
Mitochondrion as a Target of Astaxanthin Therapy in Heart Failure

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
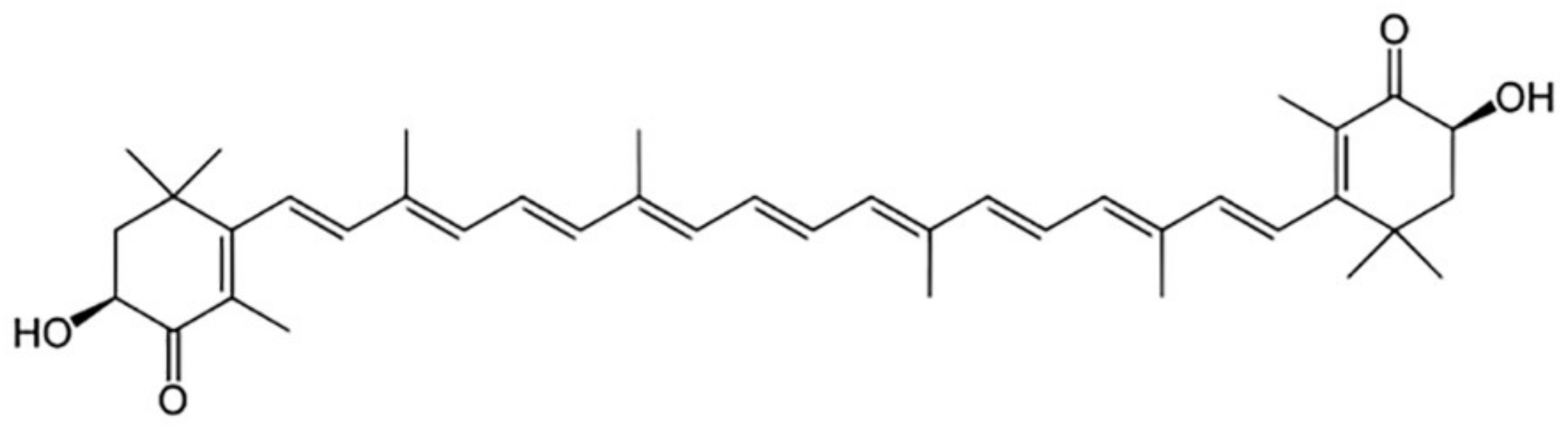
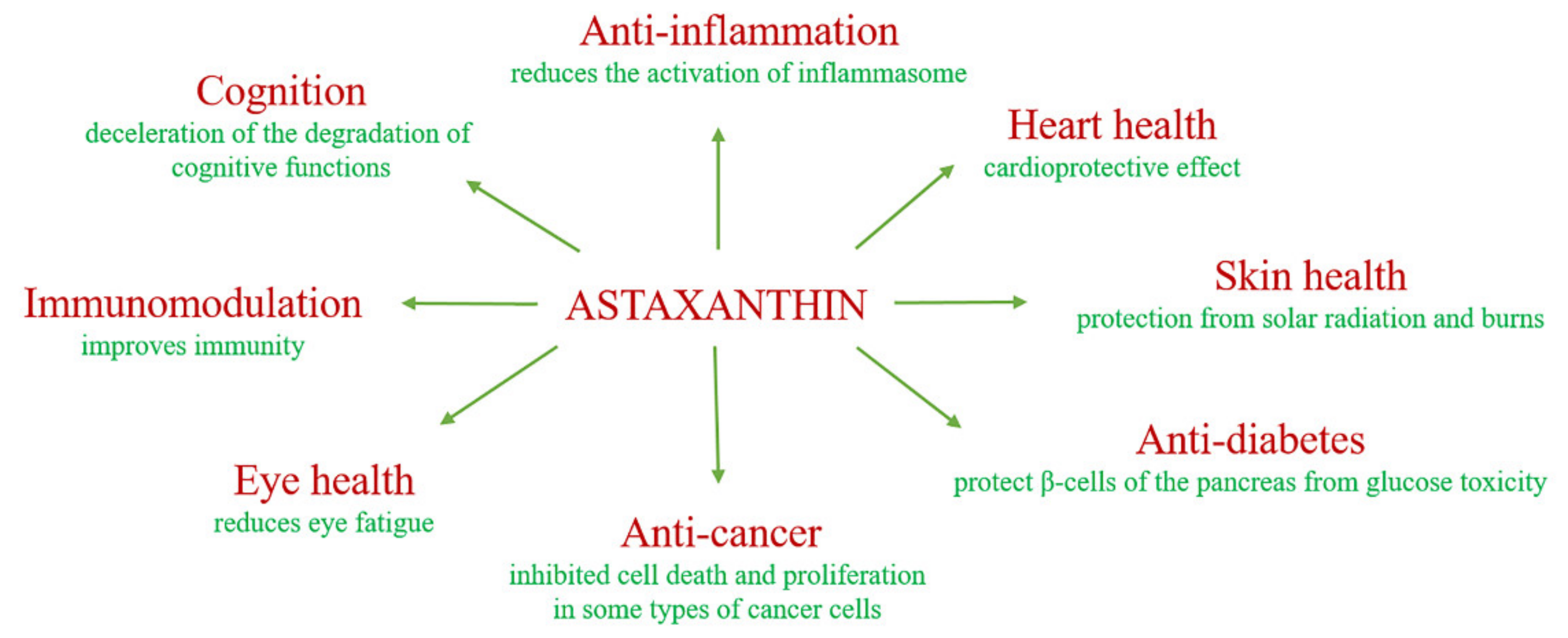
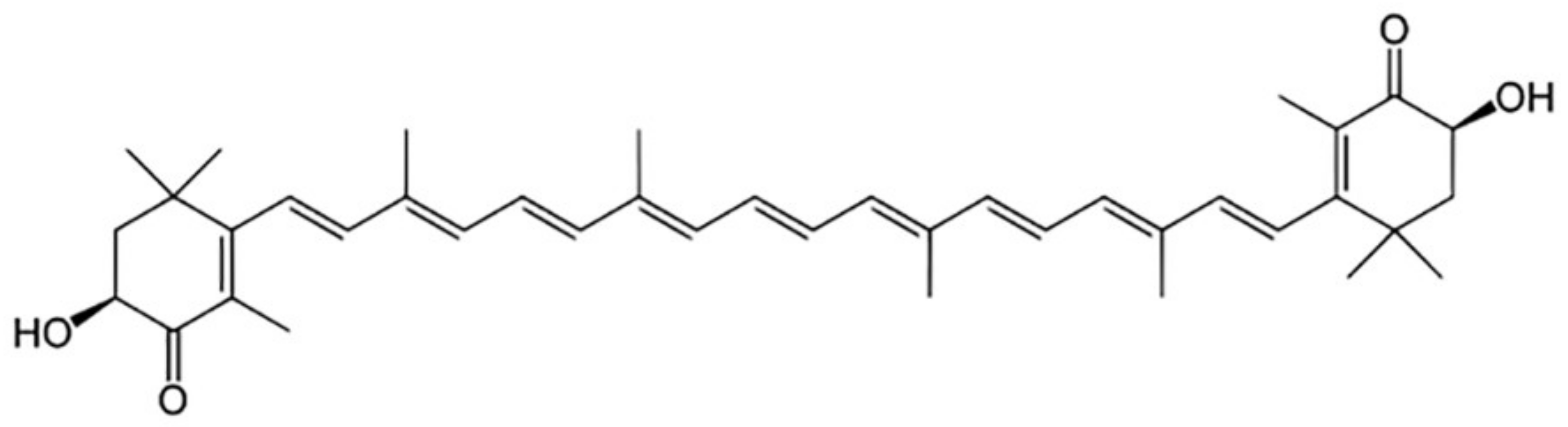
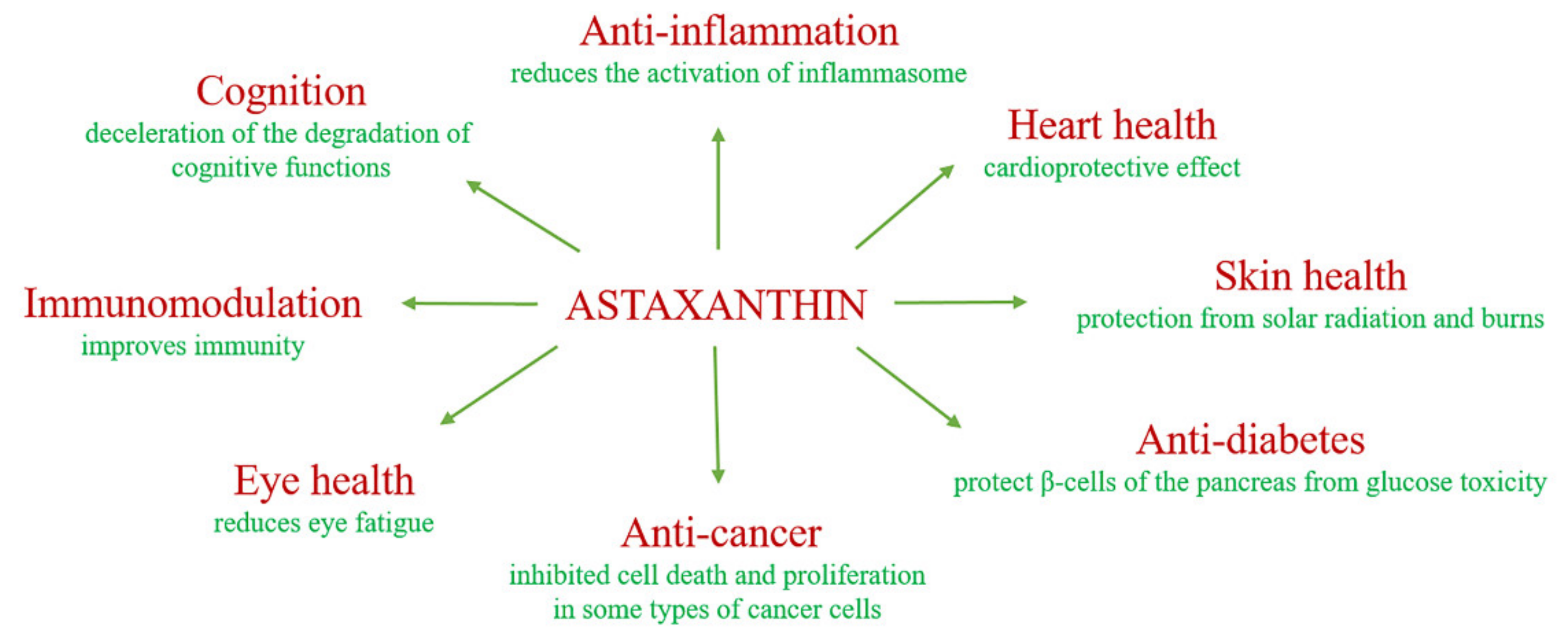
2. The Biological Role of Astaxanthin
3. Astaxanthin and Mitochondrial Permeability Transition Pore Opening (mPTP)
3.1. What Is mPTP?
3.2. The Involvement of AST in the Protection of Mitochondria from Ca2+-Induced Oxidative Stress
3.3. The Effect of Chronic Administration of AST on the Change in the Content of Proteins-Regulators of mPTP
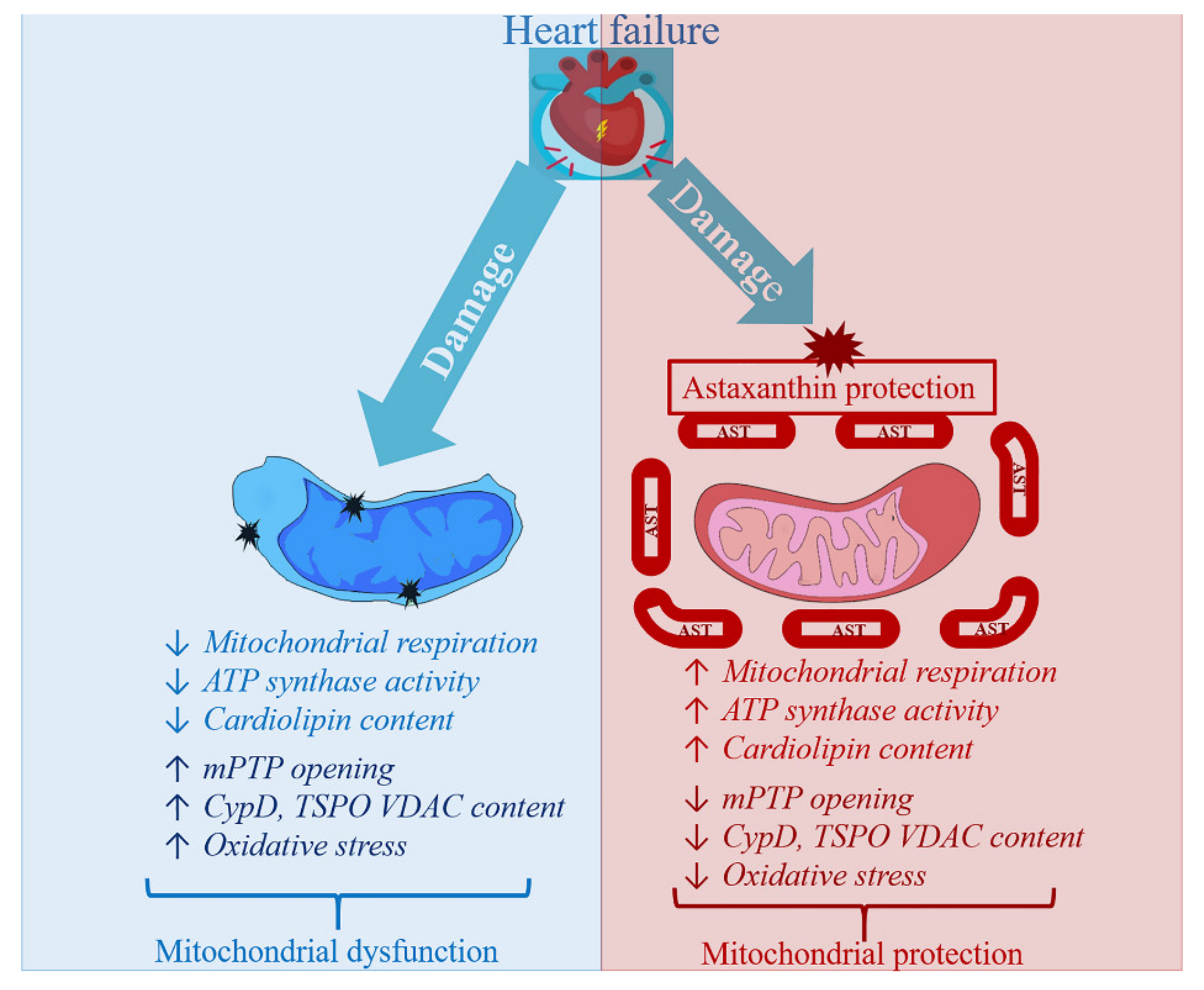
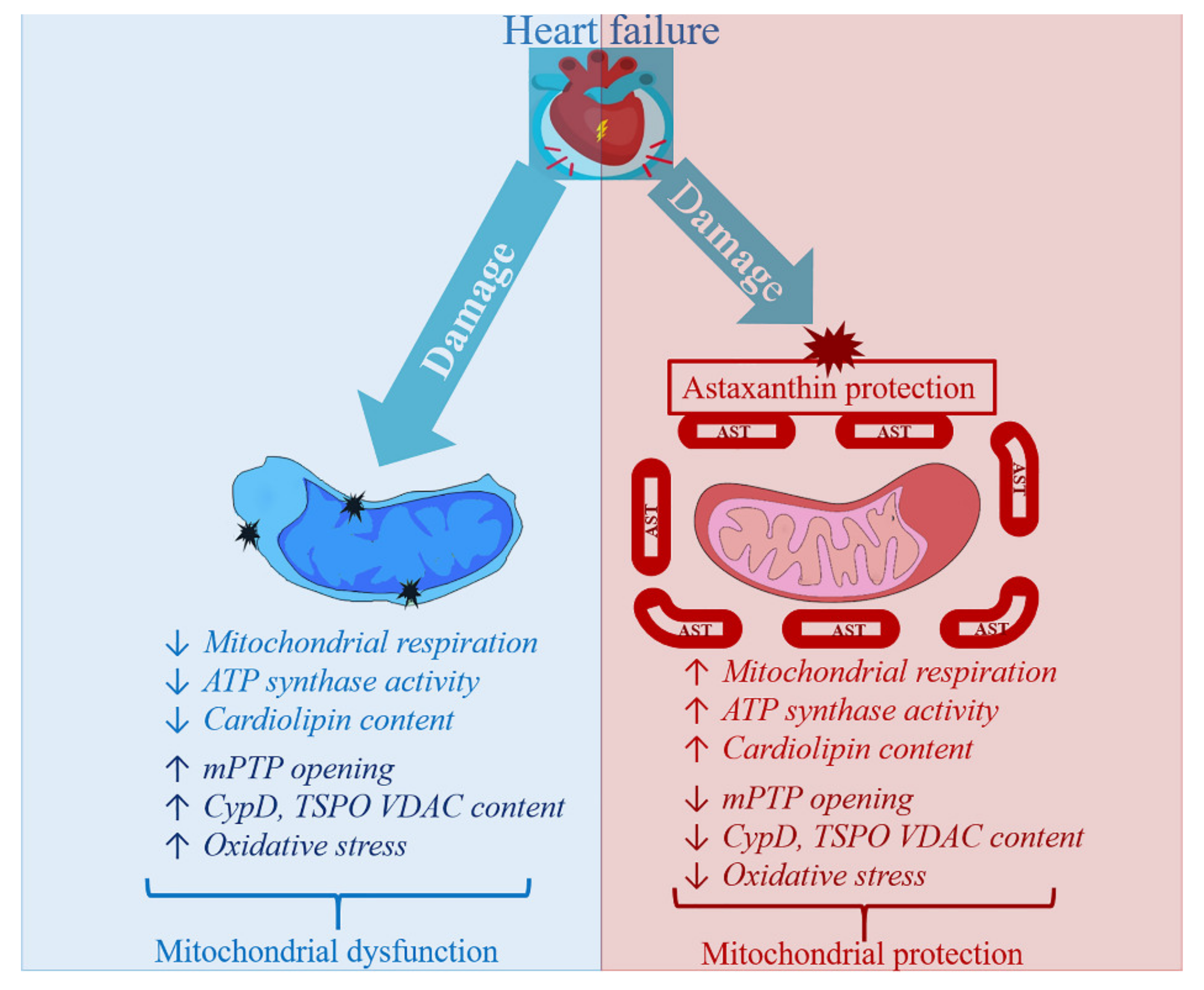
4. Astaxanthin Administration and Heart Failure
5. Summary
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Griffiths, E.J. Mitochondria and Heart Disease. Adv. Exp. Med. Biol. 2011, 942, 249–267. [Google Scholar] [CrossRef]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.H.; Kim, H. Inhibitory Effect of Astaxanthin on Oxidative Stress-Induced Mitochondrial Dysfunction—A Mini-Review. Nutrients 2018, 10, 1137. [Google Scholar] [CrossRef] [Green Version]
- Bullon, P.; Newman, H.N.; Battino, M. Obesity, diabetes mellitus, atherosclerosis and chronic periodontitis: A shared pathology via oxidative stress and mitochondrial dysfunction? Periodontol. 2000 2014, 64, 139–153. [Google Scholar] [CrossRef]
- Hernandez-Aguilera, A.; Rull, A.; Rodriguez-Gallego, E.; Riera-Borrull, M.; Luciano-Mateo, F.; Camps, J.; Menendez, J.A.; Joven, J. Mitochondrial dysfunction: A basic mechanism in inflammation-related non-communicable diseases and therapeutic opportunities. Mediators Inflamm. 2013, 2013, 135698. [Google Scholar] [CrossRef] [Green Version]
- Tsutsui, H.; Kinugawa, S.; Matsushima, S. Oxidative Stress and Mitochondrial DNA Damage in Heart Failure. Circ. J. 2008, 72 (Suppl. A), A31–A37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- James, A.M.; Cocheme, H.M.; Smith, R.A.; Murphy, M.P. Interactions of mitochondria-targeted and untargeted ubiquinones with the mitochondrial respiratory chain and reactive oxygen species. Implications for the use of exogenous ubiquinones as therapies and experimental tools. J. Biol. Chem. 2005, 280, 21295–21312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murphy, M.P.; Smith, R.A. Targeting Antioxidants to Mitochondria by Conjugation to Lipophilic Cations. Annu. Rev. Pharmacol. Toxicol. 2007, 47, 629–656. [Google Scholar] [CrossRef] [PubMed]
- Adlam, V.J.; Harrison, J.C.; Porteous, C.M.; James, A.M.; Smith, R.A.; Murphy, M.P.; Sammut, I. Targeting an antioxidant to mitochondria decreases cardiac ischemia-reperfusion injury. FASEB J. 2005, 19, 1088–1095. [Google Scholar] [CrossRef] [PubMed]
- Graham, D.; Huynh, N.N.; Hamilton, C.A.; Beattie, E.; Smith, R.A.; Cochemeé, H.M.; Murphy, M.P.; Dominiczak, A.F. Mitochondria-Targeted Antioxidant MitoQ 10 Improves Endothelial Function and Attenuates Cardiac Hypertrophy. Hypertension 2009, 54, 322–328. [Google Scholar] [CrossRef] [Green Version]
- Supinski, G.S.; Murphy, M.P.; Callahan, L.A. MitoQ administration prevents endotoxin-induced cardiac dysfunction. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2009, 297, R1095–R1102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shiomi, T.; Tsutsui, H.; Matsusaka, H.; Murakami, K.; Hayashidani, S.; Ikeuchi, M.; Wen, J.; Kubota, T.; Utsumi, H.; Takeshita, A. Overexpression of Glutathione Peroxidase Prevents Left Ventricular Remodeling and Failure After Myocardial Infarction in Mice. Circulation 2004, 109, 544–549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hill, M.F.; Palace, V.P.; Kaur, K.; Kumar, D.; Khaper, N.; Singal, P.K. Reduction in oxidative stress and modulation of heart failure subsequent to myocardial infarction in rats. Exp. Clin. Cardiol. 2005, 10, 146–153. [Google Scholar] [PubMed]
- Li, Y.; Huang, T.-T.; Carlson, E.J.; Melov, S.; Ursell, P.C.; Olson, J.L.; Noble, L.J.; Yoshimura, M.P.; Berger, C.N.; Chan, P.H.; et al. Dilated cardiomyopathy and neonatal lethality in mutant mice lacking manganese superoxide dismutase. Nat. Genet. 1995, 11, 376–381. [Google Scholar] [CrossRef] [PubMed]
- Carpenter, K.L.; Kirkpatrick, P.J.; Weissberg, P.L.; Challis, I.R.; Dennis, I.F.; Freeman, M.A.; Mitchinson, M.J. Oral alpha-tocopherol supplementation inhibits lipid oxidation in established human atherosclerotic lesions. Free Radic. Res. 2003, 37, 1235–1244. [Google Scholar] [CrossRef] [PubMed]
- Ellingsen, I.; Seljeflot, I.; Arnesen, H.; Tonstad, S. Vitamin C consumption is associated with less progression in carotid intima media thickness in elderly men: A 3-year intervention study. Nutr. Metab. Cardiovasc. Dis. 2009, 19, 8–14. [Google Scholar] [CrossRef]
- Poljsak, B.; Suput, D.; Milisav, I. Achieving the balance between ros and antioxidants: When to use the synthetic antioxi-dants. Oxid. Med. Cell Longev. 2013, 2013, 956792. [Google Scholar] [CrossRef]
- Jackson, H.; Braun, C.L.; Ernst, H. The Chemistry of Novel Xanthophyll Carotenoids. Am. J. Cardiol. 2008, 101, S50–S57. [Google Scholar] [CrossRef]
- McNulty, H.P.; Byun, J.; Lockwood, S.F.; Jacob, R.F.; Mason, R.P. Differential effects of carotenoids on lipid peroxidation due to membrane interactions: X-ray diffraction analysis. Biochim. Biophys. Acta 2007, 1768, 167–174. [Google Scholar] [CrossRef] [Green Version]
- Chang, J.-H.; Chen, Y.; Holland, D.; Grabowski, J. Estimating spatial distribution of American lobster Homarus americanus using habitat variables. Mar. Ecol. Prog. Ser. 2010, 420, 145–156. [Google Scholar] [CrossRef]
- Choi, S.; Koo, S. Efficient Syntheses of the Keto-carotenoids Canthaxanthin, Astaxanthin, and Astacene. J. Org. Chem. 2005, 70, 3328–3331. [Google Scholar] [CrossRef] [PubMed]
- Margalith, P.Z. Production of ketocarotenoids by microalgae. Appl. Microbiol. Biotechnol. 1999, 51, 431–438. [Google Scholar] [CrossRef]
- Ambati, R.R.; Phang, S.-M.; Ravi, S.; Aswathanarayana, R.G. Astaxanthin: Sources, Extraction, Stability, Biological Activities and Its Commercial Applications—A Review. Mar. Drugs 2014, 12, 128–152. [Google Scholar] [CrossRef] [PubMed]
- Spiller, G.A.; Dewell, A. Safety of an Astaxanthin-Rich Haematococcus pluvialisAlgal Extract: A Randomized Clinical Trial. J. Med. Food 2003, 6, 51–56. [Google Scholar] [CrossRef] [PubMed]
- Vitale, G.A.; Coppola, D.; Palma Esposito, F.; Buonocore, C.; Ausuri, J.; Tortorella, E.; De Pascale, D. Antioxidant Molecules from Marine Fungi: Methodologies and Perspectives. Antioxidants (Basel) 2020, 9, 1183. [Google Scholar] [CrossRef] [PubMed]
- Hussein, G.; Sankawa, U.; Goto, H.; Matsumoto, K.; Watanabe, H. Astaxanthin, a Carotenoid with Potential in Human Health and Nutrition. J. Nat. Prod. 2006, 69, 443–449. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.B.; Osawa, T. Cis astaxanthin and especially 9-cis astaxanthin exhibits a higher antioxidant activity in vitro compared to the all-trans isomer. Biochem. Biophys. Res. Commun. 2007, 357, 187–193. [Google Scholar] [CrossRef]
- Novak, E.A.; Mollen, K.P. Mitochondrial dysfunction in inflammatory bowel disease. Front. Cell Dev. Biol. 2015, 3, 62. [Google Scholar] [CrossRef] [Green Version]
- Che, H.; Li, Q.; Zhang, T.; Wang, D.; Yang, L.; Xu, J.; Yanagita, T.; Xue, C.; Chang, Y.; Wang, Y. Effects of Astaxanthin and Docosahexaenoic-Acid-Acylated Astaxanthin on Alzheimer’s Disease in APP/PS1 Double-Transgenic Mice. J. Agric. Food Chem. 2018, 66, 4948–4957. [Google Scholar] [CrossRef]
- Wisniewska, A.; Subczynski, W.K. Effects of polar carotenoids on the shape of the hydrophobic barrier of phospholipid bilayers. Biochim. Biophys. Acta 1998, 1368, 235–246. [Google Scholar] [CrossRef] [Green Version]
- Edge, R.; Gaikwad, P.; Navaratnam, S.; Rao, B.S.; George Truscott, T. Reduction of oxidized guanosine by dietary carotenoids: A pulse radiolysis study. Arch. Biochem. Biophys. 2010, 504, 100–103. [Google Scholar] [CrossRef]
- Yuan, J.-P.; Peng, J.; Yin, K.; Wang, J.-H. Potential health-promoting effects of astaxanthin: A high-value carotenoid mostly from microalgae. Mol. Nutr. Food Res. 2011, 55, 150–165. [Google Scholar] [CrossRef]
- Rao, A.R.; Reddy, A.H.; Aradhya, S.M. Antibacterial properties of spirulina platensis, haematococcus pluvialis, botryococcus braunii micro algal extracts. Curr. Trends Biotechnol. Pharm. 2010, 4, 807–817. [Google Scholar]
- Park, J.S.; Chyun, J.H.; Kim, Y.K.; Line, L.L.; Chew, B.P. Astaxanthin decreased oxidative stress and inflammation and enhanced immune response in humans. Nutr. Metab. (Lond.) 2010, 7, 18. [Google Scholar] [CrossRef] [Green Version]
- Uchiyama, K.; Naito, Y.; Hasegawa, G.; Nakamura, N.; Takahashi, J.; Yoshikawa, T. Astaxanthin protects beta-cells against glucose toxicity in diabetic db/db mice. Redox Rep. 2002, 7, 290–293. [Google Scholar] [CrossRef] [PubMed]
- Otton, R.; Marin, D.P.; Bolin, A.P.; de Cássia Macedo dos Santos, R.; Polotow, T.G.; Sampaio, S.C.; Paes de Barros, M. Astaxanthin ameliorates the redox imbalance in lymphocytes of experimental diabetic rats. Chem. Biol. Interact. 2010, 186, 306–315. [Google Scholar] [CrossRef]
- Palozza, P.; Torelli, C.; Boninsegna, A.; Simone, R.; Catalano, A.; Mele, M.C.; Picci, N. Growth-inhibitory effects of the astaxanthin-rich alga Haematococcus pluvialis in human colon cancer cells. Cancer Lett. 2009, 283, 108–117. [Google Scholar] [CrossRef]
- Nakao, R.; Nelson, O.L.; Park, J.S.; Mathison, B.D.; Thompson, P.A.; Chew, B.P. Effect of dietary astaxanthin at different stages of mammary tumor initiation in BALB/c mice. Anticancer Res. 2010, 30, 2171–2175. [Google Scholar]
- Prabhu, P.N.; AshokKumar, P.; Sudhandiran, G. Antioxidative and antiproliferative effects of astaxanthin during the initiation stages of 1,2-dimethyl hydrazine-induced experimental colon carcinogenesis. Fundam. Clin. Pharmacol. 2009, 23, 225–234. [Google Scholar] [CrossRef]
- Satoh, A.; Tsuji, S.; Okada, Y.; Murakami, N.; Urami, M.; Nakagawa, K.; Ishikura, M.; Katagiri, M.; Koga, Y.; Shirasawa, T. Preliminary Clinical Evaluation of Toxicity and Efficacy of A New Astaxanthin-rich Haematococcus pluvialis Extract. J. Clin. Biochem. Nutr. 2009, 44, 280–284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Pan, L.; Wei, X.; Gao, H.; Liu, J. Impact of astaxanthin-enriched algal powder of Haematococcus pluvialis on memory improvement in BALB/c mice. Environ. Geochem. Health 2007, 29, 483–489. [Google Scholar] [CrossRef]
- Tominaga, K.; Hongo, N.; Karato, M.; Yamashita, E. Cosmetic benefits of astaxanthin on humans subjects. Acta Biochim. Pol. 2012, 59, 43–47. [Google Scholar] [CrossRef]
- D’Orazio, N.; Gemello, E.; Gammone, M.A.; De Girolamo, M.; Ficoneri, C.; Riccioni, G. Fucoxantin: A Treasure from the Sea. Mar. Drugs 2012, 10, 604–616. [Google Scholar] [CrossRef] [Green Version]
- Halestrap, A.P. What is the mitochondrial permeability transition pore? J. Mol. Cell. Cardiol. 2009, 46, 821–831. [Google Scholar] [CrossRef] [PubMed]
- Tsujimoto, Y.; Shimizu, S. Role of the mitochondrial membrane permeability transition in cell death. Apoptosis 2007, 12, 835–840. [Google Scholar] [CrossRef] [Green Version]
- Hurst, S.; Hoek, J.; Sheu, S.-S. Mitochondrial Ca(2+) and regulation of the permeability transition pore. J. Bioenerg. Biomembr. 2017, 49, 27–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morciano, G.; Giorgi, C.; Bonora, M.; Punzetti, S.; Pavasini, R.; Wieckowski, M.R.; Campo, G.; Pinton, P. Molecular identity of the mitochondrial permeability transition pore and its role in ischemia-reperfusion injury. J. Mol. Cell. Cardiol. 2015, 78, 142–153. [Google Scholar] [CrossRef]
- Azarashvili, T.; Krestinina, O.; Galvita, A.; Grachev, D.; Baburina, Y.; Stricker, R.; Reiser, G. Identification of phosphorylated form of 2’, 3’-cyclic nucleotide 3’-phosphodiesterase (cnpase) as 46 kda phosphoprotein in brain non-synaptic mitochondria overloaded by calcium. J. Bioenerg. Biomembr. 2014, 46, 135–145. [Google Scholar] [CrossRef]
- Azarashvili, T.; Krestinina, O.; Galvita, A.; Grachev, D.; Baburina, Y.; Stricker, R.; Evtodienko, Y.; Reiser, G. Ca2+-dependent permeability transition regulation in rat brain mitochondria by 2’,3’-cyclic nucleotides and 2’,3’-cyclic nucleotide 3’-phosphodiesterase. Am. J. Physiol Cell Physiol. 2009, 296, C1428–C1439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galvita, A.; Grachev, D.; Azarashvili, T.; Baburina, Y.; Krestinina, O.; Stricker, R.; Reiser, G. The brain-specific protein, p42(iP4)(ADAP 1) is localized in mitochondria and involved in regulation of mitochondrial Ca2+. J. Neurochem. 2009, 109, 1701–1713. [Google Scholar] [CrossRef]
- Baburina, Y.; Azarashvili, T.; Grachev, D.; Krestinina, O.; Galvita, A.; Stricker, R.; Reiser, G. Mitochondrial 2′, 3′-cyclic nucleotide 3′-phosphodiesterase (CNP) interacts with mPTP modulators and functional complexes (I–V) coupled with release of apoptotic factors. Neurochem. Int. 2015, 90, 46–55. [Google Scholar] [CrossRef]
- Célis, H. 1-Butanol extracted proteolipid. Proton conducting properties. Biochem. Biophys. Res. Commun. 1980, 92, 26–31. [Google Scholar] [CrossRef]
- Wittig, I.; Schägger, H. Structural organization of mitochondrial ATP synthase. Biochim. Biophys. Acta 2008, 1777, 592–598. [Google Scholar] [CrossRef] [Green Version]
- Azarashvili, T.; Odinokova, I.; Bakunts, A.; Ternovsky, V.; Krestinina, O.; Tyynelä, J.; Saris, N.-E.L. Potential role of subunit c of F0F1-ATPase and subunit c of storage body in the mitochondrial permeability transition. Effect of the phosphorylation status of subunit c on pore opening. Cell Calcium 2014, 55, 69–77. [Google Scholar] [CrossRef]
- Gunter, T.E.; Yule, D.I.; Gunter, K.K.; Eliseev, R.A.; Salter, J.D. Calcium and mitochondria. FEBS Lett. 2004, 567, 96–102. [Google Scholar] [CrossRef] [PubMed]
- Crompton, M.; Barksby, E.; Johnson, N.; Capano, M. Mitochondrial intermembrane junctional complexes and their involvement in cell death. Biochimie 2002, 84, 143–152. [Google Scholar] [CrossRef]
- Crompton, M.; Costi, A. Kinetic evidence for a heart mitochondrial pore activated by Ca2+, inorganic phosphate and oxidative stress. A potential mechanism for mitochondrial dysfunction during cellular Ca2+ overload. Eur. J. Biochem. 1988, 178, 489–501. [Google Scholar] [CrossRef] [PubMed]
- Crompton, M.; Costi, A.; Hayat, L. Evidence for the presence of a reversible Ca2+-dependent pore activated by oxidative stress in heart mitochondria. Biochem. J. 1987, 245, 915–918. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nazareth, W.; Yafei, N.; Crompton, M. Inhibition of anoxia-induced injury in heart myocytes by cyclosporin A. J. Mol. Cell. Cardiol. 1991, 23, 1351–1354. [Google Scholar] [CrossRef]
- Leyssens, A.; Nowicky, A.V.; Patterson, L.; Crompton, M.; Duchen, M.R. The relationship between mitochondrial state, ATP hydrolysis, [Mg2+]i and [Ca2+]i studied in isolated rat cardiomyocytes. J. Physiol. 1996, 496 Pt 1, 111–128. [Google Scholar] [CrossRef]
- Griffiths, E.J.; Halestrap, A.P. Mitochondrial non-specific pores remain closed during cardiac ischaemia, but open upon reperfusion. Biochem. J. 1995, 307, 93–98. [Google Scholar] [CrossRef] [Green Version]
- Griffiths, E.J.; Halestrap, A. Protection by Cyclosporin A of Ischemia/Reperfusion-Induced Damage in Isolated Rat Hearts. J. Mol. Cell. Cardiol. 1993, 25, 1461–1469. [Google Scholar] [CrossRef]
- Di Lisa, F.; Bernardi, P. Mitochondria and ischemia–reperfusion injury of the heart: Fixing a hole. Cardiovasc. Res. 2006, 70, 191–199. [Google Scholar] [CrossRef] [PubMed]
- Halestrap, A.P.; Clarke, S.J.; Javadov, S.A. Mitochondrial permeability transition pore opening during myocardial reperfusion—A target for cardioprotection. Cardiovasc. Res. 2004, 61, 372–385. [Google Scholar] [CrossRef] [Green Version]
- Halestrap, A.P.; Pasdois, P. The role of the mitochondrial permeability transition pore in heart disease. Biochim. Biophys. Acta 2009, 1787, 1402–1415. [Google Scholar] [CrossRef] [Green Version]
- Yellon, D.M.; Hausenloy, D.J. Myocardial Reperfusion Injury. N. Engl. J. Med. 2007, 357, 1121–1135. [Google Scholar] [CrossRef]
- Halestrap, A.P.; McStay, G.P.; Clarke, S.J. The permeability transition pore complex: Another view. Biochimie 2002, 84, 153–166. [Google Scholar] [CrossRef]
- Juhaszova, M.; Wang, S.; Zorov, D.B.; Nuss, H.B.; Gleichmann, M.; Mattson, M.P.; Sollott, S.J. The identity and regulation of the mitochondrial permeability transition pore: Where the known meets the unknown. Ann. N. Y. Acad. Sci. 2008, 1123, 197–212. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.S.; Jin, Y.; Lemasters, J.J. Reactive oxygen species, but not ca2+ overloading, trigger ph- and mitochondrial permeability transition-dependent death of adult rat myocytes after ischemia-reperfusion. Am. J. Physiol. Heart Circ. Physiol. 2006, 290, H2024–H2034. [Google Scholar] [CrossRef] [PubMed]
- Wolf, A.M.; Asoh, S.; Hiranuma, H.; Ohsawa, I.; Iio, K.; Satou, A.; Ishikura, M.; Ohta, S. Astaxanthin protects mitochondrial redox state and functional integrity against oxidative stress. J. Nutr. Biochem. 2010, 21, 381–389. [Google Scholar] [CrossRef] [PubMed]
- Kuroki, T.; Ikeda, S.; Okada, T.; Maoka, T.; Kitamura, A.; Sugimoto, M.; Kume, S. Astaxanthin ameliorates heat stress-induced impairment of blastocyst development in vitro:--astaxanthin colocalization with and action on mitochondria. J. Assist. Reprod. Genet. 2013, 30, 623–631. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.-W.; Xu, X.-C.; Liu, T.; Yuan, S. Mitochondrion-Permeable Antioxidants to Treat ROS-Burst-Mediated Acute Diseases. Oxid. Med. Cell. Longev. 2016, 2016, 6859523. [Google Scholar] [CrossRef] [Green Version]
- Park, J.S.; Mathison, B.D.; Hayek, M.G.; Zhang, J.; Reinhart, G.A.; Chew, B.P. Astaxanthin modulates age-associated mi-tochondrial dysfunction in healthy dogs. J. Anim. Sci. 2013, 91, 268–275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baburina, Y.; Krestinin, R.; Odinokova, I.; Sotnikova, L.; Kruglov, A.; Krestinina, O. Astaxanthin Inhibits Mitochondrial Permeability Transition Pore Opening in Rat Heart Mitochondria. Antioxidants (Basel) 2019, 8, 576. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.; Shamoto-Nagai, M.; Maruyama, W.; Osawa, T.; Naoi, M. Phytochemicals prevent mitochondrial membrane permeabilization and protect SH-SY5Y cells against apoptosis induced by PK11195, a ligand for outer membrane translocator protein. J. Neural Transm. (Vienna) 2016, 124, 89–98. [Google Scholar] [CrossRef] [PubMed]
- Morin, D.; Musman, J.; Pons, S.; Berdeaux, A.; Ghaleh, B. Mitochondrial translocator protein (TSPO): From physiology to cardioprotection. Biochem. Pharmacol. 2016, 105, 1–13. [Google Scholar] [CrossRef]
- McEnery, M.W.; Snowman, A.M.; Trifiletti, R.R.; Snyder, S.H. Isolation of the mitochondrial benzodiazepine receptor: Association with the voltage-dependent anion channel and the adenine nucleotide carrier. Proc. Natl. Acad. Sci. USA 1992, 89, 3170–3174. [Google Scholar] [CrossRef] [Green Version]
- Elrod, J.W.; Molkentin, J.D. Physiologic Functions of Cyclophilin D and the Mitochondrial Permeability Transition Pore. Circ. J. 2013, 77, 1111–1122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Basso, E.; Fante, L.; Fowlkes, J.; Petronilli, V.; Forte, M.A.; Bernardi, P. Properties of the Permeability Transition Pore in Mitochondria Devoid of Cyclophilin D. J. Biol. Chem. 2005, 280, 18558–18561. [Google Scholar] [CrossRef] [Green Version]
- Giorgio, V.; Bisetto, E.; Soriano, M.E.; Dabbeni-Sala, F.; Basso, E.; Petronilli, V.; Forte, M.A.; Bernardi, P.; Lippe, G. Cy-clophilin d modulates mitochondrial f0f1-atp synthase by interacting with the lateral stalk of the complex. J. Biol. Chem. 2009, 284, 33982–33988. [Google Scholar] [CrossRef] [Green Version]
- Porter, G.A., Jr.; Beutner, G. Cyclophilin d, somehow a master regulator of mitochondrial function. Biomolecules 2018, 8, 176. [Google Scholar] [CrossRef] [Green Version]
- Chinopoulos, C.; Adam-Vizi, V. Modulation of the mitochondrial permeability transition by cyclophilin D: Moving closer to f(0)–f(1) ATP synthase? Mitochondrion 2012, 12, 41–45. [Google Scholar] [CrossRef]
- Bonora, M.; Bononi, A.; De Marchi, E.; Giorgi, C.; Lebiedzinska, M.; Marchi, S.; Patergnani, S.; Rimessi, A.; Suski, J.M.; Wojtala, A.; et al. Role of the c subunit of the fo atp synthase in mitochondrial permeability transition. Cell Cycle 2013, 12, 674–683. [Google Scholar] [CrossRef] [Green Version]
- Neginskaya, M.A.; Solesio, M.E.; Berezhnaya, E.V.; Amodeo, G.F.; Mnatsakanyan, N.; Jonas, E.A.; Pavlov, E.V. Atp synthase c-subunit-deficient mitochondria have a small cyclosporine a-sensitive channel, but lack the permeability transition pore. Cell Rep. 2019, 26, 11–17e12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krestinin, R.; Baburina, Y.; Odinokova, I.; Kruglov, A.; Fadeeva, I.; Zvyagina, A.; Sotnikova, L.; Krestinina, O. Isoproterenol-induced permeability transition pore-related dysfunction of heart mitochondria is attenuated by astaxanthin. Biomedicines 2020, 8, 437. [Google Scholar] [CrossRef] [PubMed]
- Feng, W.; Li, W. The study of ISO induced heart failure rat model. Exp. Mol. Pathol. 2010, 88, 299–304. [Google Scholar] [CrossRef]
- Guo, R.; Zong, S.; Wu, M.; Gu, J.; Yang, M. Architecture of Human Mitochondrial Respiratory Megacomplex I2III2IV2. Cell 2017, 170, 1247–1257.e12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iwata, S.; Lee, J.W.; Okada, K.; Lee, J.K.; Iwata, M.; Rasmussen, B.; Link, T.A.; Ramaswamy, S.; Jap, B.K. Complete Structure of the 11-Subunit Bovine Mitochondrial Cytochrome bc1 Complex. Science 1998, 281, 64–71. [Google Scholar] [CrossRef]
- Zhao, R.-Z.; Jiang, S.; Zhang, L.; Yu, Z.-B. Mitochondrial electron transport chain, ROS generation and uncoupling (Review). Int. J. Mol. Med. 2019, 44, 3–15. [Google Scholar] [CrossRef] [Green Version]
- Schlame, M.; Rua, D.; Greenberg, M.L. The biosynthesis and functional role of cardiolipin. Prog. Lipid Res. 2000, 39, 257–288. [Google Scholar] [CrossRef]
- Chicco, A.J.; Sparagna, G.C. Role of cardiolipin alterations in mitochondrial dysfunction and disease. Am. J. Physiol. Cell Physiol. 2007, 292, C33–C44. [Google Scholar] [CrossRef] [Green Version]
- Dolinsky, V.W.; Cole, L.K.; Sparagna, G.C.; Hatch, G.M. Cardiac mitochondrial energy metabolism in heart failure: Role of cardiolipin and sirtuins. Biochim. Biophys. Acta 2016, 1861, 1544–1554. [Google Scholar] [CrossRef]
- Fiedorczuk, K.; Letts, J.A.; Degliesposti, G.; Kaszuba, K.; Skehel, M.; Sazanov, L.A. Atomic structure of the entire mammalian mitochondrial complex i. Nature 2016, 538, 406–410. [Google Scholar] [CrossRef] [Green Version]
- Palsdottir, H.; Lojero, C.G.; Trumpower, B.L.; Hunte, C. Structure of the yeast cytochrome bc1 complex with a hydroxyquinone anion qo site inhibitor bound. J. Biol. Chem. 2003, 278, 31303–31311. [Google Scholar] [CrossRef] [Green Version]
- Shinzawa-Itoh, K.; Aoyama, H.; Muramoto, K.; Terada, H.; Kurauchi, T.; Tadehara, Y.; Yamasaki, A.; Sugimura, T.; Kurono, S.; Tsujimoto, K.; et al. Structures and physiological roles of 13 integral lipids of bovine heart cytochrome c oxidase. EMBO J. 2007, 26, 1713–1725. [Google Scholar] [CrossRef] [PubMed]
- Dudek, J.; Cheng, I.F.; Chowdhury, A.; Wozny, K.; Balleininger, M.; Reinhold, R.; Grunau, S.D.; Callegari, S.; Toischer, K.; Wanders, R.J.; et al. Cardiac-specific succinate dehydrogenase deficiency in Barth syndrome. EMBO Mol. Med. 2016, 8, 139–154. [Google Scholar] [CrossRef] [PubMed]
- Dudek, J. Role of Cardiolipin in Mitochondrial Signaling Pathways. Front. Cell Dev. Biol. 2017, 5, 90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodrigo, R.; Libuy, M.; Feliu, F.; Hasson, D. Oxidative stress-related biomarkers in essential hypertension and ischemia-reperfusion myocardial damage. Dis. Mark. 2013, 35, 773–790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Long, Q.; Yang, K.; Yang, Q. Regulation of mitochondrial ATP synthase in cardiac pathophysiology. Am. J. Cardiovasc. Dis. 2015, 5, 19–32. [Google Scholar]
- Sinatra, S.T. Metabolic cardiology: An integrative strategy in the treatment of congestive heart failure. Altern. Ther. Health Med. 2009, 15, 44–52. [Google Scholar] [PubMed]
- Chen, C.; Ko, Y.; Delannoy, M.; Ludtke, S.J.; Chiu, W.; Pedersen, P.L. Mitochondrial atp synthasome: Three-dimensional structure by electron microscopy of the atp synthase in complex formation with carriers for pi and adp/atp. J. Biol. Chem. 2004, 279, 31761–31768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, C.; Saxena, A.K.; Simcoke, W.N.; Garboczi, D.N.; Pedersen, P.L.; Ko, Y.H. Mitochondrial atp synthase. Crystal structure of the catalytic f1 unit in a vanadate-induced transition-like state and implications for mechanism. J. Biol. Chem. 2006, 281, 13777–13783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Krestinina, O.; Baburina, Y.; Krestinin, R. Mitochondrion as a Target of Astaxanthin Therapy in Heart Failure. Int. J. Mol. Sci. 2021, 22, 7964. https://doi.org/10.3390/ijms22157964
Krestinina O, Baburina Y, Krestinin R. Mitochondrion as a Target of Astaxanthin Therapy in Heart Failure. International Journal of Molecular Sciences. 2021; 22(15):7964. https://doi.org/10.3390/ijms22157964
Chicago/Turabian StyleKrestinina, Olga, Yulia Baburina, and Roman Krestinin. 2021. "Mitochondrion as a Target of Astaxanthin Therapy in Heart Failure" International Journal of Molecular Sciences 22, no. 15: 7964. https://doi.org/10.3390/ijms22157964
APA StyleKrestinina, O., Baburina, Y., & Krestinin, R. (2021). Mitochondrion as a Target of Astaxanthin Therapy in Heart Failure. International Journal of Molecular Sciences, 22(15), 7964. https://doi.org/10.3390/ijms22157964