
Regulatory Macrophages and Tolerogenic Dendritic Cells in Myeloid Regulatory Cell-Based Therapies


Abstract
1. Introduction
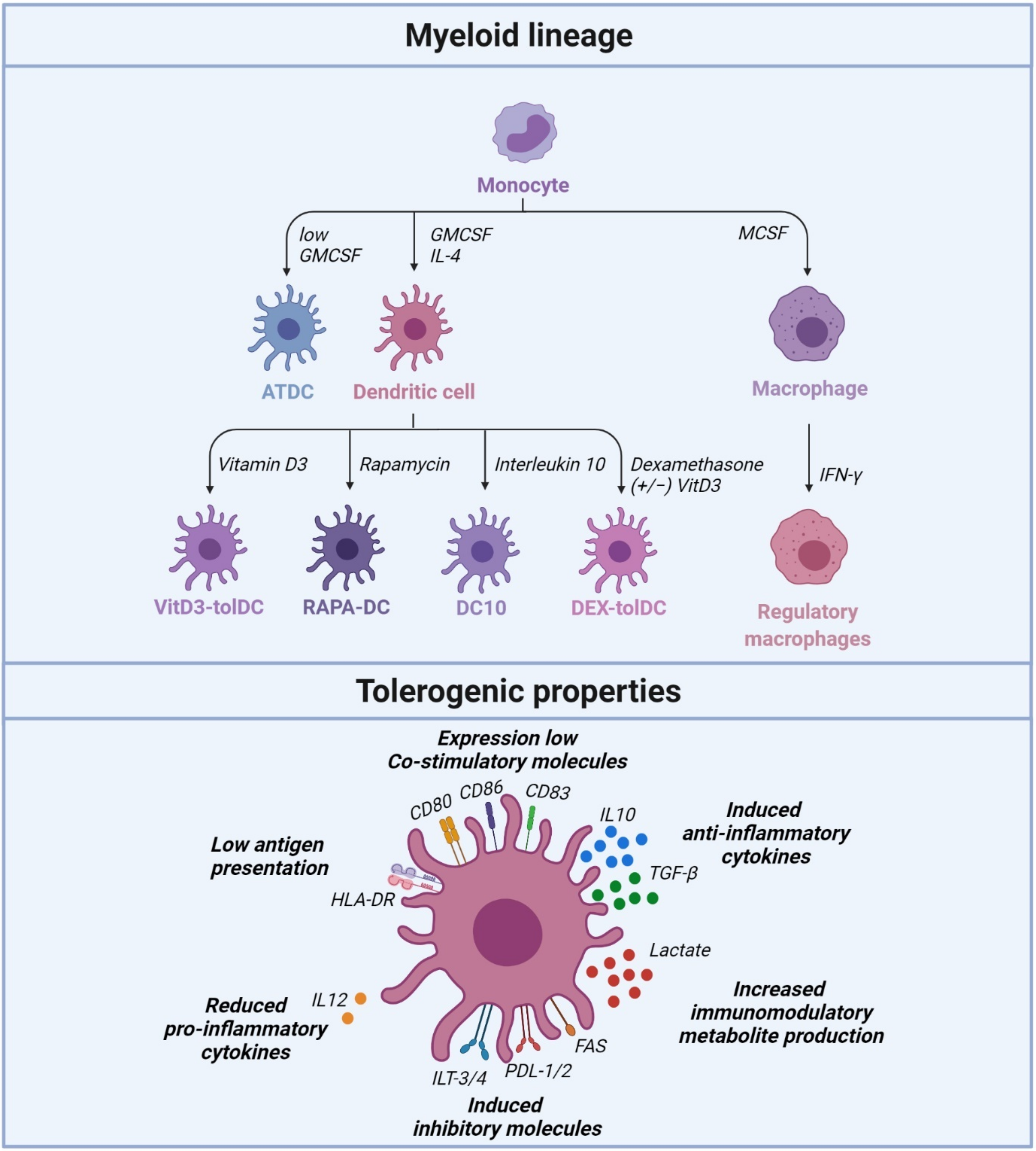
2. Tolerogenic Dendritic Cells Regulate Peripheral Tolerance
2.1. Vitamin D3-Induced Tolerogenic Dendritic Cells (VITD3-tolDC)
2.2. Dexamethasone/VitaminD3-Tolerogenic Dendritic Cells (DEX/VITD3-tolDC)
2.3. Rapamycin-Treated Dendritic Cells (RAPA-DC)
2.4. Interleukin 10-Dendritic Cells (DC10)
2.5. Autologous Tolerogenic Dendritic Cells (ATDC)
3. Regulatory Macrophages Regulating Peripheral Tolerance
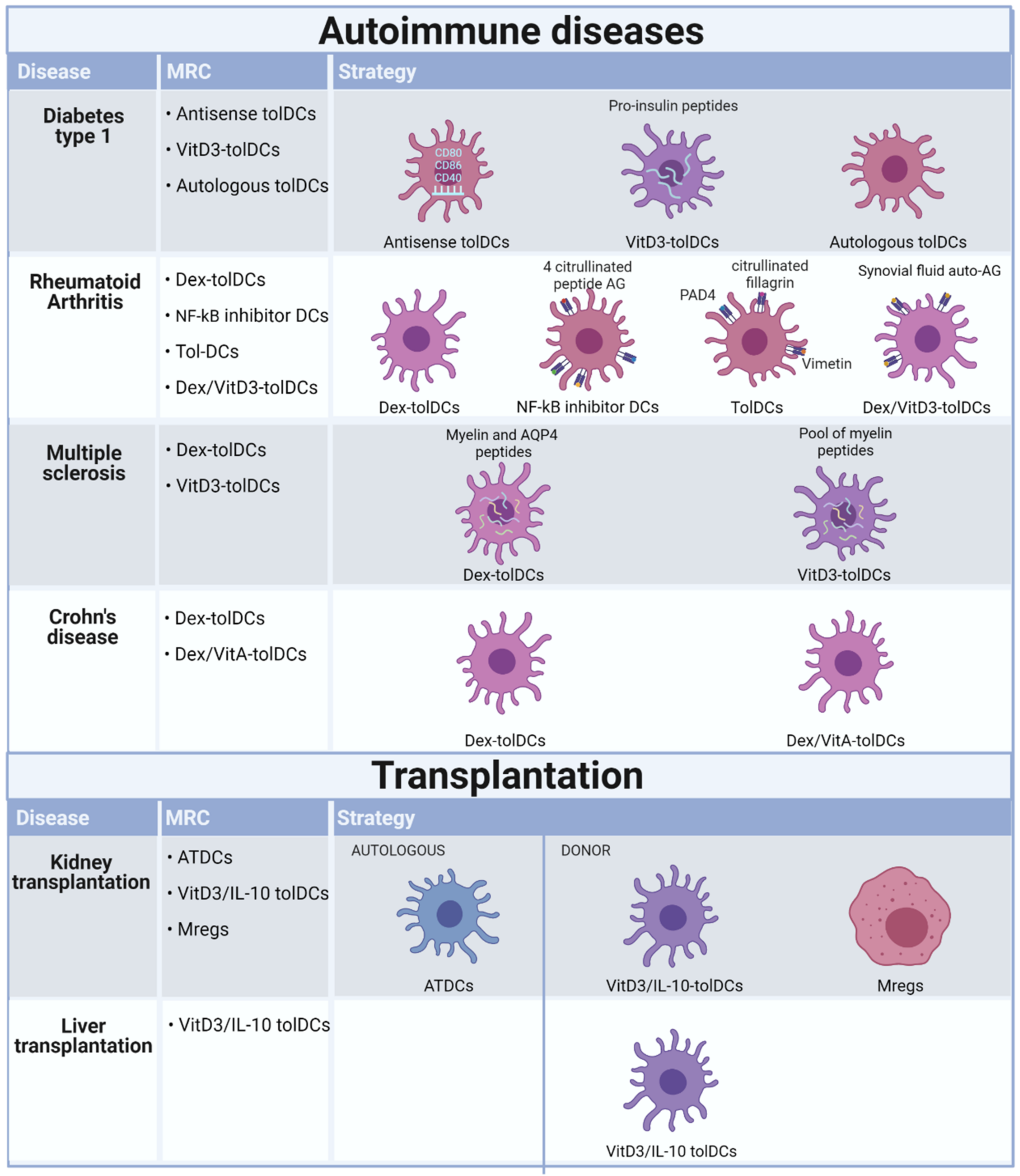
4. Completed and Ongoing Clinical Trials Investigating Tolerogenic Application of Myeloid Regulatory Cells in Autoimmune Diseases
5. Completed and Ongoing Clinical Trials Investigating Tolerogenic Application of Myeloid Regulatory Cells in Transplantation
6. Future Perspectives of a Myeloid-Regulated Tolerogenic Environment in Immunological Therapies
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Park, H.W. Managing failure: Sir peter brian medawar’s transplantation research. Notes Rec. 2018, 72, 75–100. [Google Scholar] [CrossRef]
- Billingham, R.E.; Brent, L.; Medawar, P.B. Actively acquired tolerance of foreign cells. Nature 1953, 172, 603–606. [Google Scholar] [CrossRef]
- Starlz TE, S.T. Peter Brian Medawar: Father of transplantation. J. Am. Coll. Surg. 1995, 180, 332–336. [Google Scholar]
- Morante-Palacios, O.; Fondelli, F.; Ballestar, E.; Martínez-Cáceres, E.M. Tolerogenic Dendritic Cells in Autoimmunity and Inflammatory Diseases. Trends Immunol. 2021, 42, 59–75. [Google Scholar] [CrossRef]
- Hutchinson, J.A.; Ahrens, N.; Geissler, E.K. MITAP-compliant characterization of human regulatory macrophages. Transpl. Int. 2017, 30, 765–775. [Google Scholar] [CrossRef]
- Wekerle, T.; Segev, D.; Lechler, R.; Oberbauer, R. Strategies for long-term preservation of kidney graft function. Lancet 2017, 389, 2152–2162. [Google Scholar] [CrossRef]
- Kono, H.; Rock, K.L. How dying cells alert the immune system to danger. Nat. Rev. Immunol. 2008, 8, 279–289. [Google Scholar] [CrossRef] [PubMed]
- Erwig, L.-P.; Henson, P.M. Immunological consequences of apoptotic cell phagocytosis. Am. J. Pathol. 2007, 171, 2–8. [Google Scholar] [CrossRef]
- Mosser, D.M.; Edwards, J.P. Exploring the full spectrum of macrophage activation. Nat. Rev. Immunol. 2008, 8, 958–969. [Google Scholar] [CrossRef] [PubMed]
- Gordon, S. Alternative activation of macrophages. Nat. Rev. Immunol. 2003, 3, 23–35. [Google Scholar] [CrossRef]
- Edwards, J.P.; Zhang, X.; Frauwirth, K.A.; Mosser, D.M. Biochemical and functional characterization of three activated macrophage populations. J. Leukoc. Biol. 2006, 80, 1298–1307. [Google Scholar] [CrossRef]
- Banchereau, J.; Steinman, R.M. Dendritic cells and the control of immunity. Nature 1998, 392, 245–252. [Google Scholar] [CrossRef]
- Steinbrink, K.; Wölfl, M.; Jonuleit, H.; Knop, J.; Enk, A.H. Induction of tolerance by IL-10-treated dendritic cells. J. Immunol. 1997, 159, 4772–4780. [Google Scholar]
- Fu, F.; Li, Y.; Qian, S.; Lu, L.; Chambers, F.; Starzl, T.E.; Fung, J.J.; Thomson, A.W. Costimulatory molecule-deficient dendritic cell progenitors (MHC class II+, CD80dim, CD86-) prolong cardiac allograft survival in nonimmunosuppressed recipients. Transplantation 1996, 62, 659–665. [Google Scholar] [CrossRef]
- Hawiger, D.; Inaba, K.; Dorsett, Y.; Guo, M.; Mahnke, K.; Rivera, M.; Ravetch, J.V.; Steinman, R.M.; Nussenzweig, M.C. Dendritic cells induce peripheral T cell unresponsiveness under steady state conditions in vivo. J. Exp. Med. 2001, 194, 769–779. [Google Scholar] [CrossRef]
- Hutchinson, J.A.; Geissler, E.K. Now or never? The case for cell-based immunosuppression in kidney transplantation. Kidney Int. 2015, 87, 1116–1124. [Google Scholar] [CrossRef] [PubMed]
- Marín, E.; Cuturi, M.C.; Moreau, A. Tolerogenic dendritic cells in solid organ transplantation: Where do we stand? Front. Immunol. 2018, 9, 274. [Google Scholar] [CrossRef] [PubMed]
- Sandri, S.; De Sanctis, F.; Lamolinara, A.; Boschi, F.; Poffe, O.; Trovato, R.; Fiore, A.; Sartori, S.; Sbarbati, A.; Bondanza, A.; et al. Effective control of acute myeloid leukaemia and acute lymphoblastic leukaemia progression by telomerase specific adoptive T-cell therapy. Oncotarget 2017, 8, 86987–87001. [Google Scholar] [CrossRef] [PubMed]
- Dahlberg, C.I.M.; Sarhan, D.; Chrobok, M.; Duru, A.D.; Alici, E. Natural killer cell-based therapies targeting cancer: Possible strategies to gain and sustain anti-tumor activity. Front. Immunol. 2015, 6, 605. [Google Scholar] [CrossRef]
- De Sanctis, F.; Sandri, S.; Martini, M.; Mazzocco, M.; Fiore, A.; Trovato, R.; Garetto, S.; Brusa, D.; Ugel, S.; Sartoris, S. Hyperthermic treatment at 56 °C induces tumour-specific immune protection in a mouse model of prostate cancer in both prophylactic and therapeutic immunization regimens. Vaccine 2018, 36, 3708–3716. [Google Scholar] [CrossRef] [PubMed]
- Banchereau, J.; Palucka, A.K. Dendritic cells as therapeutic vaccines against cancer. Nat. Rev. Immunol. 2005, 5, 296–306. [Google Scholar] [CrossRef] [PubMed]
- Saxena, M.; van der Burg, S.H.; Melief, C.J.M.; Bhardwaj, N. Therapeutic cancer vaccines. Nat. Rev. Cancer. 2021, 21, 360–378. [Google Scholar] [CrossRef]
- Dáňová, K.; Grohová, A.; Strnadová, P.; Funda, D.P.; Šumník, Z.; Lebl, J.; Cinek, O.; Průhová, Š.; Koloušková, S.; Obermannová, B.; et al. Tolerogenic dendritic cells from poorly compensated type 1 diabetes patients have decreased ability to induce stable antigen-specific T cell hyporesponsiveness and generation of suppressive regulatory T cells. J. Immunol. 2017, 198, 729–740. [Google Scholar] [CrossRef]
- Waldmann, H.; Graca, L. Infectious tolerance. What are we missing? Cell. Immunol. 2020, 354, 104152. [Google Scholar] [CrossRef]
- Kendal, A.R.; Chen, Y.; Regateiro, F.S.; Ma, J.; Adams, E.; Cobbold, S.P.; Hori, S.; Waldmann, H. Sustained suppression by Foxp3+ regulatory T cells is vital for infectious transplantation tolerance. J. Exp. Med. 2011, 208, 2043–2053. [Google Scholar] [CrossRef]
- Giannoukakis, N.; Phillips, B.; Finegold, D.; Harnaha, J.; Trucco, M. Phase I (safety) study of autologous Tolerogenic dendritic cells in type 1 diabetic patients. Diabetes Care 2011, 34, 2026–2032. [Google Scholar] [CrossRef]
- Kurochkina, Y.; Tikhonova, M.; Tyrinova, T.; Leplina, O.; Sizikov, A.; Sulutian, A.; Chumasova, O.; Ostanin, A.; Chernykh, E. SAT0212 The safety and tolerability of intra-articular injection of tolerogenic dendritic cells in patients with rheumatoid arthritis: The preliminary results. Ann. Rheum. Dis. 2018, 77, 966–967. [Google Scholar]
- Benham, H.; Nel, H.J.; Law, S.C.; Mehdi, A.M.; Street, S.; Ramnoruth, N.; Pahau, H.; Lee, B.T.; Ng, J.; Brunck, M.E.G.; et al. Citrullinated peptide dendritic cell immunotherapy in HLA risk genotype-positive rheumatoid arthritis patients. Sci. Transl. Med. 2015, 7, 290ra87. [Google Scholar] [CrossRef] [PubMed]
- Bell, G.M.; Anderson, A.E.; Diboll, J.; Reece, R.; Eltherington, O.; Harry, R.A.; Fouweather, T.; MacDonald, C.; Chadwick, T.; McColl, E.; et al. Autologous tolerogenic dendritic cells for rheumatoid and inflammatory arthritis. Ann. Rheum. Dis. 2017, 76, 227–234. [Google Scholar] [CrossRef] [PubMed]
- Zubizarreta, I.; Flórez-Grau, G.; Vila, G.; Cabezón, R.; España, C.; Andorra, M.; Saiz, A.; Llufriu, S.; Sepulveda, M.; Sola-Valls, N.; et al. Immune tolerance in multiple sclerosis and neuromyelitis optica with peptide-loaded tolerogenic dendritic cells in a phase 1b trial. Proc. Natl. Acad. Sci. USA 2019, 116, 8463–8470. [Google Scholar] [CrossRef] [PubMed]
- Willekens, B.; Presas-Rodríguez, S.; Mansilla, M.; Derdelinckx, J.; Lee, W.-P.; Nijs, G.; De Laere, M.; Wens, I.; Cras, P.; Parizel, P.; et al. Tolerogenic dendritic cell-based treatment for multiple sclerosis (MS): A harmonised study protocol for two phase I clinical trials comparing intradermal and intranodal cell administration. BMJ Open 2019, 9, e030309. [Google Scholar] [CrossRef]
- Sawitzki, B.; Harden, P.N.; Reinke, P.; Moreau, A.; Hutchinson, J.A.; Game, D.S.; Tang, Q.; Guinan, E.C.; Battaglia, M.; Burlingham, W.J.; et al. Regulatory cell therapy in kidney transplantation (The ONE Study): A harmonised design and analysis of seven non-randomised, single-arm, phase 1/2A trials. Lancet 2020, 395, 1627–1639. [Google Scholar] [CrossRef]
- Jauregui-Amezaga, A.; Cabezón, R.; Ramírez-Morros, A.; España, C.; Rimola, J.; Bru, C.; Pinó-Donnay, S.; Gallego, M.; Masamunt, M.C.; Ordás, I.; et al. Intraperitoneal administration of autologous tolerogenic dendritic cells for refractory crohn’s disease: A phase I study. J. Crohn’s Colitis 2015, 9, 1071–1078. [Google Scholar] [CrossRef]
- Comi, M.; Avancini, D.; Santoni de Sio, F.; Villa, M.; Uyeda, M.J.; Floris, M.; Tomasoni, D.; Bulfone, A.; Roncarolo, M.G.; Gregori, S. Coexpression of CD163 and CD141 identifies human circulating IL-10-producing dendritic cells (DC-10). Cell. Mol. Immunol. 2020, 17, 95–107. [Google Scholar] [CrossRef]
- Marin, E.; Bouchet-Delbos, L.; Renoult, O.; Louvet, C.; Nerriere-Daguin, V.; Managh, A.J.; Even, A.; Giraud, M.; Vu Manh, T.P.; Aguesse, A.; et al. Human tolerogenic dendritic cells regulate immune responses through lactate synthesis. Cell Metab. 2019, 30, 1075–1090.e8. [Google Scholar] [CrossRef] [PubMed]
- Bouchet-Delbos, L.; Even, A.; Varey, E.; Saïagh, S.; Bercegeay, S.; Braudeau, C.; Dréno, B.; Blancho, G.; Josien, R.; Cuturi, M.-C.; et al. Preclinical assessment of autologous tolerogenic dendritic cells from end-stage renal disease patients. Transplantation 2021, 105, 832–841. [Google Scholar] [CrossRef] [PubMed]
- Grohmann, U.; Fallarino, F.; Bianchi, R.; Belladonna, M.L.; Vacca, C.; Orabona, C.; Uyttenhove, C.; Fioretti, M.C.; Puccetti, P. IL-6 inhibits the tolerogenic function of CD8 alpha+ dendritic cells expressing indoleamine 2,3-dioxygenase. J. Immunol. 2001, 167, 708–714. [Google Scholar] [CrossRef] [PubMed]
- Süss, G.; Shortman, K. A subclass of dendritic cells kills CD4 T cells via Fas/Fas-ligand-induced apoptosis. J. Exp. Med. 1996, 183, 1789–1796. [Google Scholar] [CrossRef]
- Jansen, M.A.A.; Spiering, R.; Ludwig, I.S.; van Eden, W.; Hilkens, C.M.U.; Broere, F. Matured tolerogenic dendritic cells effectively inhibit autoantigen specific CD4(+) T cells in a murine arthritis model. Front. Immunol. 2019, 10, 2068. [Google Scholar] [CrossRef] [PubMed]
- Comi, M.; Amodio, G.; Passeri, L.; Fortunato, M.; Santoni de Sio, F.R.; Andolfi, G.; Kajaste-Rudnitski, A.; Russo, F.; Cesana, L.; Gregori, S. Generation of powerful human tolerogenic dendritic cells by lentiviral-mediated IL-10 gene transfer. Front. Immunol. 2020, 11, 1260. [Google Scholar] [CrossRef]
- Svajger, U.; Obermajer, N.; Jeras, M. Novel findings in drug-induced dendritic cell tolerogenicity. Int. Rev. Immunol. 2010, 29, 574–607. [Google Scholar] [CrossRef] [PubMed]
- Gordon, J.R.; Ma, Y.; Churchman, L.; Gordon, S.A.; Dawicki, W. Regulatory dendritic cells for immunotherapy in immunologic diseases. Front. Immunol. 2014, 5, 7. [Google Scholar] [CrossRef]
- Piemonti, L.; Monti, P.; Allavena, P.; Sironi, M.; Soldini, L.; Leone, B.E.; Socci, C.; Di Carlo, V. Glucocorticoids affect human dendritic cell differentiation and maturation. J. Immunol. 1999, 162, 6473–6481. [Google Scholar]
- Lagaraine, C.; Hoarau, C.; Chabot, V.; Velge-Roussel, F.; Lebranchu, Y. Mycophenolic acid-treated human dendritic cells have a mature migratory phenotype and inhibit allogeneic responses via direct and indirect pathways. Int. Immunol. 2005, 17, 351–363. [Google Scholar] [CrossRef] [PubMed]
- Woltman, A.M.; de Fijter, J.W.; Kamerling, S.W.; Paul, L.C.; Daha, M.R.; van Kooten, C. The effect of calcineurin inhibitors and corticosteroids on the differentiation of human dendritic cells. Eur. J. Immunol. 2000, 30, 1807–1812. [Google Scholar] [CrossRef]
- Li, X.-L.; Liu, Y.; Cao, L.-L.; Li, H.; Yue, L.-T.; Wang, S.; Zhang, M.; Li, X.-H.; Dou, Y.-C.; Duan, R.-S. Atorvastatin-modified dendritic cells in vitro ameliorate experimental autoimmune myasthenia gravis by up-regulated Treg cells and shifted Th1/Th17 to Th2 cytokines. Mol. Cell. Neurosci. 2013, 56, 85–95. [Google Scholar] [CrossRef] [PubMed]
- Bhatt, S.; Qin, J.; Bennett, C.; Qian, S.; Fung, J.J.; Hamilton, T.A.; Lu, L. All-trans retinoic acid induces arginase-1 and inducible nitric oxide synthase-producing dendritic cells with T cell inhibitory function. J. Immunol. 2014, 192, 5098–5108. [Google Scholar] [CrossRef]
- Kim, N.; Park, C.-S.; Im, S.-A.; Kim, J.-W.; Lee, J.-H.; Park, Y.-J.; Song, S.; Lee, C.-K. Minocycline promotes the generation of dendritic cells with regulatory properties. Oncotarget 2016, 7, 52818–52831. [Google Scholar] [CrossRef]
- Torres-Aguilar, H.; Aguilar-Ruiz, S.R.; González-Pérez, G.; Munguía, R.; Bajaña, S.; Meraz-Ríos, M.A.; Sánchez-Torres, C. Tolerogenic dendritic cells generated with different immunosuppressive cytokines induce antigen-specific anergy and regulatory properties in memory CD4+ T cells. J. Immunol. 2010, 184, 1765–1775. [Google Scholar] [CrossRef] [PubMed]
- Nayyar, A.; Dawicki, W.; Huang, H.; Lu, M.; Zhang, X.; Gordon, J.R. Induction of prolonged asthma tolerance by IL-10-differentiated dendritic cells: Differential impact on airway hyperresponsiveness and the Th2 immunoinflammatory response. J. Immunol. 2012, 189, 72–79. [Google Scholar] [CrossRef]
- Moreau, A.; Varey, E.; Bouchet-Delbos, L.; Cuturi, M.-C. Cell therapy using tolerogenic dendritic cells in transplantation. Transplant. Res. 2012, 1, 13. [Google Scholar] [CrossRef] [PubMed]
- Gregori, S.; Tomasoni, D.; Pacciani, V.; Scirpoli, M.; Battaglia, M.; Magnani, C.F.; Hauben, E.; Roncarolo, M.-G. Differentiation of type 1 T regulatory cells (Tr1) by tolerogenic DC-10 requires the IL-10-dependent ILT4/HLA-G pathway. Blood 2010, 116, 935–944. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.H.; Jung, H.H.; Lee, C.K. Generation, characteristics and clinical trials of ex vivo generated tolerogenic dendritic cells. Yonsei Med. J. 2018, 59, 807–815. [Google Scholar] [CrossRef]
- Menges, M.; Rössner, S.; Voigtländer, C.; Schindler, H.; Kukutsch, N.A.; Bogdan, C.; Erb, K.; Schuler, G.; Lutz, M.B. Repetitive injections of dendritic cells matured with tumor necrosis factor alpha induce antigen-specific protection of mice from autoimmunity. J. Exp. Med. 2002, 195, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Eljaafari, A.; Li, Y.-P.; Miossec, P. IFN-gamma, as secreted during an alloresponse, induces differentiation of monocytes into tolerogenic dendritic cells, resulting in FoxP3+ regulatory T cell promotion. J. Immunol. 2009, 183, 2932–2945. [Google Scholar] [CrossRef]
- Rutella, S.; Bonanno, G.; Procoli, A.; Mariotti, A.; de Ritis, D.G.; Curti, A.; Danese, S.; Pessina, G.; Pandolfi, S.; Natoni, F.; et al. Hepatocyte growth factor favors monocyte differentiation into regulatory interleukin (IL)-10++IL-12low/neg accessory cells with dendritic-cell features. Blood 2006, 108, 218–227. [Google Scholar] [CrossRef]
- Brandt, K.; Bulfone-Paus, S.; Foster, D.C.; Rückert, R. Interleukin-21 inhibits dendritic cell activation and maturation. Blood 2003, 102, 4090–4098. [Google Scholar] [CrossRef] [PubMed]
- Vanherwegen, A.-S.; Cook, D.P.; Ferreira, G.B.; Gysemans, C.; Mathieu, C. Vitamin D-modulated dendritic cells delay lethal graft-versus-host disease through induction of regulatory T cells. J. Steroid Biochem. Mol. Biol. 2019, 188, 103–110. [Google Scholar] [CrossRef]
- Adorini, L.; Penna, G. Dendritic cell tolerogenicity: A key mechanism in immunomodulation by vitamin D receptor agonists. Hum. Immunol. 2009, 70, 345–352. [Google Scholar] [CrossRef]
- Canning, M.O.; Grotenhuis, K.; de Wit, H.; Ruwhof, C.; Drexhage, H.A. 1-alpha,25-dihydroxyvitamin D3 (1,25(OH)(2)D(3)) hampers the maturation of fully active immature dendritic cells from monocytes. Eur. J. Endocrinol. 2001, 145, 351–357. [Google Scholar] [CrossRef]
- Penna, G.; Adorini, L. 1 Alpha, 25-dihydroxyvitamin D3 inhibits differentiation, maturation, activation, and survival of dendritic cells leading to impaired alloreactive T cell activation. J. Immunol. 2000, 164, 2405–2411. [Google Scholar] [CrossRef]
- Piemonti, L.; Monti, P.; Sironi, M.; Fraticelli, P.; Leone, B.E.; Dal Cin, E.; Allavena, P.; Di Carlo, V. Vitamin D3 affects differentiation, maturation, and function of human monocyte-derived dendritic cells. J. Immunol. 2000, 164, 4443–4451. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, G.B.; van Etten, E.; Verstuyf, A.; Waer, M.; Overbergh, L.; Gysemans, C.; Mathieu, C. 1,25-Dihydroxyvitamin D3 alters murine dendritic cell behaviour in vitro and in vivo. Diabetes/Metab. Res. Rev. 2011, 27, 933–941. [Google Scholar] [CrossRef]
- Ferreira, G.B.; Kleijwegt, F.S.; Waelkens, E.; Lage, K.; Nikolic, T.; Hansen, D.A.; Workman, C.T.; Roep, B.O.; Overbergh, L.; Mathieu, C. Differential protein pathways in 1,25-dihydroxyvitamin D3 and Dexamethasone modulated tolerogenic human dendritic cells. J. Proteome Res. 2012, 11, 941–971. [Google Scholar] [CrossRef]
- Széles, L.; Keresztes, G.; Töröcsik, D.; Balajthy, Z.; Krenács, L.; Póliska, S.; Steinmeyer, A.; Zuegel, U.; Pruenster, M.; Rot, A.; et al. 1,25-dihydroxyvitamin D3 is an autonomous regulator of the transcriptional changes leading to a tolerogenic dendritic cell phenotype. J. Immunol. 2009, 182, 2074. [Google Scholar] [CrossRef] [PubMed]
- Gauzzi, M.C.; Purificato, C.; Donato, K.; Jin, Y.; Wang, L.; Daniel, K.C.; Maghazachi, A.A.; Belardelli, F.; Adorini, L.; Gessani, S. Suppressive effect of 1α, 25-dihydroxyvitamin D3 on type I IFN-Mediated monocyte differentiation into dendritic cells: Impairment of functional activities and chemotaxis. J. Immunol. 2005, 174, 270. [Google Scholar] [CrossRef]
- Pedersen, A.E.; Gad, M.; Walter, M.R.; Claesson, M.H. Induction of regulatory dendritic cells by dexamethasone and 1alpha,25-dihydroxyvitamin D(3). Immunol. Lett. 2004, 91, 63–69. [Google Scholar] [CrossRef]
- Penna, G.; Roncari, A.; Amuchastegui, S.; Daniel, K.C.; Berti, E.; Colonna, M.; Adorini, L. Expression of the inhibitory receptor ILT3 on dendritic cells is dispensable for induction of CD4+Foxp3+ regulatory T cells by 1,25-dihydroxyvitamin D3. Blood 2005, 106, 3490–3497. [Google Scholar] [CrossRef]
- Xing, N.; Maldonado, M.L.; Bachman, L.A.; McKean, D.J.; Kumar, R.; Griffin, M.D. Distinctive dendritic cell modulation by vitamin D3 and glucocorticoid pathways. Biochem. Biophys. Res. Commun. 2002, 297, 645–652. [Google Scholar] [CrossRef]
- Anderson, A.E.; Swan, D.J.; Sayers, B.L.; Harry, R.A.; Patterson, A.M.; von Delwig, A.; Robinson, J.H.; Isaacs, J.D.; Hilkens, C.M.U. LPS activation is required for migratory activity and antigen presentation by tolerogenic dendritic cells. J. Leukoc. Biol. 2009, 85, 243–250. [Google Scholar] [CrossRef]
- Raϊch-Regué, D.; Grau-López, L.; Naranjo-Gómez, M.; Ramo-Tello, C.; Pujol-Borrell, R.; Martínez-Cáceres, E.; Borràs, F.E. Stable antigen-specific T-cell hyporesponsiveness induced by tolerogenic dendritic cells from multiple sclerosis patients. Eur. J. Immunol. 2012, 42, 771–782. [Google Scholar] [CrossRef] [PubMed]
- Hench, P.S.; Kendall, E.C.; Slocumb, C.H.; Polley, H.F. Effects of cortisone acetate and pituitary acth on rheumatoid arthritis, rheumatic fever and certain other conditions: A study in clinical physiology. Arch. Intern. Med. 1950, 85, 545–666. [Google Scholar] [CrossRef] [PubMed]
- Coutinho, A.E.; Chapman, K.E. The anti-inflammatory and immunosuppressive effects of glucocorticoids, recent developments and mechanistic insights. Mol. Cell. Endocrinol. 2011, 335, 2–13. [Google Scholar] [CrossRef] [PubMed]
- Falcón-Beas, C.; Tittarelli, A.; Mora-Bau, G.; Tempio, F.; Pérez, C.; Hevia, D.; Behrens, C.; Flores, I.; Falcón-Beas, F.; Garrido, P.; et al. Dexamethasone turns tumor antigen-presenting cells into tolerogenic dendritic cells with T cell inhibitory functions. Immunobiology 2019, 224, 697–705. [Google Scholar] [CrossRef] [PubMed]
- Osorio, F.; Fuentes, C.; López, M.N.; Salazar-Onfray, F.; González, F.E. Role of dendritic cells in the induction of lymphocyte tolerance. Front. Immunol. 2015, 6, 535. [Google Scholar] [CrossRef] [PubMed]
- Harry, R.A.; Anderson, A.E.; Isaacs, J.D.; Hilkens, C.M.U. Generation and characterisation of therapeutic tolerogenic dendritic cells for rheumatoid arthritis. Ann. Rheum. Dis. 2010, 69, 2042–2050. [Google Scholar] [CrossRef]
- Anderson, A.E.; Sayers, B.L.; Haniffa, M.A.; Swan, D.J.; Diboll, J.; Wang, X.-N.; Isaacs, J.D.; Hilkens, C.M.U. Differential regulation of naïve and memory CD4+ T cells by alternatively activated dendritic cells. J. Leukoc. Biol. 2008, 84, 124–133. [Google Scholar] [CrossRef]
- Stenger, E.O.; Rosborough, B.R.; Mathews, L.R.; Ma, H.; Mapara, M.Y.; Thomson, A.W.; Turnquist, H.R. IL-12hi rapamycin-conditioned dendritic cells mediate IFN-γ-dependent apoptosis of alloreactive CD4+ T cells in vitro and reduce lethal graft-versus-host disease. Biol. Blood Marrow Transplant. 2014, 20, 192–201. [Google Scholar] [CrossRef] [PubMed]
- Turnquist, H.R.; Raimondi, G.; Zahorchak, A.F.; Fischer, R.T.; Wang, Z.; Thomson, A.W. Rapamycin-conditioned dendritic cells are poor stimulators of allogeneic CD4+ T cells, but enrich for antigen-specific Foxp3+ T regulatory cells and promote organ transplant tolerance. J. Immunol. 2007, 178, 7018–7031. [Google Scholar] [CrossRef] [PubMed]
- Naranjo-Gómez, M.; Raïch-Regué, D.; Oñate, C.; Grau-López, L.; Ramo-Tello, C.; Pujol-Borrell, R.; Martínez-Cáceres, E.; Borràs, F.E. Comparative study of clinical grade human tolerogenic dendritic cells. J. Transl. Med. 2011, 9, 89. [Google Scholar] [CrossRef]
- Zhu, J.; Paul, W.E. CD4 T cells: Fates, functions, and faults. Blood 2008, 112, 1557–1569. [Google Scholar] [CrossRef]
- Singer, S.J.; Tiernan, R.; Sullivan, E.J. Interstitial pneumonitis associated with sirolimus therapy in renal-transplant recipients. N. Engl. J. Med. 2000, 343, 1815–1816. [Google Scholar]
- Dittrich, E.; Schmaldienst, S.; Soleiman, A.; Hörl, W.H.; Pohanka, E. Rapamycin-associated post-transplantation glomerulonephritis and its remission after reintroduction of calcineurin-inhibitor therapy. Transpl. Int. 2004, 17, 215–220. [Google Scholar] [CrossRef]
- Ferrer, I.R.; Wagener, M.E.; Robertson, J.M.; Turner, A.P.; Araki, K.; Ahmed, R.; Kirk, A.D.; Larsen, C.P.; Ford, M.L. Cutting edge: Rapamycin augments pathogen-specific but not graft-reactive CD8+ T cell responses. J. Immunol. 2010, 185, 2004–2008. [Google Scholar] [CrossRef] [PubMed]
- Mosser, D.M.; Zhang, X. Interleukin-10: New perspectives on an old cytokine. Immunol. Rev. 2008, 226, 205–218. [Google Scholar] [CrossRef]
- Glocker, E.-O.; Kotlarz, D.; Boztug, K.; Gertz, E.M.; Schäffer, A.A.; Noyan, F.; Perro, M.; Diestelhorst, J.; Allroth, A.; Murugan, D.; et al. Inflammatory bowel disease and mutations affecting the interleukin-10 receptor. N. Engl. J. Med. 2009, 361, 2033–2045. [Google Scholar] [CrossRef]
- Engelhardt, K.R.; Grimbacher, B. IL-10 in humans: Lessons from the gut, IL-10/IL-10 receptor deficiencies, and IL-10 polymorphisms. Curr. Top. Microbiol. Immunol. 2014, 380, 1–18. [Google Scholar]
- Comi, M.; Amodio, G.; Gregori, S. Interleukin-10-producing DC-10 is a unique tool to promote tolerance via antigen-specific T regulatory type 1 cells. Front. Immunol. 2018, 9, 682. [Google Scholar] [CrossRef]
- Buelens, C.; Verhasselt, V.; De Groote, D.; Thielemans, K.; Goldman, M.; Willems, F. Human dendritic cell responses to lipopolysaccharide and CD40 ligation are differentially regulated by interleukin-10. Eur. J. Immunol. 1997, 27, 1848–1852. [Google Scholar] [CrossRef] [PubMed]
- Buelens, C.; Willems, F.; Delvaux, A.; Piérard, G.; Delville, J.P.; Velu, T.; Goldman, M. Interleukin-10 differentially regulates B7-1 (CD80) and B7-2 (CD86) expression on human peripheral blood dendritic cells. Eur. J. Immunol. 1995, 25, 2668–2672. [Google Scholar] [CrossRef] [PubMed]
- Steinbrink, K.; Jonuleit, H.; Müller, G.; Schuler, G.; Knop, J.; Enk, A.H. Interleukin-10-treated human dendritic cells induce a melanoma-antigen-specific anergy in CD8(+) T cells resulting in a failure to lyse tumor cells. Blood 1999, 93, 1634–1642. [Google Scholar] [CrossRef]
- Steinbrink, K.; Graulich, E.; Kubsch, S.; Knop, J.; Enk, A.H. CD4(+) and CD8(+) anergic T cells induced by interleukin-10-treated human dendritic cells display antigen-specific suppressor activity. Blood 2002, 99, 2468–2476. [Google Scholar] [CrossRef]
- Zahorchak, A.F.; Kean, L.S.; Tokita, D.; Turnquist, H.R.; Abe, M.; Finke, J.; Hamby, K.; Rigby, M.R.; Larsen, C.P.; Thomson, A.W. Infusion of stably immature monocyte-derived dendritic cells plus CTLA4Ig modulates alloimmune reactivity in rhesus macaques. Transplantation 2007, 84, 196–206. [Google Scholar] [CrossRef] [PubMed]
- Levings, M.K.; Gregori, S.; Tresoldi, E.; Cazzaniga, S.; Bonini, C.; Roncarolo, M.G. Differentiation of Tr1 cells by immature dendritic cells requires IL-10 but not CD25+CD4+ Tr cells. Blood 2005, 105, 1162–1169. [Google Scholar] [CrossRef] [PubMed]
- Takayama, T.; Morelli, A.E.; Onai, N.; Hirao, M.; Matsushima, K.; Tahara, H.; Thomson, A.W. Mammalian and viral IL-10 enhance C-C chemokine receptor 5 but down-regulate C-C chemokine receptor 7 expression by myeloid dendritic cells: Impact on chemotactic responses and in vivo homing ability. J. Immunol. 2001, 166, 7136–7143. [Google Scholar] [CrossRef]
- Garrod, K.R.; Chang, C.K.; Liu, F.-C.; Brennan, T.V.; Foster, R.D.; Kang, S.-M. Targeted lymphoid homing of dendritic cells is required for prolongation of allograft survival. J. Immunol. 2006, 177, 863–868. [Google Scholar] [CrossRef] [PubMed]
- Lutz, M.B.; Suri, R.M.; Niimi, M.; Ogilvie, A.L.J.; Rößner, S.; Schuler, G.; Austyn, J.M. Immature dendritic cells generated with low doses of GM-CSF in the absence of IL-4 are maturation resistant and prolong allograft survival in vivo. Eur. J. Immunol. 2000, 30, 1813–1822. [Google Scholar] [CrossRef]
- Peche, H.; Trinite, B.; Martinet, B.; Cuturi, M.C. Prolongation of heart allograft survival by immature dendritic cells Generated from recipient type bone marrow progenitors. Am. J. Transplant. 2005, 5, 255–267. [Google Scholar] [CrossRef]
- Segovia, M.; Louvet, C.; Charnet, P.; Savina, A.; Tilly, G.; Gautreau, L.; Carretero-Iglesia, L.; Beriou, G.; Cebrian, I.; Cens, T.; et al. Autologous dendritic cells prolong allograft survival through Tmem176b-dependent antigen cross-presentation: Immunoregulatory mechanisms of autologous DCs. Am. J. Transplant. 2014, 14, 1021–1031. [Google Scholar] [CrossRef]
- Baas, M.C.; Kuhn, C.; Valette, F.; Mangez, C.; Duarte, M.S.; Hill, M.; Besançon, A.; Chatenoud, L.; Cuturi, M.-C.; You, S. Combining autologous dendritic cell therapy with CD3 antibodies promotes regulatory T cells and permanent islet allograft acceptance. J. Immunol. 2014, 193, 4696–4703. [Google Scholar] [CrossRef] [PubMed]
- Bériou, G.; Pêche, H.; Guillonneau, C.; Merieau, E.; Cuturi, M.-C. Donor-specific allograft tolerance by administration of recipient-derived immature dendritic cells and suboptimal immunosuppression. Transplantation 2005, 79, 969–972. [Google Scholar] [CrossRef]
- Moreau, A.; Hill, M.; Thébault, P.; Deschamps, J.Y.; Chiffoleau, E.; Chauveau, C.; Moullier, P.; Anegon, I.; Alliot-Licht, B.; Cuturi, M.C. Tolerogenic dendritic cells actively inhibit T cells through heme oxygenase-1 in rodents and in nonhuman primates. FASEB J. 2009, 23, 3070–3077. [Google Scholar] [CrossRef]
- Moreau, A.; Vandamme, C.; Segovia, M.; Devaux, M.; Guilbaud, M.; Tilly, G.; Jaulin, N.; Le Duff, J.; Cherel, Y.; Deschamps, J.-Y.; et al. Generation and in vivo evaluation of IL10-treated dendritic cells in a nonhuman primate model of AAV-based gene transfer. Mol. Ther.Methods Clin. Dev. 2014, 1, 14028. [Google Scholar] [CrossRef]
- Chitta, S.; Santambrogio, L.; Stern, L.J. GMCSF in the absence of other cytokines sustains human dendritic cell precursors with T cell regulatory activity and capacity to differentiate into functional dendritic cells. Immunol. Lett. 2008, 116, 41–54. [Google Scholar] [CrossRef]
- Epelman, S.; Lavine, K.J.; Randolph, G.J. Origin and functions of tissue macrophages. Immunity 2014, 41, 21–35. [Google Scholar] [CrossRef] [PubMed]
- Nahrendorf, M.; Swirski, F.K.; Aikawa, E.; Stangenberg, L.; Wurdinger, T.; Figueiredo, J.-L.; Libby, P.; Weissleder, R.; Pittet, M.J. The healing myocardium sequentially mobilizes two monocyte subsets with divergent and complementary functions. J. Exp. Med. 2007, 204, 3037–3047. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, L.A.J.; Artyomov, M.N. Itaconate: The poster child of metabolic reprogramming in macrophage function. Nat. Rev. Immunol. 2019, 19, 273–281. [Google Scholar] [CrossRef]
- De Paoli, F.; Staels, B.; Chinetti-Gbaguidi, G. Macrophage phenotypes and their modulation in atherosclerosis. Circ. J 2014, 78, 1775–1781. [Google Scholar] [CrossRef] [PubMed]
- Ochando, J.; Ordikhani, F.; Boros, P.; Jordan, S. The innate immune response to allotransplants: Mechanisms and therapeutic potentials. Cell. Mol. Immunol. 2019, 16, 350–356. [Google Scholar] [CrossRef] [PubMed]
- Martinez, F.O.; Sica, A.; Mantovani, A.; Locati, M. Macrophage activation and polarization. Front. Biosci. 2008, 13, 453–461. [Google Scholar] [CrossRef]
- Mills, C.D.; Kincaid, K.; Alt, J.M.; Heilman, M.J.; Hill, A.M. M-1/M-2 macrophages and the Th1/Th2 paradigm. J. Immunol. 2000, 164, 6166–6173. [Google Scholar] [CrossRef]
- Nathan, C.F.; Murray, H.W.; Wiebe, M.E.; Rubin, B.Y. Identification of interferon-gamma as the lymphokine that activates human macrophage oxidative metabolism and antimicrobial activity. J. Exp. Med. 1983, 158, 670–689. [Google Scholar] [CrossRef] [PubMed]
- Glass, C.K.; Natoli, G. Molecular control of activation and priming in macrophages. Nat. Immunol. 2016, 17, 26–33. [Google Scholar] [CrossRef]
- O’Shea, J.J.; Murray, P.J. Cytokine signaling modules in inflammatory responses. Immunity 2008, 28, 477–487. [Google Scholar] [CrossRef]
- Dale, D.C.; Boxer, L.; Liles, W.C. The phagocytes: Neutrophils and monocytes. Blood 2008, 112, 935–945. [Google Scholar] [CrossRef] [PubMed]
- Kroner, A.; Greenhalgh, A.D.; Zarruk, J.G.; Passos Dos Santos, R.; Gaestel, M.; David, S. TNF and increased intracellular iron alter macrophage polarization to a detrimental M1 phenotype in the injured spinal cord. Neuron 2014, 83, 1098–1116. [Google Scholar] [CrossRef]
- Ordikhani, F.; Pothula, V.; Sanchez-Tarjuelo, R.; Jordan, S.; Ochando, J. Macrophages in organ transplantation. Front. Immunol. 2020, 11, 582939. [Google Scholar] [CrossRef]
- Mantovani, A.; Sica, A.; Sozzani, S.; Allavena, P.; Vecchi, A.; Locati, M. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. 2004, 25, 677–686. [Google Scholar] [CrossRef] [PubMed]
- Rőszer, T. Understanding the mysterious M2 macrophage through activation markers and effector mechanisms. Mediat. Inflamm. 2015, 2015, 816460. [Google Scholar] [CrossRef]
- Loke, P.; Gallagher, I.; Nair, M.G.; Zang, X.; Brombacher, F.; Mohrs, M.; Allison, J.P.; Allen, J.E. Alternative activation is an innate response to injury that requires CD4+ T cells to be sustained during chronic infection. J. Immunol. 2007, 179, 3926–3936. [Google Scholar] [CrossRef] [PubMed]
- Wynn, T.A.; Vannella, K.M. Macrophages in tissue repair, regeneration, and fibrosis. Immunity 2016, 44, 450–462. [Google Scholar] [CrossRef]
- Cordeiro-da-Silva, A.; Tavares, J.; Araújo, N.; Cerqueira, F.; Tomás, A.; Lin, P.K.T.; Ouaissi, A. Immunological alterations induced by polyamine derivatives on murine splenocytes and human mononuclear cells. Int. Immunopharmacol. 2004, 4, 547–556. [Google Scholar] [CrossRef]
- Campbell, L.; Saville, C.R.; Murray, P.J.; Cruickshank, S.M.; Hardman, M.J. Local arginase 1 activity is required for cutaneous wound healing. J. Investig. Dermatol. 2013, 133, 2461–2470. [Google Scholar] [CrossRef]
- Sternberg, E.M. Neural regulation of innate immunity: A coordinated nonspecific host response to pathogens. Nat. Rev. Immunol. 2006, 6, 318–328. [Google Scholar] [CrossRef]
- O’Neill, L.A.J.; Pearce, E.J. Immunometabolism governs dendritic cell and macrophage function. J. Exp. Med. 2016, 213, 15–23. [Google Scholar] [CrossRef]
- Mills, E.L.; Kelly, B.; O’Neill, L.A.J. Mitochondria are the powerhouses of immunity. Nat. Immunol. 2017, 18, 488–498. [Google Scholar] [CrossRef]
- Strelko, C.L.; Lu, W.; Dufort, F.J.; Seyfried, T.N.; Chiles, T.C.; Rabinowitz, J.D.; Roberts, M.F. Itaconic acid is a mammalian metabolite induced during macrophage activation. J. Am. Chem. Soc. 2011, 133, 16386–16389. [Google Scholar] [CrossRef]
- Mills, E.L.; Ryan, D.G.; Prag, H.A.; Dikovskaya, D.; Menon, D.; Zaslona, Z.; Jedrychowski, M.P.; Costa, A.S.H.; Higgins, M.; Hams, E.; et al. Itaconate is an anti-inflammatory metabolite that activates Nrf2 via alkylation of KEAP1. Nature 2018, 556, 113–117. [Google Scholar] [CrossRef]
- Nelson, V.L.; Nguyen, H.C.B.; Garcìa-Cañaveras, J.C.; Briggs, E.R.; Ho, W.Y.; DiSpirito, J.R.; Marinis, J.M.; Hill, D.A.; Lazar, M.A. PPARγ is a nexus controlling alternative activation of macrophages via glutamine metabolism. Genes. Dev. 2018, 32, 1035–1044. [Google Scholar] [CrossRef]
- Rožman, P.; Švajger, U. The tolerogenic role of IFN-γ. Cytokine Growth Factor Rev. 2018, 41, 40–53. [Google Scholar] [CrossRef]
- Xiao, B.; Wu, X.; Yang, J.; Xu, L.; Liu, X.; Huang, Y.; Bjelke, B.; Link, H. Therapeutic potential of IFN-γ-modified dendritic cells in acute and chronic experimental allergic encephalomyelitis. Int. Immunol. 2004, 16, 13–22. [Google Scholar] [CrossRef]
- Švajger, U.; Obermajer, N.; Jeras, M. IFN-γ-rich environment programs dendritic cells toward silencing of cytotoxic immune responses. J. Leukoc. Biol. 2014, 95, 33–46. [Google Scholar] [CrossRef]
- Munn, D.H.; Mellor, A.L. Indoleamine 2,3 dioxygenase and metabolic control of immune responses. Trends Immunol. 2013, 34, 137–143. [Google Scholar] [CrossRef]
- Murray, P.J.; Allen, J.E.; Biswas, S.K.; Fisher, E.A.; Gilroy, D.W.; Goerdt, S.; Gordon, S.; Hamilton, J.A.; Ivashkiv, L.B.; Lawrence, T.; et al. Macrophage activation and polarization: Nomenclature and experimental guidelines. Immunity 2014, 41, 14–20. [Google Scholar] [CrossRef]
- Anderson, C.F.; Mosser, D.M. A novel phenotype for an activated macrophage: The type 2 activated macrophage. J. Leukoc. Biol. 2002, 72, 101–106. [Google Scholar]
- Riquelme, P.; Tomiuk, S.; Kammler, A.; Fändrich, F.; Schlitt, H.J.; Geissler, E.K.; Hutchinson, J.A. IFN-γ-induced iNOS expression in mouse regulatory macrophages prolongs allograft survival in fully immunocompetent recipients. Mol. Ther. 2013, 21, 409–422. [Google Scholar] [CrossRef]
- Hutchinson, J.A.; Riquelme, P.; Sawitzki, B.; Tomiuk, S.; Miqueu, P.; Zuhayra, M.; Oberg, H.H.; Pascher, A.; Lützen, U.; Janssen, U.; et al. Cutting edge: Immunological consequences and trafficking of human regulatory macrophages administered to renal transplant recipients. J. Immunol. 2011, 187, 2072–2078. [Google Scholar] [CrossRef]
- Riquelme, P.; Hutchinson, J.A. Novel molecules mediate specialized functions of human regulatory macrophages. Curr. Opin. Organ. Transplant. 2018, 23, 533–537. [Google Scholar] [CrossRef]
- Riquelme, P.; Haarer, J.; Kammler, A.; Walter, L.; Tomiuk, S.; Ahrens, N.; Wege, A.K.; Goecze, I.; Zecher, D.; Banas, B.; et al. TIGIT(+) iTregs elicited by human regulatory macrophages control T cell immunity. Nat. Commun. 2018, 9, 2858. [Google Scholar] [CrossRef] [PubMed]
- Hutchinson, J.A.; Riquelme, P.; Geissler, E.K.; Fändrich, F. Human regulatory macrophages. Methods Mol. Biol. 2011, 677, 181–192. [Google Scholar]
- Riquelme, P.; Amodio, G.; Macedo, C.; Moreau, A.; Obermajer, N.; Brochhausen, C.; Ahrens, N.; Kekarainen, T.; Fändrich, F.; Cuturi, C.; et al. DHRS9 Is a stable marker of human regulatory macrophages. Transplantation 2017, 101, 2731–2738. [Google Scholar] [CrossRef]
- Conde, P.; Rodriguez, M.; van der Touw, W.; Jimenez, A.; Burns, M.; Miller, J.; Brahmachary, M.; Chen, H.; Boros, P.; Rausell-Palamos, F.; et al. DC-SIGN(+) macrophages control the induction of transplantation tolerance. Immunity 2015, 42, 1143–1158. [Google Scholar] [CrossRef]
- Redman, B.G.; Chang, A.E.; Whitfield, J.; Esper, P.; Jiang, G.; Braun, T.; Roessler, B.; Mulé, J.J. Phase Ib trial assessing autologous, tumor-pulsed dendritic cells as a vaccine administered with or without IL-2 in patients with metastatic melanoma. J. Immunother. 2008, 31, 591–598. [Google Scholar] [CrossRef]
- Dhodapkar, M.V.; Steinman, R.M.; Krasovsky, J.; Munz, C.; Bhardwaj, N. Antigen-specific inhibition of effector T cell function in humans after injection of immature dendritic cells. J. Exp. Med. 2001, 193, 233–238. [Google Scholar] [CrossRef]
- Dhodapkar, M.V.; Steinman, R.M. Antigen-bearing immature dendritic cells induce peptide-specific CD8(+) regulatory T cells in vivo in humans. Blood 2002, 100, 174–177. [Google Scholar] [CrossRef]
- Marin-Gallen, S.; Clemente-Casares, X.; Planas, R.; Pujol-Autonell, I.; Carrascal, J.; Carrillo, J.; Ampudia, R.; Verdaguer, J.; Pujol-Borrell, R.; Borràs, F.E.; et al. Dendritic cells pulsed with antigen-specific apoptotic bodies prevent experimental type 1 diabetes: Tolerogenic DCs prevent T1D. Clin. Exp. Immunol. 2009, 160, 207–214. [Google Scholar] [CrossRef]
- Machen, J.; Harnaha, J.; Lakomy, R.; Styche, A.; Trucco, M.; Giannoukakis, N. Antisense oligonucleotides down-regulating costimulation confer diabetes-preventive properties to nonobese diabetic mouse dendritic cells. J. Immunol. 2004, 173, 4331–4341. [Google Scholar] [CrossRef]
- Nikolic, T.; Zwaginga, J.; Uitbeijerse, B.; Woittiez, N.; Koning, E.; Aanstoot, H.-J.; Roep, B. Safety and feasibility of intradermal injection with tolerogenic dendritic cells pulsed with proinsulin peptide—for type 1 diabetes. Lancet Diabetes Endocrinol. 2020, 8, 470–472. [Google Scholar] [CrossRef]
- Martin, E.; Capini, C.; Duggan, E.; Lutzky, V.P.; Stumbles, P.; Pettit, A.R.; O’Sullivan, B.; Thomas, R. Antigen-specific suppression of established arthritis in mice by dendritic cells deficient in NF-kappaB. Arthritis Rheum. 2007, 56, 2255–2266. [Google Scholar] [CrossRef]
- Van Duivenvoorde, L.M.; Louis-Plence, P.; Apparailly, F.; van der Voort, E.I.H.; Huizinga, T.W.J.; Jorgensen, C.; Toes, R.E.M. Antigen-specific immunomodulation of collagen-induced arthritis with tumor necrosis factor-stimulated dendritic cells. Arthritis Rheum. 2004, 50, 3354–3364. [Google Scholar] [CrossRef] [PubMed]
- Stoop, J.N.; Harry, R.A.; von Delwig, A.; Isaacs, J.D.; Robinson, J.H.; Hilkens, C.M.U. Therapeutic effect of tolerogenic dendritic cells in established collagen-induced arthritis is associated with a reduction in Th17 responses. Arthritis Rheum. 2010, 62, 3656–3665. [Google Scholar] [CrossRef]
- Klareskog, L.; Catrina, A.I.; Paget, S. Rheumatoid arthritis. Lancet 2009, 373, 659–672. [Google Scholar] [CrossRef]
- Law, S.C.; Street, S.; Yu, C.-H.A.; Capini, C.; Ramnoruth, S.; Nel, H.J.; van Gorp, E.; Hyde, C.; Lau, K.; Pahau, H.; et al. T-cell autoreactivity to citrullinated autoantigenic peptides in rheumatoid arthritis patients carrying HLA-DRB1 shared epitope alleles. Arthritis Res. Ther. 2012, 14, R118. [Google Scholar] [CrossRef]
- Hill, M.; Thebault, P.; Segovia, M.; Louvet, C.; Bériou, G.; Tilly, G.; Merieau, E.; Anegon, I.; Chiffoleau, E.; Cuturi, M.-C. Cell therapy with autologous tolerogenic dendritic cells induces allograft tolerance through interferon-gamma and epstein-barr virus-induced gene 3: Cell therapy with TolDC. Am. J. Transplant. 2011, 11, 2036–2045. [Google Scholar] [CrossRef]
- Von Delwig, A.; Locke, J.; Robinson, J.H.; Ng, W.-F. Response of Th17 cells to a citrullinated arthritogenic aggrecan peptide in patients with rheumatoid arthritis. Arthritis Rheum. 2010, 62, 143–149. [Google Scholar] [CrossRef] [PubMed]
- Diogo, D.; Okada, Y.; Plenge, R.M. Genome-wide association studies to advance our understanding of critical cell types and pathways in rheumatoid arthritis: Recent findings and challenges. Curr. Opin. Rheumatol. 2014, 26, 85–92. [Google Scholar] [CrossRef]
- Martin, E.; O’Sullivan, B.; Low, P.; Thomas, R. Antigen-specific suppression of a primed immune response by dendritic cells mediated by regulatory T cells secreting interleukin-10. Immunity 2003, 18, 155–167. [Google Scholar] [CrossRef]
- Li, M.; Zhang, X.; Zheng, X.; Lian, D.; Zhang, Z.-X.; Ge, W.; Yang, J.; Vladau, C.; Suzuki, M.; Chen, D.; et al. Immune Modulation and Tolerance Induction by RelB-Silenced Dendritic Cells through RNA Interference. J. Immunol. 2007, 178, 5480. [Google Scholar] [CrossRef] [PubMed]
- Wallström, E.; Khademi, M.; Andersson, M.; Weissert, R.; Linington, C.; Olsson, T. Increased reactivity to myelin oligodendrocyte glycoprotein peptides and epitope mapping in HLA DR2(15)+ multiple sclerosis. Eur. J. Immunol. 1998, 28, 3329–3335. [Google Scholar] [CrossRef]
- Bielekova, B.; Goodwin, B.; Richert, N.; Cortese, I.; Kondo, T.; Afshar, G.; Gran, B.; Eaton, J.; Antel, J.; Frank, J.A.; et al. Encephalitogenic potential of the myelin basic protein peptide (amino acids 83-99) in multiple sclerosis: Results of a phase II clinical trial with an altered peptide ligand. Nat. Med. 2000, 6, 1167–1175. [Google Scholar] [CrossRef] [PubMed]
- Villoslada, P.; Abel, K.; Heald, N.; Goertsches, R.; Hauser, S.L.; Genain, C.P. Frequency, heterogeneity and encephalitogenicity of T cells specific for myelin oligodendrocyte glycoprotein in naive outbred primates. Eur. J. Immunol. 2001, 31, 2942–2950. [Google Scholar] [CrossRef]
- Bielekova, B.; Sung, M.-H.; Kadom, N.; Simon, R.; McFarland, H.; Martin, R. Expansion and Functional Relevance of High-Avidity Myelin-Specific CD4+ T Cells in Multiple Sclerosis. J. Immunol. 2004, 172, 3893. [Google Scholar] [CrossRef]
- Lutterotti, A.; Yousef, S.; Sputtek, A.; Stürner, K.H.; Stellmann, J.-P.; Breiden, P.; Reinhardt, S.; Schulze, C.; Bester, M.; Heesen, C.; et al. Antigen-specific tolerance by autologous myelin peptide–coupled cells: A phase 1 trial in multiple sclerosis. Sci. Transl. Med. 2013, 5, 188ra75. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.-P.; Willekens, B.; Cras, P.; Goossens, H.; Martínez-Cáceres, E.; Berneman, Z.N.; Cools, N. Immunomodulatory effects of 1,25-dihydroxyvitamin D3 on dendritic cells promote induction of T cell hyporesponsiveness to myelin-derived antigens. J. Immunol. Res. 2016, 2016, 5392623. [Google Scholar] [CrossRef]
- Jauregui-Amezaga, A.; Rovira, M.; Pinó Donnay, S.; Marín, P.J.; Feu, F.; Elizalde, J.I.; Fernández-Aviles, F.; Martínez, C.; Rosiñol, L.; Suarez-Lledó, M.; et al. P471 Hematopoietic stem cell transplantation in refractory Crohn’s disease: Feasibility and toxicity. J. Crohn’s Colitis 2014, 8, S263. [Google Scholar] [CrossRef]
- Kühn, R.; Löhler, J.; Rennick, D.; Rajewsky, K.; Müller, W. Interleukin-10-deficient mice develop chronic enterocolitis. Cell 1993, 75, 263–274. [Google Scholar] [CrossRef]
- Delgado, M.; Gonzalez-Rey, E.; Ganea, D. The neuropeptide vasoactive intestinal peptide generates tolerogenic dendritic cells. J. Immunol. 2005, 175, 7311–7324. [Google Scholar] [CrossRef]
- Chorny, A.; Gonzalez-Rey, E.; Fernandez-Martin, A.; Pozo, D.; Ganea, D.; Delgado, M. Vasoactive intestinal peptide induces regulatory dendritic cells with therapeutic effects on autoimmune disorders. Proc. Natl. Acad. Sci. USA 2005, 102, 13562–13567. [Google Scholar] [CrossRef]
- Cabezón, R.; Ricart, E.; España, C.; Panés, J.; Benitez-Ribas, D. Gram-negative enterobacteria induce tolerogenic maturation in dexamethasone conditioned dendritic cells. PLoS ONE 2012, 7, e52456. [Google Scholar] [CrossRef]
- Boks, M.A.; Kager-Groenland, J.R.; Haasjes, M.S.P.; Zwaginga, J.J.; van Ham, S.M.; ten Brinke, A. IL-10-generated tolerogenic dendritic cells are optimal for functional regulatory T cell induction—A comparative study of human clinical-applicable DC. Clin. Immunol. 2012, 142, 332–342. [Google Scholar] [CrossRef] [PubMed]
- Raïch-Regué, D.; Naranjo-Gómez, M.; Grau-López, L.; Ramo, C.; Pujol-Borrell, R.; Martínez-Cáceres, E.; Borràs, F.E. Differential effects of monophosphoryl lipid A and cytokine cocktail as maturation stimuli of immunogenic and tolerogenic dendritic cells for immunotherapy. Vaccine 2012, 30, 378–387. [Google Scholar] [CrossRef] [PubMed]
- Morelli, A.E.; Thomson, A.W. Tolerogenic dendritic cells and the quest for transplant tolerance. Nat. Rev. Immunol. 2007, 7, 610–621. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.; Li, W.; Fu, F.; Chambers, F.G.; Qian, S.; Fung, J.J.; Thomson, A.W. Blockade of the CD40-CD40 ligand pathway potentiates the capacity of donor-derived dendritic cell progenitors to induce long-term cardiac allograft survival. Transplantation 1997, 64, 1808–1815. [Google Scholar] [CrossRef] [PubMed]
- Qi, F.; Adair, A.; Ferenbach, D.; Vass, D.G.; Mylonas, K.J.; Kipari, T.; Clay, M.; Kluth, D.C.; Hughes, J.; Marson, L.P. Depletion of cells of monocyte lineage prevents loss of renal microvasculature in murine kidney transplantation. Transplantation 2008, 86, 1267–1274. [Google Scholar] [CrossRef] [PubMed]
- Macedo, C.; Tran, L.M.; Zahorchak, A.F.; Dai, H.; Gu, X.; Ravichandran, R.; Mohanakumar, T.; Elinoff, B.; Zeevi, A.; Styn, M.A.; et al. Donor-derived regulatory dendritic cell infusion results in host cell cross-dressing and T cell subset changes in prospective living donor liver transplant recipients. Am. J. Transplant. 2020, 21, 2372–2386. [Google Scholar] [CrossRef] [PubMed]
- Efremova, M.; Vento-Tormo, M.; Teichmann, S.A.; Vento-Tormo, R. CellPhoneDB: Inferring cell–cell communication from combined expression of multi-subunit ligand–receptor complexes. Nat. Protoc. 2020, 15, 1484–1506. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| ID | Status | Autoimmune Disease | Differentiation Protocol | Cohorts | Administration | Outcome | Ref. |
|---|---|---|---|---|---|---|---|
| NCT00445913 | Completed | Type 1 diabetes | Tolerogenic monocyte-derived dendritic cells (Tol-MoDC) modified with anti-CD40, CD80, CD86 oligonucleotides (ODN) | Unmodified tol-monocyte derived DC (10 million cells) and ODN tol-MoDC (10 million cells) | Four intradermal injections, bi-weekly scheme | Increase in B220+CD11c B cells in blood and no adverse effects | [26] |
| NTR5542 | Completed | Type 1 diabetes | Proinsuline-loaded vitamin D3 generated tolDCs | 5, 10, 20 million cells | Two intradermal injections with a 28-day interval | unknown | |
| NCT02354911 | Unknown | Type 1 diabetes | Tolerogenic monocyte-derived dendritic cells (Tol-MoDC) modified with anti-CD40, CD80, CD86 oligonucleotides (ODN) | 10 million cells | Four intradermal injections, bi-weekly scheme | ||
| NCT03895996 | Recruiting | Type 1 diabetes | Autologous dendritic cell therapy | 7–10 million cells | Three monthly intravenous injection | ||
| NCT03337165 | Completed | Rheumatoid arthritis | Dexamethasone generated tolDCs | 1, 3, 5, 8, 10 million cells | Single intra-arterial infusion and dose escalation | Decrease in DAS28, HAQ improvement and no adverse effect | [27] |
| Rheumavax | Completed | Rheumatoid arthritis | Nuclear factor kappa-light chain enhancer of activated B (NF-kB) inhibitor tolDCs loaded with citrullinated peptides | low dose (1 million cells) and high dose (5 million cells) | One dose of 2 progressive levels was injected intradermally | Increase in Treg levels, decrease in T-cell response to vimentin 447–455, reduced serum level of proinflammatory cytokines/chemokines and no adverse effect | [28] |
| CreaVax-RA | Completed | Rheumatoid arthritis | DCs pulsed with PAD4, HNRNPA2B1, citrullinated filaggrin and vimentin antigens | 0.5 or 1 million cells | Five injections times in a two-dose regimen | Decrease of IFNy-producing T cells and autoantibody levels and no adverse effect. | |
| AutoDECRA | Completed | Rheumatoid arthritis | Dexamethasone/vitaminD3 generated tolDCs loaded with autologous synovial fluid | 1, 3, 10 million cells | Injection intralesional in a dose-escalation single injection | No biological effect in blood and no adverse effect | [29] |
| NCT02283671 | Completed | Multiple sclerosis and Neuromyelitisoptica | Dexamethasone generated tolDCs loaded with myelin peptides or aquaporine-4- derived peptides | 50, 100, 150, 300 million cells | Three injections administered bi-weekly | Decrease in frequency of CD8, NK, and CD14+ CD56+ cells and no adverse effect | [30] |
| NCT02903537 | Recruiting | Multiple sclerosis | Vitamin D3 generated tolDCs loaded with a pool of myelin peptides | 5, 10, 15 million cells | Intranodal injection in a dose escalation of six injections, four bi-weekly and two monthly injections | [31] | |
| NCT02618902 | Recruiting | Multiple sclerosis | Vitamin D3 generated tolDCs loaded with a pool of myelin peptides | 5, 10, 15 million cells | Intradermal injection in a dose escalation of six injections, four bi-weekly and two monthly injections | [31] | |
| 2007-003469-42 | Completed | Crohn’s disease | Dexamethasone/vitamin A generated tolDCs | 2, 5, 10 million cells | Intralesional injection, frequency unkown | Patients withdrew due to worsening of symptoms and no adverse effect | [33] |
| NCT02622763 | Terminated | Crohn’s disease | dexamethasone generated tolDCs | 10, 100 million cells | Intraperitioneal injection in a dose escalation of single injections vs three injections biweekly |
| ID | Status | Transplantation | Differentiation Protocol | Cohorts | Administration | Outcome | Ref. |
|---|---|---|---|---|---|---|---|
| NCT02252055 | Completed | Kidney | Granulocyte macrophage colony stimulating factor-stimulated tolDCs | 1 million cells/kg | Intravenous injection in a single injection one day before transplantation | Administration of ATDCs are safe and feasible | [32] |
| NCT02252055 | Active | Liver | Vitamin D3 and interleukin-10 generated tolDCs | 2.5–10 million cells | Intravenous injection in a single infusion one week before transplantation | ||
| NCT03726307 | Recruiting | Kidney | Vitamin D3 and interleukin-10 generated tolDCs | 0.5, 1.2, 2.5 million cells/kg | Intravenous injection in a single injection one week before transplantation | ||
| ONE study | terminated | Kidney | Regulatory macrophages stimulated with macrophage colony stimulating factor and interferon-γ | 2.5–7.5 million cells/kg | Intravenous injection in a single injection one week before transplantation | Administration of Mregs are safe and feasible | [32] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Suuring, M.; Moreau, A. Regulatory Macrophages and Tolerogenic Dendritic Cells in Myeloid Regulatory Cell-Based Therapies. Int. J. Mol. Sci. 2021, 22, 7970. https://doi.org/10.3390/ijms22157970
Suuring M, Moreau A. Regulatory Macrophages and Tolerogenic Dendritic Cells in Myeloid Regulatory Cell-Based Therapies. International Journal of Molecular Sciences. 2021; 22(15):7970. https://doi.org/10.3390/ijms22157970
Chicago/Turabian StyleSuuring, Maaike, and Aurélie Moreau. 2021. "Regulatory Macrophages and Tolerogenic Dendritic Cells in Myeloid Regulatory Cell-Based Therapies" International Journal of Molecular Sciences 22, no. 15: 7970. https://doi.org/10.3390/ijms22157970
APA StyleSuuring, M., & Moreau, A. (2021). Regulatory Macrophages and Tolerogenic Dendritic Cells in Myeloid Regulatory Cell-Based Therapies. International Journal of Molecular Sciences, 22(15), 7970. https://doi.org/10.3390/ijms22157970