
Rapamycin Plus Doxycycline Combination Affects Growth Arrest and Selective Autophagy-Dependent Cell Death in Breast Cancer Cells

 , , , , and
, , , , and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
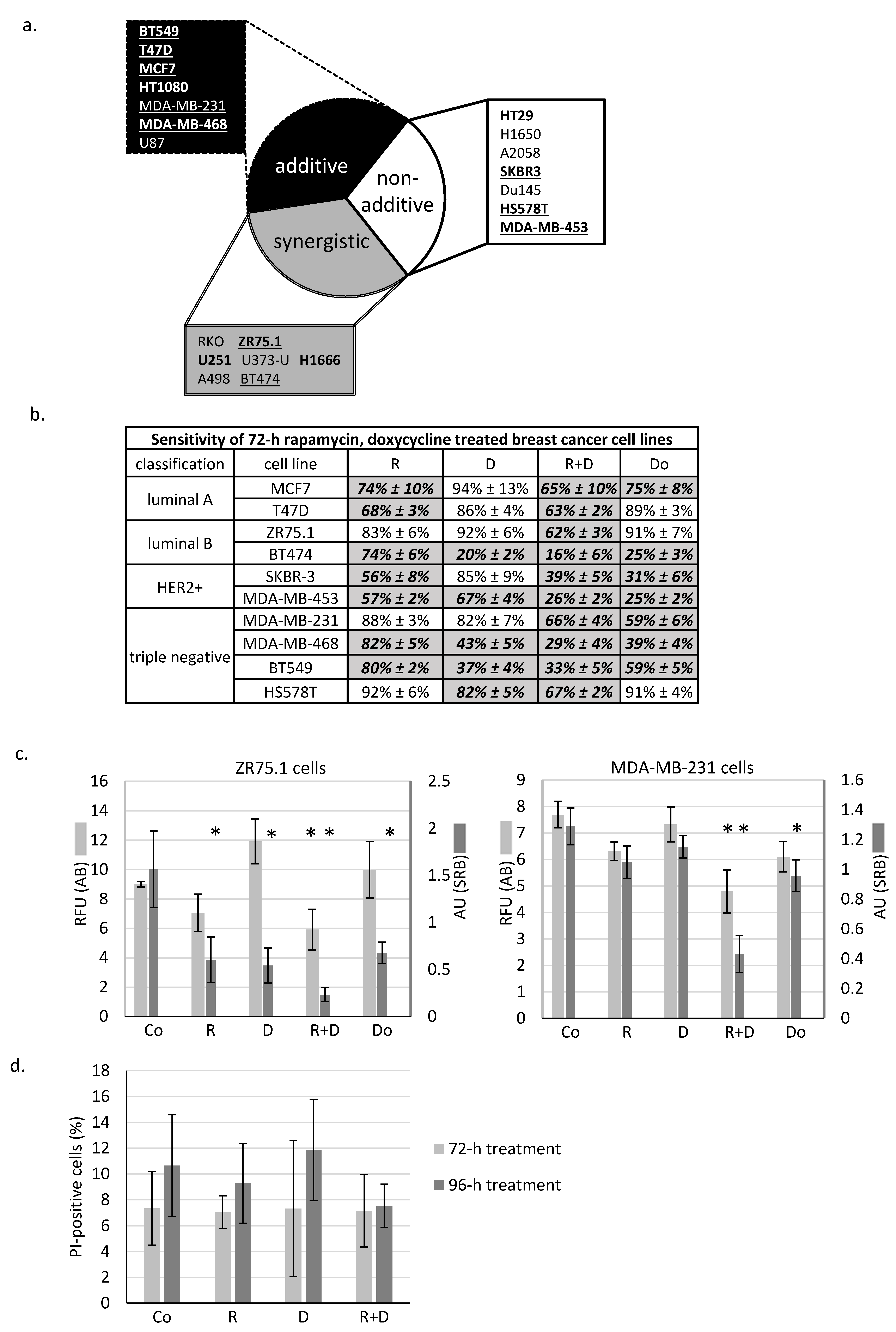
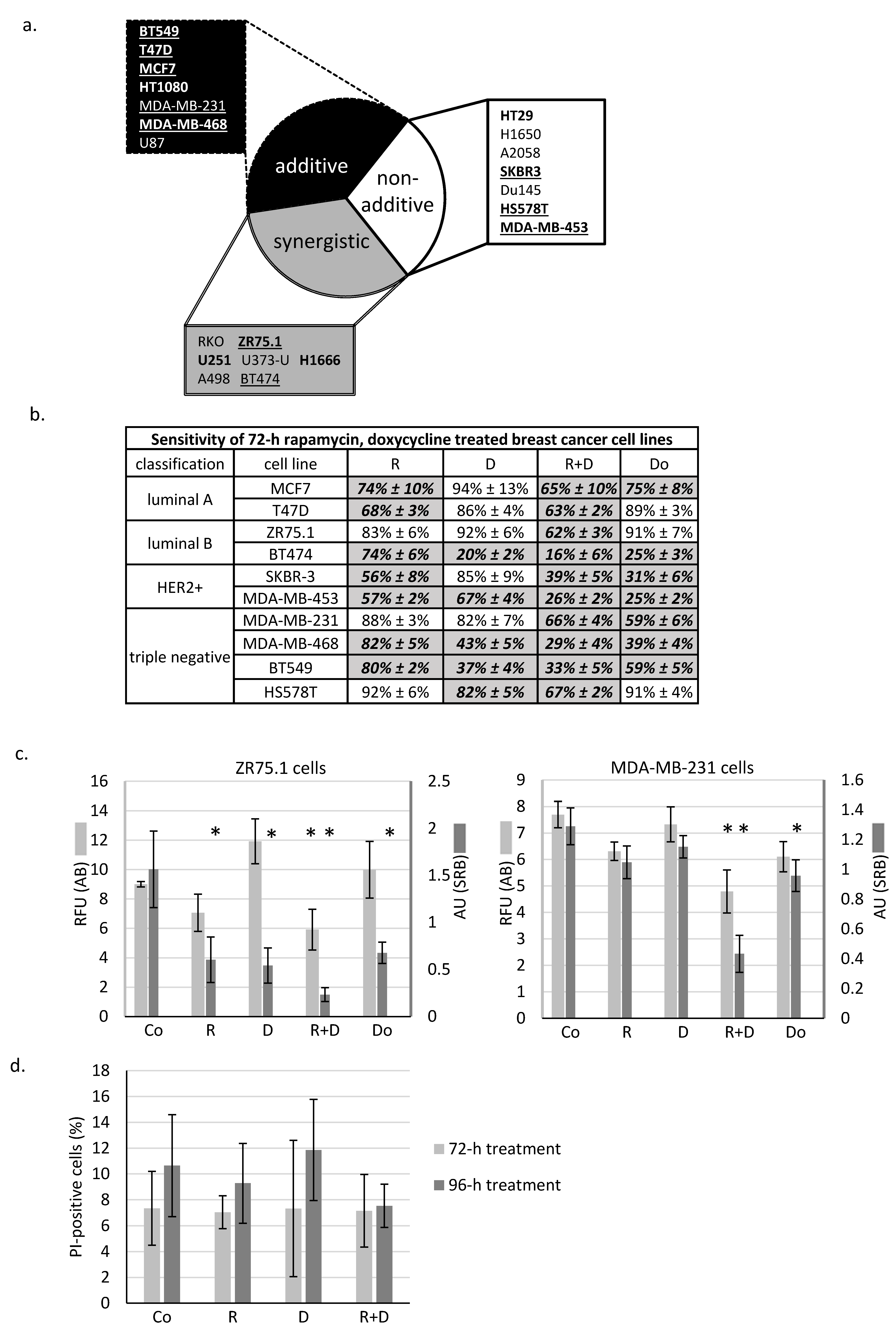
2.1. Rapamycin + Doxycycline Combination Inhibits Cell Proliferation in Human Breast Cancer Cells
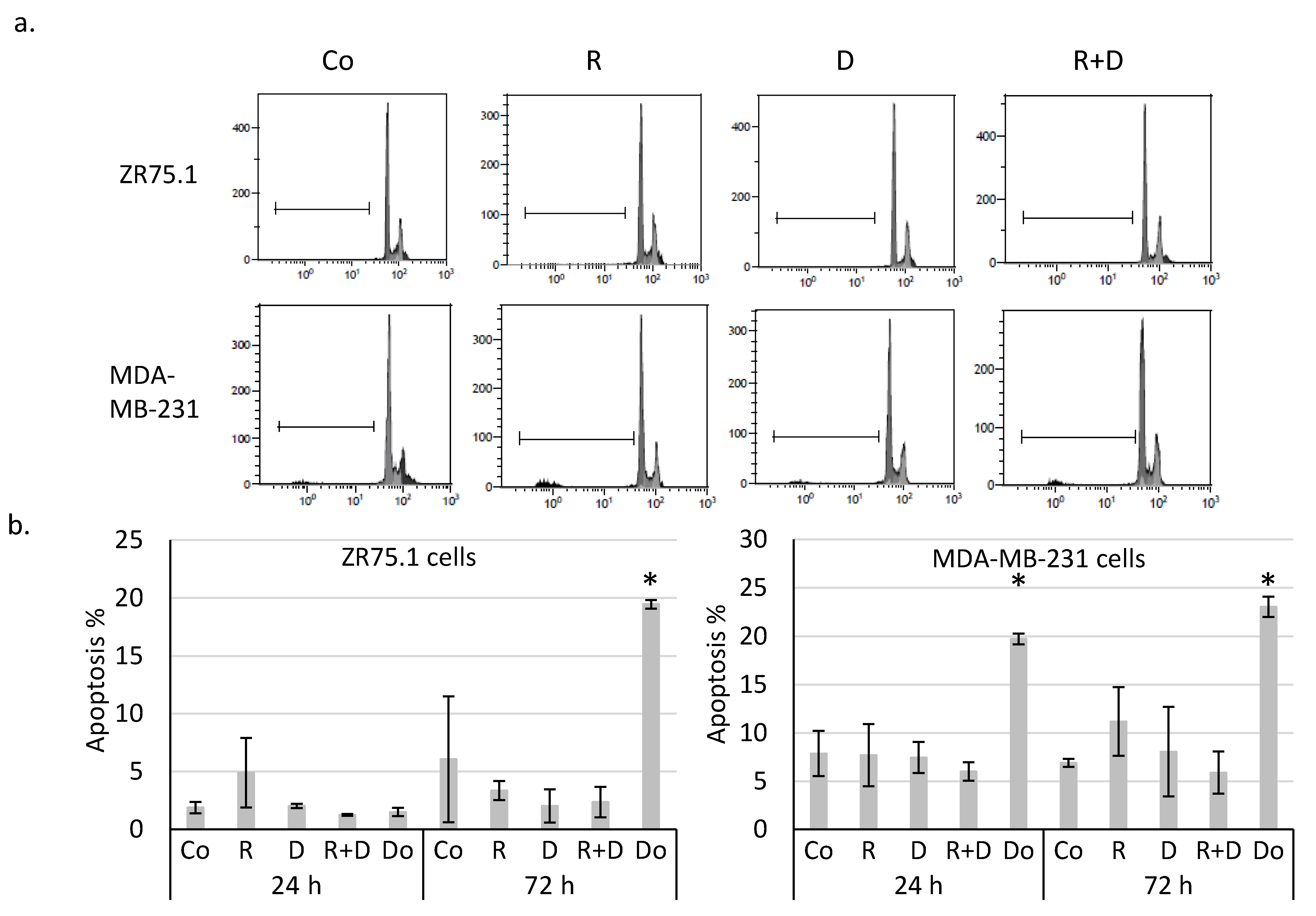
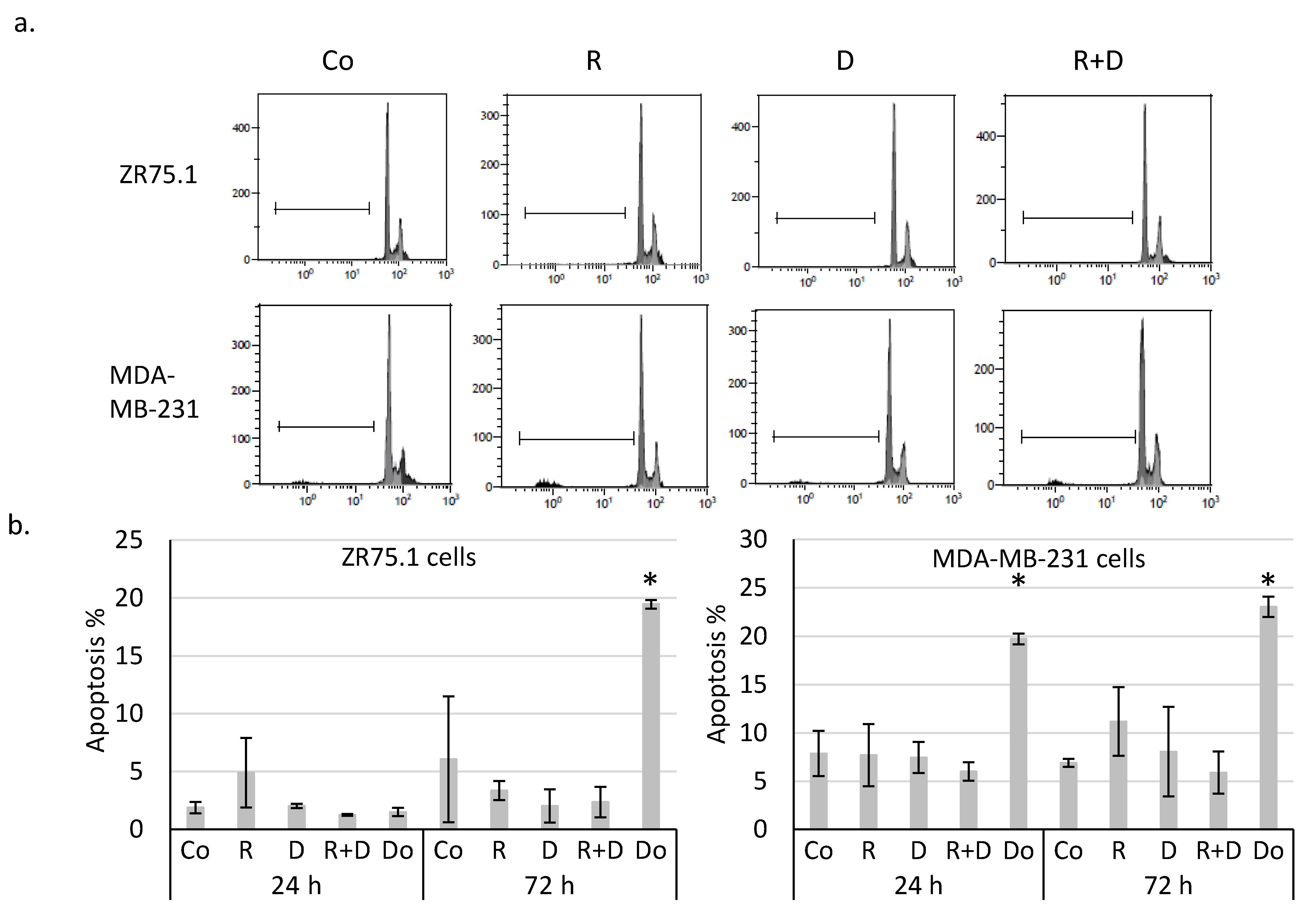
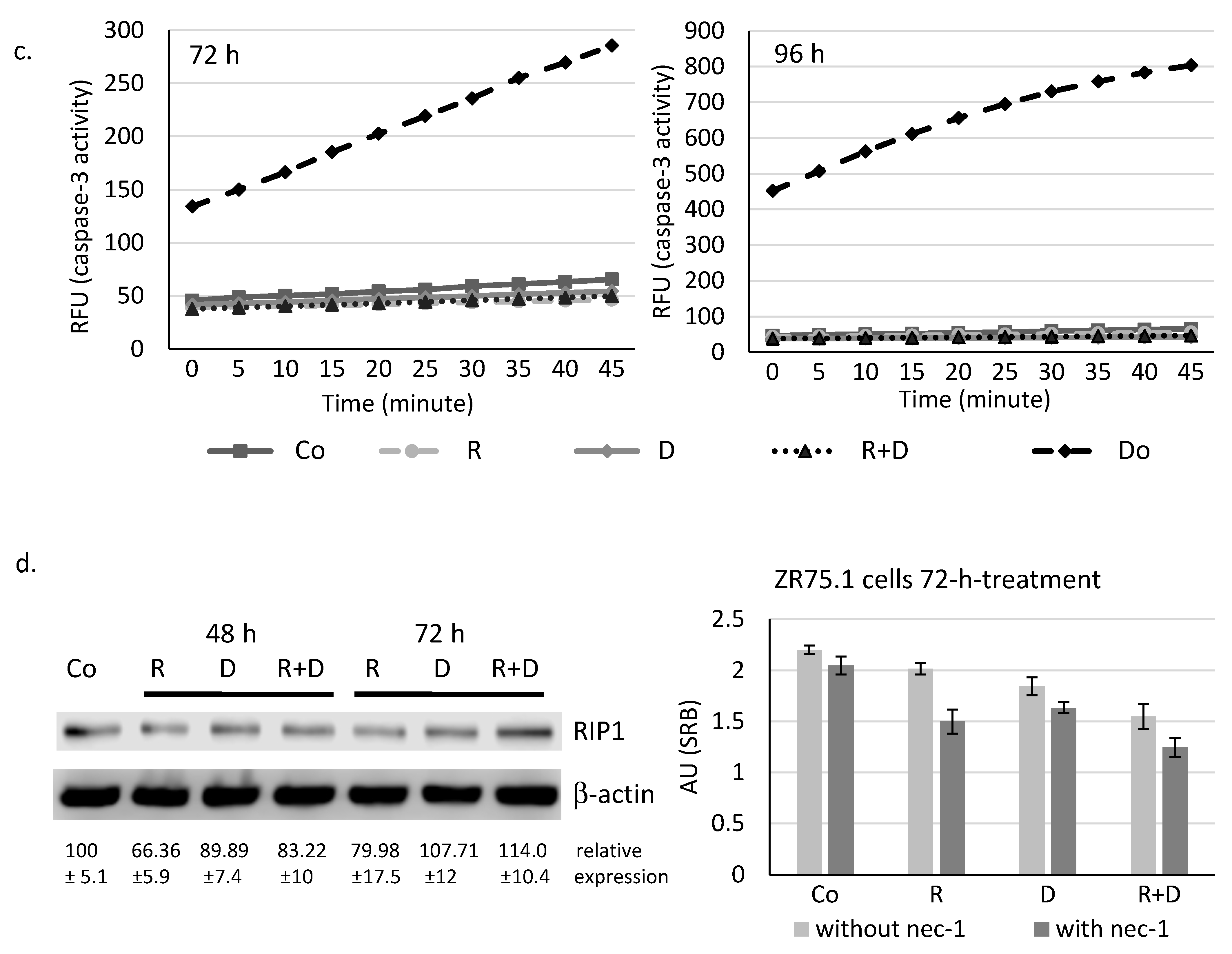
2.2. Rapamycin + Doxycycline Combination Did Not Induce Apoptosis or Necrosis after 72-h or 96-h Treatments
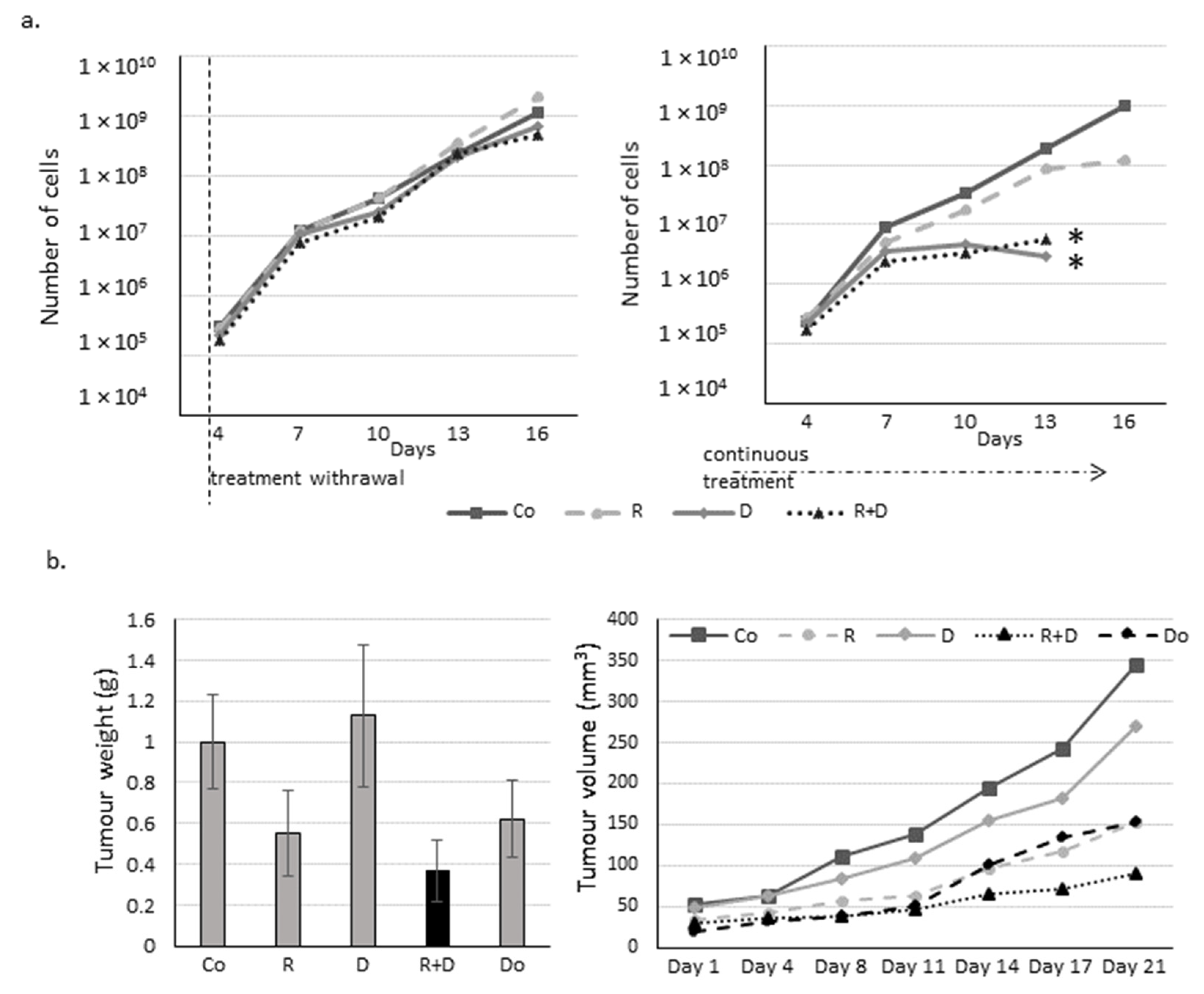
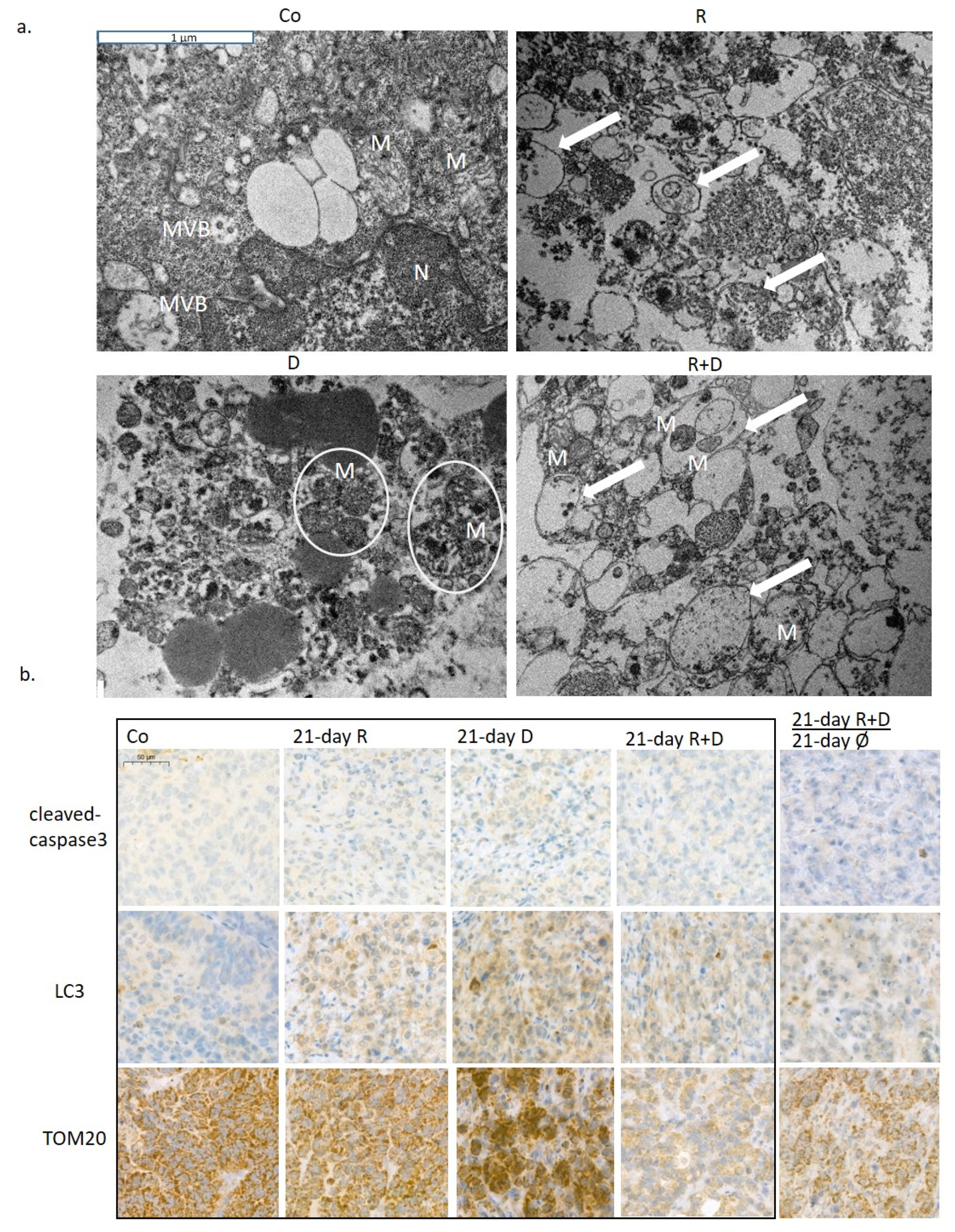
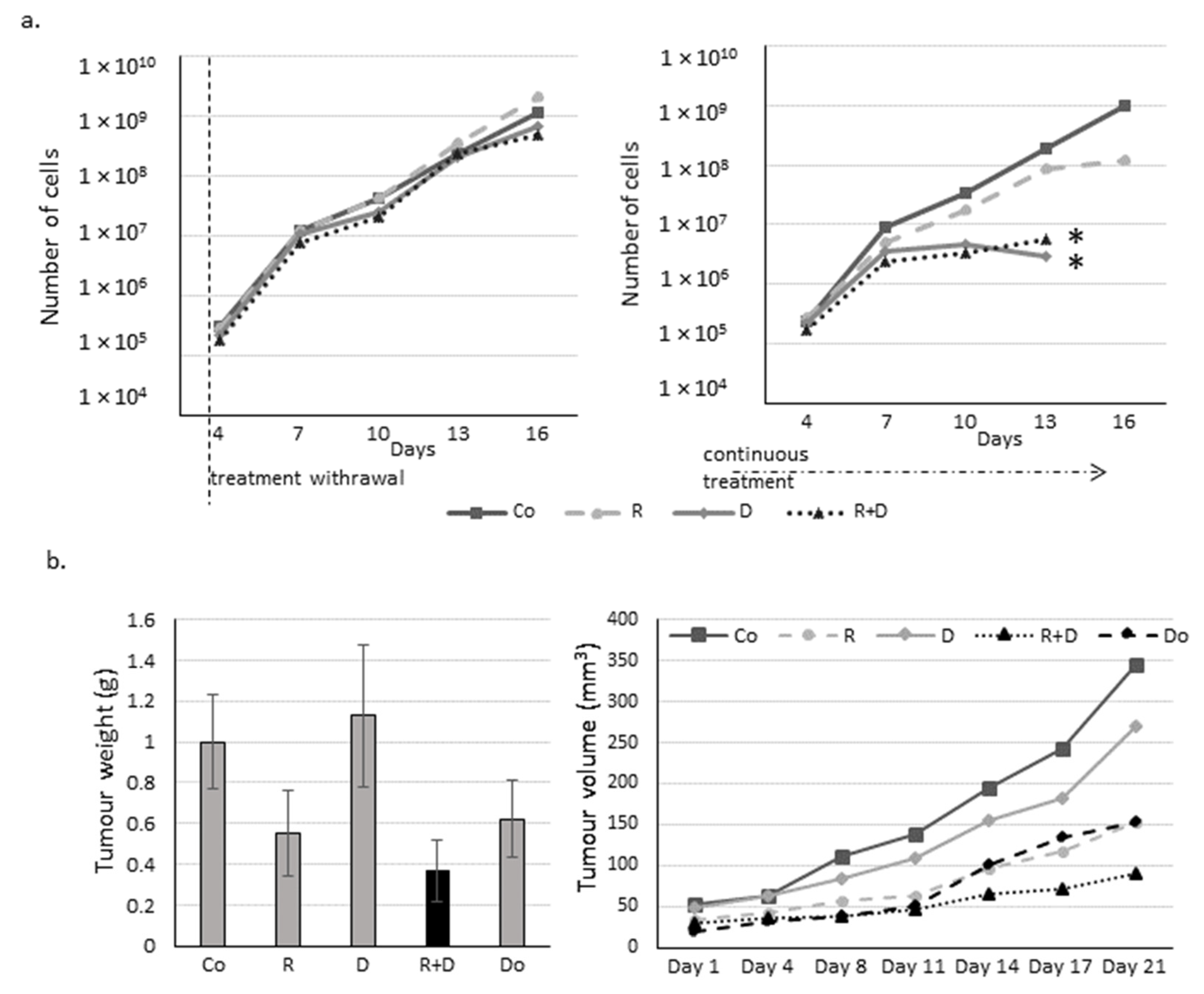
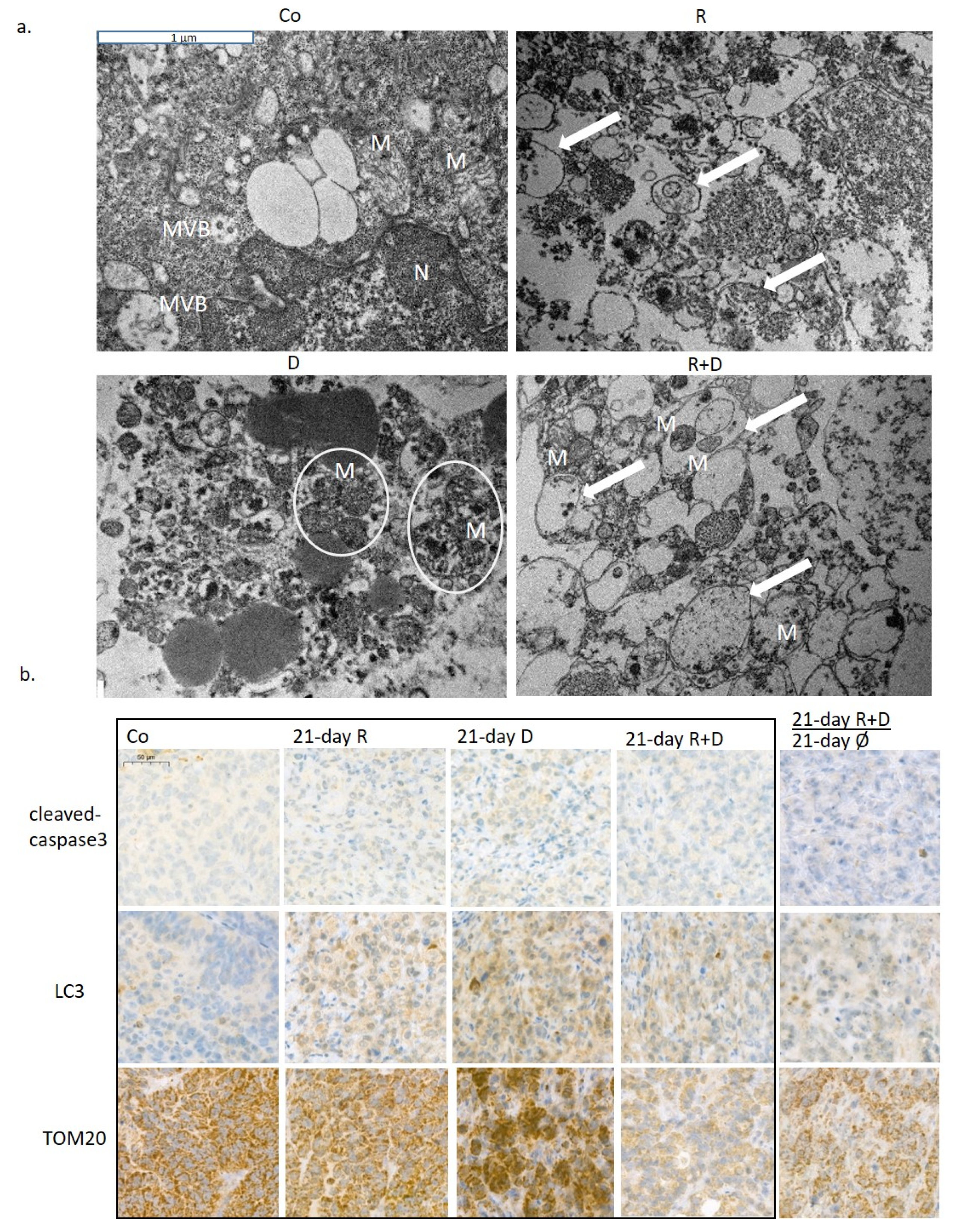
2.3. The Effect of Long-Term Treatment and Treatment Withdrawal in ZR75.1 Breast Cancer Cells In Vitro and In Vivo
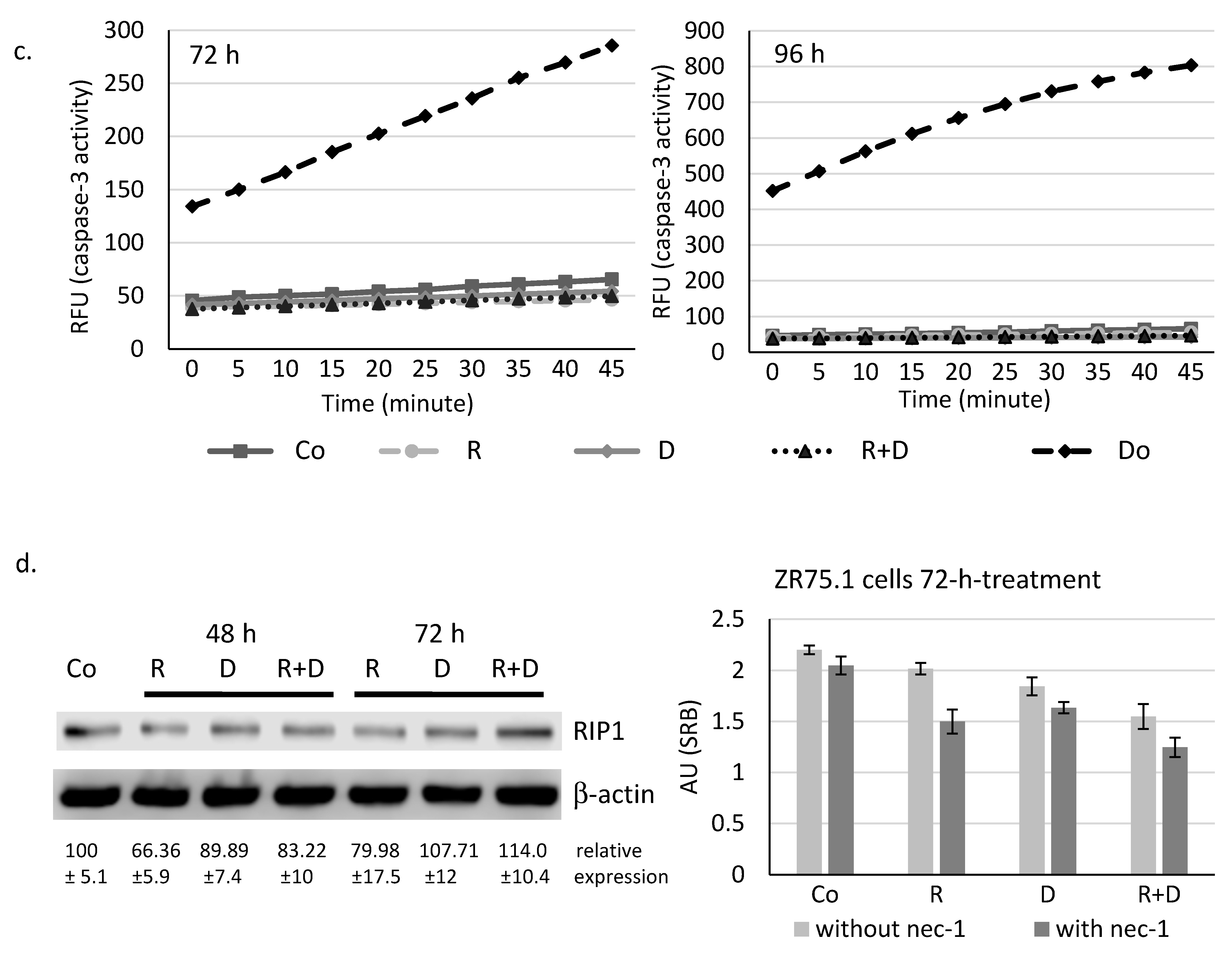
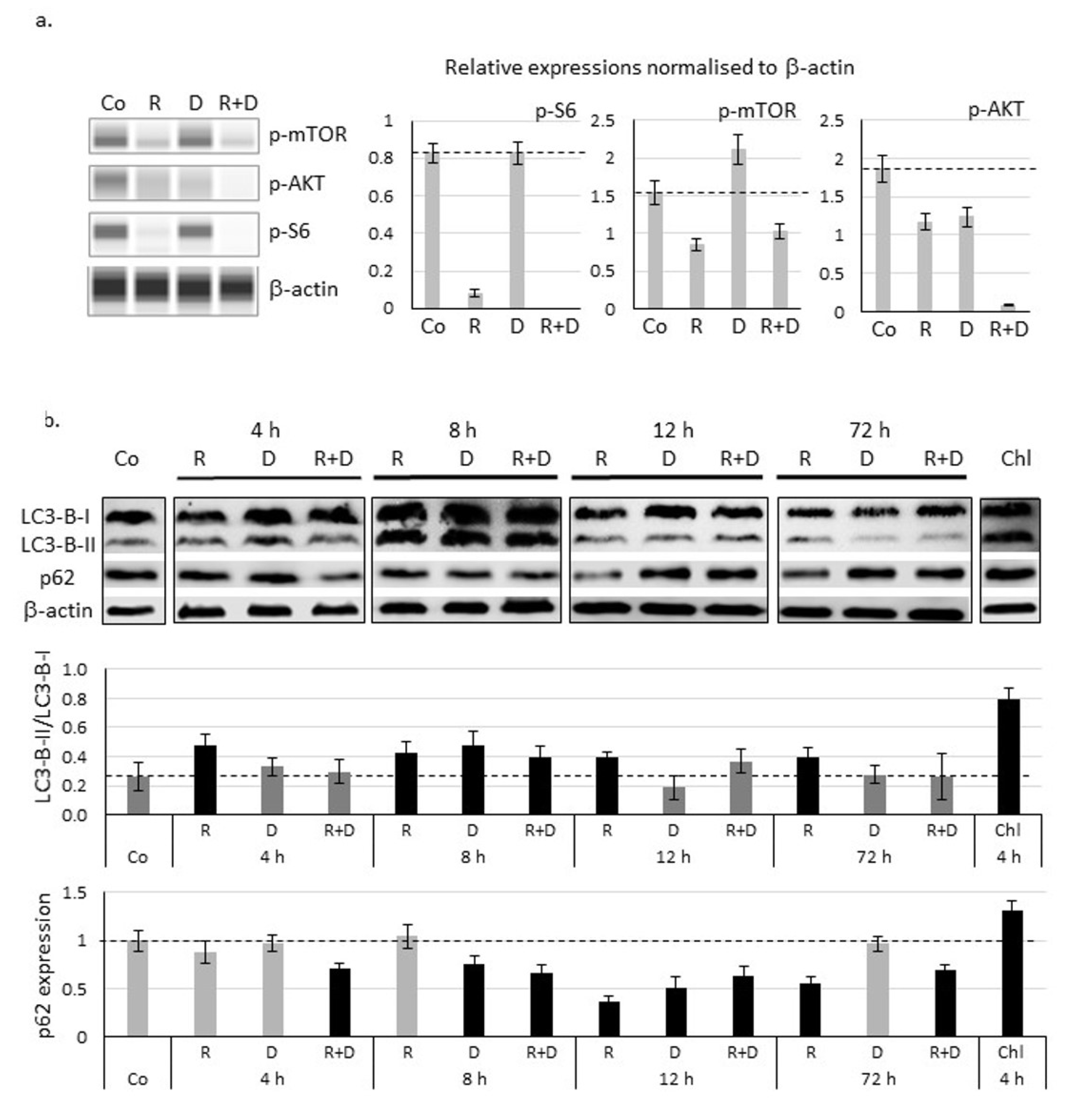
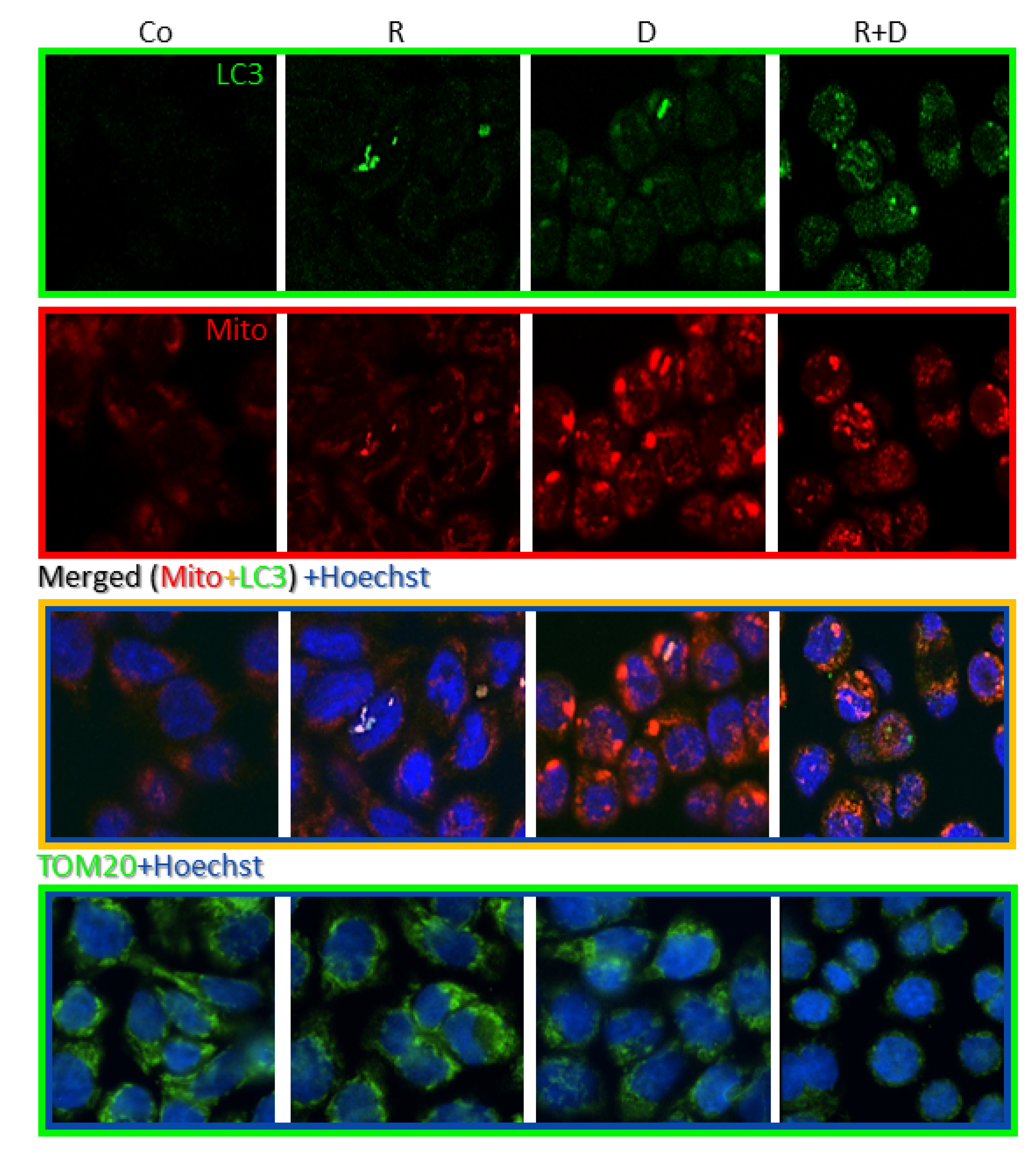
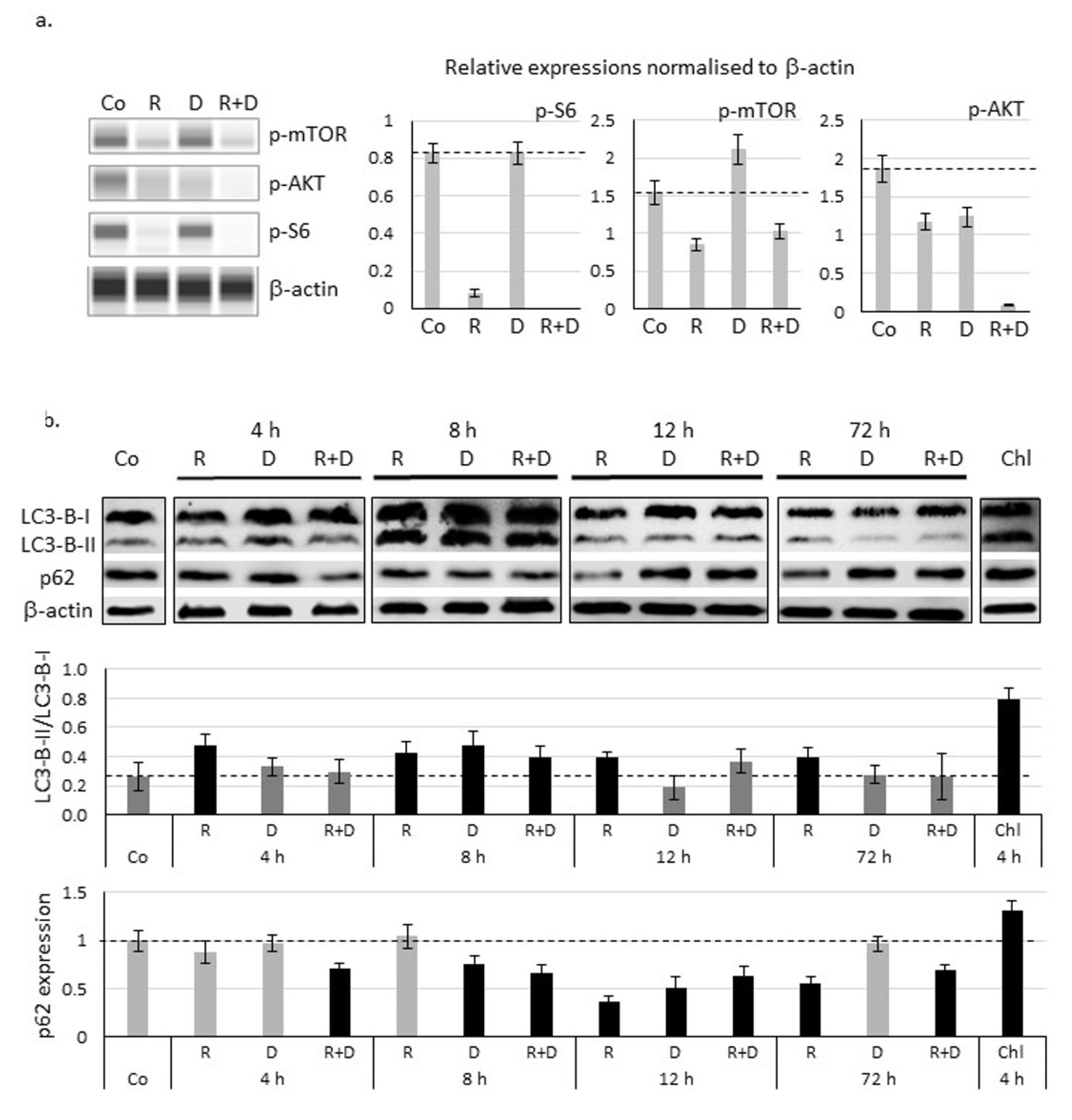
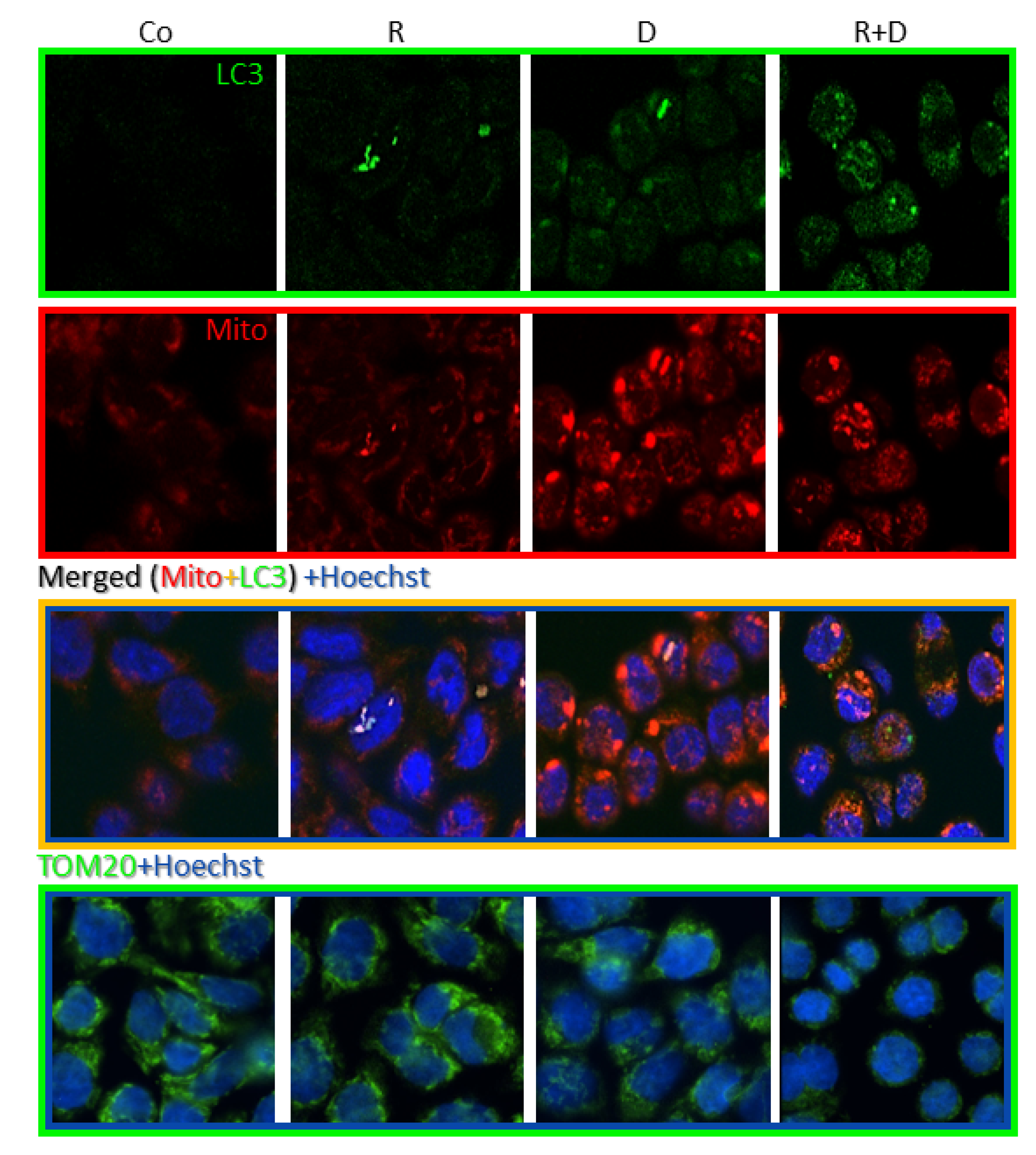
2.4. Induced Autophagy and Mitophagy in Rapamycin + Doxycycline Treatments
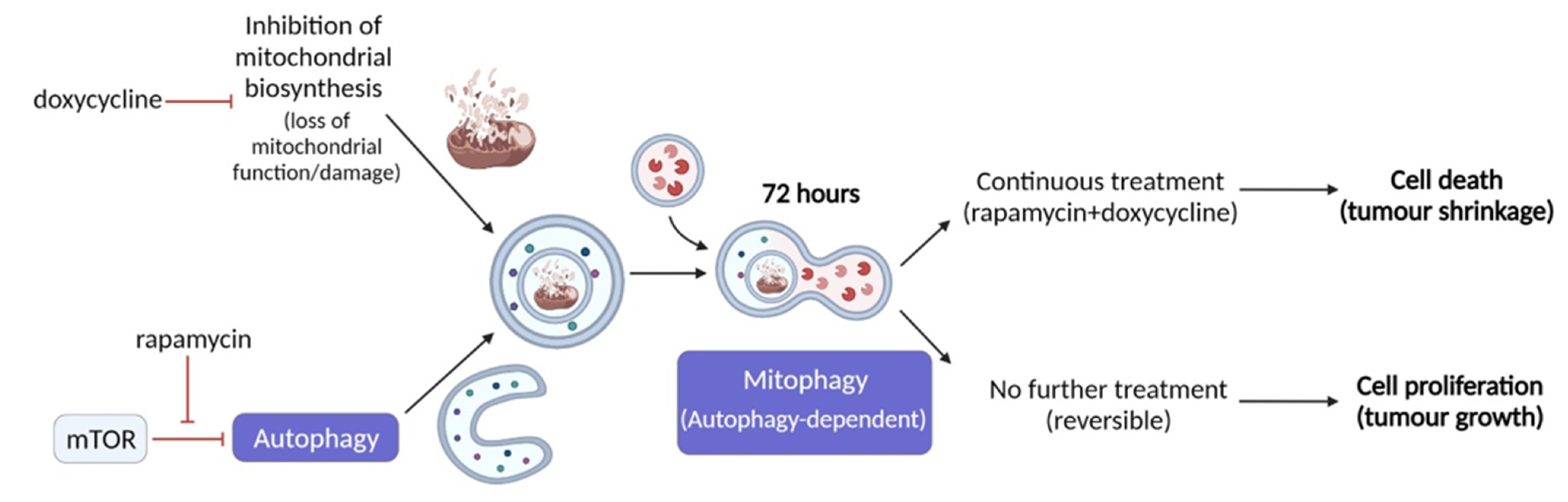
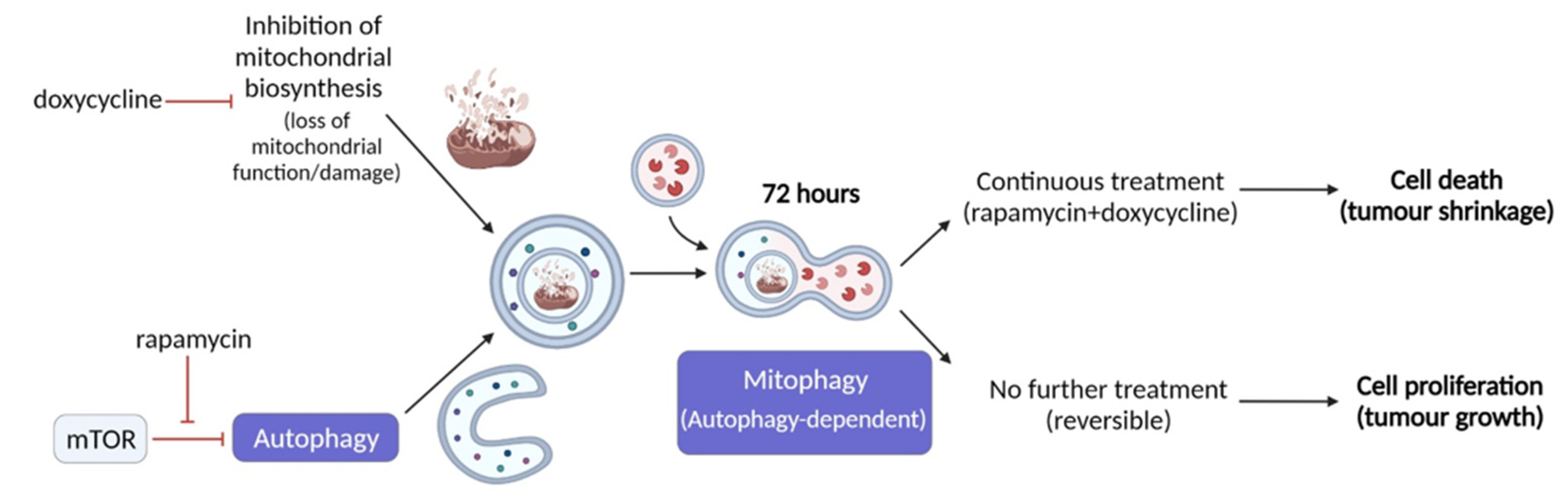
3. Discussion
4. Materials and Methods
4.1. Cell Culturing and In Vitro Treatments
4.2. In Vitro Proliferation Assays—Alamar Blue and Sulforhodamine B Assays
4.3. Protein Expression Studies: Western Blot, WesTM Simple and Immunohistochemical Analyses
4.4. Immunostainings
4.5. Flow Cytometry Measurements
4.6. In Vitro Fluorogenic Caspase-3 Assay
4.7. In Vivo Xenograft Model
4.8. Transmission Electron Microscopy
4.9. Statistical Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Harbeck, N.; Gnant, M. Breast cancer. Lancet 2017, 389, 1134–1150. [Google Scholar] [CrossRef]
- Liu, G.Y.; Sabatini, D.M. mTOR at the nexus of nutrition, growth, ageing and disease. Nat. Rev. Mol. Cell Biol. 2020, 21, 183–203. [Google Scholar] [CrossRef]
- Mossmann, D.; Park, S.; Hall, M.N. mTOR signalling and cellular metabolism are mutual determinants in cancer. Nat. Rev. Cancer 2018, 18, 744–757. [Google Scholar] [CrossRef] [PubMed]
- Teng, Q.-X.; Ashar, Y.V.; Gupta, P.; Gadee, E.; Fan, Y.-F.; Reznik, S.E.; Wurpel, J.N.; Chen, Z.-S. Revisiting mTOR inhibitors as anticancer agents. Drug Discov. Today 2019, 24, 2086–2095. [Google Scholar] [CrossRef]
- Desbats, M.A.; Giacomini, I.; Prayer-Galetti, T.; Montopoli, M. Metabolic Plasticity in Chemotherapy Resistance. Front. Oncol. 2020, 10, 281. [Google Scholar] [CrossRef] [PubMed]
- McGuirk, S.; Audet-Delage, Y.; St-Pierre, J. Metabolic Fitness and Plasticity in Cancer Progression. Trends Cancer 2020, 6, 49–61. [Google Scholar] [CrossRef] [PubMed]
- DeBerardinis, R.J.; Chandel, N.S. Fundamentals of cancer metabolism. Sci. Adv. 2016, 2, e1600200. [Google Scholar] [CrossRef] [Green Version]
- Gupta, P.B.; Pastushenko, I.; Skibinski, A.; Blanpain, C.; Kuperwasser, C. Phenotypic Plasticity: Driver of Cancer Initiation, Progression, and Therapy Resistance. Cell Stem Cell 2019, 24, 65–78. [Google Scholar] [CrossRef] [Green Version]
- Porporato, P.E.; Filigheddu, N.; Pedro, J.M.B.-S.; Kroemer, G.; Galluzzi, L. Mitochondrial metabolism and cancer. Cell Res. 2018, 28, 265–280. [Google Scholar] [CrossRef]
- Tarragó-Celada, J.; Cascante, M. Targeting the Metabolic Adaptation of Metastatic Cancer. Cancers 2021, 13, 1641. [Google Scholar] [CrossRef]
- Schiliro, C.; Firestein, B. Mechanisms of Metabolic Reprogramming in Cancer Cells Supporting Enhanced Growth and Proliferation. Cells 2021, 10, 1056. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Outschoorn, U.; Peiris-Pagés, M.; Pestell, R.G.; Sotgia, F.; Lisanti, M. Cancer metabolism: A therapeutic perspective. Nat. Rev. Clin. Oncol. 2017, 14, 11–31. [Google Scholar] [CrossRef] [PubMed]
- Zou, Z.; Tao, T.; Li, H.; Zhu, X. mTOR signaling pathway and mTOR inhibitors in cancer: Progress and challenges. Cell Biosci. 2020, 10, 1–11. [Google Scholar] [CrossRef]
- Magaway, C.; Kim, E.; Jacinto, E. Targeting mTOR and Metabolism in Cancer: Lessons and Innovations. Cells 2019, 8, 1584. [Google Scholar] [CrossRef] [Green Version]
- Anttila, J.V.; Shubin, M.; Cairns, J.; Borse, F.; Guo, Q.; Mononen, T.; Vázquez-García, I.; Pulkkinen, O.; Mustonen, V. Contrasting the impact of cytotoxic and cytostatic drug therapies on tumour progression. PLoS Comput. Biol. 2019, 15, e1007493. [Google Scholar] [CrossRef] [Green Version]
- Su, Z.; Yang, Z.; Xu, Y.; Chen, Y.; Yu, Q. Apoptosis, autophagy, necroptosis, and cancer metastasis. Mol. Cancer 2015, 14, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Denton, D.; Kumar, S. Autophagy-dependent cell death. Cell Death Differ. 2019, 26, 605–616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doherty, J.; Baehrecke, E.H. Life, death and autophagy. Nat. Cell Biol. 2018, 20, 1110–1117. [Google Scholar] [CrossRef]
- Kriel, J.; Loos, B. The good, the bad and the autophagosome: Exploring unanswered questions of autophagy-dependent cell death. Cell Death Differ. 2019, 26, 640–652. [Google Scholar] [CrossRef] [Green Version]
- Condon, K.J.; Sabatini, D.M. Nutrient regulation of mTORC1 at a glance. J. Cell Sci. 2019, 132. [Google Scholar] [CrossRef]
- Koromilas, A.E. M(en)TORship lessons on life and death by the integrated stress response. Biochim. Biophys. Acta Gen. Subj. 2019, 1863, 644–649. [Google Scholar] [CrossRef]
- Sticz, T.; Molnár, A.; Dankó, T.; Hujber, Z.; Petővári, G.; Nagy, N.; Végső, G.; Kopper, L.; Sebestyén, A. The Effects of Different mTOR Inhibitors in EGFR Inhibitor Resistant Colon Carcinoma Cells. Pathol. Oncol. Res. 2019, 25, 1379–1386. [Google Scholar] [CrossRef]
- Márk, Á.; Hajdu, M.; Váradi, Z.; Sticz, T.B.; Nagy, N.; Csomor, J.; Berczi, L.; Varga, V.; Csóka, M.; Kopper, L.; et al. Characteristic mTOR activity in Hodgkin-lymphomas offers a potential therapeutic target in high risk disease–a combined tissue microarray, in vitro and in vivo study. BMC Cancer 2013, 13, 250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petővári, G.; Dankó, T.; Krencz, I.; Hujber, Z.; Rajnai, H.; Vetlényi, E.; Raffay, R.; Pápay, J.; Jeney, A.; Sebestyén, A. Inhibition of Metabolic Shift can Decrease Therapy Resistance in Human High-Grade Glioma Cells. Pathol. Oncol. Res. 2020, 26, 23–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ózsvári, B.; Sotgia, F.; Lisanti, M.P. First-in-class candidate therapeutics that target mitochondria and effectively prevent cancer cell metastasis: Mitoriboscins and TPP compounds. Aging 2020, 12, 10162–10179. [Google Scholar] [CrossRef]
- Kim, M.-Y.; Kruger, A.J.; Jeong, J.-Y.; Kim, J.; Shin, P.-K.; Kim, S.Y.; Cho, J.Y.; Hahm, K.B.; Hong, S.P. Combination Therapy with a PI3K/mTOR Dual Inhibitor and Chloroquine Enhances Synergistic Apoptotic Cell Death in Epstein-Barr Virus-Infected Gastric Cancer Cells. Mol. Cells 2019, 42, 448–459. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.; Murthy, A. Targeting Autophagy to Treat Cancer: Challenges and Opportunities. Front. Pharmacol. 2020, 11. [Google Scholar] [CrossRef]
- Petővári, G.; Dankó, T.; Tőkés, A.-M.; Vetlényi, E.; Krencz, I.; Raffay, R.; Hajdu, M.; Sztankovics, D.; Németh, K.; Vellai-Takács, K.; et al. In Situ Metabolic Characterisation of Breast Cancer and Its Potential Impact on Therapy. Cancers 2020, 12, 2492. [Google Scholar] [CrossRef]
- Lamming, D.; Sabatini, D.M. A Central Role for mTOR in Lipid Homeostasis. Cell Metab. 2013, 18, 465–469. [Google Scholar] [CrossRef] [Green Version]
- Ma, L.; Zong, X. Metabolic Symbiosis in Chemoresistance: Refocusing the Role of Aerobic Glycolysis. Front. Oncol. 2020, 10, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kezic, A.; Popovic, L.; Lalic, K. mTOR Inhibitor Therapy and Metabolic Consequences: Where Do We Stand? Oxid. Med. Cell. Longev. 2018, 2018, 1–8. [Google Scholar] [CrossRef]
- Zhang, X.; Li, X.-R.; Zhang, J. Current status and future perspectives of PI3K and mTOR inhibitor as anticancer drugs in breast cancer. Curr. Cancer Drug Targets 2013, 13, 175–187. [Google Scholar] [CrossRef] [PubMed]
- Patel, J.; Nguyen, S.A.; Ogretmen, B.; Gutkind, J.S.; Nathan, C.-A.; Day, T. mTOR inhibitor use in head and neck squamous cell carcinoma: A meta-analysis on survival, tumor response, and toxicity. Laryngoscope 2020, 5, 243–255. [Google Scholar] [CrossRef] [Green Version]
- Zhu, M.; Molina, J.R.; Dy, G.K.; Croghan, G.A.; Qi, Y.; Glockner, J.; Hanson, L.J.; Roos, M.M.; Tan, A.D.; Adjei, A.A. A phase I study of the VEGFR kinase inhibitor vatalanib in combination with the mTOR inhibitor, everolimus, in patients with advanced solid tumors. Investig. New Drugs 2020, 38, 1755–1762. [Google Scholar] [CrossRef]
- Lorenzen, S.; Knorrenschild, J.R.; Pauligk, C.; Hegewisch-Becker, S.; Seraphin, J.; Thuss-Patience, P.; Kopp, H.G.; Dechow, T.; Vogel, A.; Luley, K.B.; et al. Phase III randomized, double-blind study of paclitaxel with and without everolimus in patients with advanced gastric or esophagogastric junction carcinoma who have progressed after therapy with a fluoropyrimidine/platinum-containing regimen (RADPAC). Int. J. Cancer 2020, 147, 2493–2502. [Google Scholar] [CrossRef]
- Li, J.; Kim, S.G.; Blenis, J. Rapamycin: One Drug, Many Effects. Cell Metab. 2014, 19, 373–379. [Google Scholar] [CrossRef] [Green Version]
- Nunes, T.; Hamdan, D.; Leboeuf, C.; El Bouchtaoui, M.; Gapihan, G.; Nguyen, T.T.; Meles, S.; Angeli, E.; Ratajczak, P.; Lu, H.; et al. Targeting Cancer Stem Cells to Overcome Chemoresistance. Int. J. Mol. Sci. 2018, 19, 4036. [Google Scholar] [CrossRef] [Green Version]
- Ilagan, E.; Manning, B.D. Emerging Role of mTOR in the Response to Cancer Therapeutics. Trends Cancer 2016, 2, 241–251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guri, Y.; Hall, M.N. mTOR Signaling Confers Resistance to Targeted Cancer Drugs. Trends Cancer 2016, 2, 688–697. [Google Scholar] [CrossRef]
- Chen, X.; Chen, S.; Yu, D. Metabolic Reprogramming of Chemoresistant Cancer Cells and the Potential Significance of Metabolic Regulation in the Reversal of Cancer Chemoresistance. Metabolites 2020, 10, 289. [Google Scholar] [CrossRef] [PubMed]
- Götting, I.; Jendrossek, V.; Matschke, J. A New Twist in Protein Kinase B/Akt Signaling: Role of Altered Cancer Cell Metabolism in Akt-Mediated Therapy Resistance. Int. J. Mol. Sci. 2020, 21, 8563. [Google Scholar] [CrossRef] [PubMed]
- Garcia, V.; López-Urrutia, E.; Villanueva-Sánchez, O.; Ávila-Rodríguez, M.Á.; Zentella-Dehesa, A.; Cortés-González, C.; López-Camarillo, C.; Jacobo-Herrera, N.J.; Perez-Plasencia, C. Targeting Metabolic Remodeling in Triple Negative Breast Cancer in a Murine Model. J. Cancer 2017, 8, 178–189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Bai, W. Repression of phosphoglycerate dehydrogenase sensitizes triple-negative breast cancer to doxorubicin. Cancer Chemother. Pharmacol. 2016, 78, 655–659. [Google Scholar] [CrossRef]
- Varghese, E.; Samuel, S.M.; Líšková, A.; Samec, M.; Kubatka, P.; Büsselberg, D. Targeting Glucose Metabolism to Overcome Resistance to Anticancer Chemotherapy in Breast Cancer. Cancers 2020, 12, 2252. [Google Scholar] [CrossRef]
- Fiorillo, M.; Tóth, F.; Sotgia, F.; Lisanti, M.P. Doxycycline, Azithromycin and Vitamin C (DAV): A potent combination therapy for targeting mitochondria and eradicating cancer stem cells (CSCs). Aging 2019, 11, 2202–2216. [Google Scholar] [CrossRef] [PubMed]
- Dong, L.; Gopalan, V.; Holland, O.; Neuzil, J. Mitocans Revisited: Mitochondrial Targeting as Efficient Anti-cancer Therapy. Int. J. Mol. Sci. 2020, 21, 7941. [Google Scholar] [CrossRef]
- Lee, N.; Woodrum, C.L.; Nobil, A.M.; Rauktys, A.E.; Messina, M.P.; Dabora, S.L. Rapamycin weekly maintenance dosing and the potential efficacy of combination sorafenib plus rapamycin but not atorvastatin or doxycycline in tuberous sclerosis preclinical models. BMC Pharmacol. 2009, 9, 8. [Google Scholar] [CrossRef] [Green Version]
- Scatena, C.; Roncella, M.; Di Paolo, A.; Aretini, P.; Menicagli, M.; Fanelli, G.; Marini, C.; Mazzanti, C.M.; Ghilli, M.; Sotgia, F.; et al. Doxycycline, an Inhibitor of Mitochondrial Biogenesis, Effectively Reduces Cancer Stem Cells (CSCs) in Early Breast Cancer Patients: A Clinical Pilot Study. Front. Oncol. 2018, 8, 452. [Google Scholar] [CrossRef]
- Helmink, B.A.; Khan, M.A.W.; Hermann, A.; Gopalakrishnan, V.; Wargo, J.A. The microbiome, cancer, and cancer therapy. Nat. Med. 2019, 25, 377–388. [Google Scholar] [CrossRef]
- Yoshida, G.J. Therapeutic strategies of drug repositioning targeting autophagy to induce cancer cell death: From pathophysiology to treatment. J. Hematol. Oncol. 2017, 10, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Gremke, N.; Polo, P.; Dort, A.; Schneikert, J.; Elmshäuser, S.; Brehm, C.; Klingmüller, U.; Schmitt, A.; Reinhardt, H.C.; Timofeev, O.; et al. mTOR-mediated cancer drug resistance suppresses autophagy and generates a druggable metabolic vulnerability. Nat. Commun. 2020, 11, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Levine, B.L. Autosis and autophagic cell death: The dark side of autophagy. Cell Death Differ. 2015, 22, 367–376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, D.R.; Thorburn, A. Autophagy and organelle homeostasis in cancer. Dev. Cell 2021, 56, 906–918. [Google Scholar] [CrossRef] [PubMed]
- Bellot, G.; Garcia-Medina, R.; Gounon, P.; Chiche, J.; Roux, D.; Pouysségur, J.; Mazure, N.M. Hypoxia-Induced Autophagy Is Mediated through Hypoxia-Inducible Factor Induction of BNIP3 and BNIP3L via Their BH3 Domains. Mol. Cell. Biol. 2009, 29, 2570–2581. [Google Scholar] [CrossRef] [Green Version]
- Hui, L.; Wu, H.; Wang, T.-W.; Yang, N.; Guo, X.; Jang, X.-J. Hydrogen peroxide-induced mitophagy contributes to laryngeal cancer cells survival via the upregulation of FUNDC1. Clin. Transl. Oncol. 2019, 21, 596–606. [Google Scholar] [CrossRef]
- Egan, D.F.; Shackelford, D.B.; Mihaylova, M.M.; Gelino, S.; Kohnz, R.A.; Mair, W.; Vasquez, D.S.; Joshi, A.; Gwinn, D.M.; Taylor, R.; et al. Phosphorylation of ULK1 (hATG1) by AMP-Activated Protein Kinase Connects Energy Sensing to Mitophagy. Science 2011, 331, 456–461. [Google Scholar] [CrossRef] [Green Version]
- Yan, C.; Luo, L.; Guo, C.-Y.; Goto, S.; Urata, Y.; Shao, J.-H.; Li, T.-S. Doxorubicin-induced mitophagy contributes to drug resistance in cancer stem cells from HCT8 human colorectal cancer cells. Cancer Lett. 2017, 388, 34–42. [Google Scholar] [CrossRef]
- Gazzano, E.; Lazzarato, L.; Rolando, B.; Kopecka, J.; Guglielmo, S.; Costamagna, C.; Chegaev, K.; Riganti, C. Mitochondrial Delivery of Phenol Substructure Triggers Mitochondrial Depolarization and Apoptosis of Cancer Cells. Front. Pharmacol. 2018, 9, 580. [Google Scholar] [CrossRef] [PubMed]
- Biel, T.G.; Rao, V.A. Mitochondrial dysfunction activates lysosomal-dependent mitophagy selectively in cancer cells. Oncotarget 2017, 9, 995–1011. [Google Scholar] [CrossRef] [Green Version]
- Ney, P.A. Mitochondrial autophagy: Origins, significance, and role of BNIP3 and NIX. Biochim. Biophys. Acta Gen. Subj. 2015, 1853, 2775–2783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foucquier, J.; Guedj, M. Analysis of drug combinations: Current methodological landscape. Pharmacol. Res. Perspect. 2015, 3, e00149. [Google Scholar] [CrossRef] [PubMed]
- Adami, R.; Pagano, J.; Colombo, M.; Platonova, N.; Recchia, D.; Chiaramonte, R.; Bottinelli, R.; Canepari, M.; Bottai, D. Reduction of Movement in Neurological Diseases: Effects on Neural Stem Cells Characteristics. Front. Neurosci. 2018, 12, 336. [Google Scholar] [CrossRef] [PubMed]
- Nagy, N.; Hajdu, M.; Márk, Á.; Király, P.A.; Tóth, M.; Dankó, T.; Csóka, M.; Sebestyén, A. Growth inhibitory effect of rapamycin in Hodgkin-lymphoma cell lines characterized by constitutive NOTCH1 activation. Tumor Biol. 2016, 37, 13695–13704. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dankó, T.; Petővári, G.; Sztankovics, D.; Moldvai, D.; Raffay, R.; Lőrincz, P.; Visnovitz, T.; Zsiros, V.; Barna, G.; Márk, Á.; et al. Rapamycin Plus Doxycycline Combination Affects Growth Arrest and Selective Autophagy-Dependent Cell Death in Breast Cancer Cells. Int. J. Mol. Sci. 2021, 22, 8019. https://doi.org/10.3390/ijms22158019
Dankó T, Petővári G, Sztankovics D, Moldvai D, Raffay R, Lőrincz P, Visnovitz T, Zsiros V, Barna G, Márk Á, et al. Rapamycin Plus Doxycycline Combination Affects Growth Arrest and Selective Autophagy-Dependent Cell Death in Breast Cancer Cells. International Journal of Molecular Sciences. 2021; 22(15):8019. https://doi.org/10.3390/ijms22158019
Chicago/Turabian StyleDankó, Titanilla, Gábor Petővári, Dániel Sztankovics, Dorottya Moldvai, Regina Raffay, Péter Lőrincz, Tamás Visnovitz, Viktória Zsiros, Gábor Barna, Ágnes Márk, and et al. 2021. "Rapamycin Plus Doxycycline Combination Affects Growth Arrest and Selective Autophagy-Dependent Cell Death in Breast Cancer Cells" International Journal of Molecular Sciences 22, no. 15: 8019. https://doi.org/10.3390/ijms22158019
APA StyleDankó, T., Petővári, G., Sztankovics, D., Moldvai, D., Raffay, R., Lőrincz, P., Visnovitz, T., Zsiros, V., Barna, G., Márk, Á., Krencz, I., & Sebestyén, A. (2021). Rapamycin Plus Doxycycline Combination Affects Growth Arrest and Selective Autophagy-Dependent Cell Death in Breast Cancer Cells. International Journal of Molecular Sciences, 22(15), 8019. https://doi.org/10.3390/ijms22158019