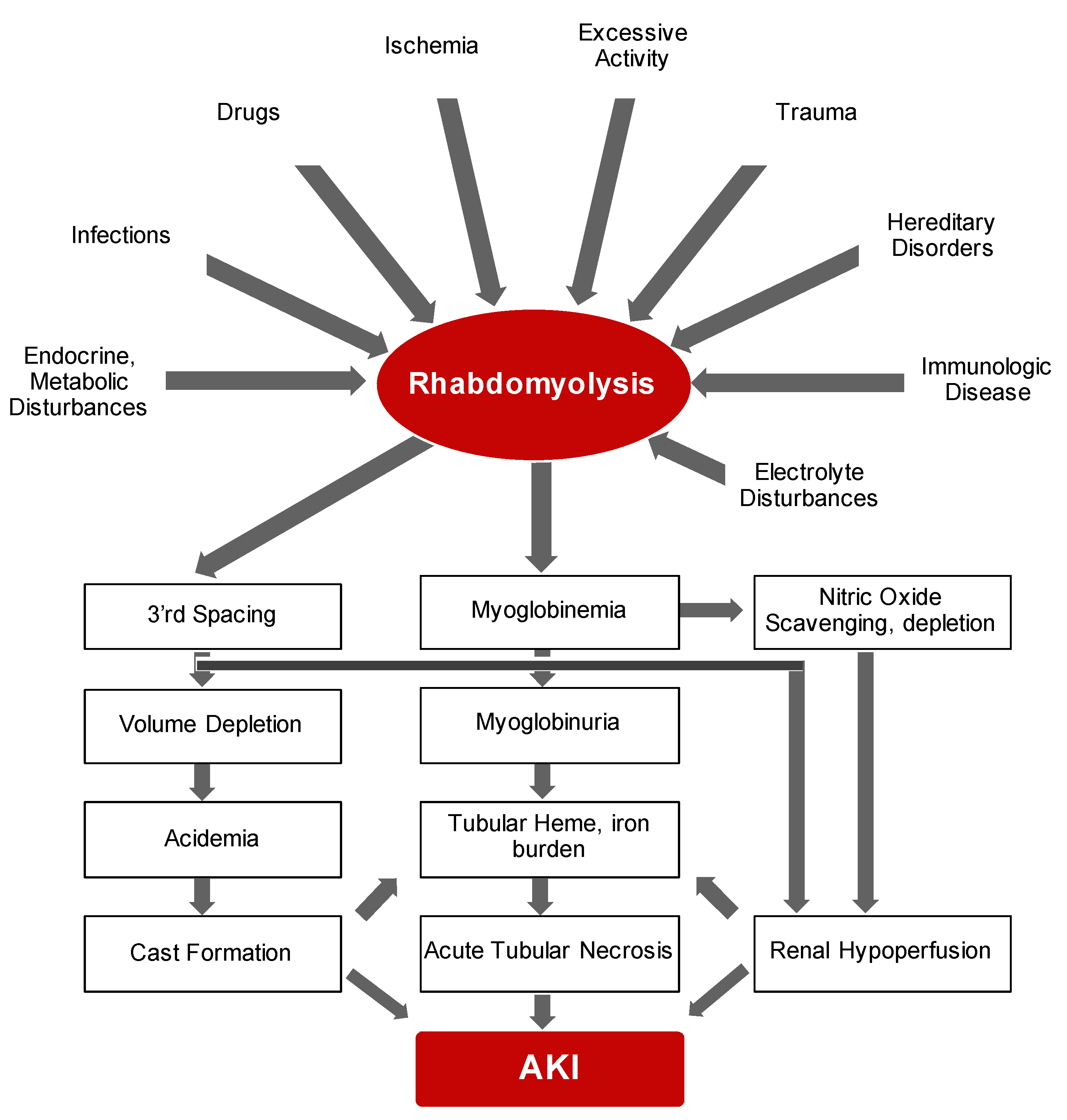
2.1. Rhabdomyolysis
2.2. Hemolytic Anemias
2.3. Paroxysmal Nocturnal Hemoglobinuria (PNH)
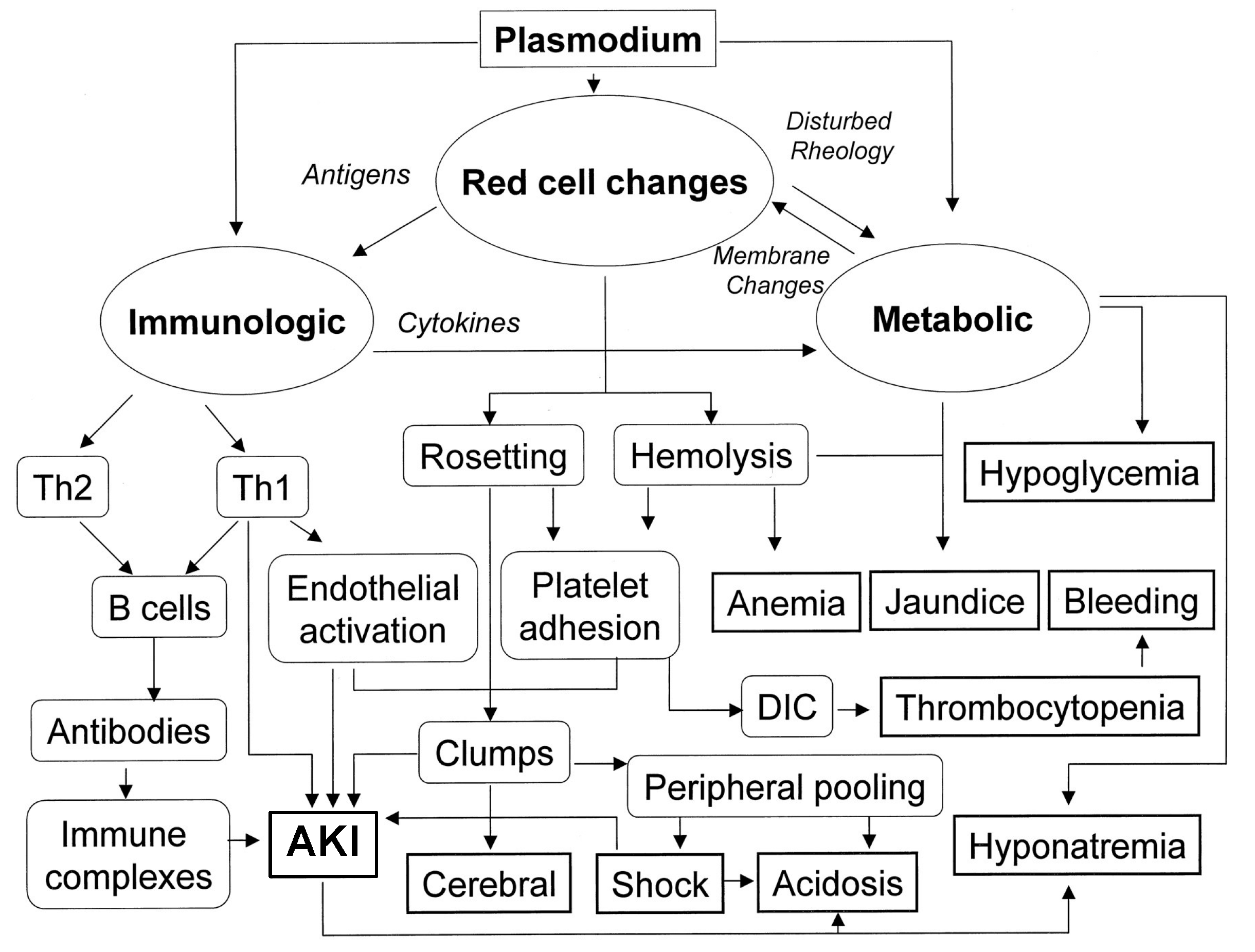
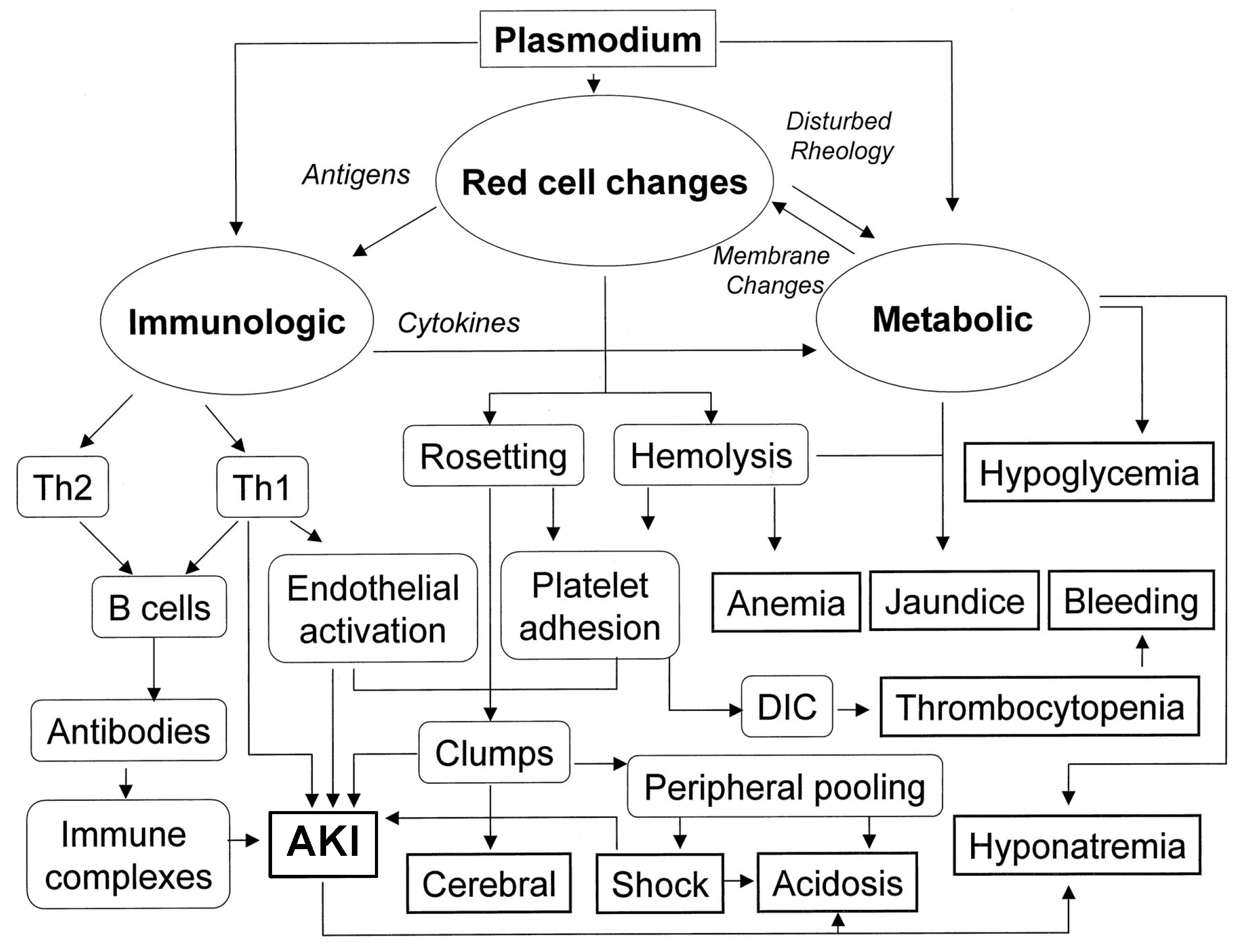
2.4. Malaria
3. Molecular Insights
3.1. Heme: The Requisite for Aerobic Life
3.2. Heme Oxygenase-1/Ferritin
4. Evidence to Support Salutary Effects of HO-1/Ferritin System against Heme-Protein Induced Kidney Disease
4.1. Rhabdomyolysis
4.2. Malaria
4.3. Sickle Cell Disease (SCD)
4.4. Other Models of Hemolytic Anemias and Protective Role of HO-1
4.5. Ferritin and Nephroprotection
5. Conclusions and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Paoli, M.; Marles-Wright, J.; Smith, A. Structure-function relationships in heme-proteins. DNA Cell Biol. 2002, 21, 271–280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gozzelino, R.; Jeney, V.; Soares, M.P. Mechanisms of cell protection by heme oxygenase-1. Annu. Rev. Pharmacol. Toxicol. 2010, 50, 323–354. [Google Scholar] [CrossRef] [Green Version]
- Balla, J.; Vercellotti, G.M.; Nath, K.; Yachie, A.; Nagy, E.; Eaton, J.W.; Balla, G. Haem, haem oxygenase and ferritin in vascular endothelial cell injury. Nephrol. Dial. Transplant. 2003, 18 (Suppl. 5), v8–v12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bozza, M.T.; Jeney, V. Pro-inflammatory actions of heme and other hemoglobin-derived DAMPs. Front. Immunol. 2020, 11, 1323. [Google Scholar] [CrossRef]
- Bolisetty, S.; Zarjou, A.; Agarwal, A. Heme oxygenase 1 as a therapeutic target in acute kidney injury. Am. J. Kidney Dis. 2017, 69, 531–545. [Google Scholar] [CrossRef] [Green Version]
- Zarjou, A.; Agarwal, A. Heme oxygenase-1 as a target for TGF-beta in kidney disease. Semin. Nephrol. 2012, 32, 277–286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tracz, M.J.; Alam, J.; Nath, K.A. Physiology and pathophysiology of heme: Implications for kidney disease. J. Am. Soc. Nephrol. 2007, 18, 414–420. [Google Scholar] [CrossRef] [PubMed]
- Balla, J.; Balla, G.; Zarjou, A. Ferritin in kidney and vascular related diseases: Novel roles for an old player. Pharmaceuticals 2019, 12, 96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nath, K.A. Heme oxygenase-1 and acute kidney injury. Curr. Opin. Nephrol. Hypertens. 2014, 23, 17–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nath, K.A.; Balla, G.; Vercellotti, G.M.; Balla, J.; Jacob, H.S.; Levitt, M.D.; Rosenberg, M.E. Induction of heme oxygenase is a rapid, protective response in rhabdomyolysis in the rat. J. Clin. Investig. 1992, 90, 267–270. [Google Scholar] [CrossRef] [Green Version]
- Balla, G.; Jacob, H.S.; Balla, J.; Rosenberg, M.; Nath, K.; Apple, F.; Eaton, J.W.; Vercellotti, G.M. Ferritin: A cytoprotective antioxidant strategem of endothelium. J. Biol. Chem. 1992, 267, 18148–18153. [Google Scholar] [CrossRef]
- Ayer, A.; Zarjou, A.; Agarwal, A.; Stocker, R. Heme Oxygenases in cardiovascular health and disease. Physiol. Rev. 2016, 96, 1449–1508. [Google Scholar] [CrossRef]
- Bywaters, E.G.; Beall, D. Crush injuries with impairment of renal function. Br. Med. J. 1941, 1, 427–432. [Google Scholar] [CrossRef] [Green Version]
- Bywaters, E.G.L.; Stead, J.K. The production of renal failure following injection of solutions containing myohæmoglobin. Q. J. Exp. Physiol. Cogn. Med Sci. 1944, 33, 53–70. [Google Scholar] [CrossRef]
- Bosch, X.; Poch, E.; Grau, J.M. Rhabdomyolysis and acute kidney injury. N. Engl. J. Med. 2009, 361, 62–72. [Google Scholar] [CrossRef] [Green Version]
- Aleckovic-Halilovic, M.; Pjanic, M.; Mesic, E.; Storrar, J.; Woywodt, A. From quail to earthquakes and human conflict: A historical perspective of rhabdomyolysis. Clin. Kidney J. 2021, 14, 1088–1096. [Google Scholar] [CrossRef]
- Cote, D.R.; Fuentes, E.; Elsayes, A.H.; Ross, J.J.; Quraishi, S.A. A “crush” course on rhabdomyolysis: Risk stratification and clinical management update for the perioperative clinician. J. Anesth. 2020, 34, 585–598. [Google Scholar] [CrossRef] [PubMed]
- Cabral, B.M.I.; Edding, S.N.; Portocarrero, J.P.; Lerma, E.V. Rhabdomyolysis. Dis. Mon. 2020, 66, 101015. [Google Scholar] [CrossRef] [PubMed]
- Gabow, P.A.; Kaehny, W.D.; Kelleher, S.P. The spectrum of rhabdomyolysis. Medicine 1982, 61, 141–152. [Google Scholar] [CrossRef] [PubMed]
- Garry, D.J.; Mammen, P.P. Molecular insights into the functional role of myoglobin. Adv. Exp. Med. Biol. 2007, 618, 181–193. [Google Scholar] [CrossRef] [PubMed]
- Brancaccio, P.; Lippi, G.; Maffulli, N. Biochemical markers of muscular damage. Clin. Chem. Lab. Med. 2010, 48, 757–767. [Google Scholar] [CrossRef]
- Huerta-Alardin, A.L.; Varon, J.; Marik, P.E. Bench-to-bedside review: Rhabdomyolysis—An overview for clinicians. Crit. Care 2005, 9, 158–169. [Google Scholar] [CrossRef] [Green Version]
- Tabbara, I.A. Hemolytic anemias. Diagnosis and management. Med. Clin. N. Am. 1992, 76, 649–668. [Google Scholar] [CrossRef]
- Van Avondt, K.; Nur, E.; Zeerleder, S. Mechanisms of haemolysis-induced kidney injury. Nat. Rev. Nephrol. 2019, 15, 671–692. [Google Scholar] [CrossRef]
- Rother, R.P.; Bell, L.; Hillmen, P.; Gladwin, M.T. The clinical sequelae of intravascular hemolysis and extracellular plasma hemoglobin: A novel mechanism of human disease. JAMA 2005, 293, 1653–1662. [Google Scholar] [CrossRef] [PubMed]
- Sundd, P.; Gladwin, M.T.; Novelli, E.M. Pathophysiology of sickle cell disease. Annu. Rev. Pathol. 2019, 14, 263–292. [Google Scholar] [CrossRef] [PubMed]
- Nath, K.A.; Hebbel, R.P. Sickle cell disease: Renal manifestations and mechanisms. Nat. Rev. Nephrol. 2015, 11, 161–171. [Google Scholar] [CrossRef] [Green Version]
- Hebbel, R.P.; Belcher, J.D.; Vercellotti, G.M. The multifaceted role of ischemia/reperfusion in sickle cell anemia. J. Clin. Investig. 2020, 130, 1062–1072. [Google Scholar] [CrossRef]
- Fattizzo, B.; Serpenti, F.; Giannotta, J.A.; Barcellini, W. Difficult cases of paroxysmal nocturnal hemoglobinuria: Diagnosis and therapeutic novelties. J. Clin. Med. 2021, 10, 948. [Google Scholar] [CrossRef] [PubMed]
- Risitano, A.M. Paroxysmal nocturnal hemoglobinuria and the complement system: Recent insights and novel anticomplement strategies. Adv. Exp. Med. Biol. 2013, 735, 155–172. [Google Scholar] [CrossRef]
- Savage, W.J.; Brodsky, R.A. New insights into paroxysmal nocturnal hemoglobinuria. Hematology 2007, 12, 371–376. [Google Scholar] [CrossRef]
- Hill, A.; DeZern, A.E.; Kinoshita, T.; Brodsky, R.A. Paroxysmal nocturnal haemoglobinuria. Nat. Rev. Dis. Primers 2017, 3, 17028. [Google Scholar] [CrossRef] [Green Version]
- Brodsky, R.A. Paroxysmal nocturnal hemoglobinuria. Blood 2014, 124, 2804–2811. [Google Scholar] [CrossRef] [PubMed]
- Rubin, H. Paroxysmal nocturnal hemoglobinuria with renal failure. JAMA 1971, 215, 433–436. [Google Scholar] [CrossRef] [PubMed]
- Clark, D.A.; Butler, S.A.; Braren, V.; Hartmann, R.C.; Jenkins, D.E., Jr. The kidneys in paroxysmal nocturnal hemoglobinuria. Blood 1981, 57, 83–89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chow, K.M.; Lai, F.M.; Wang, A.Y.; Chan, Y.L.; Tang, N.L.; Li, P.K. Reversible renal failure in paroxysmal nocturnal hemoglobinuria. Am. J. Kidney Dis. 2001, 37, E17. [Google Scholar] [CrossRef] [PubMed]
- Ram, R.; Adiraju, K.P.; Gudithi, S.; Dakshinamurty, K.V. Renal manifestations in paroxysmal nocturnal hemoglobinuria. Indian J. Nephrol. 2017, 27, 289–293. [Google Scholar] [CrossRef] [PubMed]
- Parker, C.; Omine, M.; Richards, S.; Nishimura, J.; Bessler, M.; Ware, R.; Hillmen, P.; Luzzatto, L.; Young, N.; Kinoshita, T.; et al. Diagnosis and management of paroxysmal nocturnal hemoglobinuria. Blood 2005, 106, 3699–3709. [Google Scholar] [CrossRef] [Green Version]
- Schrezenmeier, H.; Muus, P.; Socie, G.; Szer, J.; Urbano-Ispizua, A.; Maciejewski, J.P.; Brodsky, R.A.; Bessler, M.; Kanakura, Y.; Rosse, W.; et al. Baseline characteristics and disease burden in patients in the International Paroxysmal Nocturnal Hemoglobinuria Registry. Haematologica 2014, 99, 922–929. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borowitz, M.J.; Craig, F.E.; Digiuseppe, J.A.; Illingworth, A.J.; Rosse, W.; Sutherland, D.R.; Wittwer, C.T.; Richards, S.J.; Clinical Cytometry, S. Guidelines for the diagnosis and monitoring of paroxysmal nocturnal hemoglobinuria and related disorders by flow cytometry. Cytometry B Clin. Cytom. 2010, 78, 211–230. [Google Scholar] [CrossRef] [PubMed]
- Mace, K.E.; Lucchi, N.W.; Tan, K.R. Malaria surveillance—United States, 2017. MMWR Surveill. Summ. 2021, 70, 1–35. [Google Scholar] [CrossRef]
- Cotter, C.; Sturrock, H.J.; Hsiang, M.S.; Liu, J.; Phillips, A.A.; Hwang, J.; Gueye, C.S.; Fullman, N.; Gosling, R.D.; Feachem, R.G. The changing epidemiology of malaria elimination: New strategies for new challenges. Lancet 2013, 382, 900–911. [Google Scholar] [CrossRef]
- Clark, I.A.; Alleva, L.M.; Mills, A.C.; Cowden, W.B. Pathogenesis of malaria and clinically similar conditions. Clin. Microbiol. Rev. 2004, 17, 509–539. [Google Scholar] [CrossRef] [Green Version]
- Milner, D.A., Jr. Malaria pathogenesis. Cold Spring Harb. Perspect. Med. 2018, 8, a025569. [Google Scholar] [CrossRef] [Green Version]
- Brown, G.V.; Beck, H.P.; Molyneux, M.; Marsh, K. Molecular approaches to epidemiology and clinical aspects of malaria. Parasitol. Today 2000, 16, 448–451. [Google Scholar] [CrossRef]
- Griffith, K.S.; Lewis, L.S.; Mali, S.; Parise, M.E. Treatment of malaria in the United States: A systematic review. JAMA 2007, 297, 2264–2277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mbanefo, A.; Kumar, N. Evaluation of malaria diagnostic methods as a key for successful control and elimination programs. Trop. Med. Infect. Dis. 2020, 5, 102. [Google Scholar] [CrossRef] [PubMed]
- Barsoum, R.S. Malarial acute renal failure. J. Am. Soc. Nephrol. 2000, 11, 2147–2154. [Google Scholar] [CrossRef]
- Silva, G.B.D.J.; Pinto, J.R.; Barros, E.J.G.; Farias, G.M.N.; Daher, E.F. Kidney involvement in malaria: An update. Rev. Inst. Med. Trop. Sao Paulo 2017, 59, e53. [Google Scholar] [CrossRef] [Green Version]
- Kaur, C.; Pramanik, A.; Kumari, K.; Mandage, R.; Dinda, A.K.; Sankar, J.; Bagga, A.; Agarwal, S.K.; Sinha, A.; Singh, G.; et al. Renal detection of Plasmodium falciparum, Plasmodium vivax and Plasmodium knowlesi in malaria associated acute kidney injury: A retrospective case-control study. BMC Res. Notes 2020, 13, 37. [Google Scholar] [CrossRef] [PubMed]
- Nguansangiam, S.; Day, N.P.; Hien, T.T.; Mai, N.T.; Chaisri, U.; Riganti, M.; Dondorp, A.M.; Lee, S.J.; Phu, N.H.; Turner, G.D.; et al. A quantitative ultrastructural study of renal pathology in fatal Plasmodium falciparum malaria. Trop. Med. Int. Health 2007, 12, 1037–1050. [Google Scholar] [CrossRef]
- Amoura, A.; Moktefi, A.; Halfon, M.; Karras, A.; Rafat, C.; Gibier, J.B.; Gleeson, P.J.; Servais, A.; Argy, N.; Maille, P.; et al. Malaria, collapsing glomerulopathy, and focal and segmental glomerulosclerosis. Clin. J. Am. Soc. Nephrol. 2020, 15, 964–972. [Google Scholar] [CrossRef]
- Barber, B.E.; Grigg, M.J.; Piera, K.A.; William, T.; Cooper, D.J.; Plewes, K.; Dondorp, A.M.; Yeo, T.W.; Anstey, N.M. Intravascular haemolysis in severe Plasmodium knowlesi malaria: Association with endothelial activation, microvascular dysfunction, and acute kidney injury. Emerg. Microbes Infect. 2018, 7, 106. [Google Scholar] [CrossRef] [Green Version]
- Badiane, A.S.; Diongue, K.; Diallo, S.; Ndongo, A.A.; Diedhiou, C.K.; Deme, A.B.; Ma, D.; Ndiaye, M.; Seck, M.C.; Dieng, T.; et al. Acute kidney injury associated with Plasmodium malariae infection. Malar. J. 2014, 13, 226. [Google Scholar] [CrossRef] [Green Version]
- Ponka, P. Cell biology of heme. Am. J. Med. Sci. 1999, 318, 241–256. [Google Scholar] [CrossRef]
- Fujiwara, T.; Harigae, H. Biology of heme in mammalian erythroid cells and related disorders. Biomed. Res. Int. 2015, 2015, 278536. [Google Scholar] [CrossRef] [Green Version]
- Soares, M.P.; Bozza, M.T. Red alert: Labile heme is an alarmin. Curr. Opin. Immunol. 2016, 38, 94–100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balla, J.; Vercellotti, G.M.; Jeney, V.; Yachie, A.; Varga, Z.; Jacob, H.S.; Eaton, J.W.; Balla, G. Heme, heme oxygenase, and ferritin: How the vascular endothelium survives (and dies) in an iron-rich environment. Antioxid. Redox Signal. 2007, 9, 2119–2137. [Google Scholar] [CrossRef]
- Nagy, E.; Eaton, J.W.; Jeney, V.; Soares, M.P.; Varga, Z.; Galajda, Z.; Szentmiklosi, J.; Mehes, G.; Csonka, T.; Smith, A.; et al. Red cells, hemoglobin, heme, iron, and atherogenesis. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 1347–1353. [Google Scholar] [CrossRef] [PubMed]
- Jeney, V.; Balla, J.; Yachie, A.; Varga, Z.; Vercellotti, G.M.; Eaton, J.W.; Balla, G. Pro-oxidant and cytotoxic effects of circulating heme. Blood 2002, 100, 879–887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balla, J.; Potor, L.; Hendrik, Z.; Patsalos, A.; Katona, E.; Mehes, G.; Poliska, S.; Csosz, E.; Kallo, G.; Komaromi, I.; et al. Oxidation of hemoglobin drives a pro-atherogenic polarization of macrophages in human atherosclerosis. Antioxid. Redox Signal. 2021. [Google Scholar] [CrossRef]
- Kassa, T.; Jana, S.; Meng, F.; Alayash, A.I. Differential heme release from various hemoglobin redox states and the upregulation of cellular heme oxygenase-1. FEBS Open Bio 2016, 6, 876–884. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.; McCulloh, R.J. Hemopexin and haptoglobin: Allies against heme toxicity from hemoglobin not contenders. Front. Physiol. 2015, 6, 187. [Google Scholar] [CrossRef]
- Vinchi, F.; Costa da Silva, M.; Ingoglia, G.; Petrillo, S.; Brinkman, N.; Zuercher, A.; Cerwenka, A.; Tolosano, E.; Muckenthaler, M.U. Hemopexin therapy reverts heme-induced proinflammatory phenotypic switching of macrophages in a mouse model of sickle cell disease. Blood 2016, 127, 473–486. [Google Scholar] [CrossRef] [PubMed]
- Ashouri, R.; Fangman, M.; Burris, A.; Ezenwa, M.O.; Wilkie, D.J.; Dore, S. Critical role of hemopexin mediated cytoprotection in the pathophysiology of sickle cell disease. Int. J. Mol. Sci. 2021, 22, 6408. [Google Scholar] [CrossRef] [PubMed]
- Vinchi, F.; De Franceschi, L.; Ghigo, A.; Townes, T.; Cimino, J.; Silengo, L.; Hirsch, E.; Altruda, F.; Tolosano, E. Hemopexin therapy improves cardiovascular function by preventing heme-induced endothelial toxicity in mouse models of hemolytic diseases. Circulation 2013, 127, 1317–1329. [Google Scholar] [CrossRef] [Green Version]
- Ferreira, A.; Balla, J.; Jeney, V.; Balla, G.; Soares, M.P. A central role for free heme in the pathogenesis of severe malaria: The missing link? J. Mol. Med. 2008, 86, 1097–1111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haines, D.D.; Tosaki, A. Heme degradation in pathophysiology of and countermeasures to inflammation-associated disease. Int. J. Mol. Sci. 2020, 21, 9698. [Google Scholar] [CrossRef] [PubMed]
- Vincent, S.H. Oxidative effects of heme and porphyrins on proteins and lipids. Semin. Hematol. 1989, 26, 105–113. [Google Scholar]
- Tappel, A.L. Unsaturated lipide oxidation catalyzed by hematin compounds. J. Biol. Chem. 1955, 217, 721–733. [Google Scholar] [CrossRef]
- Zarjou, A.; Balla, J.; Balla, G.; Agarwal, A. Iron metabolism and oxidative stress. In Studies on Renal Disorders; Miyata, T., Eckardt, K.-U., Nangaku, M., Eds.; Humana Press: Totowa, NJ, USA, 2011; pp. 205–228. [Google Scholar]
- Nath, K.A.; Grande, J.P.; Croatt, A.J.; Likely, S.; Hebbel, R.P.; Enright, H. Intracellular targets in heme protein-induced renal injury. Kidney Int. 1998, 53, 100–111. [Google Scholar] [CrossRef] [Green Version]
- Dutra, F.F.; Alves, L.S.; Rodrigues, D.; Fernandez, P.L.; de Oliveira, R.B.; Golenbock, D.T.; Zamboni, D.S.; Bozza, M.T. Hemolysis-induced lethality involves inflammasome activation by heme. Proc. Natl. Acad. Sci. USA 2014, 111, E4110–E4118. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, S.; Adisa, O.A.; Chappa, P.; Tan, F.; Jackson, K.A.; Archer, D.R.; Ofori-Acquah, S.F. Extracellular hemin crisis triggers acute chest syndrome in sickle mice. J. Clin. Investig. 2013, 123, 4809–4820. [Google Scholar] [CrossRef] [Green Version]
- Nath, K.A.; Vercellotti, G.M.; Grande, J.P.; Miyoshi, H.; Paya, C.V.; Manivel, J.C.; Haggard, J.J.; Croatt, A.J.; Payne, W.D.; Alam, J. Heme protein-induced chronic renal inflammation: Suppressive effect of induced heme oxygenase-1. Kidney Int. 2001, 59, 106–117. [Google Scholar] [CrossRef] [Green Version]
- Wagener, F.A.; Eggert, A.; Boerman, O.C.; Oyen, W.J.; Verhofstad, A.; Abraham, N.G.; Adema, G.; van Kooyk, Y.; de Witte, T.; Figdor, C.G. Heme is a potent inducer of inflammation in mice and is counteracted by heme oxygenase. Blood 2001, 98, 1802–1811. [Google Scholar] [CrossRef]
- Figueiredo, R.T.; Fernandez, P.L.; Mourao-Sa, D.S.; Porto, B.N.; Dutra, F.F.; Alves, L.S.; Oliveira, M.F.; Oliveira, P.L.; Graca-Souza, A.V.; Bozza, M.T. Characterization of heme as activator of Toll-like receptor 4. J. Biol. Chem. 2007, 282, 20221–20229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wagener, F.A.; Feldman, E.; de Witte, T.; Abraham, N.G. Heme induces the expression of adhesion molecules ICAM-1, VCAM-1, and E selectin in vascular endothelial cells. Proc. Soc. Exp. Biol. Med. 1997, 216, 456–463. [Google Scholar] [CrossRef]
- Balla, G.; Vercellotti, G.; Eaton, J.W.; Jacob, H.S. Heme uptake by endothelium synergizes polymorphonuclear granulocyte-mediated damage. Trans. Assoc. Am. Phys. 1990, 103, 174–179. [Google Scholar] [PubMed]
- Balla, G.; Vercellotti, G.M.; Muller-Eberhard, U.; Eaton, J.; Jacob, H.S. Exposure of endothelial cells to free heme potentiates damage mediated by granulocytes and toxic oxygen species. Lab. Investig. 1991, 64, 648–655. [Google Scholar]
- Okubo, K.; Kurosawa, M.; Kamiya, M.; Urano, Y.; Suzuki, A.; Yamamoto, K.; Hase, K.; Homma, K.; Sasaki, J.; Miyauchi, H.; et al. Macrophage extracellular trap formation promoted by platelet activation is a key mediator of rhabdomyolysis-induced acute kidney injury. Nat. Med. 2018, 24, 232–238. [Google Scholar] [CrossRef] [PubMed]
- Boudhabhay, I.; Poillerat, V.; Grunenwald, A.; Torset, C.; Leon, J.; Daugan, M.V.; Lucibello, F.; El Karoui, K.; Ydee, A.; Chauvet, S.; et al. Complement activation is a crucial driver of acute kidney injury in rhabdomyolysis. Kidney Int. 2021, 99, 581–597. [Google Scholar] [CrossRef]
- Arruda, M.A.; Rossi, A.G.; de Freitas, M.S.; Barja-Fidalgo, C.; Graca-Souza, A.V. Heme inhibits human neutrophil apoptosis: Involvement of phosphoinositide 3-kinase, MAPK, and NF-kappaB. J. Immunol. 2004, 173, 2023–2030. [Google Scholar] [CrossRef]
- Gbotosho, O.T.; Kapetanaki, M.G.; Kato, G.J. The worst things in life are free: The role of free heme in sickle cell disease. Front. Immunol. 2020, 11, 561917. [Google Scholar] [CrossRef] [PubMed]
- Poss, K.D.; Tonegawa, S. Heme oxygenase 1 is required for mammalian iron reutilization. Proc. Natl. Acad. Sci. USA 1997, 94, 10919–10924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yachie, A.; Niida, Y.; Wada, T.; Igarashi, N.; Kaneda, H.; Toma, T.; Ohta, K.; Kasahara, Y.; Koizumi, S. Oxidative stress causes enhanced endothelial cell injury in human heme oxygenase-1 deficiency. J. Clin. Investig. 1999, 103, 129–135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yachie, A. Heme oxygenase-1 deficiency and oxidative stress: A review of 9 independent human cases and animal models. Int. J. Mol. Sci. 2021, 22, 1514. [Google Scholar] [CrossRef]
- Sun, J.; Hoshino, H.; Takaku, K.; Nakajima, O.; Muto, A.; Suzuki, H.; Tashiro, S.; Takahashi, S.; Shibahara, S.; Alam, J.; et al. Hemoprotein Bach1 regulates enhancer availability of heme oxygenase-1 gene. EMBO J. 2002, 21, 5216–5224. [Google Scholar] [CrossRef]
- Lin, Q.; Weis, S.; Yang, G.; Weng, Y.H.; Helston, R.; Rish, K.; Smith, A.; Bordner, J.; Polte, T.; Gaunitz, F.; et al. Heme oxygenase-1 protein localizes to the nucleus and activates transcription factors important in oxidative stress. J. Biol. Chem. 2007, 282, 20621–20633. [Google Scholar] [CrossRef] [Green Version]
- Kovtunovych, G.; Eckhaus, M.A.; Ghosh, M.C.; Ollivierre-Wilson, H.; Rouault, T.A. Dysfunction of the heme recycling system in heme oxygenase 1-deficient mice: Effects on macrophage viability and tissue iron distribution. Blood 2010, 116, 6054–6062. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tzima, S.; Victoratos, P.; Kranidioti, K.; Alexiou, M.; Kollias, G. Myeloid heme oxygenase-1 regulates innate immunity and autoimmunity by modulating IFN-beta production. J. Exp. Med. 2009, 206, 1167–1179. [Google Scholar] [CrossRef] [PubMed]
- Gullotta, F.; di Masi, A.; Ascenzi, P. Carbon monoxide: An unusual drug. IUBMB Life 2012, 64, 378–386. [Google Scholar] [CrossRef]
- Ryter, S.W.; Alam, J.; Choi, A.M. Heme oxygenase-1/carbon monoxide: From basic science to therapeutic applications. Physiol. Rev. 2006, 86, 583–650. [Google Scholar] [CrossRef]
- Lee, S.; Lee, S.J.; Coronata, A.A.; Fredenburgh, L.E.; Chung, S.W.; Perrella, M.A.; Nakahira, K.; Ryter, S.W.; Choi, A.M. Carbon monoxide confers protection in sepsis by enhancing beclin 1-dependent autophagy and phagocytosis. Antioxid. Redox Signal. 2014, 20, 432–442. [Google Scholar] [CrossRef] [PubMed]
- Jung, S.S.; Moon, J.S.; Xu, J.F.; Ifedigbo, E.; Ryter, S.W.; Choi, A.M.; Nakahira, K. Carbon monoxide negatively regulates NLRP3 inflammasome activation in macrophages. Am. J. Physiol. Lung Cell. Mol. Physiol. 2015, 308, L1058–L1067. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pecorella, S.R.; Potter, J.V.; Cherry, A.D.; Peacher, D.F.; Welty-Wolf, K.E.; Moon, R.E.; Piantadosi, C.A.; Suliman, H.B. The HO-1/CO system regulates mitochondrial-capillary density relationships in human skeletal muscle. Am. J. Physiol. Lung Cell. Mol. Physiol. 2015, 309, L857–L871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryter, S.W. Heme oxygenase-1/carbon monoxide as modulators of autophagy and inflammation. Arch. Biochem. Biophys. 2019, 678, 108186. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Liu, J.; Tetzlaff, W.; Paty, D.W.; Cynader, M.S. Biliverdin reductase, a major physiologic cytoprotectant, suppresses experimental autoimmune encephalomyelitis. Free Radic. Biol. Med. 2006, 40, 960–967. [Google Scholar] [CrossRef]
- Stocker, R.; Yamamoto, Y.; McDonagh, A.F.; Glazer, A.N.; Ames, B.N. Bilirubin is an antioxidant of possible physiological importance. Science 1987, 235, 1043–1046. [Google Scholar] [CrossRef] [PubMed]
- Ryter, S.W. Bile pigments in pulmonary and vascular disease. Front. Pharmacol. 2012, 3, 39. [Google Scholar] [CrossRef] [Green Version]
- Ollinger, R.; Wang, H.; Yamashita, K.; Wegiel, B.; Thomas, M.; Margreiter, R.; Bach, F.H. Therapeutic applications of bilirubin and biliverdin in transplantation. Antioxid. Redox Signal. 2007, 9, 2175–2185. [Google Scholar] [CrossRef]
- Arosio, P.; Ingrassia, R.; Cavadini, P. Ferritins: A family of molecules for iron storage, antioxidation and more. Biochim. Biophys. Acta 2009, 1790, 589–599. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, C.; Bucchini, D.; Martin, M.E.; Levi, S.; Arosio, P.; Grandchamp, B.; Beaumont, C. Early embryonic lethality of H ferritin gene deletion in mice. J. Biol. Chem. 2000, 275, 3021–3024. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zarjou, A.; Black, L.M.; McCullough, K.R.; Hull, T.D.; Esman, S.K.; Boddu, R.; Varambally, S.; Chandrashekar, D.S.; Feng, W.; Arosio, P.; et al. Ferritin light chain confers protection against sepsis-induced inflammation and organ injury. Front. Immunol. 2019, 10, 131. [Google Scholar] [CrossRef] [Green Version]
- Sikura, K.E.; Potor, L.; Szerafin, T.; Zarjou, A.; Agarwal, A.; Arosio, P.; Poli, M.; Hendrik, Z.; Mehes, G.; Oros, M.; et al. Potential role of H-ferritin in mitigating valvular mineralization. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 413–431. [Google Scholar] [CrossRef] [Green Version]
- Zarjou, A.; Jeney, V.; Arosio, P.; Poli, M.; Antal-Szalmas, P.; Agarwal, A.; Balla, G.; Balla, J. Ferritin prevents calcification and osteoblastic differentiation of vascular smooth muscle cells. J. Am. Soc. Nephrol. 2009, 20, 1254–1263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zarjou, A.; Bolisetty, S.; Joseph, R.; Traylor, A.; Apostolov, E.O.; Arosio, P.; Balla, J.; Verlander, J.; Darshan, D.; Kuhn, L.C.; et al. Proximal tubule H-ferritin mediates iron trafficking in acute kidney injury. J. Clin. Investig. 2013, 123, 4423–4434. [Google Scholar] [CrossRef] [Green Version]
- Bolisetty, S.; Zarjou, A.; Hull, T.D.; Traylor, A.M.; Perianayagam, A.; Joseph, R.; Kamal, A.I.; Arosio, P.; Soares, M.P.; Jeney, V.; et al. Macrophage and epithelial cell H-ferritin expression regulates renal inflammation. Kidney Int. 2015, 88, 95–108. [Google Scholar] [CrossRef] [Green Version]
- Zarjou, A.; Kim, J.; Traylor, A.M.; Sanders, P.W.; Balla, J.; Agarwal, A.; Curtis, L.M. Paracrine effects of mesenchymal stem cells in cisplatin-induced renal injury require heme oxygenase-1. Am. J. Physiol. Renal Physiol. 2011, 300, F254–F262. [Google Scholar] [CrossRef] [Green Version]
- Bolisetty, S.; Traylor, A.; Joseph, R.; Zarjou, A.; Agarwal, A. Proximal tubule-targeted heme oxygenase-1 in cisplatin-induced acute kidney injury. Am. J. Physiol. Renal Physiol. 2016, 310, F385–F94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nath, K.A.; Haggard, J.J.; Croatt, A.J.; Grande, J.P.; Poss, K.D.; Alam, J. The indispensability of heme oxygenase-1 in protecting against acute heme protein-induced toxicity in vivo. Am. J. Pathol. 2000, 156, 1527–1535. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Zarjou, A.; Traylor, A.M.; Bolisetty, S.; Jaimes, E.A.; Hull, T.D.; George, J.F.; Mikhail, F.M.; Agarwal, A. In vivo regulation of the heme oxygenase-1 gene in humanized transgenic mice. Kidney Int. 2012, 82, 278–291. [Google Scholar] [CrossRef] [Green Version]
- Wei, Q.; Hill, W.D.; Su, Y.; Huang, S.; Dong, Z. Heme oxygenase-1 induction contributes to renoprotection by G-CSF during rhabdomyolysis-associated acute kidney injury. Am. J. Physiol. Renal Physiol. 2011, 301, F162–F170. [Google Scholar] [CrossRef] [PubMed]
- Boddu, R.; Hull, T.D.; Bolisetty, S.; Hu, X.; Moehle, M.S.; Daher, J.P.; Kamal, A.I.; Joseph, R.; George, J.F.; Agarwal, A.; et al. Leucine-rich repeat kinase 2 deficiency is protective in rhabdomyolysis-induced kidney injury. Hum. Mol. Genet. 2015, 24, 4078–4093. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Pan, X.; Fu, H.; Zheng, Y.; Dai, Y.; Yin, Y.; Chen, Q.; Hao, Q.; Bao, D.; Hou, D. Effect of curcumin on glycerol-induced acute kidney injury in rats. Sci. Rep. 2017, 7, 10114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shah, S.V.; Walker, P.D. Evidence suggesting a role for hydroxyl radical in glycerol-induced acute renal failure. Am. J. Physiol. 1988, 255, F438–F443. [Google Scholar] [CrossRef] [PubMed]
- Paller, M.S. Hemoglobin- and myoglobin-induced acute renal failure in rats: Role of iron in nephrotoxicity. Am. J. Physiol. 1988, 255, F539–F544. [Google Scholar] [CrossRef]
- Pamplona, A.; Ferreira, A.; Balla, J.; Jeney, V.; Balla, G.; Epiphanio, S.; Chora, A.; Rodrigues, C.D.; Gregoire, I.P.; Cunha-Rodrigues, M.; et al. Heme oxygenase-1 and carbon monoxide suppress the pathogenesis of experimental cerebral malaria. Nat. Med. 2007, 13, 703–710. [Google Scholar] [CrossRef]
- Pereira, M.L.; Ortolan, L.S.; Sercundes, M.K.; Debone, D.; Murillo, O.; Lima, F.A.; Marinho, C.R.; Epiphanio, S. Association of heme oxygenase 1 with lung protection in malaria-associated ALI/ARDS. Mediat. Inflamm. 2016, 2016, 4158698. [Google Scholar] [CrossRef] [Green Version]
- Aubouy, A.; Olagnier, D.; Bertin, G.; Ezinmegnon, S.; Majorel, C.; Mimar, S.; Massougbodji, A.; Deloron, P.; Pipy, B.; Coste, A. Nrf2-driven CD36 and HO-1 gene expression in circulating monocytes correlates with favourable clinical outcome in pregnancy-associated malaria. Malar. J. 2015, 14, 358. [Google Scholar] [CrossRef] [Green Version]
- Ramos, S.; Carlos, A.R.; Sundaram, B.; Jeney, V.; Ribeiro, A.; Gozzelino, R.; Bank, C.; Gjini, E.; Braza, F.; Martins, R.; et al. Renal control of disease tolerance to malaria. Proc. Natl. Acad. Sci. USA 2019, 116, 5681–5686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muller-Eberhard, U.; Javid, J.; Liem, H.H.; Hanstein, A.; Hanna, M. Plasma concentrations of hemopexin, haptoglobin and heme in patients with various hemolytic diseases. Blood 1968, 32, 811–815. [Google Scholar] [CrossRef] [PubMed]
- Saraf, S.L.; Zhang, X.; Shah, B.; Kanias, T.; Gudehithlu, K.P.; Kittles, R.; Machado, R.F.; Arruda, J.A.; Gladwin, M.T.; Singh, A.K.; et al. Genetic variants and cell-free hemoglobin processing in sickle cell nephropathy. Haematologica 2015, 100, 1275–1284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nath, K.A.; Grande, J.P.; Haggard, J.J.; Croatt, A.J.; Katusic, Z.S.; Solovey, A.; Hebbel, R.P. Oxidative stress and induction of heme oxygenase-1 in the kidney in sickle cell disease. Am. J. Pathol. 2001, 158, 893–903. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, S.; Tan, F.; Yu, T.; Li, Y.; Adisa, O.; Mosunjac, M.; Ofori-Acquah, S.F. Global gene expression profiling of endothelium exposed to heme reveals an organ-specific induction of cytoprotective enzymes in sickle cell disease. PLoS ONE 2011, 6, e18399. [Google Scholar] [CrossRef] [Green Version]
- Camus, S.M.; De Moraes, J.A.; Bonnin, P.; Abbyad, P.; Le Jeune, S.; Lionnet, F.; Loufrani, L.; Grimaud, L.; Lambry, J.C.; Charue, D.; et al. Circulating cell membrane microparticles transfer heme to endothelial cells and trigger vasoocclusions in sickle cell disease. Blood 2015, 125, 3805–3814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belcher, J.D.; Mahaseth, H.; Welch, T.E.; Otterbein, L.E.; Hebbel, R.P.; Vercellotti, G.M. Heme oxygenase-1 is a modulator of inflammation and vaso-occlusion in transgenic sickle mice. J. Clin. Investig. 2006, 116, 808–816. [Google Scholar] [CrossRef] [Green Version]
- Nath, K.A.; Katusic, Z.S. Vasculature and kidney complications in sickle cell disease. J. Am. Soc. Nephrol. 2012, 23, 781–784. [Google Scholar] [CrossRef] [Green Version]
- Payan-Pernia, S.; Ruiz Llobet, A.; Remacha Sevilla, A.F.; Egido, J.; Ballarin Castan, J.A.; Moreno, J.A. Sickle cell nephropathy. Clinical manifestations and new mechanisms involved in kidney injury. Nefrologia 2021. [Google Scholar] [CrossRef]
- Reiter, C.D.; Wang, X.; Tanus-Santos, J.E.; Hogg, N.; Cannon, R.O., 3rd; Schechter, A.N.; Gladwin, M.T. Cell-free hemoglobin limits nitric oxide bioavailability in sickle-cell disease. Nat. Med. 2002, 8, 1383–1389. [Google Scholar] [CrossRef] [PubMed]
- Taha, H.; Skrzypek, K.; Guevara, I.; Nigisch, A.; Mustafa, S.; Grochot-Przeczek, A.; Ferdek, P.; Was, H.; Kotlinowski, J.; Kozakowska, M.; et al. Role of heme oxygenase-1 in human endothelial cells: Lesson from the promoter allelic variants. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 1634–1641. [Google Scholar] [CrossRef] [Green Version]
- Grunenwald, A.; Roumenina, L.T.; Frimat, M. Heme oxygenase 1: A defensive mediator in kidney diseases. Int. J. Mol. Sci. 2021, 22, 9. [Google Scholar] [CrossRef] [PubMed]
- Johnson, A.C.; Becker, K.; Zager, R.A. Parenteral iron formulations differentially affect MCP-1, HO-1, and NGAL gene expression and renal responses to injury. Am. J. Physiol. Renal Physiol. 2010, 299, F426–F435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leaf, D.E.; Rajapurkar, M.; Lele, S.S.; Mukhopadhyay, B.; Rawn, J.D.; Frendl, G.; Waikar, S.S. Increased plasma catalytic iron in patients may mediate acute kidney injury and death following cardiac surgery. Kidney Int. 2015, 87, 1046–1054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leaf, D.E.; Rajapurkar, M.; Lele, S.S.; Mukhopadhyay, B.; Waikar, S.S. Plasma catalytic iron, AKI, and death among critically ill patients. Clin. J. Am. Soc. Nephrol. 2014, 9, 1849–1856. [Google Scholar] [CrossRef] [PubMed]
- Hatcher, H.C.; Tesfay, L.; Torti, S.V.; Torti, F.M. Cytoprotective Effect of Ferritin H in Renal Ischemia Reperfusion Injury. PLoS ONE 2015, 10, e0138505. [Google Scholar] [CrossRef] [Green Version]
- Scindia, Y.; Dey, P.; Thirunagari, A.; Liping, H.; Rosin, D.L.; Floris, M.; Okusa, M.D.; Swaminathan, S. Hepcidin mitigates renal ischemia-reperfusion injury by modulating systemic iron homeostasis. J. Am. Soc. Nephrol. 2015, 26, 2800–2814. [Google Scholar] [CrossRef] [Green Version]
- Van Swelm, R.P.; Wetzels, J.F.; Verweij, V.G.; Laarakkers, C.M.; Pertijs, J.C.; van der Wijst, J.; Thevenod, F.; Masereeuw, R.; Swinkels, D.W. Renal handling of circulating and renal-synthesized hepcidin and its protective effects against hemoglobin-mediated kidney injury. J. Am. Soc. Nephrol. 2016, 27, 2720–2732. [Google Scholar] [CrossRef] [Green Version]

{kind=link}
{kind=link}
| Causes of Hemolytic Anemias |
|---|
| Intrinsic (intracorpuscular) A. Genetic defects
Extrinsic (extracorpuscular) A. Antibody mediated
D. Chemical agents: Arsine, Glycerol, Benzene, Sodium chlorate, Methyl chloride, etc. E. Venoms: Rattle snake, Coral snake, Brown recluse spider |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Balla, J.; Zarjou, A. Heme Burden and Ensuing Mechanisms That Protect the Kidney: Insights from Bench and Bedside. Int. J. Mol. Sci. 2021, 22, 8174. https://doi.org/10.3390/ijms22158174
Balla J, Zarjou A. Heme Burden and Ensuing Mechanisms That Protect the Kidney: Insights from Bench and Bedside. International Journal of Molecular Sciences. 2021; 22(15):8174. https://doi.org/10.3390/ijms22158174
Chicago/Turabian StyleBalla, József, and Abolfazl Zarjou. 2021. "Heme Burden and Ensuing Mechanisms That Protect the Kidney: Insights from Bench and Bedside" International Journal of Molecular Sciences 22, no. 15: 8174. https://doi.org/10.3390/ijms22158174
APA StyleBalla, J., & Zarjou, A. (2021). Heme Burden and Ensuing Mechanisms That Protect the Kidney: Insights from Bench and Bedside. International Journal of Molecular Sciences, 22(15), 8174. https://doi.org/10.3390/ijms22158174