
Gene Expression Profile of Human Mesenchymal Stromal Cells Exposed to Hypoxic and Pseudohypoxic Preconditioning—An Analysis by RNA Sequencing

 , and
, and 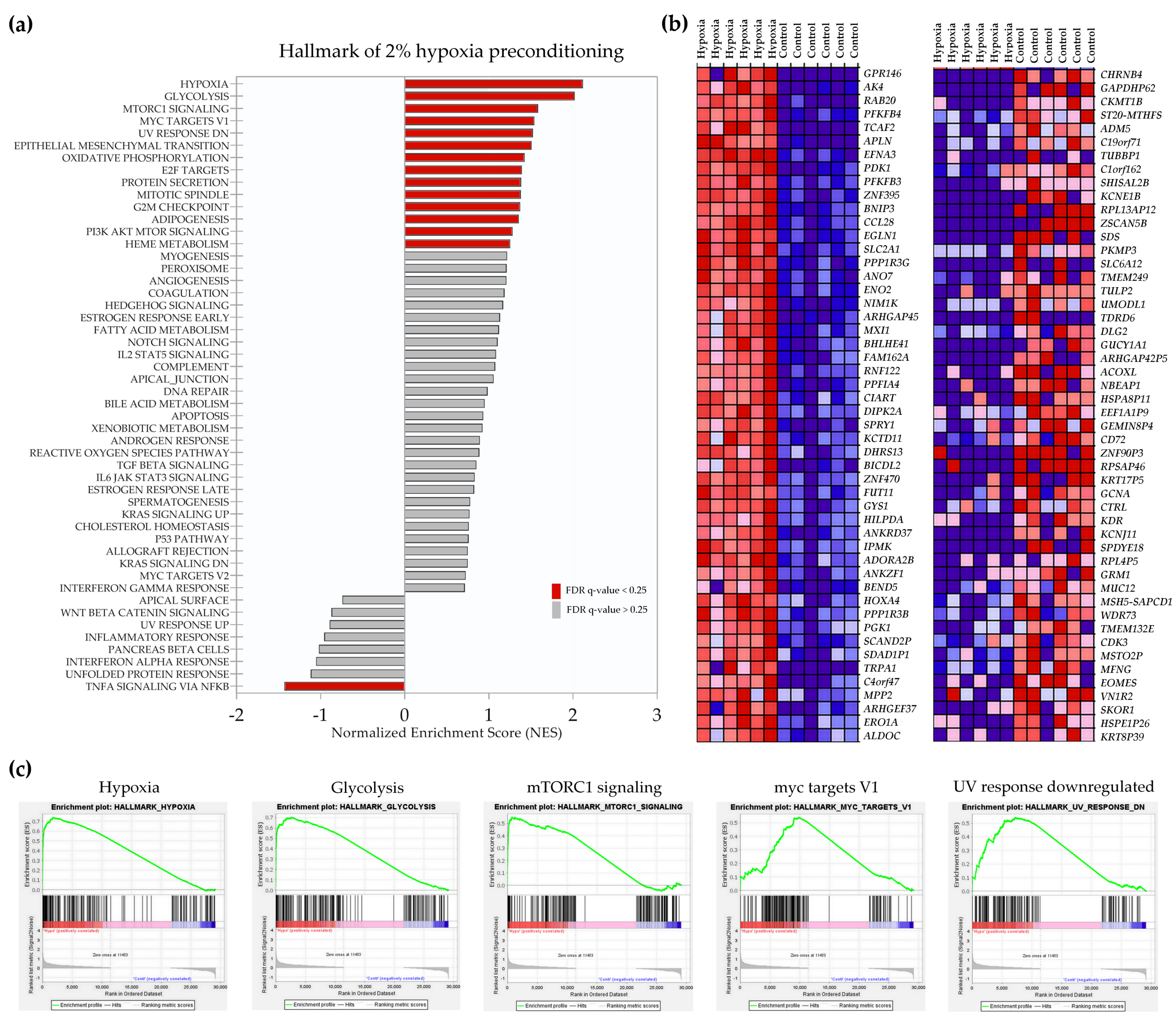{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Isolation and Characterization of Human BM-MSCs
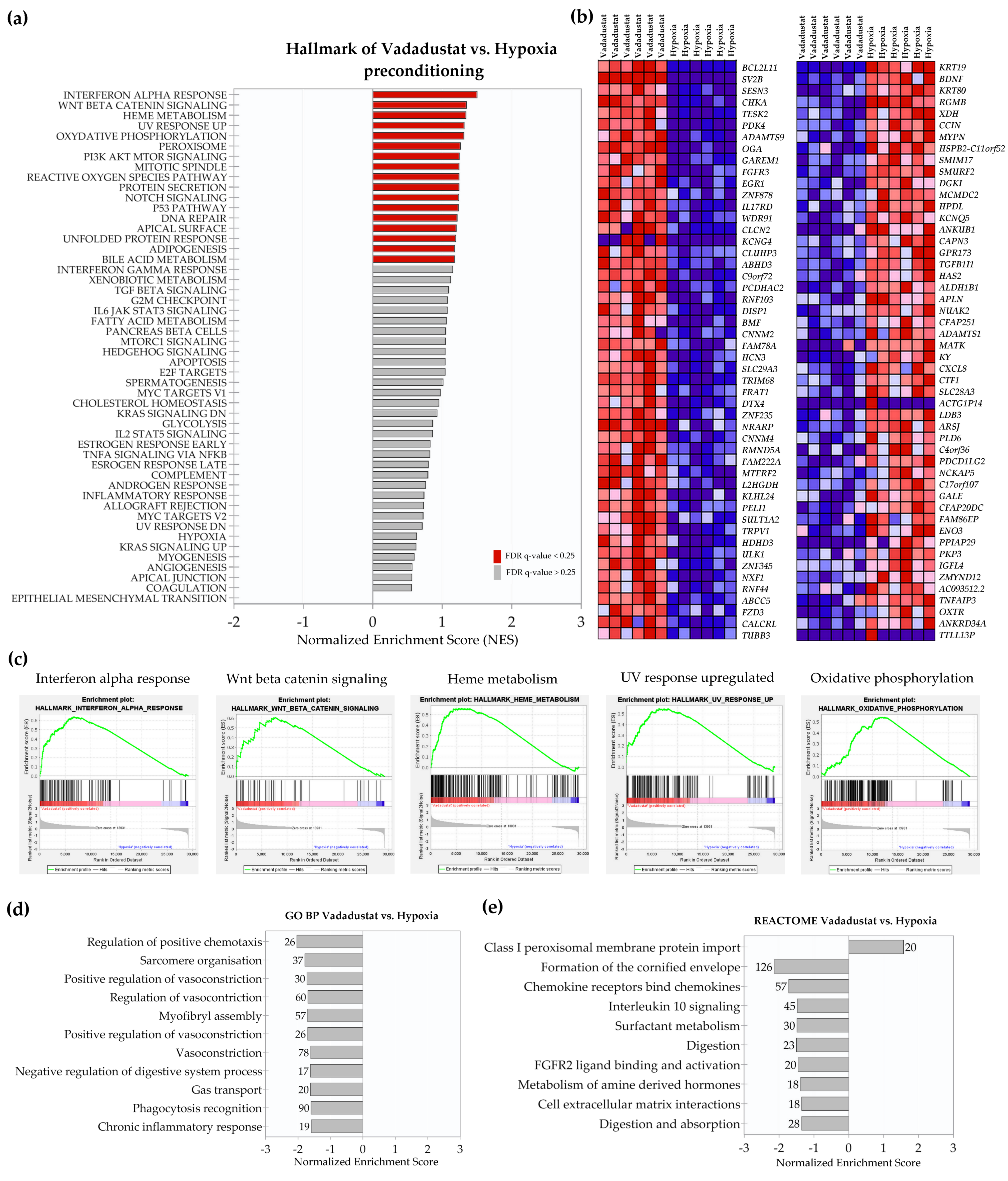
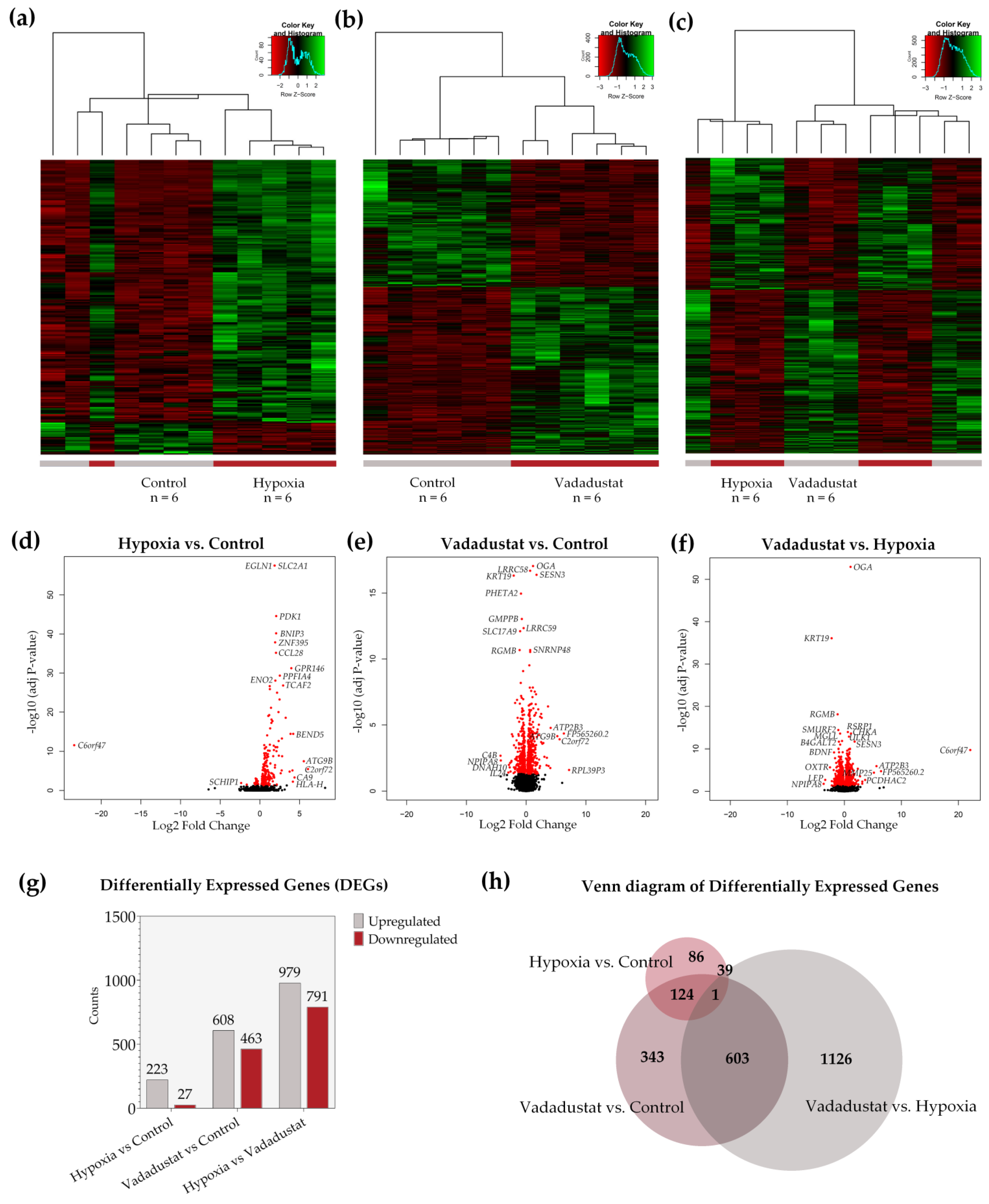
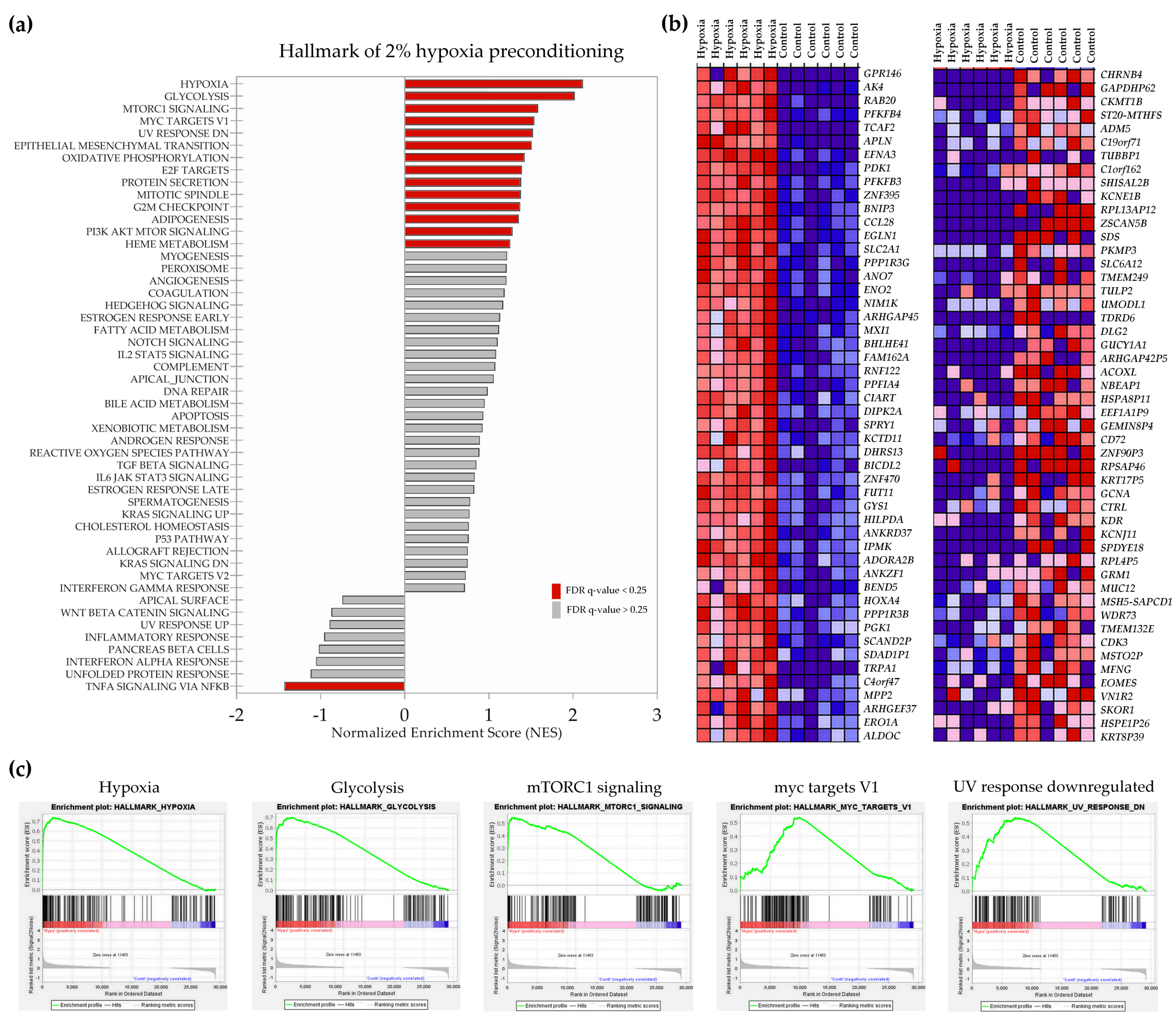
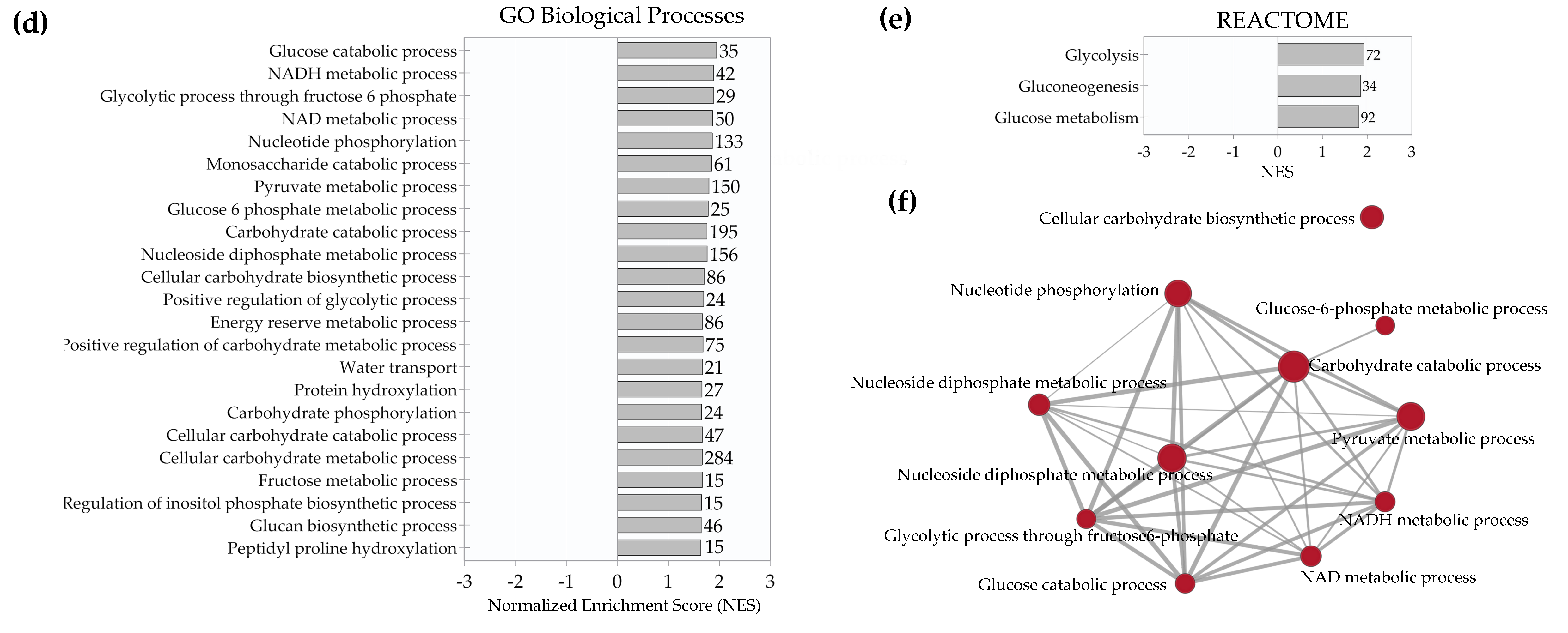
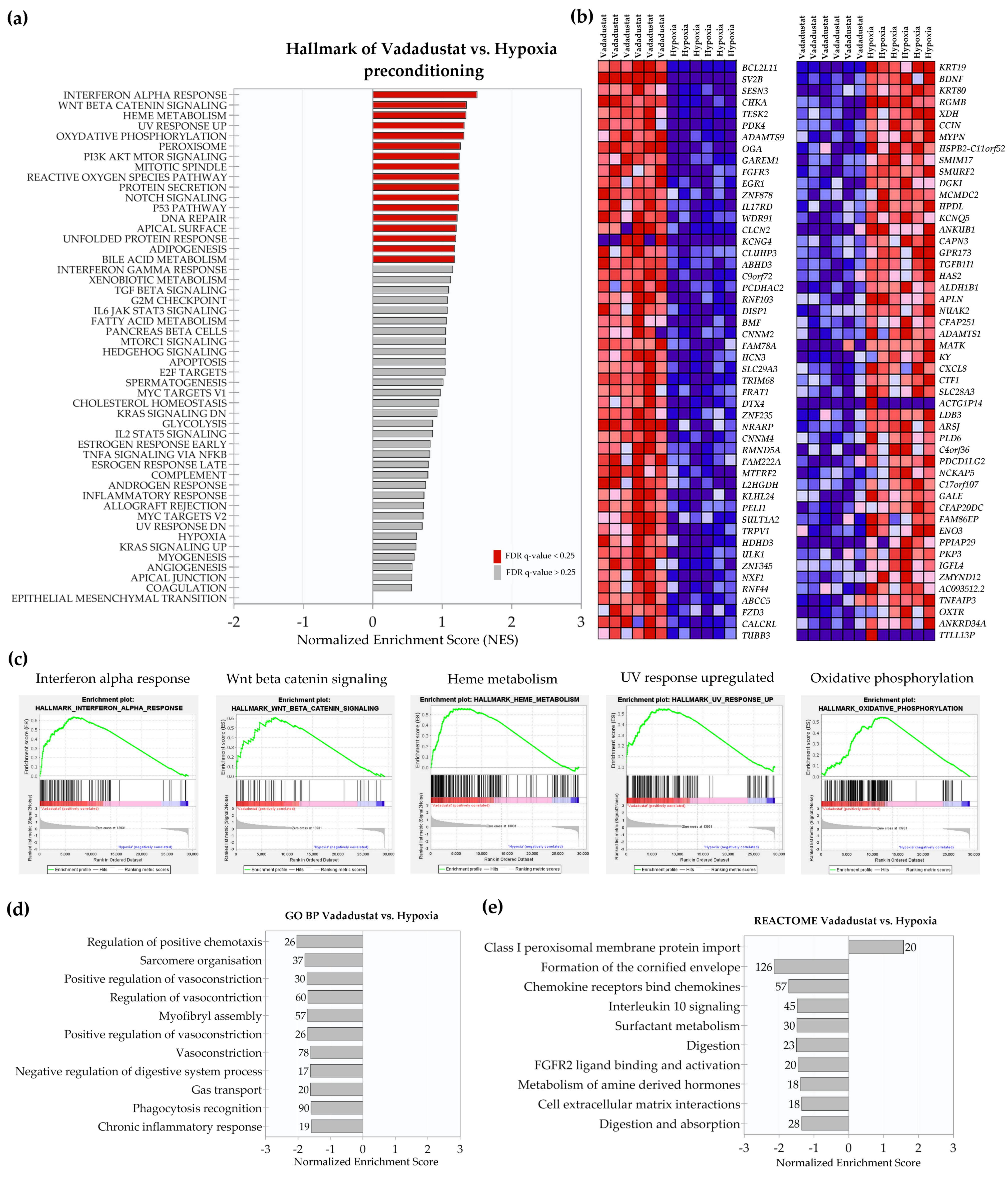
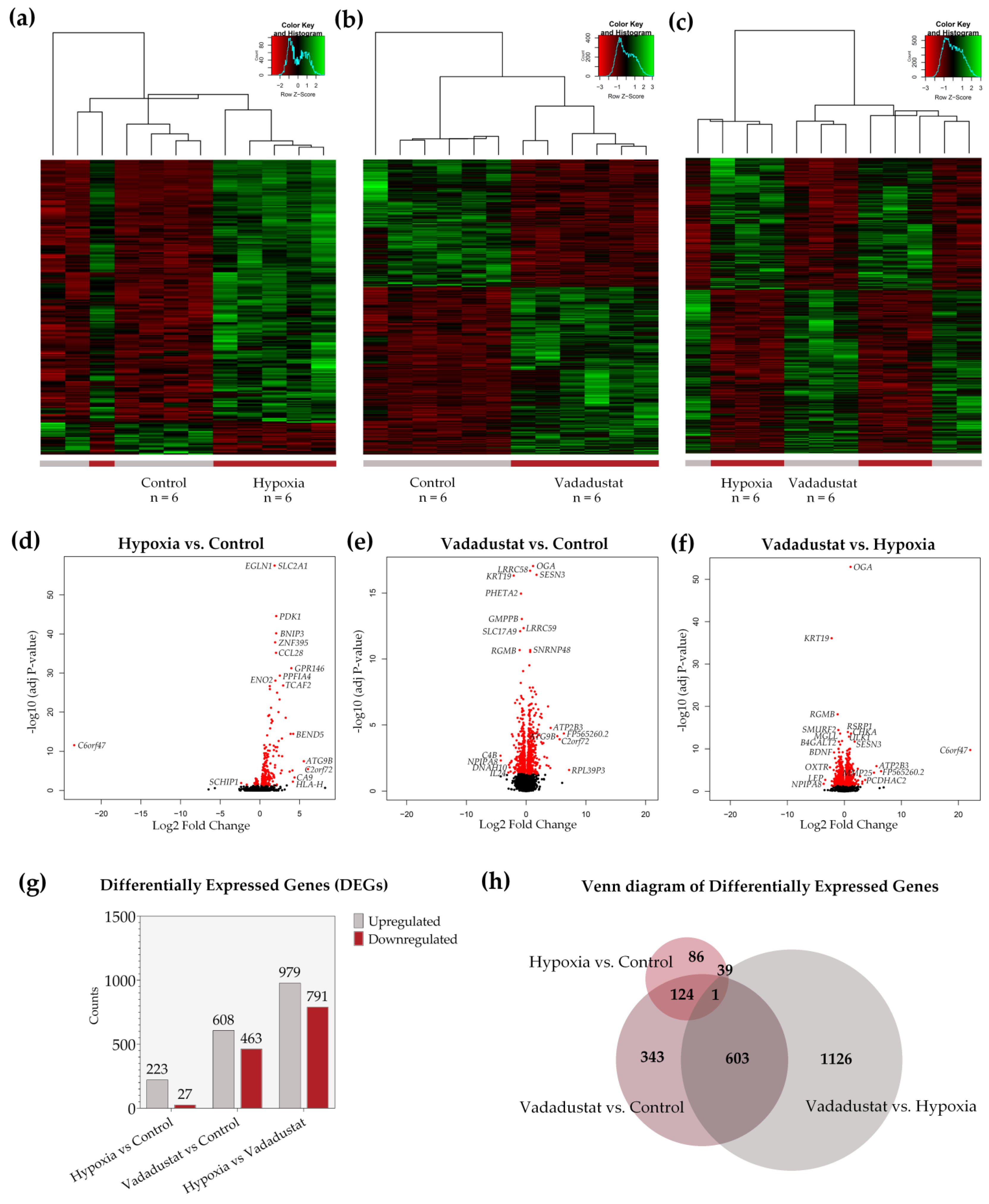
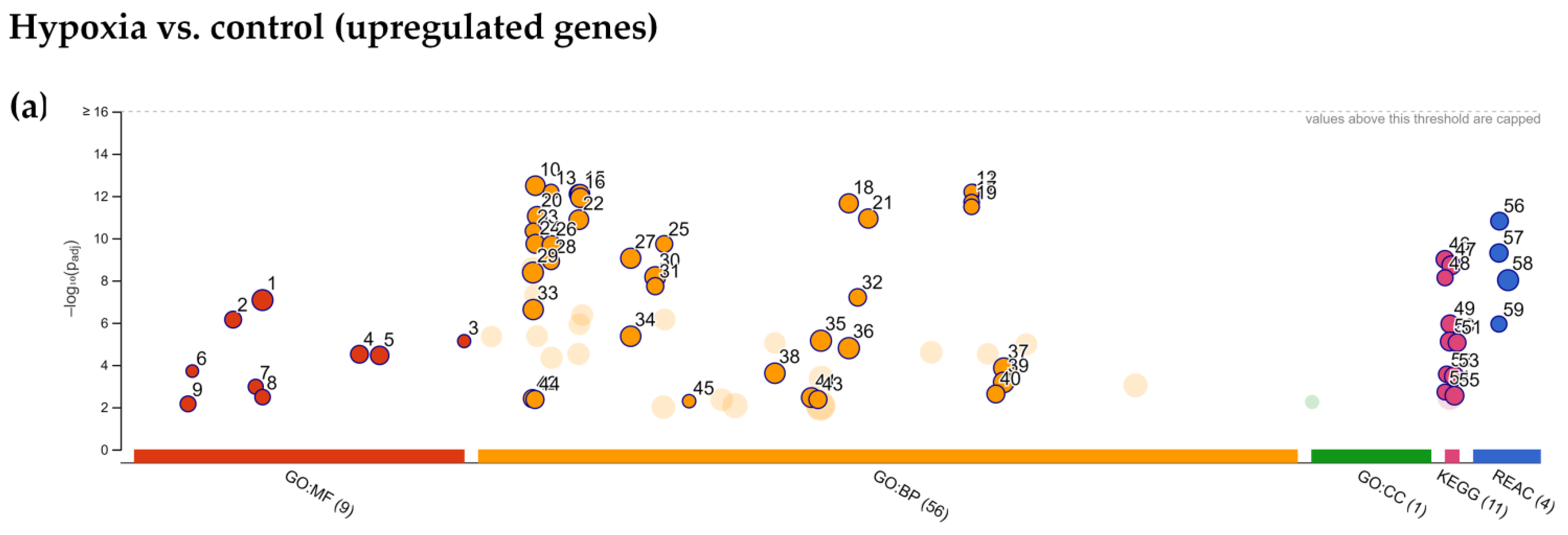
2.2. Transcriptional Signatures and Major Differences between the Response of MSCs to Two Hypoxic Preconditioning Methods
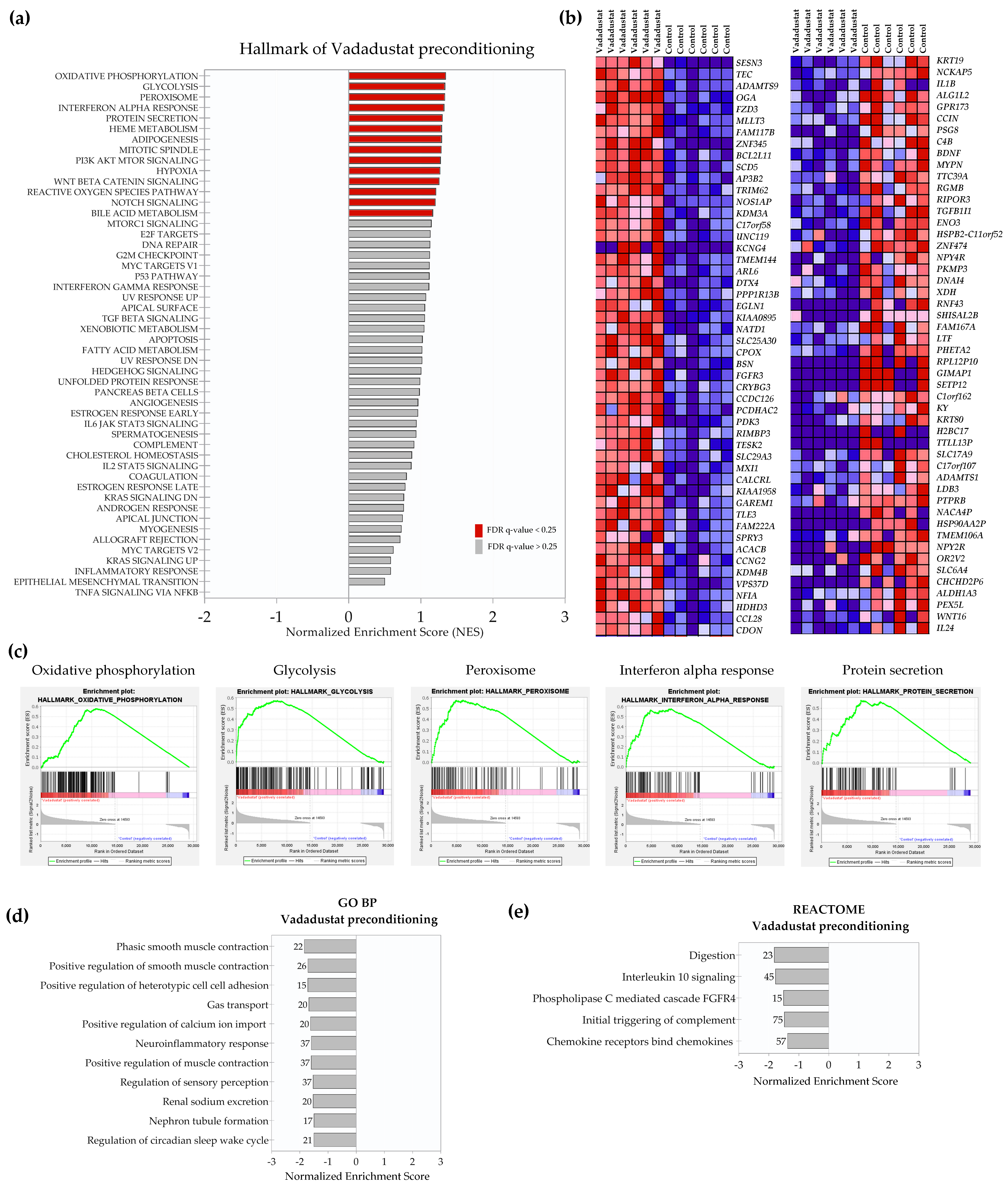
2.3. Preconditioning with Vadadustat Modulated BM-MSCs Gene Expression Profile to a Much Greater Extent Than Hypoxia
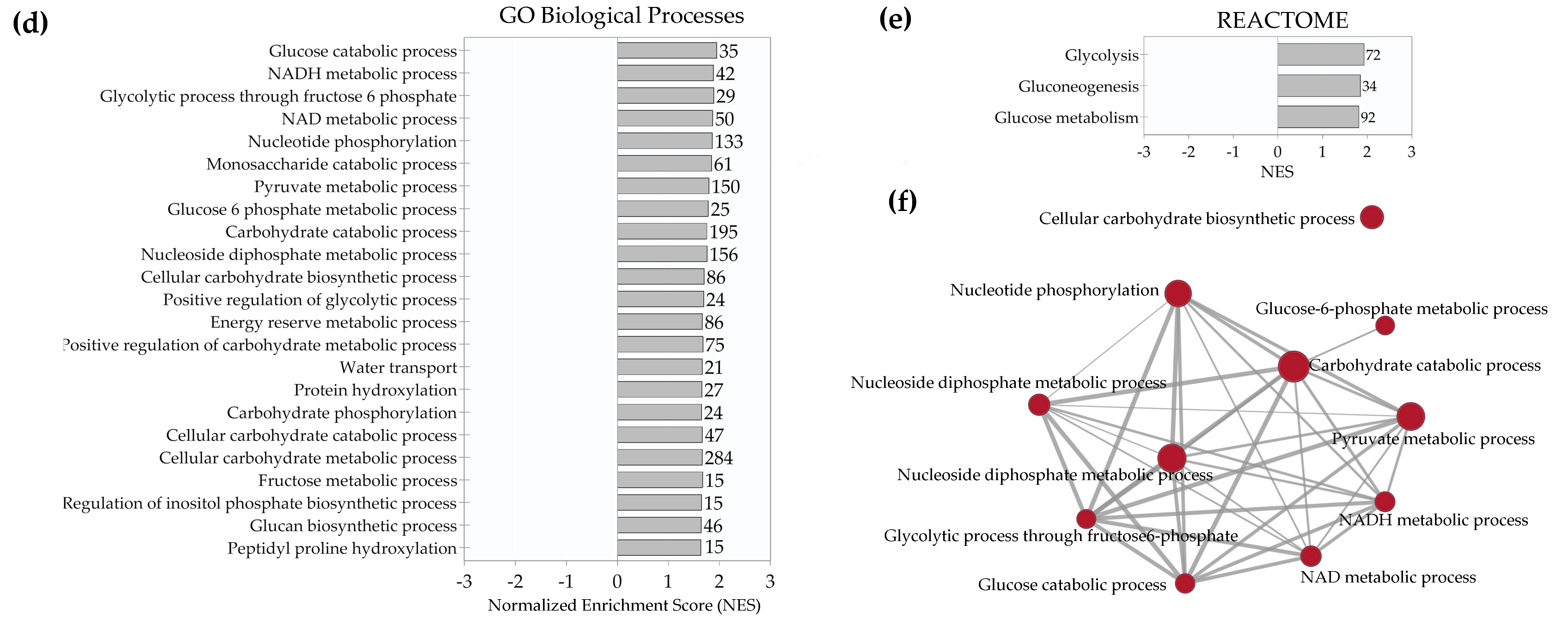
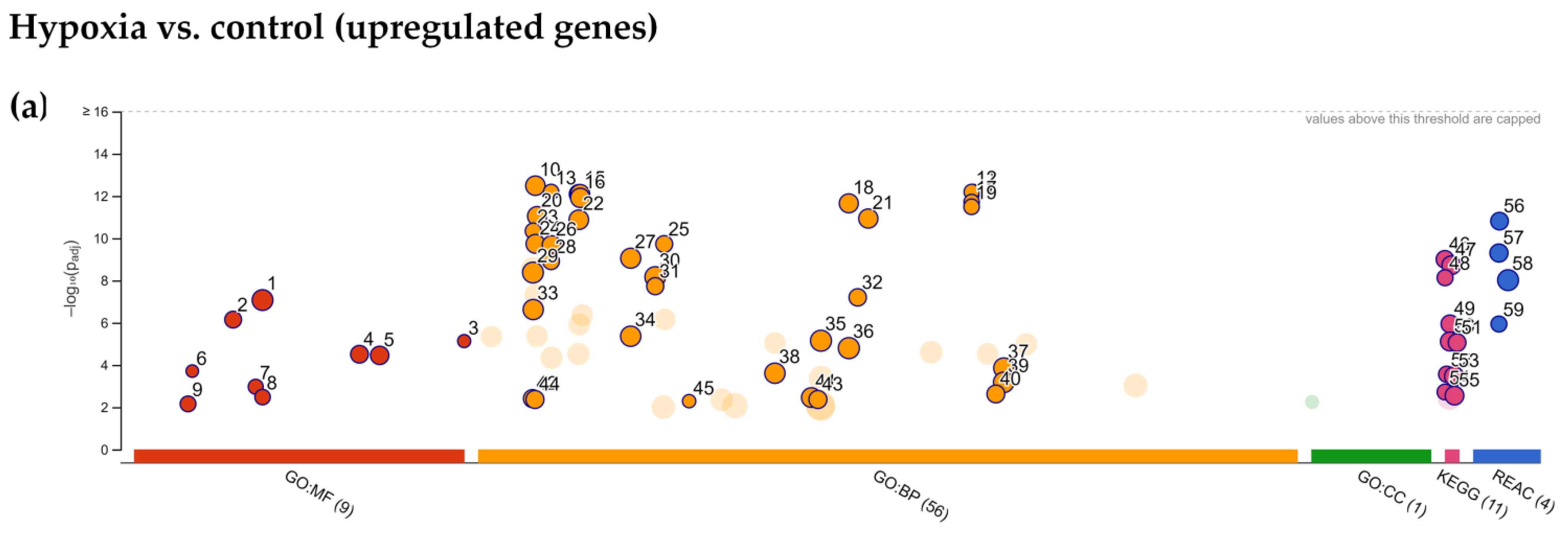
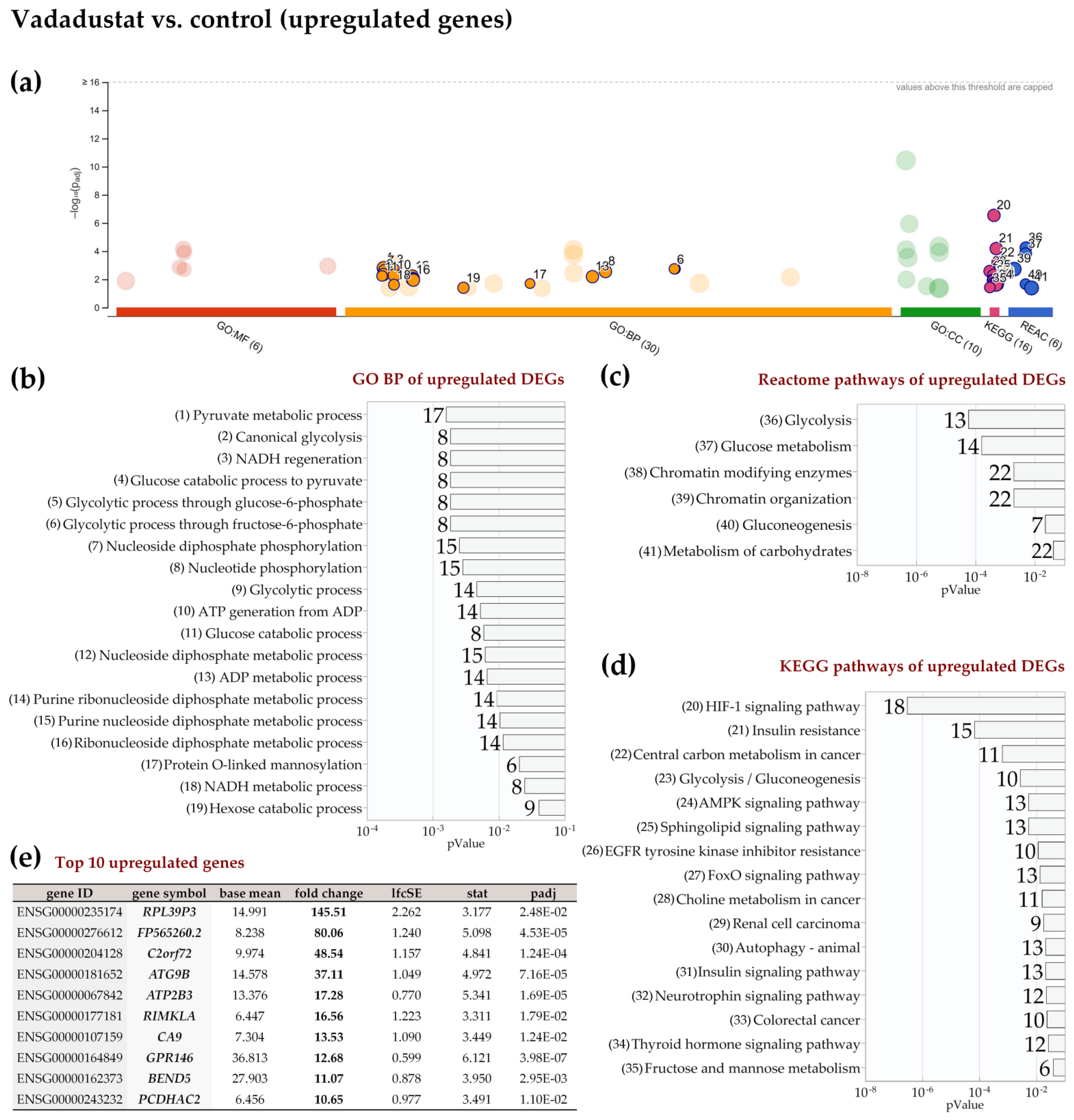
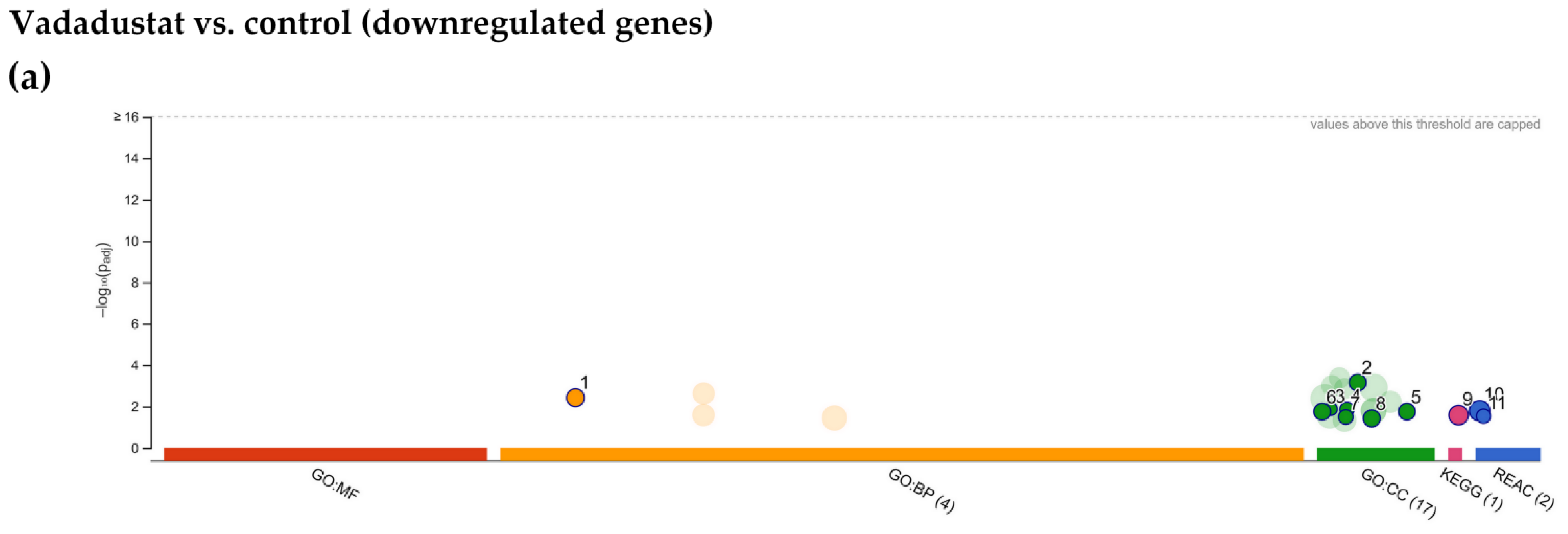
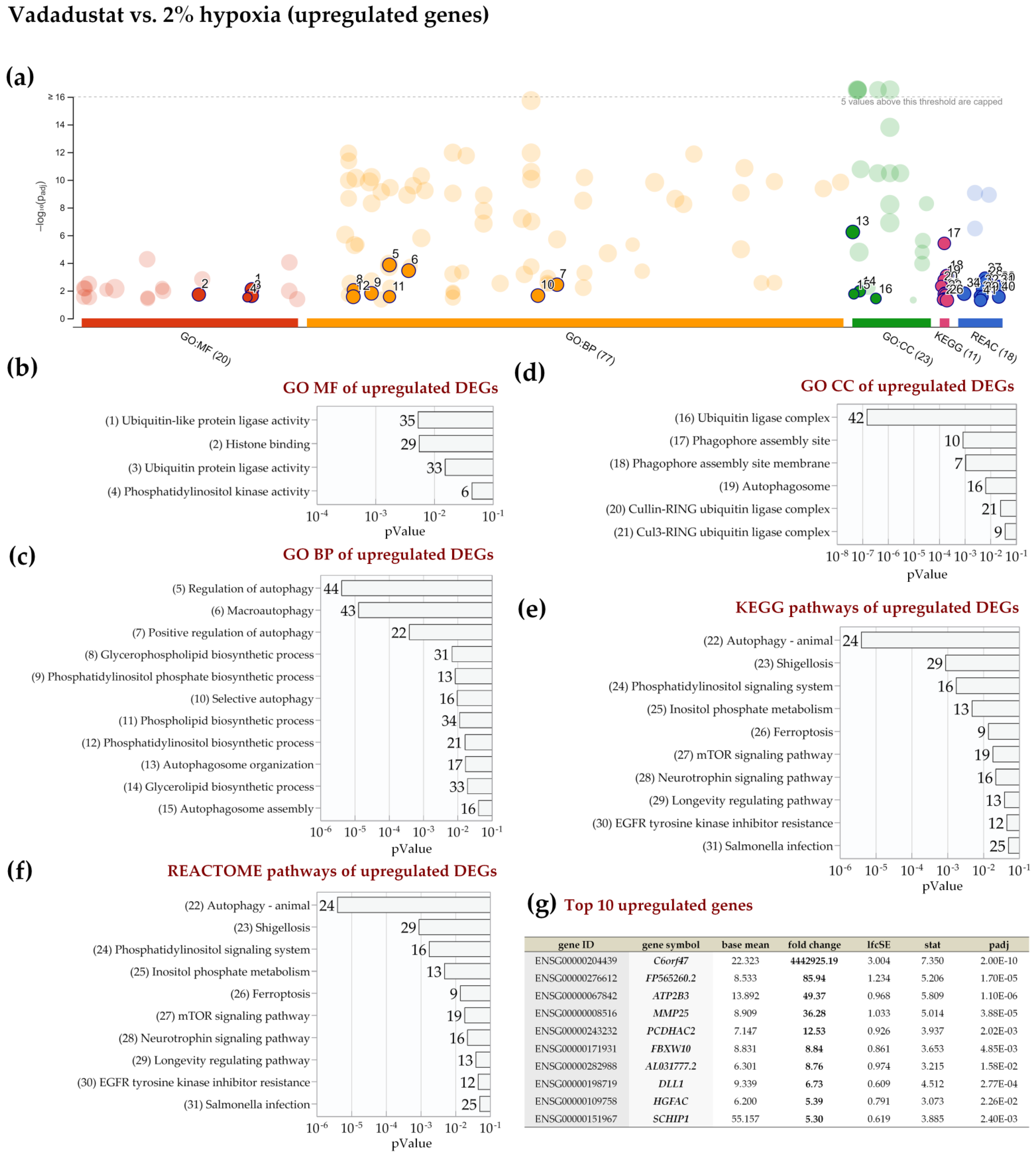
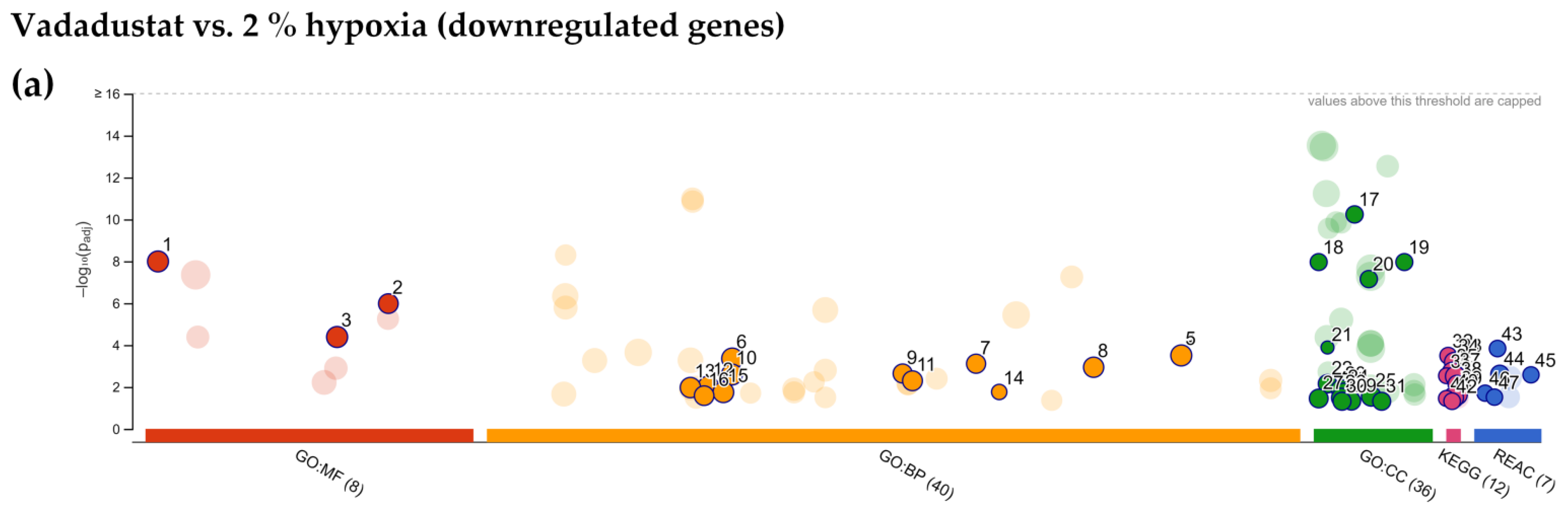
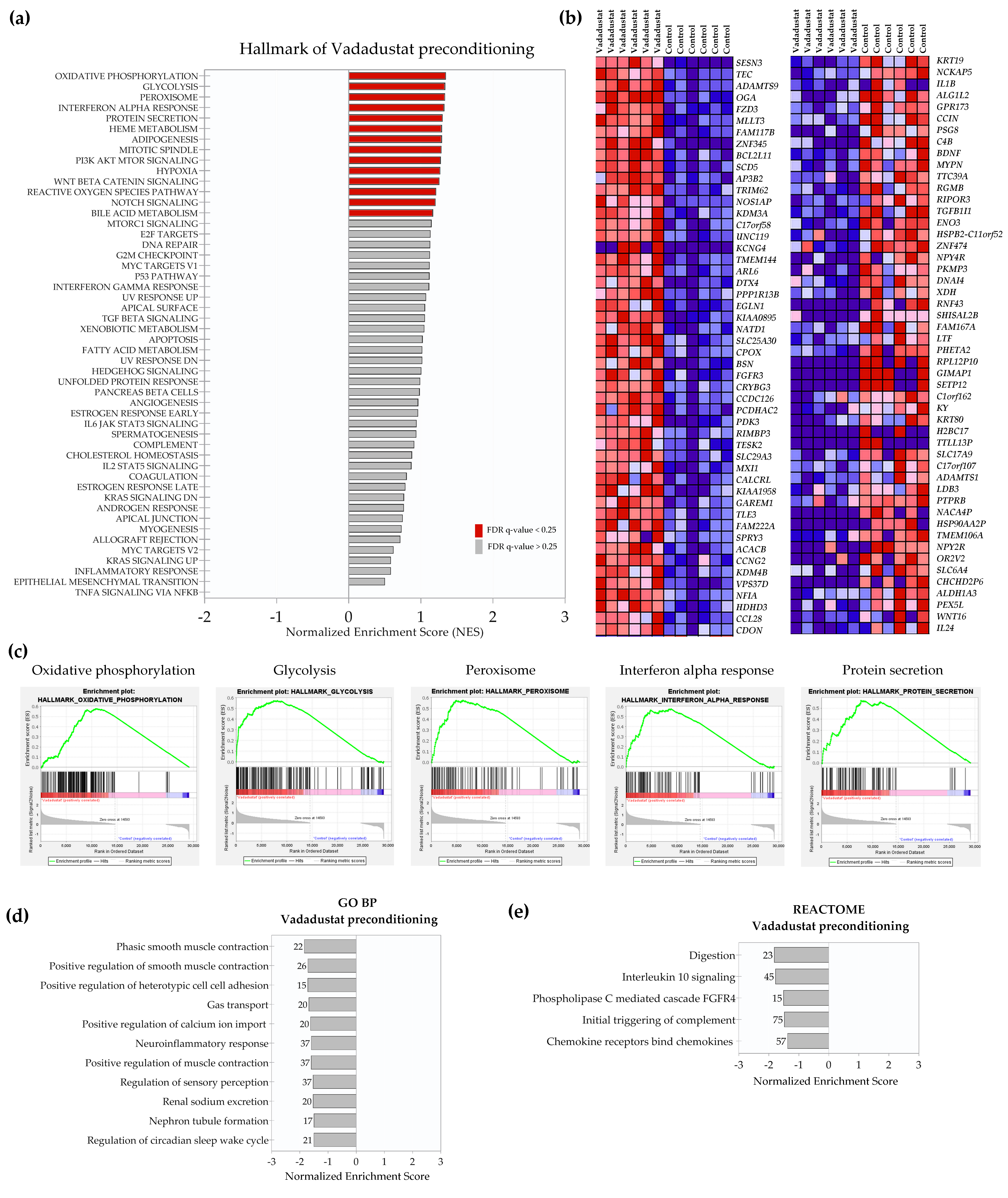
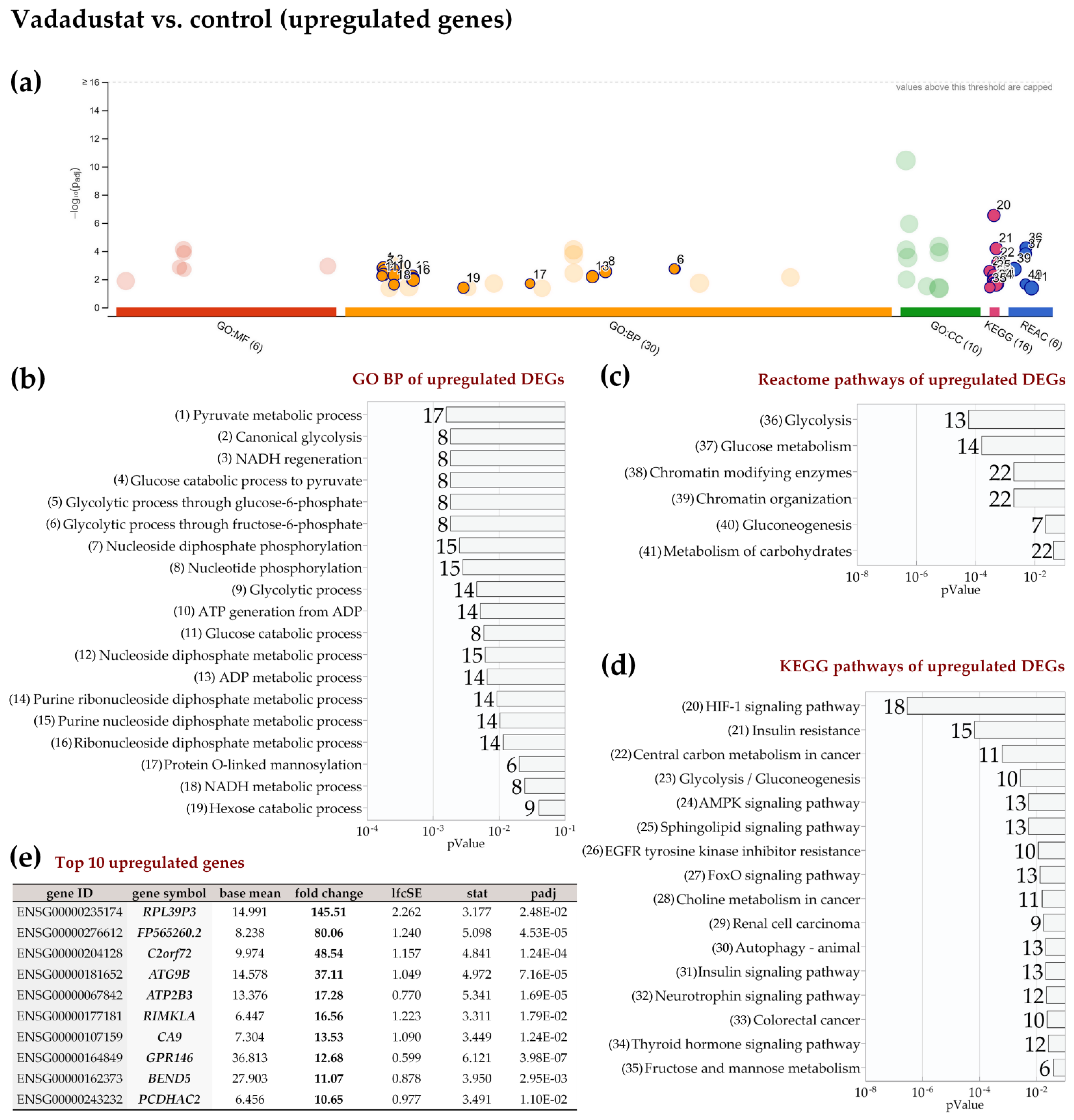
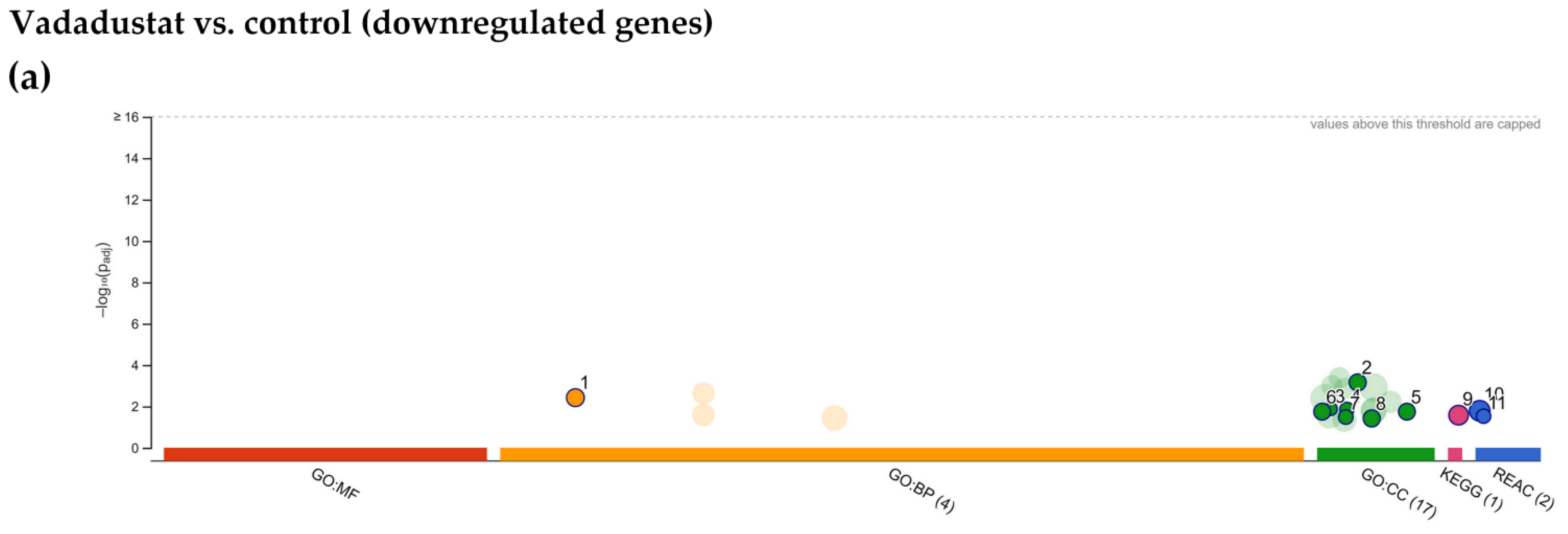
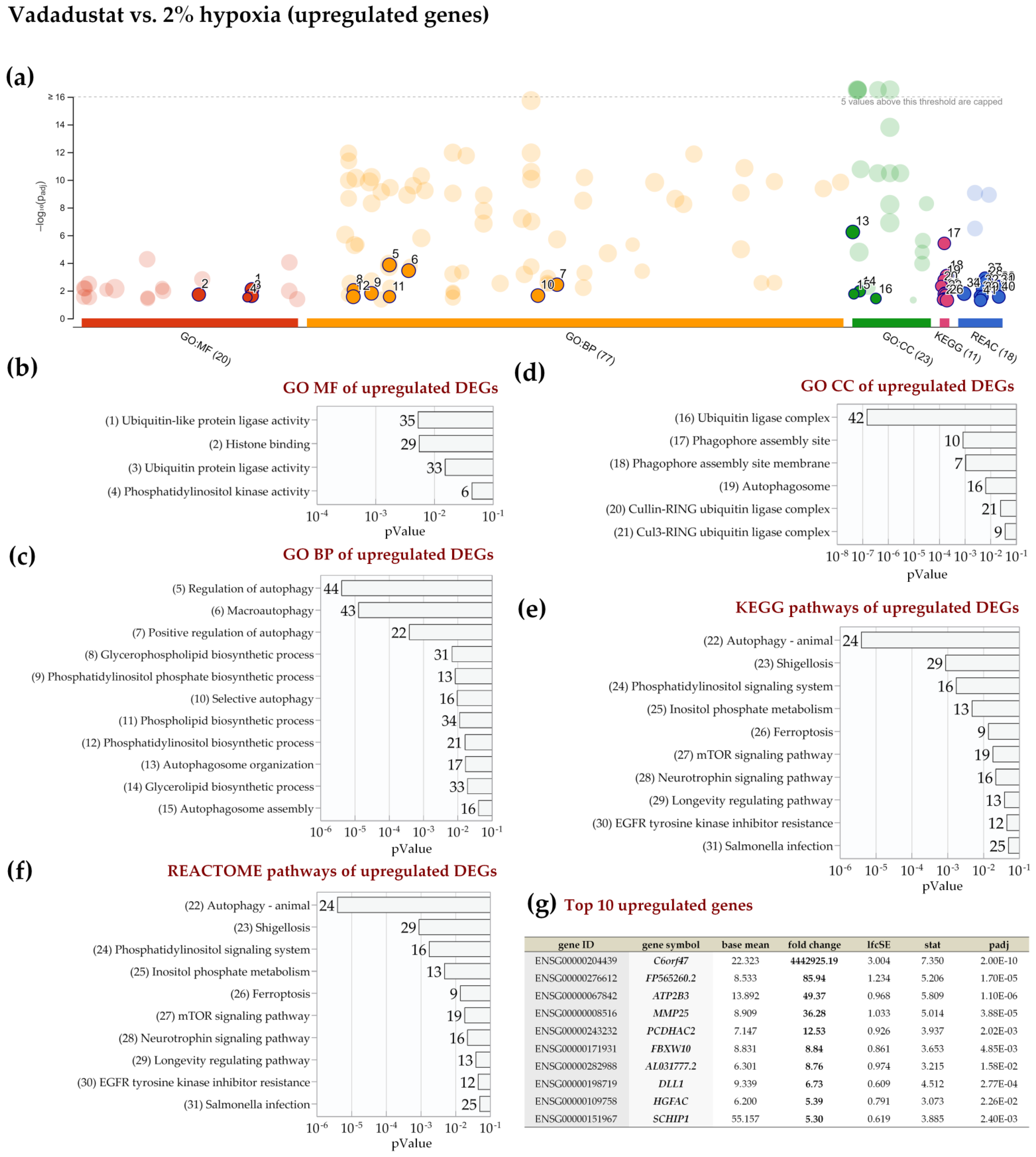
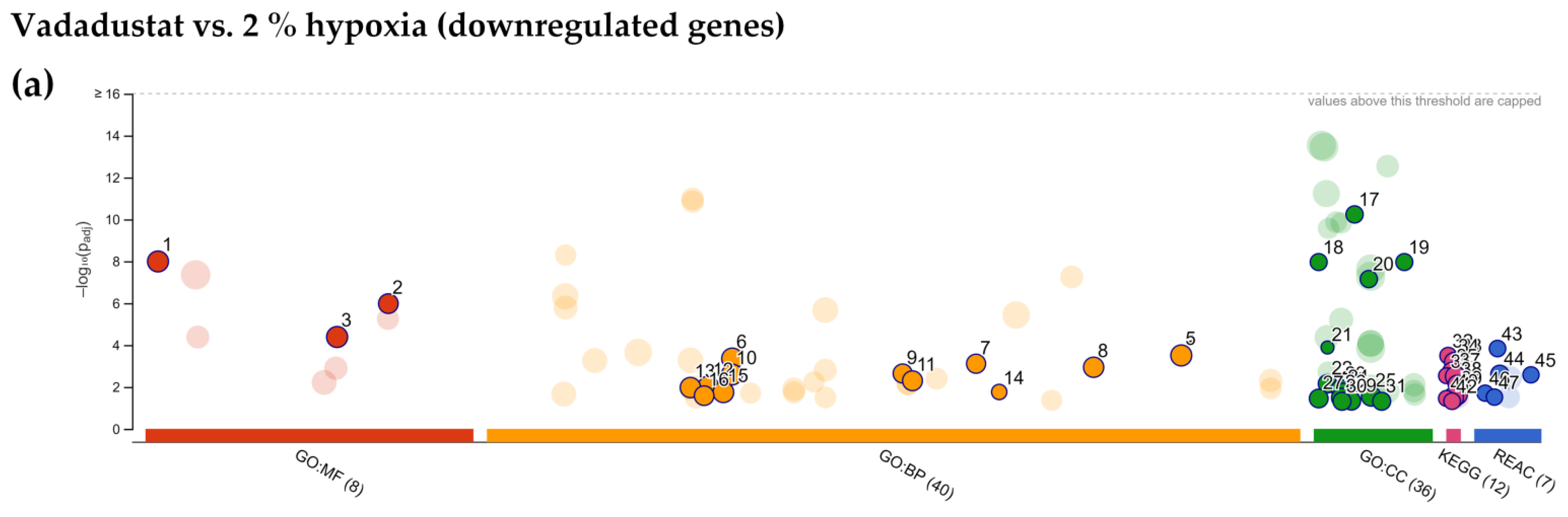
2.4. Enrichment Analysis of Biological Pathways Indicated Differences in the Effects of the Two Methods of Hypoxic Preconditioning of MSCs
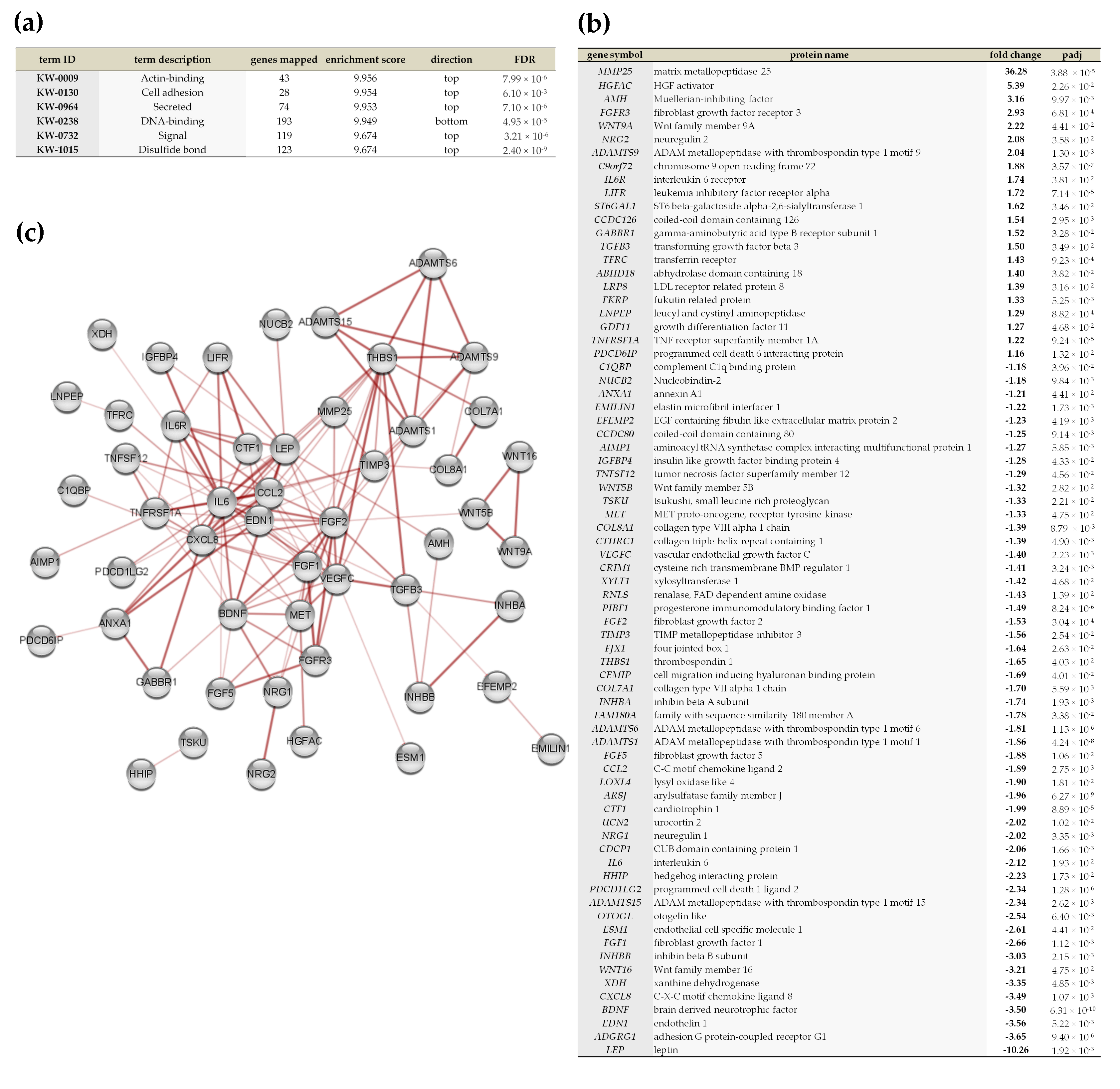
2.5. Vadadustat Preconditioning of BM-MSCs Affected the Expression of Genes Encoding Secreted Proteins
3. Discussion
4. Materials and Methods
4.1. Isolation and Culture of Human Bone Marrow-Derived Mesenchymal Stromal Cells (BM-MSCs)
4.2. Methods for the Identification of Human BM-MSCs
4.3. Two Methods Used for Hypoxic Preconditioning of BM-MSCs
4.4. RNA Isolation from BM-MSCs
4.5. Differential Gene Expression Analysis by RNA Sequencing (RNA-Seq) in BM-MSCs
4.6. Pathway Enrichment Analysis and Data Visualization
4.7. Analysis of Genes Encoding Secretory Proteins and Data Visualization
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Levy, O.; Kuai, R.; Siren, E.M.J.; Bhere, D.; Milton, Y.; Nissar, N.; De Biasio, M.; Heinelt, M.; Reeve, B.; Abdi, R.; et al. Shattering barriers toward clinically meaningful MSC therapies. Sci. Adv. 2020, 6, eaba6884. [Google Scholar] [CrossRef]
- Sagaradze, G.D.; Basalova, N.A.; Efimenko, A.Y.; Tkachuk, V.A. Mesenchymal Stromal Cells as Critical Contributors to Tissue Regeneration. Front. Cell Dev. Biol. 2020, 8, 8. [Google Scholar] [CrossRef]
- Wu, X.; Jiang, J.; Gu, Z.; Zhang, J.; Chen, Y.; Liu, X. Mesenchymal stromal cell therapies: Immunomodulatory properties and clinical progress. Stem Cell Res. Ther. 2020, 11, 345. [Google Scholar] [CrossRef] [PubMed]
- Pittenger, M.F.; Discher, D.E.; Péault, B.M.; Phinney, D.G.; Hare, J.M.; Caplan, A.I. Mesenchymal stem cell perspective: Cell biology to clinical progress. NPJ Regen. Med. 2019, 4, 22. [Google Scholar] [CrossRef] [Green Version]
- Davies, L.C.; Heldring, N.; Kadri, N.; Le Blanc, K. Mesenchymal Stromal Cell Secretion of Programmed Death-1 Ligands Regulates T Cell Mediated Immunosuppression. Stem Cells 2017, 35, 766–776. [Google Scholar] [CrossRef]
- Nasef, A.; Mathieu, N.; Chapel, A.; Frick, J.; François, S.; Mazurier, C.; Boutarfa, A.; Bouchet, S.; Gorin, N.C.; Thierry, D.; et al. Immunosuppressive effects of mesenchymal stem cells: Involvement of HLA-G. Transplantation 2007, 84, 231–237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.H.; Yoon, Y.M.; Lee, S.H. Hypoxic Preconditioning Promotes the Bioactivities of Mesenchymal Stem Cells via the HIF-1α-GRP78-Akt Axis. Int. J. Mol. Sci. 2017, 18, 1320. [Google Scholar] [CrossRef]
- Rosová, I.; Dao, M.; Capoccia, B.; Link, D.; Nolta, J.A. Hypoxic preconditioning results in increased motility and improved therapeutic potential of human mesenchymal stem cells. Stem Cells 2008, 26, 2173–2182. [Google Scholar] [CrossRef] [Green Version]
- Dos Santos, F.; Andrade, P.Z.; Boura, J.S.; Abecasis, M.M.; da Silva, C.L.; Cabral, J.M. Ex vivo expansion of human mesenchymal stem cells: A more effective cell proliferation kinetics and metabolism under hypoxia. J. Cell Physiol. 2010, 223, 27–35. [Google Scholar] [CrossRef] [PubMed]
- Beegle, J.; Lakatos, K.; Kalomoiris, S.; Stewart, H.; Isseroff, R.R.; Nolta, J.A.; Fierro, F.A. Hypoxic preconditioning of mesenchymal stromal cells induces metabolic changes, enhances survival, and promotes cell retention in vivo. Stem Cells 2015, 33, 1818–1828. [Google Scholar] [CrossRef]
- Zielniok, K.; Burdzinska, A.; Kaleta, B.; Zagozdzon, R.; Paczek, L. Vadadustat, a HIF Prolyl Hydroxylase Inhibitor, Improves Immunomodulatory Properties of Human Mesenchymal Stromal Cells. Cells 2020, 9, 2396. [Google Scholar] [CrossRef]
- Frost, J.; Ciulli, A.; Rocha, S. RNA-seq analysis of PHD and VHL inhibitors reveals differences and similarities to the hypoxia response. Wellcome Open Res. 2019, 4, 17. [Google Scholar] [CrossRef]
- Chan, M.C.; Ilott, N.E.; Schödel, J.; Sims, D.; Tumber, A.; Lippl, K.; Mole, D.R.; Pugh, C.W.; Ratcliffe, P.J.; Ponting, C.P.; et al. Tuning the Transcriptional Response to Hypoxia by Inhibiting Hypoxia-inducible Factor (HIF) Prolyl and Asparaginyl Hydroxylases. J. Biol. Chem. 2016, 291, 20661–20673. [Google Scholar] [CrossRef] [Green Version]
- Dominici, M.; Le Blanc, K.; Mueller, I.; Slaper-Cortenbach, I.; Marini, F.; Krause, D.; Deans, R.; Keating, A.; Prockop, D.; Horwitz, E. Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy 2006, 8, 315–317. [Google Scholar] [CrossRef]
- Elabd, C.; Ichim, T.E.; Miller, K.; Anneling, A.; Grinstein, V.; Vargas, V.; Silva, F.J. Comparing atmospheric and hypoxic cultured mesenchymal stem cell transcriptome: Implication for stem cell therapies targeting intervertebral discs. J. Transl. Med. 2018, 16, 222. [Google Scholar] [CrossRef]
- Mas-Bargues, C.; Sanz-Ros, J.; Román-Domínguez, A.; Inglés, M.; Gimeno-Mallench, L.; El Alami, M.; Viña-Almunia, J.; Gambini, J.; Viña, J.; Borrás, C. Relevance of Oxygen Concentration in Stem Cell Culture for Regenerative Medicine. Int. J. Mol. Sci. 2019, 20, 1195. [Google Scholar] [CrossRef] [Green Version]
- Bono, H.; Hirota, K. Meta-Analysis of Hypoxic Transcriptomes from Public Databases. Biomedicines 2020, 8, 10. [Google Scholar] [CrossRef] [Green Version]
- Zielniok, K.; Burdzinska, A.; Paczek, L. Roxadustat for Anemia in Patients with Chronic Kidney Disease. N. Engl. J. Med. 2020, 383, e3. [Google Scholar] [CrossRef] [PubMed]
- Nuschke, A.; Rodrigues, M.; Wells, A.W.; Sylakowski, K.; Wells, A. Mesenchymal stem cells/multipotent stromal cells (MSCs) are glycolytic and thus glucose is a limiting factor of in vitro models of MSC starvation. Stem Cell Res. Ther. 2016, 7, 179. [Google Scholar] [CrossRef] [Green Version]
- Estrada, J.C.; Albo, C.; Benguría, A.; Dopazo, A.; López-Romero, P.; Carrera-Quintanar, L.; Roche, E.; Clemente, E.P.; Enríquez, J.A.; Bernad, A.; et al. Culture of human mesenchymal stem cells at low oxygen tension improves growth and genetic stability by activating glycolysis. Cell Death Differ. 2012, 19, 743–755. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ciria, M.; García, N.A.; Ontoria-Oviedo, I.; González-King, H.; Carrero, R.; De La Pompa, J.L.; Montero, J.A.; Sepúlveda, P. Mesenchymal Stem Cell Migration and Proliferation Are Mediated by Hypoxia-Inducible Factor-1α Upstream of Notch and SUMO Pathways. Stem Cells Dev. 2017, 26, 973–985. [Google Scholar] [CrossRef] [PubMed]
- Contreras-Lopez, R.; Elizondo-Vega, R.; Paredes, M.J.; Luque-Campos, N.; Torres, M.J.; Tejedor, G.; Vega-Letter, A.M.; Figueroa-Valdes, A.; Pradenas, C.; Oyarce, K.; et al. HIF1alpha-dependent metabolic reprogramming governs mesenchymal stem/stromal cell immunoregulatory functions. FASEB J. 2020, 34, 8250–8264. [Google Scholar] [CrossRef] [Green Version]
- Ishiuchi, N.; Nakashima, A.; Doi, S.; Yoshida, K.; Maeda, S.; Kanai, R.; Yamada, Y.; Ike, T.; Doi, T.; Kato, Y.; et al. Hypoxia-preconditioned mesenchymal stem cells prevent renal fibrosis and inflammation in ischemia-reperfusion rats. Stem Cell Res. Ther. 2020, 11, 130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lan, Y.W.; Choo, K.B.; Chen, C.M.; Hung, T.H.; Chen, Y.B.; Hsieh, C.H.; Kuo, H.P.; Chong, K.Y. Hypoxia-preconditioned mesenchymal stem cells attenuate bleomycin-induced pulmonary fibrosis. Stem Cell Res. Ther. 2015, 6, 97. [Google Scholar] [CrossRef] [Green Version]
- Liu, W.; Li, L.; Rong, Y.; Qian, D.; Chen, J.; Zhou, Z.; Luo, Y.; Jiang, D.; Cheng, L.; Zhao, S.; et al. Hypoxic mesenchymal stem cell-derived exosomes promote bone fracture healing by the transfer of miR-126. Acta Biomater. 2020, 103, 196–212. [Google Scholar] [CrossRef]
- Zhang, W.; Liu, L.; Huo, Y.; Yang, Y.; Wang, Y. Hypoxia-pretreated human MSCs attenuate acute kidney injury through enhanced angiogenic and antioxidative capacities. Biomed Res. Int. 2014, 2014, 462472. [Google Scholar] [CrossRef]
- Gupta, N.; Nizet, V. Stabilization of Hypoxia-Inducible Factor-1 Alpha Augments the Therapeutic Capacity of Bone Marrow-Derived Mesenchymal Stem Cells in Experimental Pneumonia. Front. Med. 2018, 5, 131. [Google Scholar] [CrossRef] [Green Version]
- Palomäki, S.; Pietilä, M.; Laitinen, S.; Pesälä, J.; Sormunen, R.; Lehenkari, P.; Koivunen, P. HIF-1α is upregulated in human mesenchymal stem cells. Stem Cells 2013, 31, 1902–1909. [Google Scholar] [CrossRef]
- Najafi, R.; Sharifi, A.M. Deferoxamine preconditioning potentiates mesenchymal stem cell homing in vitro and in streptozotocin-diabetic rats. Expert Opin. Biol. Ther. 2013, 13, 959–972. [Google Scholar] [CrossRef]
- Elkington, P.T.; O’Kane, C.M.; Friedland, J.S. The paradox of matrix metalloproteinases in infectious disease. Clin. Exp. Immunol. 2005, 142, 12–20. [Google Scholar] [CrossRef]
- Lambrecht, B.N.; Vanderkerken, M.; Hammad, H. The emerging role of ADAM metalloproteinases in immunity. Nat. Rev. Immunol. 2018, 18, 745–758. [Google Scholar] [CrossRef]
- Soria-Valles, C.; Gutiérrez-Fernández, A.; Osorio, F.G.; Carrero, D.; Ferrando, A.A.; Colado, E.; Fernández-García, M.S.; Bonzon-Kulichenko, E.; Vázquez, J.; Fueyo, A.; et al. MMP-25 Metalloprotease Regulates Innate Immune Response through NF-κB Signaling. J. Immunol. 2016, 197, 296–302. [Google Scholar] [CrossRef]
- Kataoka, H.; Kawaguchi, M. Hepatocyte growth factor activator (HGFA): Pathophysiological functions in vivo. FEBS J. 2010, 277, 2230–2237. [Google Scholar] [CrossRef]
- Cummins, E.P.; Taylor, C.T. Hypoxia-responsive transcription factors. Pflügers Arch. 2005, 450, 363–371. [Google Scholar] [CrossRef] [PubMed]
- Hirota, K. HIF-α Prolyl Hydroxylase Inhibitors and Their Implications for Biomedicine: A Comprehensive Review. Biomedicines 2021, 9, 468. [Google Scholar] [CrossRef]
- Furlan, D.; Sahnane, N.; Carnevali, I.; Cerutti, R.; Uccella, S.; Bertolini, V.; Chiaravalli, A.M.; Capella, C. Up-regulation and stabilization of HIF-1alpha in colorectal carcinomas. Surg. Oncol. 2007, 16 (Suppl. 1), S25–S27. [Google Scholar] [CrossRef]
- Gudas, L.J.; Fu, L.; Minton, D.R.; Mongan, N.P.; Nanus, D.M. The role of HIF1α in renal cell carcinoma tumorigenesis. J. Mol. Med. 2014, 92, 825–836. [Google Scholar] [CrossRef] [PubMed]
- Burdzinska, A.; Dybowski, B.; Zarychta-Wisniewska, W.; Kulesza, A.; Butrym, M.; Zagozdzon, R.; Graczyk-Jarzynka, A.; Radziszewski, P.; Gajewski, Z.; Paczek, L. Intraurethral co-transplantation of bone marrow mesenchymal stem cells and muscle-derived cells improves the urethral closure. Stem Cell Res. Ther. 2018, 9, 239. [Google Scholar] [CrossRef] [Green Version]
- BBDUK. Available online: https://github.com/BioInfoTools/BBMap/blob/master/sh/bbduk2.sh (accessed on 8 May 2020).
- Patro, R.; Duggal, G.; Love, M.I.; Irizarry, R.A.; Kingsford, C. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 2017, 14, 417–419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.; Paggi, J.M.; Park, C.; Bennett, C.; Salzberg, S.L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 2019, 37, 907–915. [Google Scholar] [CrossRef] [PubMed]
- Soneson, C.; Love, M.I.; Robinson, M.D. Differential analyses for RNA-seq: Transcript-level estimates improve gene-level inferences. F1000Reserch 2015, 4, 1521. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reimand, J.; Isserlin, R.; Voisin, V.; Kucera, M.; Tannus-Lopes, C.; Rostamianfar, A.; Wadi, L.; Meyer, M.; Wong, J.; Xu, C.; et al. Pathway enrichment analysis and visualization of omics data using g:Profiler, GSEA, Cytoscape and EnrichmentMap. Nat. Protoc. 2019, 14, 482–517. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [Green Version]
- Raudvere, U.; Kolberg, L.; Kuzmin, I.; Arak, T.; Adler, P.; Peterson, H.; Vilo, J. g:Profiler: A web server for functional enrichment analysis and conversions of gene lists (2019 update). Nucleic Acids Res. 2019, 47, W191–W198. [Google Scholar] [CrossRef] [Green Version]
- Hong, G.; Zhang, W.; Li, H.; Shen, X.; Guo, Z. Separate enrichment analysis of pathways for up- and downregulated genes. J. R. Soc. Interface 2014, 11, 20130950. [Google Scholar] [CrossRef] [Green Version]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef] [Green Version]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res 2003, 13, 2498–2504. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zielniok, K.; Burdzinska, A.; Murcia Pienkowski, V.; Koppolu, A.; Rydzanicz, M.; Zagozdzon, R.; Paczek, L. Gene Expression Profile of Human Mesenchymal Stromal Cells Exposed to Hypoxic and Pseudohypoxic Preconditioning—An Analysis by RNA Sequencing. Int. J. Mol. Sci. 2021, 22, 8160. https://doi.org/10.3390/ijms22158160
Zielniok K, Burdzinska A, Murcia Pienkowski V, Koppolu A, Rydzanicz M, Zagozdzon R, Paczek L. Gene Expression Profile of Human Mesenchymal Stromal Cells Exposed to Hypoxic and Pseudohypoxic Preconditioning—An Analysis by RNA Sequencing. International Journal of Molecular Sciences. 2021; 22(15):8160. https://doi.org/10.3390/ijms22158160
Chicago/Turabian StyleZielniok, Katarzyna, Anna Burdzinska, Victor Murcia Pienkowski, Agnieszka Koppolu, Malgorzata Rydzanicz, Radoslaw Zagozdzon, and Leszek Paczek. 2021. "Gene Expression Profile of Human Mesenchymal Stromal Cells Exposed to Hypoxic and Pseudohypoxic Preconditioning—An Analysis by RNA Sequencing" International Journal of Molecular Sciences 22, no. 15: 8160. https://doi.org/10.3390/ijms22158160
APA StyleZielniok, K., Burdzinska, A., Murcia Pienkowski, V., Koppolu, A., Rydzanicz, M., Zagozdzon, R., & Paczek, L. (2021). Gene Expression Profile of Human Mesenchymal Stromal Cells Exposed to Hypoxic and Pseudohypoxic Preconditioning—An Analysis by RNA Sequencing. International Journal of Molecular Sciences, 22(15), 8160. https://doi.org/10.3390/ijms22158160