
Transcriptomic Analysis of Salt-Stress-Responsive Genes in Barley Roots and Leaves

 , ,
, ,
Abstract
:1. Introduction
2. Results
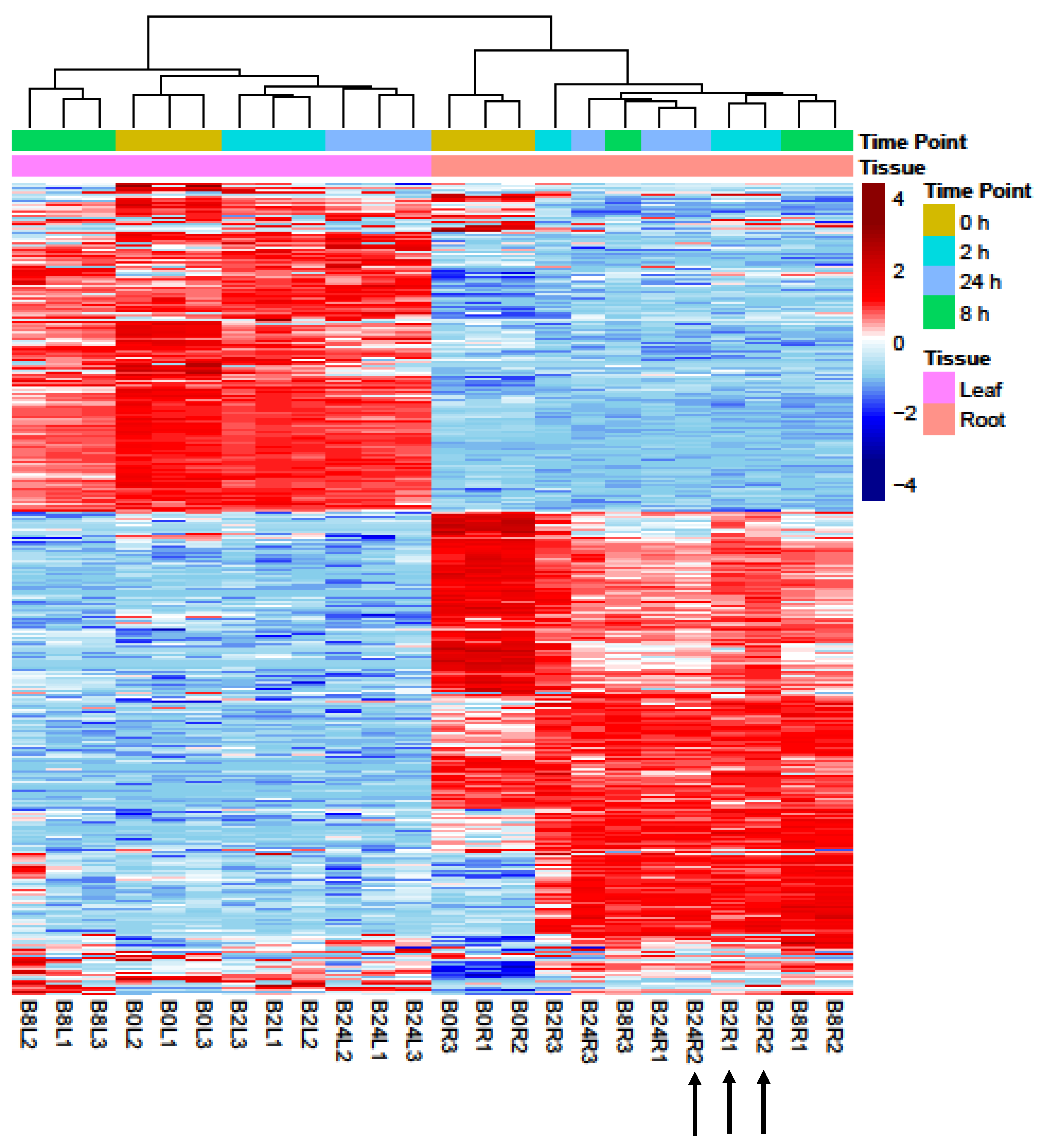
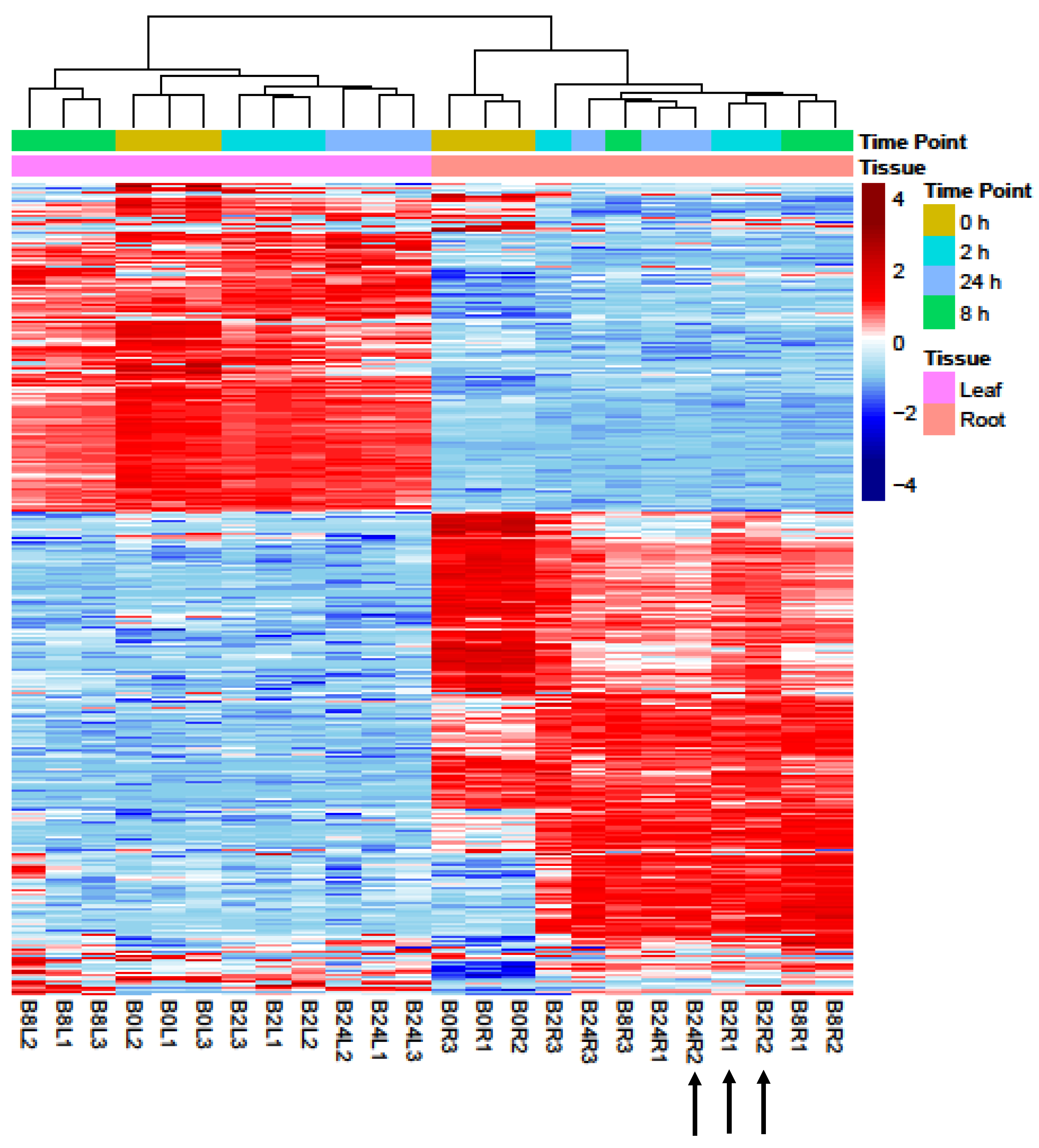
2.1. Analyses of RNA-Seq Datasets
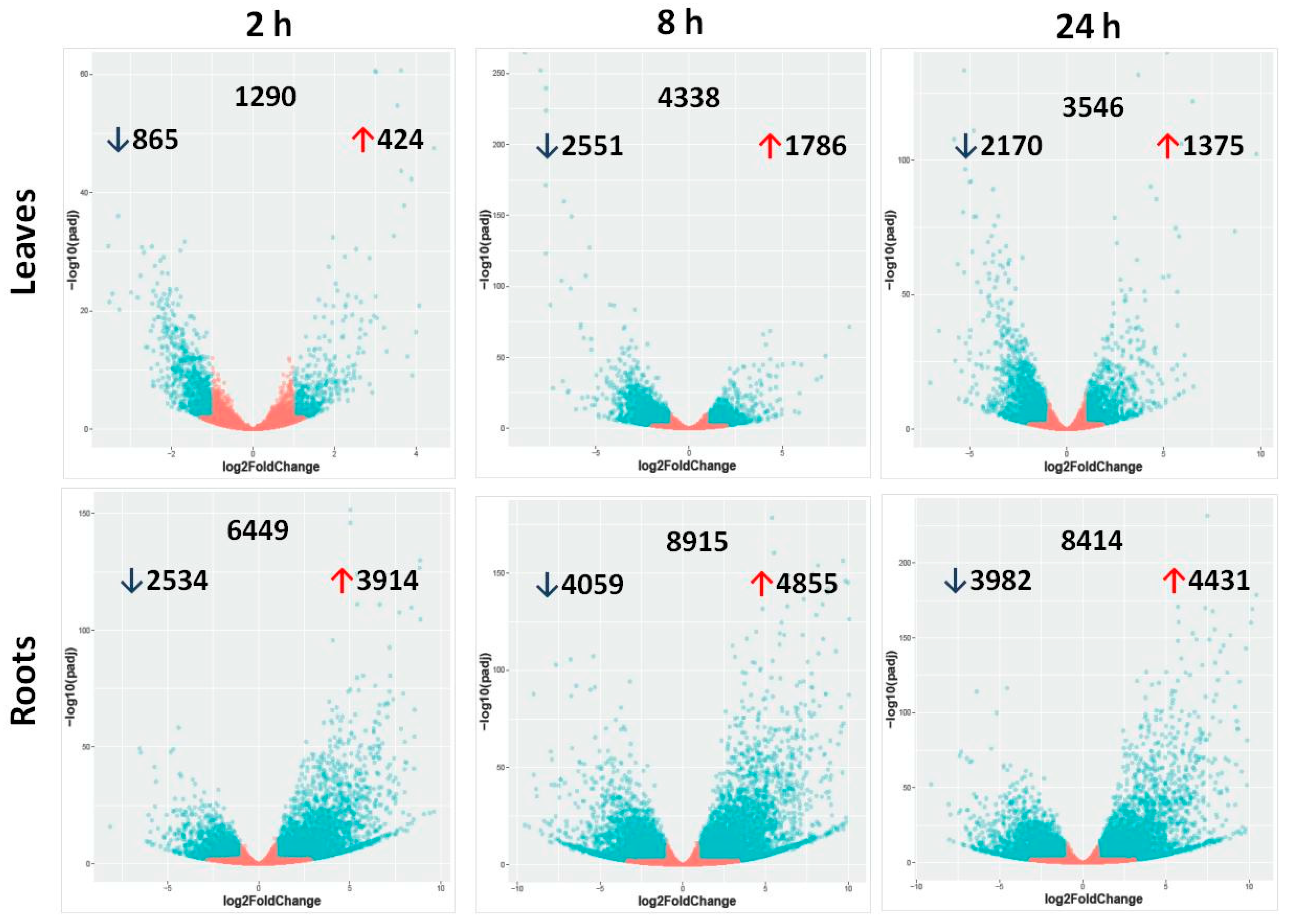
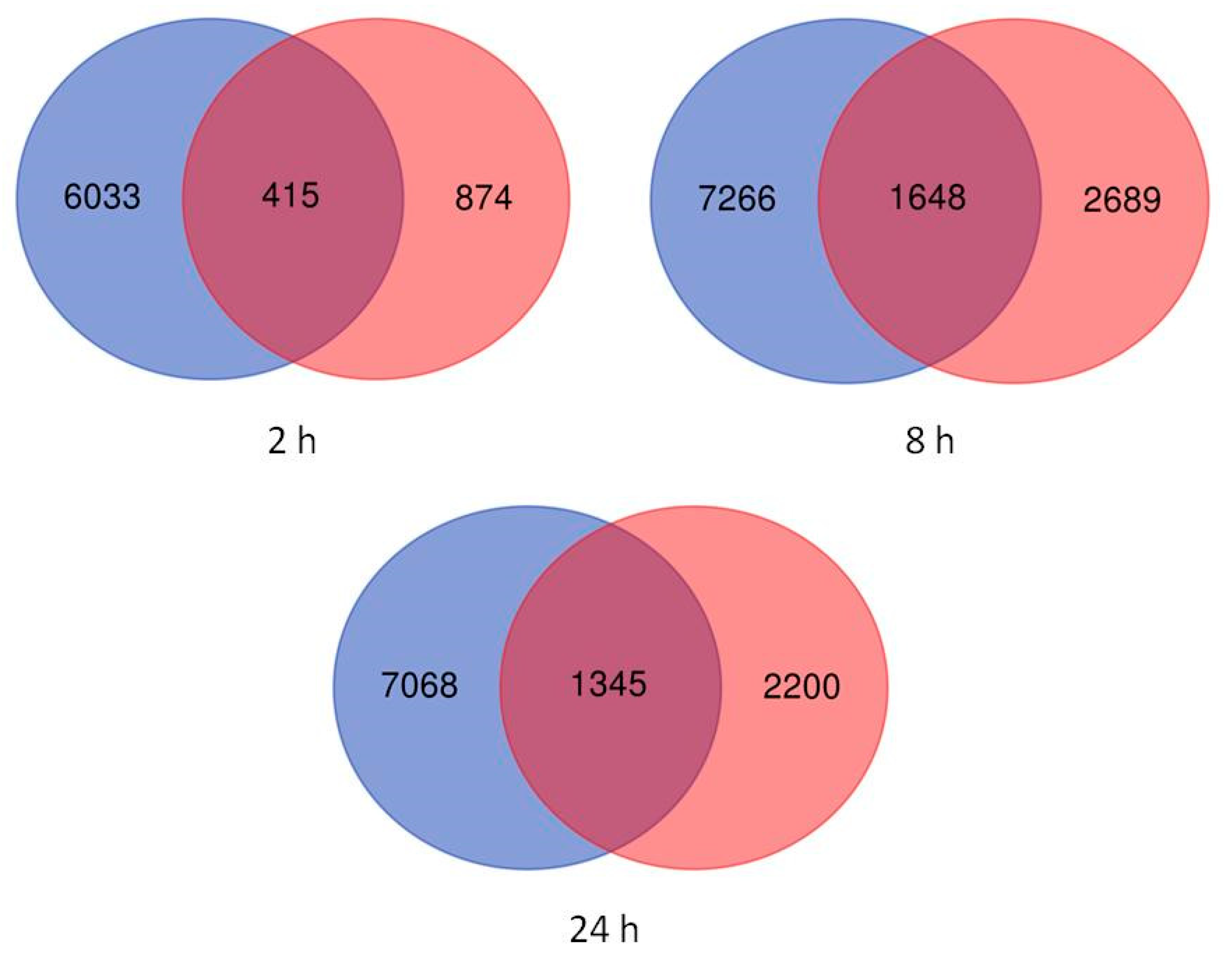
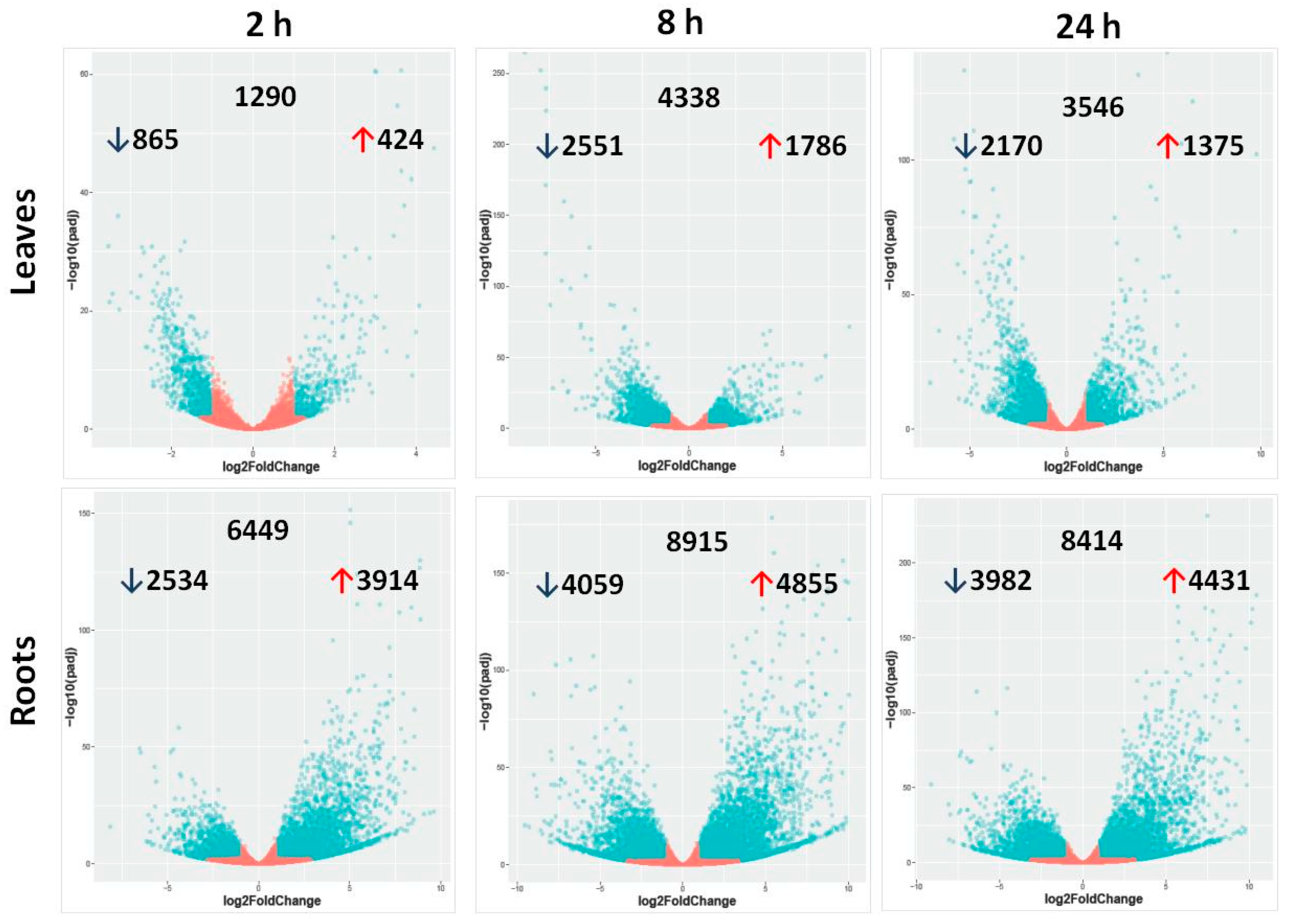
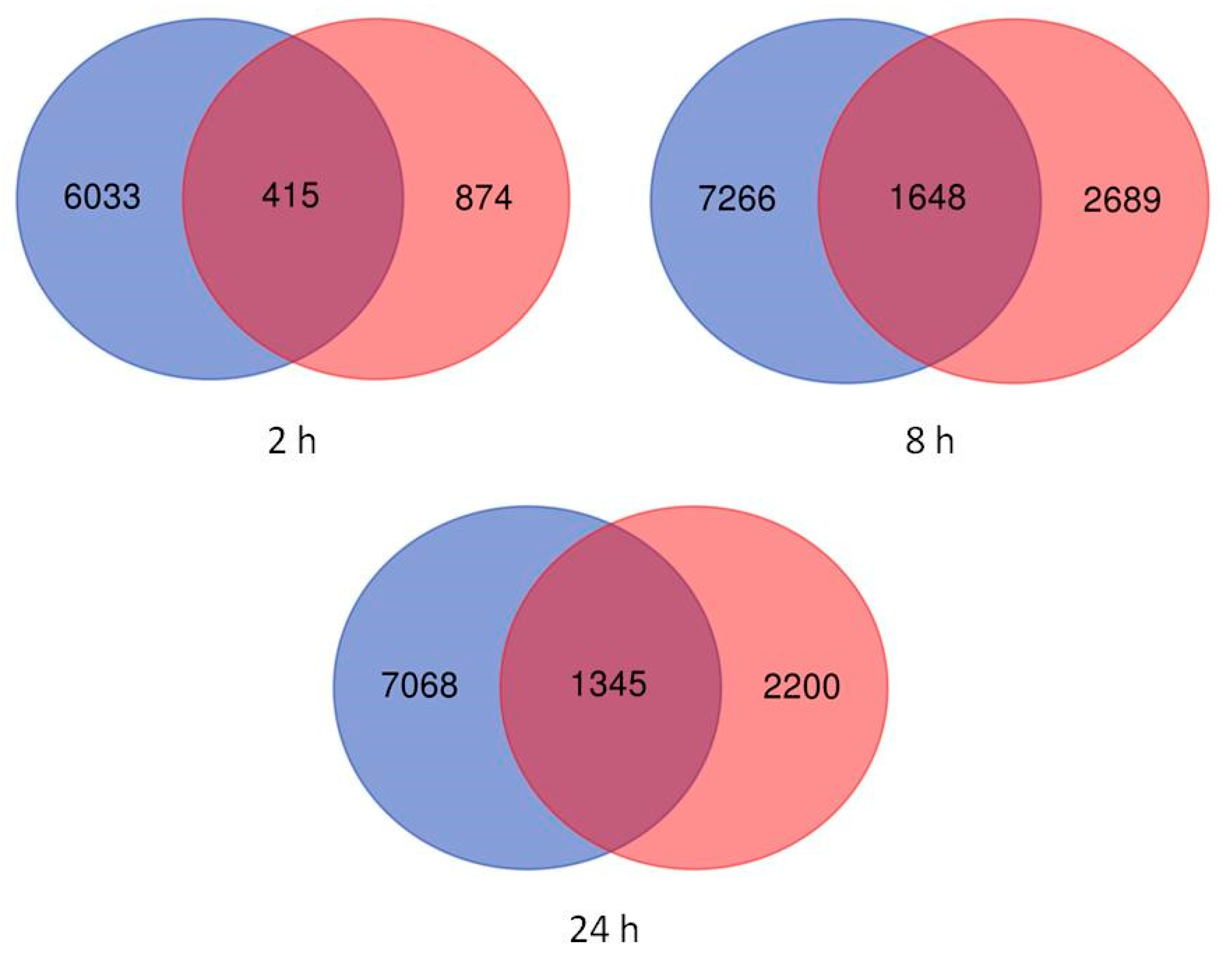
2.2. Differentially Expressed Genes in Leaves and Roots under Salt Stress
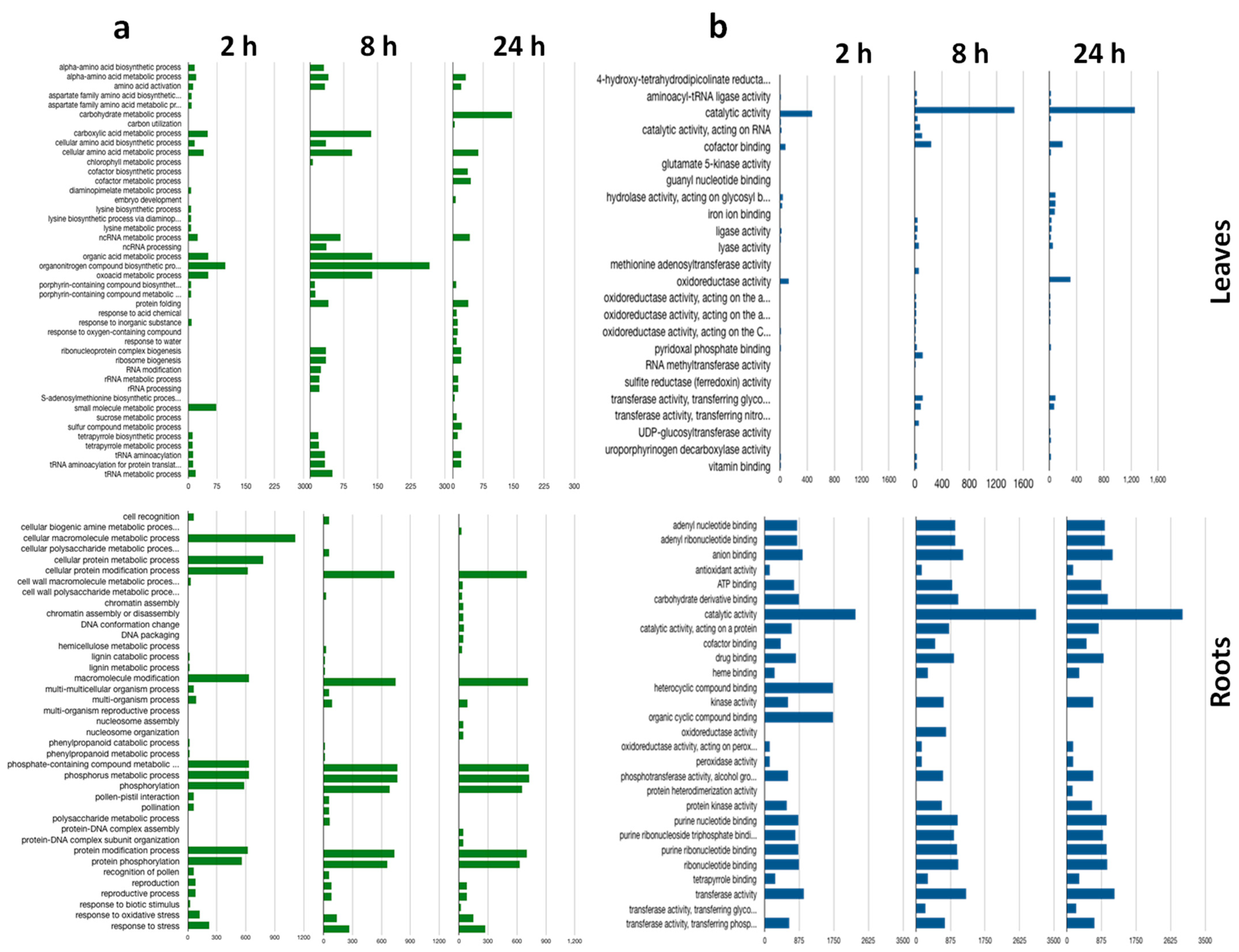
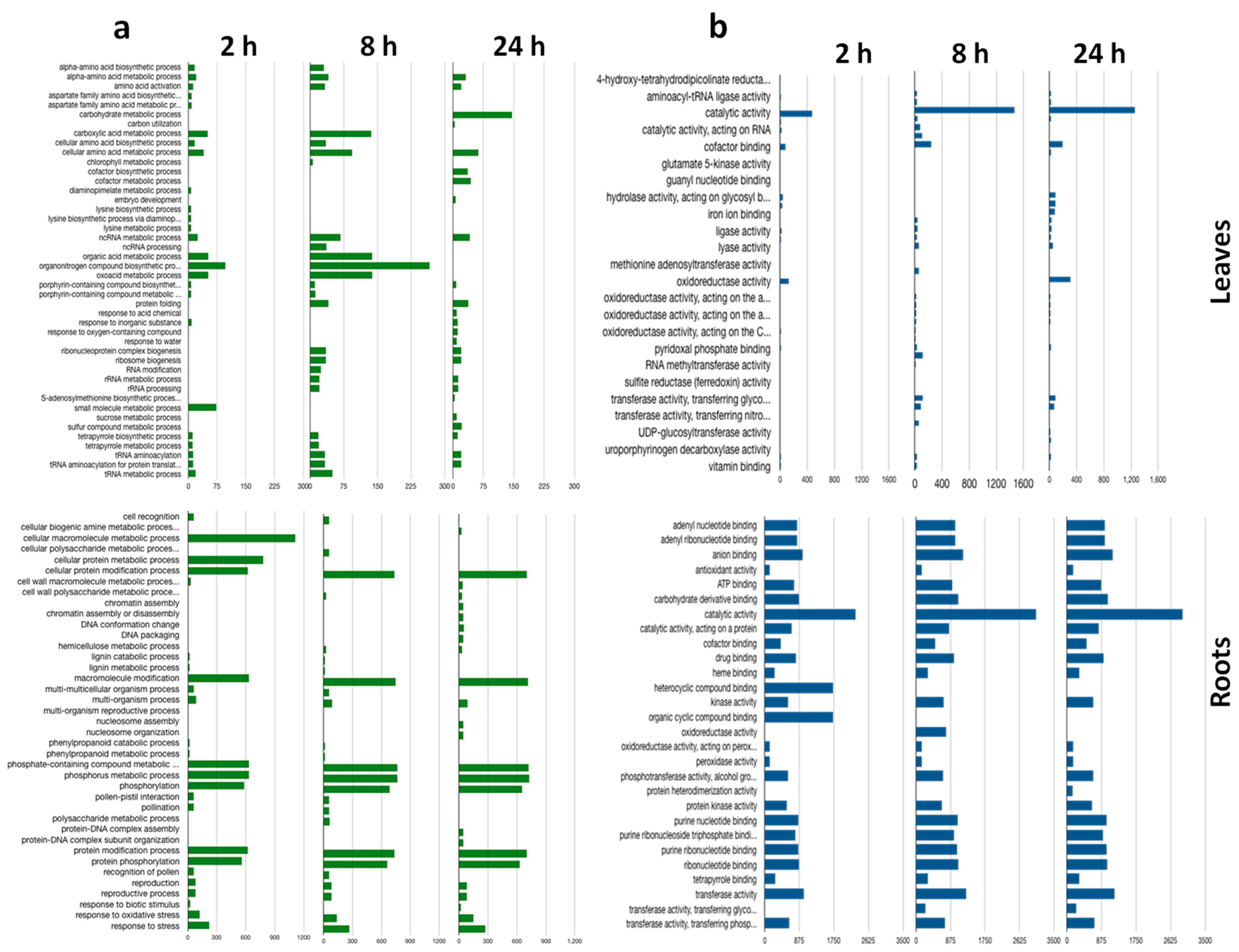
2.3. Gene Ontology Enrichment Analysis of Differentially Expressed Genes
2.4. Candidate Salt-Responsive DEGs
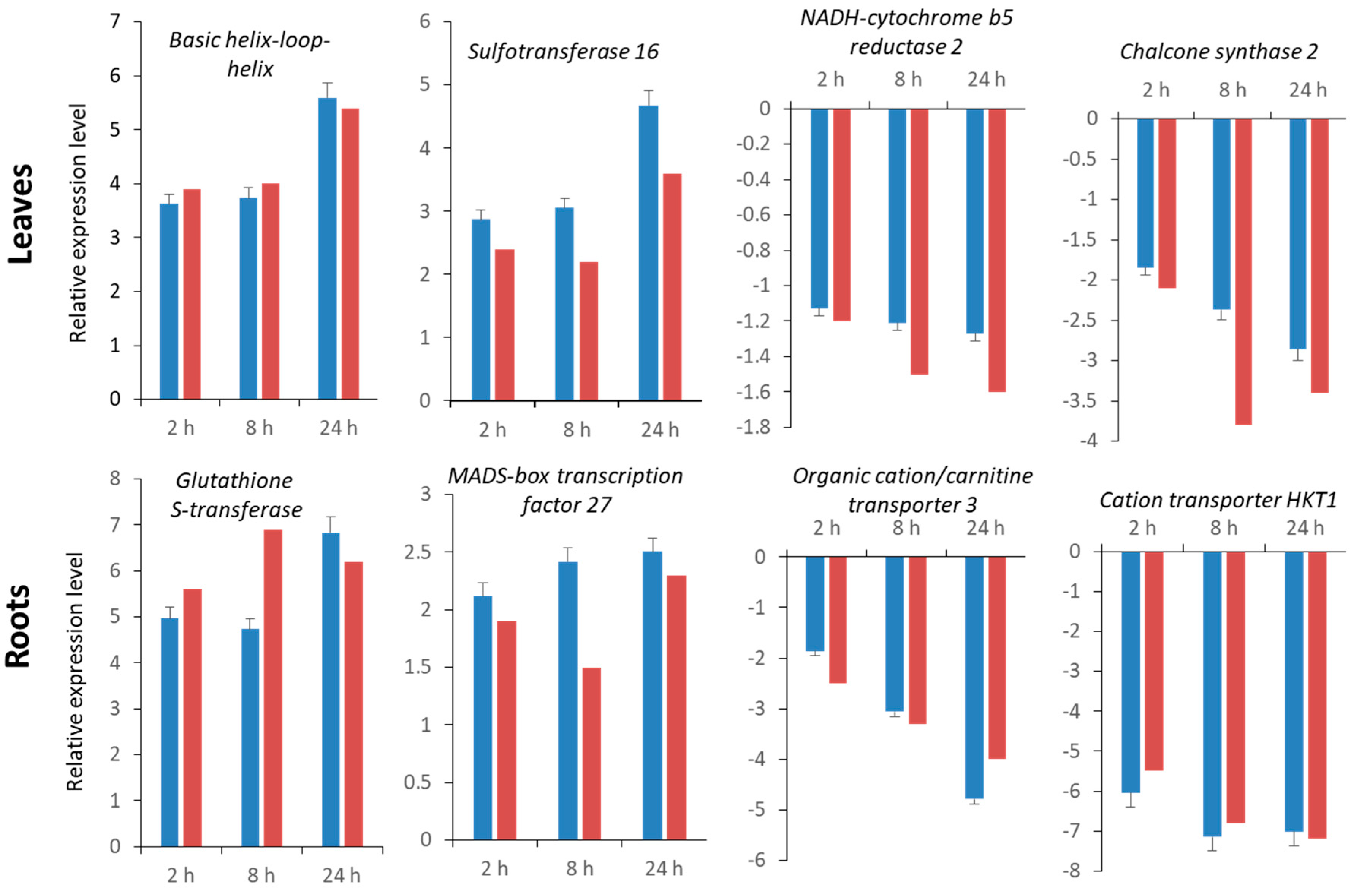
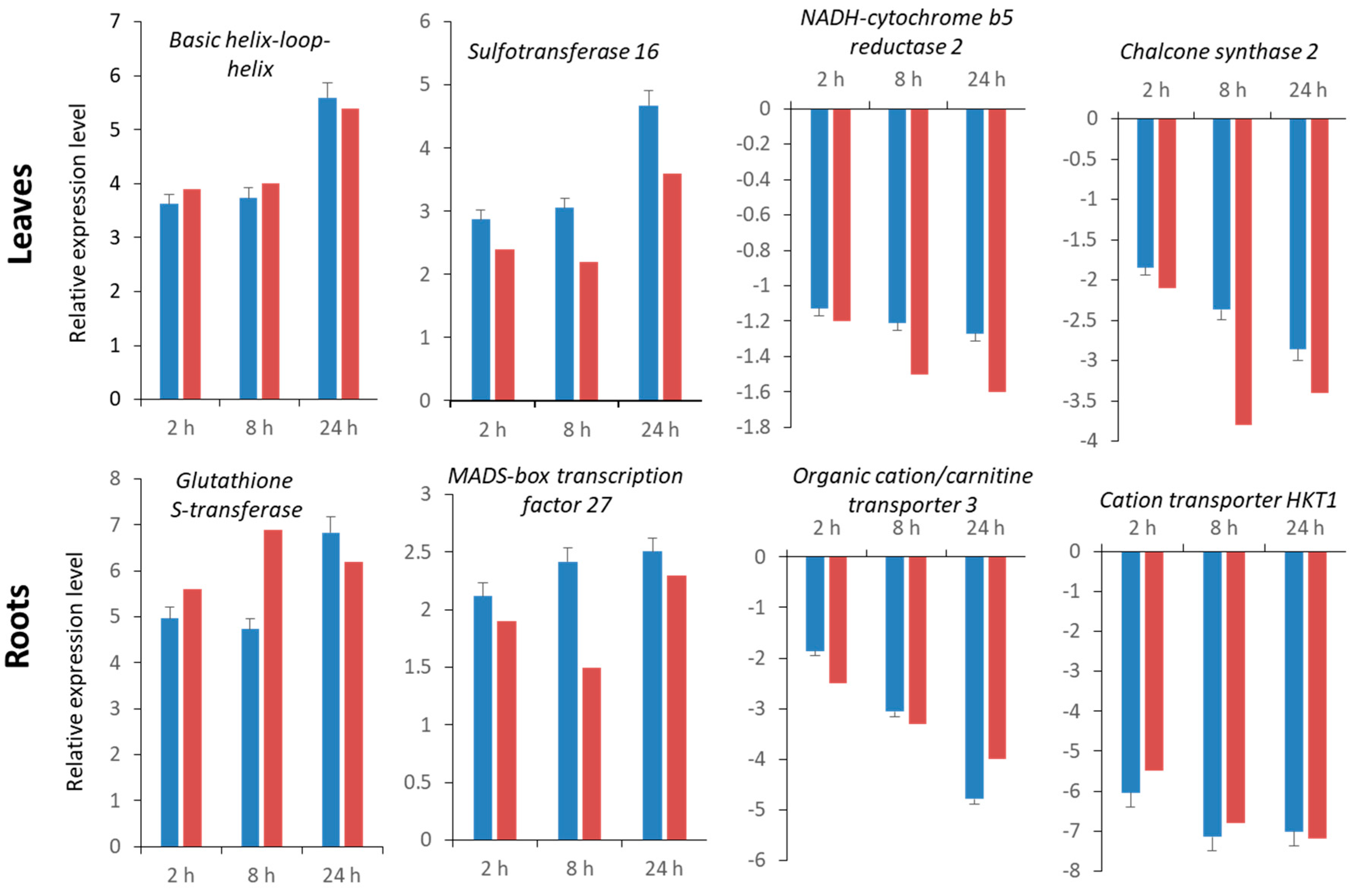
2.5. Validation of RNA-Seq Data by Quantitative Real-Time qRT-PCR
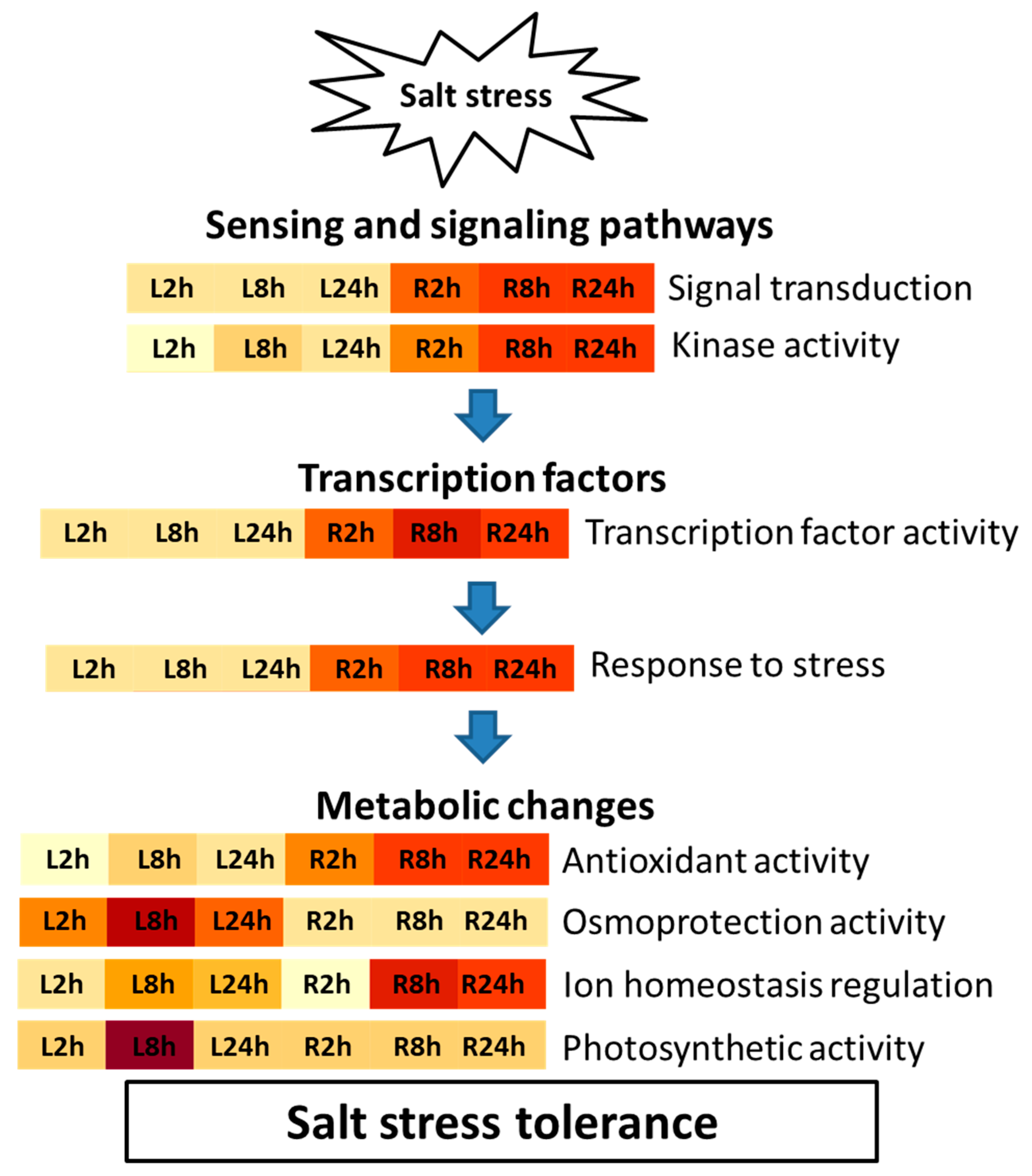
3. Discussion
4. Conclusions
5. Material and Methods
5.1. Plant Material and Hydroponic Salt Stress Treatment
5.2. Total RNA Isolation and DNase Treatment
5.3. Sequencing
5.4. Pseudoalignment and Transcript Abundance Analysis
5.5. Differential Expression Analysis
5.6. GO Enrichment Analysis and Visualization
5.7. Validation of RNA-Seq Findings by Real-Time PCR
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hernández, J.A. Salinity tolerance in plants: Trends and perspectives. Int. J. Mol. Sci. 2019, 20, 2408. [Google Scholar] [CrossRef] [Green Version]
- Zhu, J.K. Plant salt tolerance. Trends Plant Sci. 2001, 6, 66–71. [Google Scholar] [CrossRef]
- Acosta-Motos, J.R.; Ortuño, M.F.; Bernal-Vicente, A.; Diaz-Vivancos, P.; Sanchez-Blanco, M.J.; Hernández, J.A. Plant responses to salt stress: Adaptive mechanisms. Agronomy 2017, 7, 18. [Google Scholar] [CrossRef] [Green Version]
- Isayenkov, S.V.; Maathuis, F.J.M. Plant salinity stress: Many unanswered questions remain. Front. Plant Sci. 2019, 10, 80–91. [Google Scholar] [CrossRef] [Green Version]
- Krasensky, J.; Jonak, C. Drought, salt, and temperature stress-induced metabolic rearrangements and regulatory networks. J. Exp. Bot. 2012, 63, 1593–1608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwon, O.K.; Mekapogu, M.; Kim, K.S. Effect of salinity stress on photosynthesis and related physiological responses in carnation (Dianthus caryophyllus). Hortic. Environ. Biotechnol. 2019, 60, 831–839. [Google Scholar] [CrossRef]
- Bohnert, H.J.; Sheveleva, E. Plant stress adaptations—Making metabolism move. Curr. Opin. Plant Biol. 1998, 1, 267–274. [Google Scholar] [CrossRef]
- Gupta, B.; Huang, B. Mechanism of salinity tolerance in plants: Physiological, biochemical, and molecular characterization. Int. J. Genom. 2014, 2014, 701596–701614. [Google Scholar] [CrossRef]
- Blumwald, E. Sodium transport and salt tolerance in plants. Curr. Opin. Cell Biol. 2000, 12, 431–434. [Google Scholar] [CrossRef]
- Munns, R.; Tester, M. Mechanisms of salinity tolerance. Annu. Rev. Plant Biol. 2008, 59, 651–681. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Zhang, D.; Li, X.; Guan, K.; Yang, H. Novel DREB A-5 subgroup transcription factors from desert moss (Syntrichia caninervis) confers multiple abiotic stress tolerance to yeast. J. Plant Physiol. 2016, 194, 45–53. [Google Scholar] [CrossRef]
- Shinozaki, K.; Yamaguchi-Shinozaki, K. Gene expression and signal transduction in water-stress response. Plant Physiol. 1997, 115, 327–334. [Google Scholar] [CrossRef] [Green Version]
- Nakagami, H.; Pitzschke, A.; Hirt, H. Emerging MAPkinase pathways in plant stress signaling. Trends Plant Sci. 2005, 10, 339–346. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.; Gong, Q.; Bohnert, H.J. Dissecting salt stress pathways. J. Exp. Bot. 2006, 57, 1097–1107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osthoff, A.; Rose, P.D.D.; Baldauf, J.A.; Piepho, H.P.; Hochholdinger, F. Transcriptomic reprogramming of barley seminal roots by combined water deficit and salt stress. BMC Genom. 2019, 20, 325–339. [Google Scholar] [CrossRef] [PubMed]
- Wilhelm, B.T.; Marguerat, S.; Watt, S.; Schubert, F.; Wood, V.; Goodhead, I.; Penkett, C.J.; Rogers, J.; Bähler, J. Dynamic repertoire of a eukaryotic transcriptome surveyed at single-nucleotide resolution. Nature 2008, 453, 1239–1243. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Gao, S.; Muegge, K.; Zhang, W.; Zhou, B. Advanced applications of RNA sequencing and challenges. Bioinform. Biol. Insights 2015, 9, 29–46. [Google Scholar] [CrossRef] [Green Version]
- Yousefirad, S.; Soltanloo, H.; Ramezanpour, S.S.; Nezhad, K.Z.; Shariati, V. The RNA-seq transcriptomic analysis reveals genes mediating salt tolerance through rapid triggering of ion transporters in a mutant barley. PLoS ONE 2020, 15, e0229513. [Google Scholar] [CrossRef] [Green Version]
- Luo, Y.; Reid, R.; Freese, D.; Li, C.; Watkins, J.; Shi, H.; Zhang, H.; Loraine, A.; Song, B.H. Salt tolerance response revealed by RNA-Seq in a diploid halophytic wild relative of sweet potato. Sci. Rep. 2016, 7, 9624–9637. [Google Scholar] [CrossRef]
- Yong, H.Y.; Zou, Z.; Kok, E.P.; Kwan, B.H.; Chow, K.; Nasu, S.; Nanzyo, M.; Kitashiba, H.; Nishio, T. Comparative transcriptome analysis of leaves and roots in response to sudden increase in salinity in Brassica napus by RNA-seq. BioMed Res. Int. 2014, 6, 467395–467415. [Google Scholar]
- Shen, X.Y.; Wang, Z.L.; Song, X.F.; Xu, J.J.; Jiang, C.Y.; Zhao, Y.X.; Ma, C.L.; Zhang, H. Transcriptomic profiling revealed an important role of cell wall remodeling and ethylene signaling pathway during salt acclimation in Arabidopsis. Plant Mol. Biol. 2014, 86, 303–317. [Google Scholar] [CrossRef]
- Feriani, W.; Rezgui, S.; Cherif, M. Grain yield assessment of genotype by environment interaction of Tunisian doubled-haploid barley lines. J. New Sci. 2016, 27, 1507–1512. [Google Scholar]
- Adem, G.D.; Roy, S.J.; Zhou, M.; Bowman, J.P.; Shabala, S. Evaluating contribution of ionic, osmotic and oxidative stress components towards salinity tolerance in barley. BMC Plant Biol. 2014, 14, 113–126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Witzel, K.; Matros, A.; Strickert, M.; Kaspar, S.; Peukert, M.; Mühling, K.H.; Börner, A.; Mock, H.P. Salinity stress in roots of contrasting barley genotypes reveals time-distinct and genotype-specific patterns for defined proteins. Mol. Plant 2014, 7, 336–355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hasanuzzaman, M.D.; Nahar, K.; Alam, M.M.; Roychowdhury, R.; Fujita, M. Physiological, biochemical, and molecular mechanisms of heat stress tolerance in plants. Int. J. Mol. Sci. 2013, 14, 9643–9684. [Google Scholar] [CrossRef] [PubMed]
- Negrao, S.; Schmöckel, S.M.; Tester, M. Evaluating physiological responses of plants to salinity stress. Ann. Bot. 2017, 119, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Ben Chikha, M. Variability of Tolerance to Salt Stress in Local Genotypes of Barley (Hordeum vulgare L.) Depending on the Stage of Development. Ph.D. Thesis, Faculty of Sciences of Tunis, University of Tunis EL Manar, Tunis, Tunisia, 2017; p. 162. [Google Scholar]
- Ben Chikha, M.; Hessini, K.; Ourteni, R.N.; Ghorbel, A.; Zoghlami, N. Identification of barley landrace genotypes with contrasting salinity tolerance at vegetative growth stage. Plant Biotechnol. 2016, 33, 287–295. [Google Scholar] [CrossRef] [Green Version]
- Ziemann, M.; Kamboj, A.; Hove, R.M.; Loveridge, S.; El-Osta, A.; Bhave, M. Analysis of the barley leaf transcriptome under salinity stress using mRNA-Seq. Acta Physiol. Plant 2013, 35, 1915–1924. [Google Scholar] [CrossRef]
- Bahieldin, A.; Atef, A.; Sabir, J.S.; Gadalla, N.O.; Edris, S.; Alzohairy, A.M.; Radhwan, N.A.; Baeshen, M.N.; Ramadan, A.M.; Eissa, H.F.; et al. RNA-Seq analysis of the wild barley (H. spontaneum) leaf transcriptome under salt stress. C. R. Biol. 2015, 338, 285–297. [Google Scholar] [CrossRef]
- Hill, B.; Cassin, A.; Keeble-Gagnère, G.; Doblin, M.S.; Bacic, A.; Roessner, U. De novo transcriptome assembly and analysis of differentially expressed genes of two barley genotypes reveal root-zone-specific responses to salt exposure Camilla. Sci. Rep. 2016, 6, 31558–31572. [Google Scholar] [CrossRef] [Green Version]
- Baldoni, E.; Bagnaresi, P.; Locatelli, F.; Monica Mattana, M.; Genga, A. Comparative leaf and root transcriptomic analysis of two rice japonica cultivars reveals major differences in the root early response to osmotic stress. Rice 2016, 9, 25–45. [Google Scholar] [CrossRef] [Green Version]
- Ruiz, K.B.; Maldonado, J.; Biondi, S.; Silva, H. RNA-seq analysis of salt-stressed versus non salt-stressed transcriptomes of Chenopodium quinoa landrace R49. Genes 2019, 10, 1042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ksouri, N.; Jiménez, S.; Wells, C.E.; Contreras-Moreira, B.; Gogorcena, Y. Transcriptional responses in root and leaf of Prunus persica under drought stress using RNA sequencing. Front. Plant Sci. 2016, 7, 1715–1734. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, D.; Xu, J. Abiotic stress responses in plant roots: A proteomics perspective. Front. Plant Sci. 2014, 5, 6–19. [Google Scholar] [CrossRef] [Green Version]
- Agarwal, P.K.; Jha, B. Transcription factors in plants and ABA dependent and independent abiotic stress signaling. Biol. Plant. 2010, 54, 201–212. [Google Scholar] [CrossRef]
- Zhao, C.; Zhang, H.; Song, C.; Zhu, J.K.; Shabala, S. Mechanisms of plant responses and adaptation to soil salinity. Innovation 2020, 1, 1–42. [Google Scholar] [CrossRef]
- Golldack, D.; Li, C.; Mohan, H.; Probst, N. Tolerance to drought and salt stress in plants: Unraveling the signaling networks. Front. Plant Sci. 2014, 5, 151–162. [Google Scholar] [CrossRef] [Green Version]
- Seyfferth, C.; Tsuda, K. Salicylic acid signal transduction: The initiation of biosynthesis, perception and transcriptional reprogramming. Front. Plant Sci. 2014, 9, 697–707. [Google Scholar] [CrossRef] [Green Version]
- Osakabe, Y.; Yamaguchi-Shinozaki, K.; Shinozaki, K.; Tran, L.S.P. ABA control of plant macroelement membrane transport systems in response to water deficit and high salinity. New Phytol. 2014, 202, 35–49. [Google Scholar] [CrossRef]
- Zhang, L.; Li, Z.; Quan, R.; Li, G.; Wang, R.; Huang, R. An AP2 domain-containing gene, ESE1, targeted by the ethylene signaling component EIN3 is important for the salt response in Arabidopsis. Plant Physiol. 2011, 157, 854–865. [Google Scholar] [CrossRef] [Green Version]
- Dodd, A.N.; Kudla, J.; Sanders, D. The language of calcium signaling. Annu. Rev. Plant Biol. 2010, 61, 593–620. [Google Scholar] [CrossRef]
- Wolf, S. Plant cell wall signaling and receptor-like kinases. Biochem. J. 2017, 474, 471–492. [Google Scholar] [CrossRef] [PubMed]
- Sah, S.K.; Reddy, K.R.; Li, J. Abscisic acid and abiotic stress tolerance in crop plants. Front. Plant Sci. 2016, 7, 571–596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.; Zhang, Z.; Gao, J.; Wang, P.; Hu, T.; Wang, Z.; Hou, Y.J.; Wan, Y.; Liu, W.; Xie, S.; et al. Arabidopsis duodecuple mutant of PYL ABA receptors reveals PYL repression of ABA-independent SnRK2 Activity. Cell. Rep. 2018, 23, 3340–3351. [Google Scholar] [CrossRef] [PubMed]
- Hirayama, T.; Umezawa, T. The PP2C-SnRK2 complex—The central regulator of an abscisic acid signaling pathway. Plant Signal. Behav. 2010, 5, 160–163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roy, S.; Saxena, S.; Sinha, A.; Nandi, A.K. Dormancy/auxin associated family protein 2 of Arabidopsis thaliana is a negative regulator of local and systemic acquired resistance. J. Plant Res. 2020, 133, 409–417. [Google Scholar] [CrossRef] [PubMed]
- Peng, J.; Li, Z.; Wen, X.; Li, W.; Shi, H.; Yang, L.; Zhu, H.; Guo, H. Salt-induced stabilization of EIN3/EIL1 confers salinity tolerance by deterring ROS accumulation in Arabidopsis. PLoS Genet. 2014, 10, e1004664. [Google Scholar] [CrossRef] [Green Version]
- Chew, W.; Hrmova, M.; Lopato, S. Role of homeodomain leucine zipper (HD-Zip) IV transcription factors in plant development and plant protection from deleterious environmental factors. Int. J. Mol. Sci. 2013, 14, 8122–8147. [Google Scholar] [CrossRef] [Green Version]
- Khan, M.S.; Ahmad, D.; Khan, M.A. Utilization of genes encoding osmoprotectants in transgenic plants for enhanced abiotic stress tolerance. Electron. J. Biotechnol. 2015, 18, 257–266. [Google Scholar] [CrossRef] [Green Version]
- Borghi, L.; Kang, J.; Francisco, R.B. Filling the Gap: Functional clustering of ABC proteins for the investigation of hormonal transport in planta. Front. Plant Sci. 2019, 10, 422–442. [Google Scholar] [CrossRef]
- Hasegawa, P.M. Sodium (Na+) homeostasis and salt tolerance of plants. Environ. Exp. Bot. 2013, 92, 19–31. [Google Scholar] [CrossRef]
- Demidchik, V. ROS-activated ion channels in plants: Biophysical characteristics, physiological functions and molecular nature. Int. J. Mol. Sci. 2018, 19, 1263. [Google Scholar] [CrossRef] [Green Version]
- Shi, H.; Quintero, F.J.; Pardo, J.M.; Zhu, J.K. The putative plasma membrane Na+/H+ antiporter SOS1 controls long-distance Na+ transport in plants. Plant Cell. 2002, 14, 465–477. [Google Scholar] [CrossRef] [Green Version]
- Bihler, H.; Eing, C.; Hebeisen, S.; Roller, A.; Czempinski, K.; Bertl, A. TPK1 is a vacuolar ion channel different from the slow-vacuolar cation channel. Plant Physiol. 2005, 139, 417–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johansson, I.; Wulfetange, K.; Porée, F.; Michard, E.; Gajdanowicz, P.; Lacombe, B.; Sentenac, H.; Thibaud, J.B.; Mueller-Roeber, B.; Blatt, M.R.; et al. External K+ modulates the activity of the Arabidopsis potassium channel SKOR via an unusual mechanism. Plant J. 2006, 46, 269–281. [Google Scholar] [CrossRef] [PubMed]
- Stoerger, C.; Flockerzi, V. The transient receptor potential cation channel subfamily V member 6 (TRPV6): Genetics, biochemical properties, and functions of exceptional calcium channel proteins. Biochem. Cell. Biol. 2014, 92, 441–448. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Wang, H.; Yuan, Q.; Feng, Y. Structure and function of the photosystem supercomplexes. Front. Plant Sci. 2018, 20, 357–364. [Google Scholar] [CrossRef]
- Wang, J.; Qin, Q.; Pan, J.; Sun, L.; Sun, Y.; Xue, Y.; Song, K. Transcriptome analysis in roots and leaves of wheat seedlings in response to low-phosphorus stress. Sci. Rep. 2019, 9, 19802–19814. [Google Scholar] [CrossRef] [PubMed]
- Hoagland, D.R.; Arnon, D.I. The water culture method for growing plants without soil. Calif. Agric. Exp. Stn. 1950, 347, 32. [Google Scholar]
- Mascher, M.; Gundlach, H.; Himmelbach, A.; Beier, S.; Twardziok, S.O.; Wicker, T.; Radchuk, V.; Dockter, C.; Hedley, P.E.; Russell, J.; et al. A chromosome conformation capture ordered sequence of the barley genome. Nature 2017, 544, 427–433. [Google Scholar] [CrossRef] [Green Version]
- Bray, N.; Pimentel, H.; Melsted, P.; Pachter, L. Near-optimal probabilistic RNA-Seq quantification. Nat. Biotechnol. 2016, 34, 525–527. [Google Scholar] [CrossRef] [PubMed]
- Love, M.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genom. Biol. 2014, 15, 550–571. [Google Scholar] [CrossRef] [Green Version]
- Alexa, A.; Rahnenfuhrer, J. topGO: Enrichment Analysis for Gene Ontology; R Package Version 2.44.0. 2021. Available online: https://bioconductor.org/packages/release/bioc/html/topGO.html (accessed on 1 July 2021).
- Conesa, A.; Götz, S.; García-Gómez, J.M.; Terol, J.; Talón, M.; Roble, M. Blast2GO: A universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 2005, 21, 3674–3676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Untergasser, A.; Cutcutache, I.; Koressaar, T.; Ye, J.; Faircloth, B.C.; Remm, M.; Rozen, S.G. Primer3-new capabilities and interfaces. Nucleic Acids Res. 2012, 40, 115–127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmittgen, T.D.; Livak, K.J. Analyzing real-time PCR data by the comparative CT method. Nat. Protoc. 2008, 3, 1101–1108. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Leaves | Roots | ||
|---|---|---|---|
| KEGG ID | Enriched Pathway | KEGG ID | Enriched Pathway |
| Ko00860 | Porphyrin and chlorophyll metabolism | ko00980 | Metabolism of xenobiotics by cytochrome P450 |
| Ko00970 | Aminoacyl-tRNA biosynthesis | ko00982 | Drug metabolism—cytochrome P450 |
| Ko00261 | Monobactam biosynthesis (glutamate dehydrogenase (NAD(P)+)) | ko00983 | Drug metabolism—other enzymes (phospholipase) |
| Ko00300 | Lysine biosynthesis | ko00940 | Phenylpropanoid biosynthesis |
| Ko00250 | Alanine, aspartate, and glutamate metabolism | ko00460 | Cyanoamino acid metabolism |
| Ko00997 | Biosynthesis of various secondary metabolites | ko00480 | Glutathione metabolism |
| Ko00270 | Cysteine and methionine metabolism | ko00400 | Phenylalanine, tyrosine, and tryptophan biosynthesis |
| Ko00941 | Flavonoid biosynthesis | ko00140 | Steroid hormone biosynthesis |
| Ko00332 | Carbapenem biosynthesis (NADH-quinone oxidoreductase subunit C) | ko00943 | Isoflavonoid biosynthesis |
| Ko00906 | Carotenoid biosynthesis | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nefissi Ouertani, R.; Arasappan, D.; Abid, G.; Ben Chikha, M.; Jardak, R.; Mahmoudi, H.; Mejri, S.; Ghorbel, A.; Ruhlman, T.A.; Jansen, R.K. Transcriptomic Analysis of Salt-Stress-Responsive Genes in Barley Roots and Leaves. Int. J. Mol. Sci. 2021, 22, 8155. https://doi.org/10.3390/ijms22158155
Nefissi Ouertani R, Arasappan D, Abid G, Ben Chikha M, Jardak R, Mahmoudi H, Mejri S, Ghorbel A, Ruhlman TA, Jansen RK. Transcriptomic Analysis of Salt-Stress-Responsive Genes in Barley Roots and Leaves. International Journal of Molecular Sciences. 2021; 22(15):8155. https://doi.org/10.3390/ijms22158155
Chicago/Turabian StyleNefissi Ouertani, Rim, Dhivya Arasappan, Ghassen Abid, Mariem Ben Chikha, Rahma Jardak, Henda Mahmoudi, Samiha Mejri, Abdelwahed Ghorbel, Tracey A. Ruhlman, and Robert K. Jansen. 2021. "Transcriptomic Analysis of Salt-Stress-Responsive Genes in Barley Roots and Leaves" International Journal of Molecular Sciences 22, no. 15: 8155. https://doi.org/10.3390/ijms22158155
APA StyleNefissi Ouertani, R., Arasappan, D., Abid, G., Ben Chikha, M., Jardak, R., Mahmoudi, H., Mejri, S., Ghorbel, A., Ruhlman, T. A., & Jansen, R. K. (2021). Transcriptomic Analysis of Salt-Stress-Responsive Genes in Barley Roots and Leaves. International Journal of Molecular Sciences, 22(15), 8155. https://doi.org/10.3390/ijms22158155