
In Silico Structural Modeling and Analysis of Interactions of Tremellomycetes Cytochrome P450 Monooxygenases CYP51s with Substrates and Azoles

 , , ,
, , ,  and
and
Abstract
:1. Introduction
2. Results and Discussion
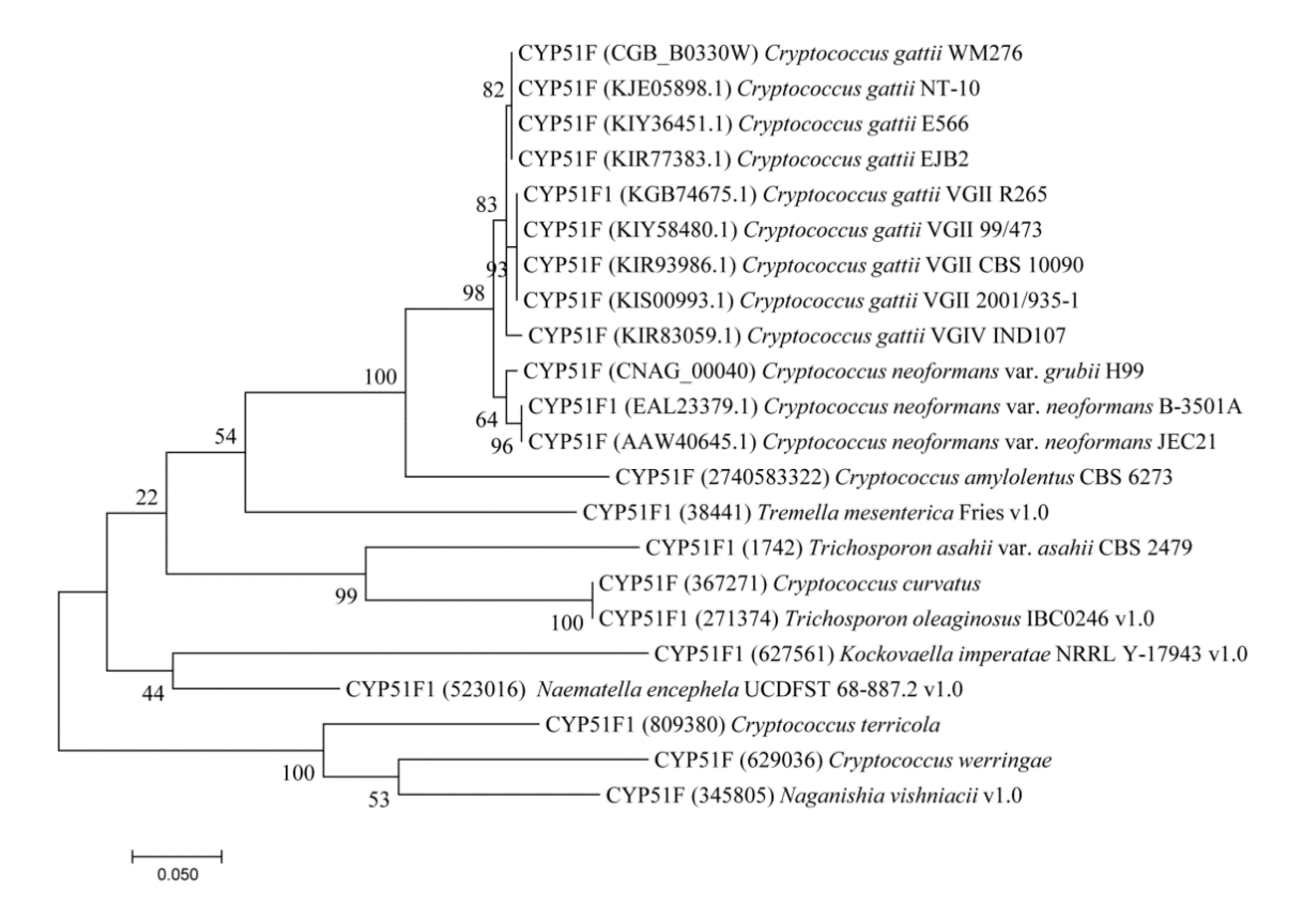
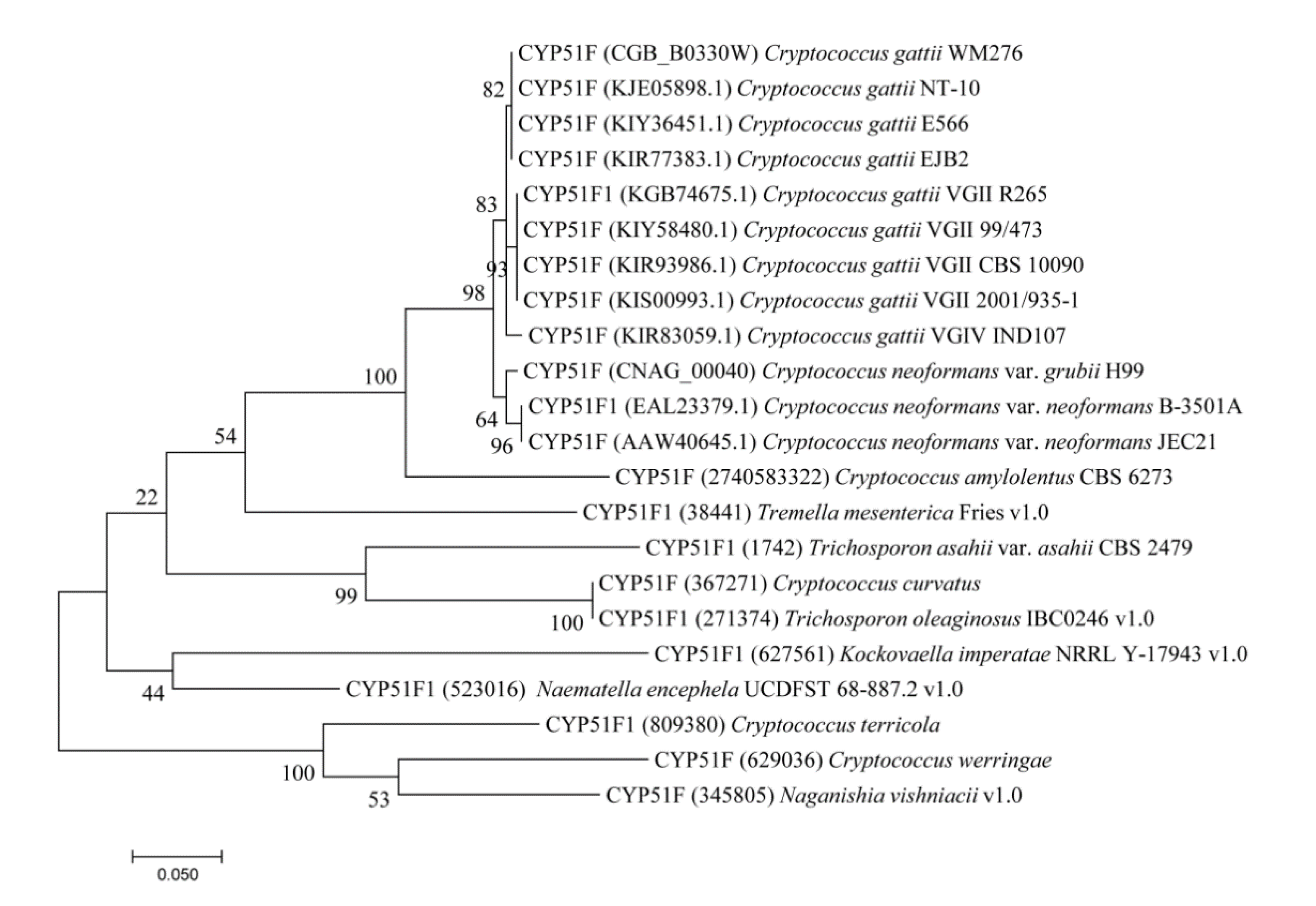
2.1. CYP51s Grouped as per Tremellomycetes Lifestyle
2.2. CYP51s of Tremellomycetes Have All the CYP Characteristic Motifs
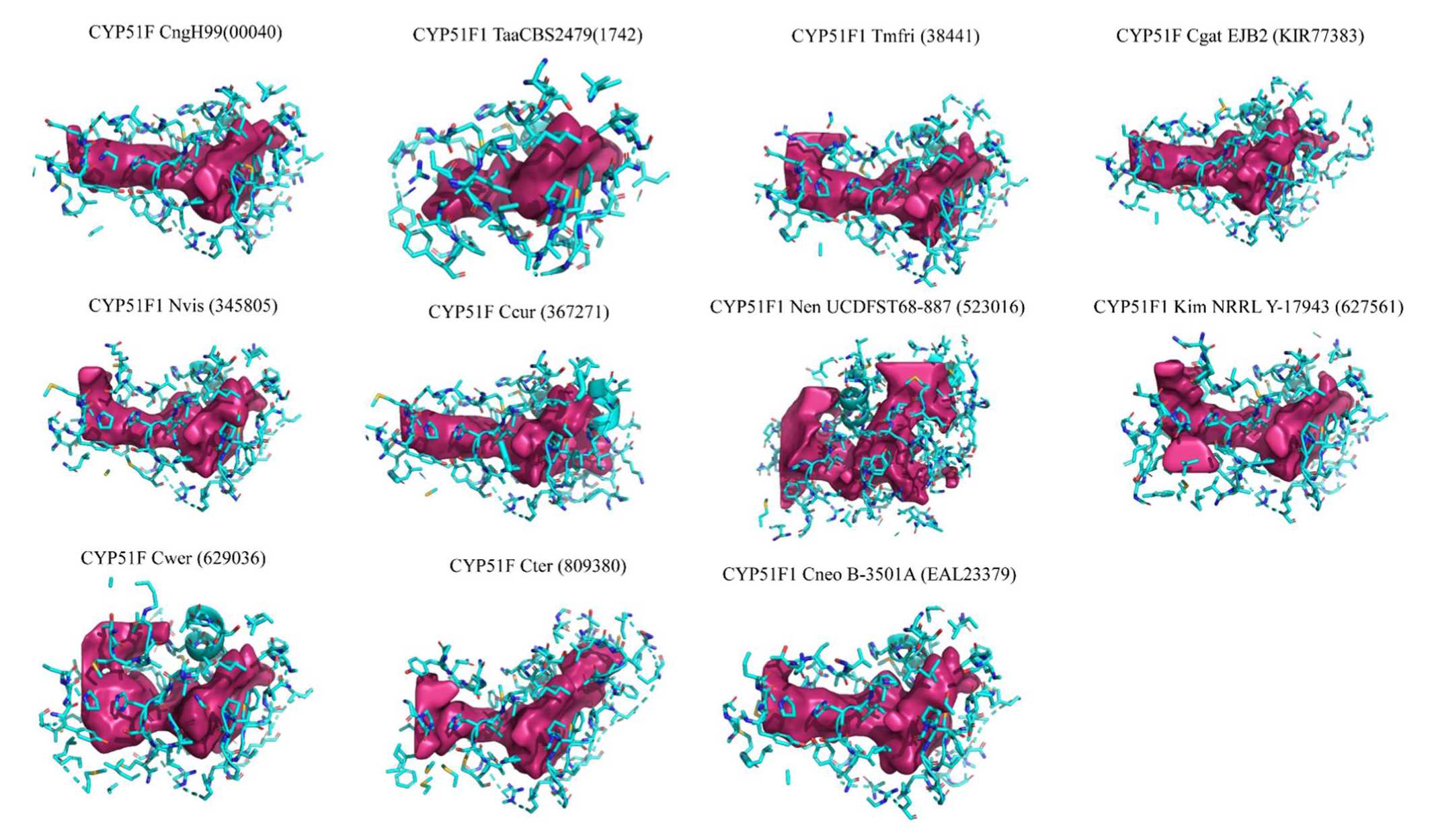
2.3. Tremellomycetes CYP51s Active Site Cavities Are Highly Hydrophobic
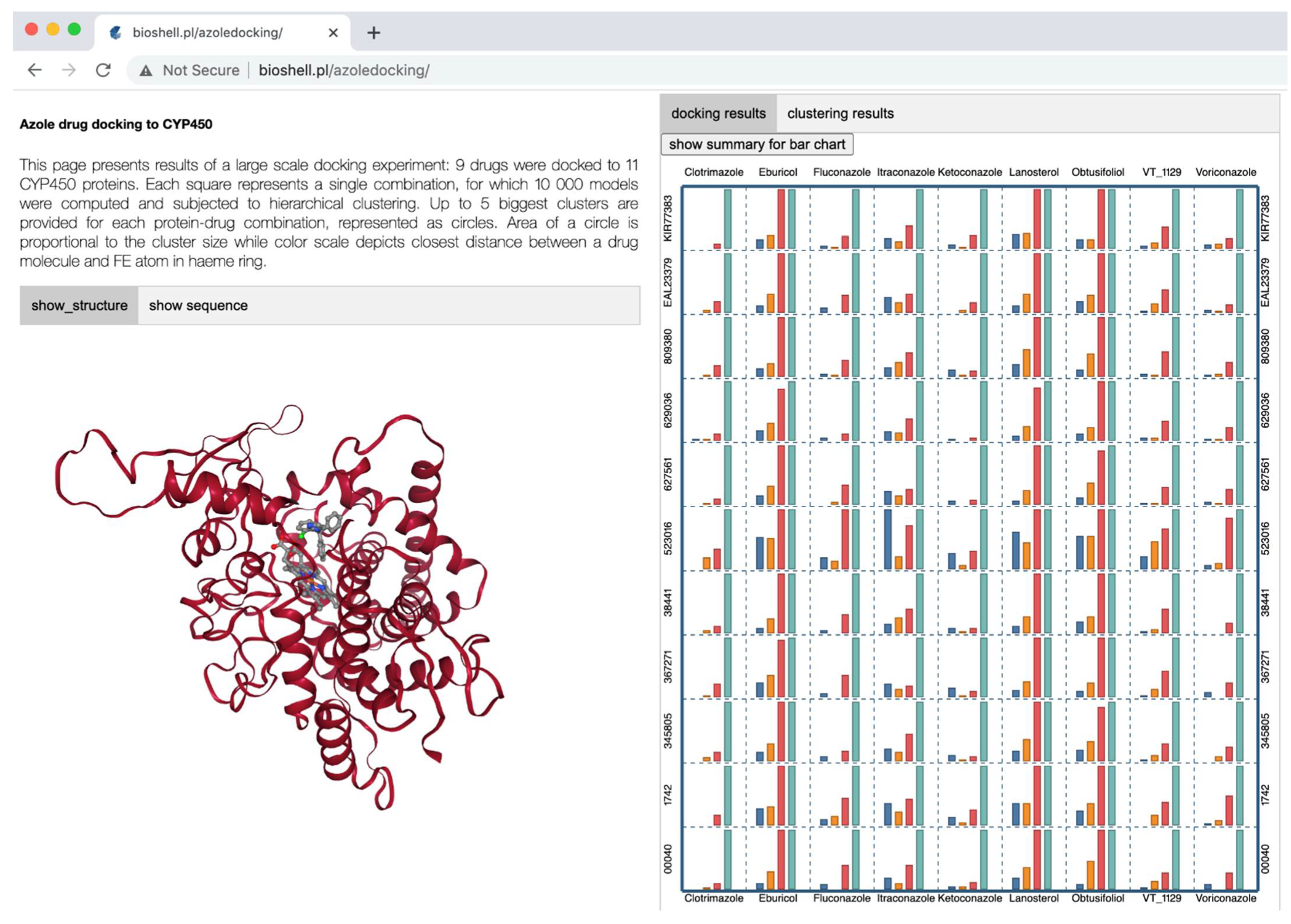
2.4. A Web Application for Visualization of Tremellomycetes CYP51s Interactions with Ligands
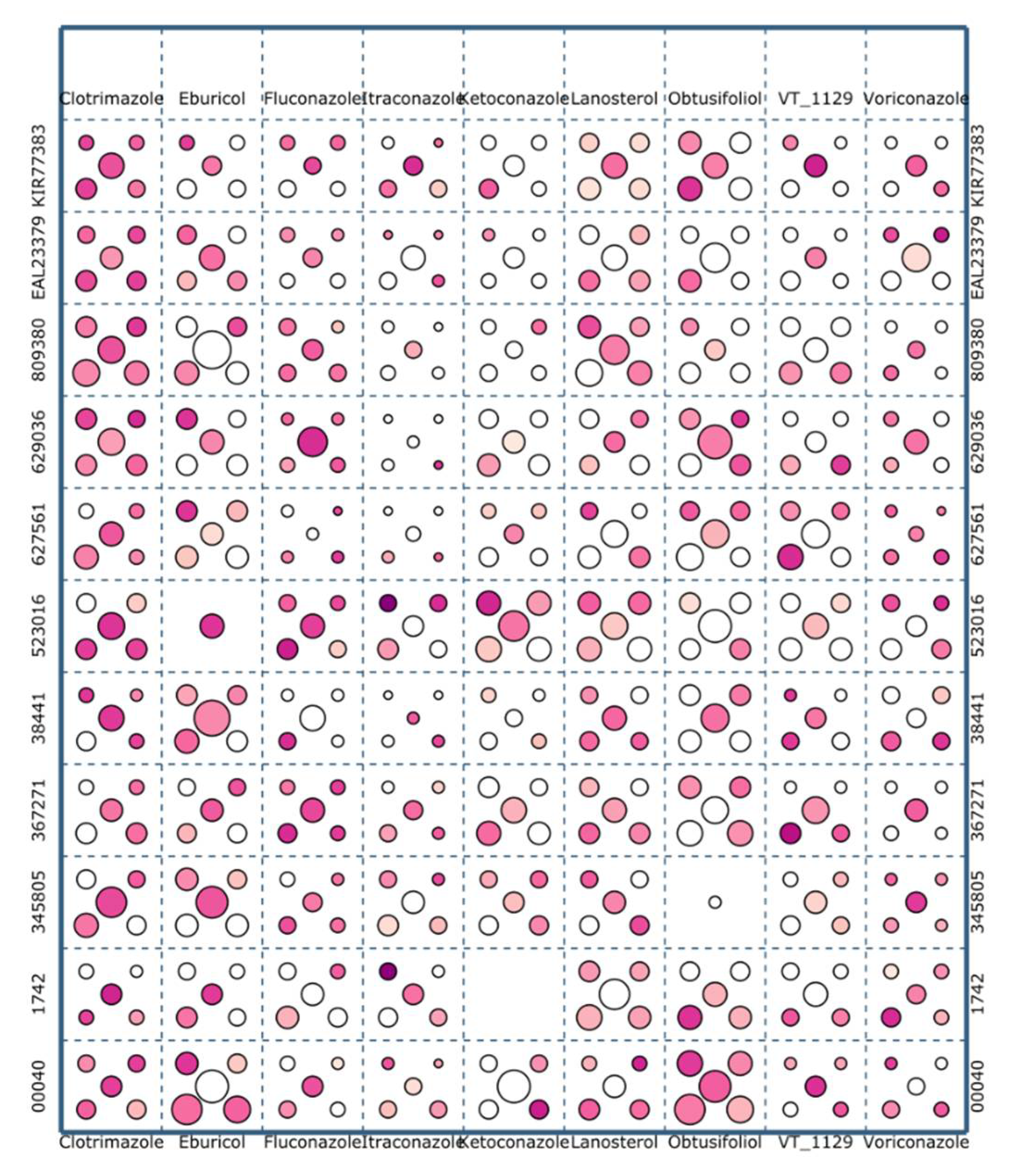
2.5. Tremellomycetes CYP51s Have the Highest Preference for Itraconazole
- ‘clashes’ where a ligand was closer to the Fe than 1.5 Å;
- ‘correct’ where dNFe was in the range 1.5–2.5 Å;
- ‘near’ with dNFe in the range 2.5–3.5 Å;
- ‘pocket’ with dNFe in the range 3.5–8.0 Å.
2.6. High Conservation Observed in Tremellomycetes CYP51s Amino Acids Interacting with Ligands
3. Materials and Methods
3.1. CYP51s Used in the Study
3.2. Phylogenetic Analysis
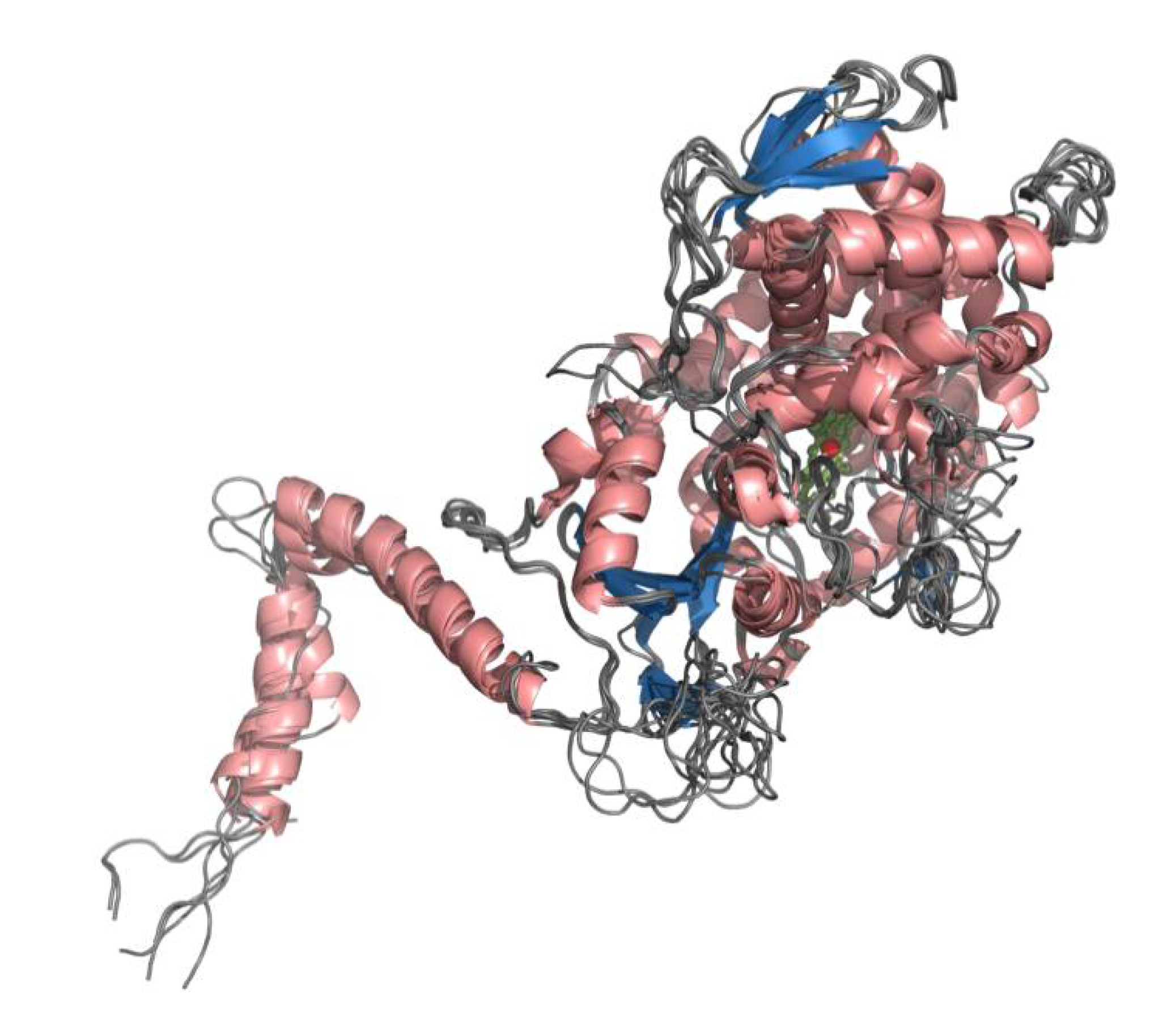
3.3. Homology Modeling of CYP51s
3.4. Preparation of CYP51s Models for Docking
3.5. Ligands and Their Docking Procedure
3.6. Hierarchical Agglomerative Clustering
3.7. Protocols
3.8. Data Visualization
3.9. Active Site Cavity and Ligand Interacting Amino Acids Detection
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rajasingham, R.; Smith, R.M.; Park, B.J.; Jarvis, J.N.; Govender, N.P.; Chiller, T.M.; Denning, D.W.; Loyse, A.; Boulware, D.R. Global burden of disease of HIV-associated cryptococcal meningitis: An updated analysis. Lancet Infect. Dis. 2017, 17, 873–881. [Google Scholar] [CrossRef] [Green Version]
- Kwon-Chung, K.J.; Fraser, J.A.; Doering, T.L.; Wang, Z.A.; Janbon, G.; Idnurm, A.; Bahn, Y.-S. Cryptococcus neoformans and Cryptococcus gattii, the etiologic agents of cryptococcosis. Cold Spring Harb. Perspect. Med. 2014, 4, a019760. [Google Scholar] [CrossRef]
- Coelho, C.; Casadevall, A. Cryptococcal therapies and drug targets: The old, the new and the promising. Cell. Microbiol. 2016, 18, 792–799. [Google Scholar] [CrossRef] [Green Version]
- WHO. Guidelines for the Diagnosis, Prevention, and Management of Cryptococcal Disease in HIV-Infected Adults, Adolescents and Children, March 2018: Supplement to the 2016 Consolidated Guidelines of the Use of Antiretroviral Drugs for Treating and Preventing HIV Infection; WHO: Geneva, Switzerland, 2018. [Google Scholar]
- Loyse, A.; Thangaraj, H.; Easterbrook, P.; Ford, N.; Roy, M.; Chiller, T.; Govender, N.; Harrison, T.S.; Bicanic, T. Cryptococcal meningitis: Improving access to essential antifungal medicines in resource-poor countries. Lancet Infect. Dis. 2013, 13, 629–637. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.-C.; Chang, T.-Y.; Liu, J.-W.; Chen, F.-J.; Chien, C.-C.; Lee, C.-H.; Lu, C.-H. Increasing trend of fluconazole-non-susceptible Cryptococcus neoformans in patients with invasive cryptococcosis: A 12-year longitudinal study. BMC Infect. Dis. 2015, 15, 277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Xiao, M.; Chen, S.C.-A.; Kong, F.; Sun, Z.-Y.; Liao, K.; Lu, J.; Shao, H.-F.; Yan, Y.; Fan, H. In vitro susceptibilities of yeast species to fluconazole and voriconazole as determined by the 2010 National China Hospital Invasive Fungal Surveillance Net (CHIF-NET) study. J. Clin. Microbiol. 2012, 50, 3952–3959. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsueh, P.-R.; Lau, Y.-J.; Chuang, Y.-C.; Wan, J.-H.; Huang, W.-K.; Shyr, J.-M.; Yan, J.-J.; Yu, K.-W.; Wu, J.-J.; Ko, W.-C. Antifungal susceptibilities of clinical isolates of Candida species, Cryptococcus neoformans, and Aspergillus species from Taiwan: Surveillance of multicenter antimicrobial resistance in Taiwan program data from 2003. Antimicrob. Agents Chemother. 2005, 49, 512–517. [Google Scholar] [CrossRef] [Green Version]
- Sar, B.; Monchy, D.; Vann, M.; Keo, C.; Sarthou, J.L.; Buisson, Y. Increasing in vitro resistance to fluconazole in Cryptococcus neoformans Cambodian isolates: April 2000 to March 2002. J. Antimicrob. Chemother. 2004, 54, 563–565. [Google Scholar] [CrossRef]
- Pan, W.; Khayhan, K.; Hagen, F.; Wahyuningsih, R.; Chakrabarti, A.; Chowdhary, A.; Ikeda, R.; Taj-Aldeen, S.J.; Khan, Z.; Imran, D. Resistance of Asian Cryptococcus neoformans serotype A is confined to few microsatellite genotypes. PLoS ONE 2012, 7, e32868. [Google Scholar] [CrossRef]
- Smith, K.D.; Achan, B.; Hullsiek, K.H.; McDonald, T.R.; Okagaki, L.H.; Alhadab, A.A.; Akampurira, A.; Rhein, J.R.; Meya, D.B.; Boulware, D.R. Increased antifungal drug resistance in clinical isolates of Cryptococcus neoformans in Uganda. Antimicrob. Agents Chemother. 2015, 59, 7197–7204. [Google Scholar] [CrossRef] [Green Version]
- Trilles, L.; Fernández-Torres, B.; dos Santos Lazéra, M.; Wanke, B.; Guarro, J. In vitro antifungal susceptibility of Cryptococcus gattii. J. Clin. Microbiol. 2004, 42, 4815–4817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thompson, G.R.; Wiederhold, N.P.; Fothergill, A.W.; Vallor, A.C.; Wickes, B.L.; Patterson, T.F. Antifungal susceptibilities among different serotypes of Cryptococcus gattii and Cryptococcus neoformans. Antimicrob. Agents Chemother. 2009, 53, 309–311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barchiesi, F.; Schimizzi, A.M.; Caselli, F.; Novelli, A.; Fallani, S.; Giannini, D.; Arzeni, D.; Di Cesare, S.; Di Francesco, L.F.; Fortuna, M. Interactions between triazoles and amphotericin B against Cryptococcus neoformans. Antimicrob. Agents Chemother. 2000, 44, 2435–2441. [Google Scholar] [CrossRef] [Green Version]
- Illnait-Zaragozi, M.-T.; Martínez, G.F.; Curfs-Breuker, I.; Fernández, C.M.; Boekhout, T.; Meis, J.F. In vitro activity of the new azole isavuconazole (BAL4815) compared with six other antifungal agents against 162 Cryptococcus neoformans isolates from Cuba. Antimicrob. Agents Chemother. 2008, 52, 1580–1582. [Google Scholar] [CrossRef] [Green Version]
- Hagen, F.; Illnait-Zaragozi, M.-T.; Bartlett, K.H.; Swinne, D.; Geertsen, E.; Klaassen, C.H.; Boekhout, T.; Meis, J.F. In vitro antifungal susceptibilities and amplified fragment length polymorphism genotyping of a worldwide collection of 350 clinical, veterinary, and environmental Cryptococcus gattii isolates. Antimicrob. Agents Chemother. 2010, 54, 5139–5145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Armengou, A. Possible development of resistance to fluconazole during suppressive therapy for AIDS-associated cryptococcal meningitis. Clin. Infect. Dis. Off. Publ. Infect. Dis. Soc. Am. 1996, 23, 1337–1338. [Google Scholar] [CrossRef]
- Hoekstra, W.J.; Garvey, E.P.; Moore, W.R.; Rafferty, S.W.; Yates, C.M.; Schotzinger, R.J. Design and optimization of highly-selective fungal CYP51 inhibitors. Bioorganic Med. Chem. Lett. 2014, 24, 3455–3458. [Google Scholar] [CrossRef]
- Warrilow, A.; Hull, C.; Parker, J.; Garvey, E.; Hoekstra, W.; Moore, W.; Schotzinger, R.; Kelly, D.; Kelly, S. The clinical candidate VT-1161 is a highly potent inhibitor of Candida albicans CYP51 but fails to bind the human enzyme. Antimicrob. Agents Chemother. 2014, 58, 7121–7127. [Google Scholar] [CrossRef] [Green Version]
- Lockhart, S.R.; Fothergill, A.W.; Iqbal, N.; Bolden, C.B.; Grossman, N.T.; Garvey, E.P.; Brand, S.R.; Hoekstra, W.J.; Schotzinger, R.J.; Ottinger, E. The investigational fungal Cyp51 inhibitor VT-1129 demonstrates potent in vitro activity against Cryptococcus neoformans and Cryptococcus gattii. Antimicrob. Agents Chemother. 2016, 60, 2528–2531. [Google Scholar] [CrossRef] [Green Version]
- Nielsen, K.; Vedula, P.; Smith, K.D.; Meya, D.B.; Garvey, E.P.; Hoekstra, W.J.; Schotzinger, R.J.; Boulware, D.R. Activity of VT-1129 against Cryptococcus neoformans clinical isolates with high fluconazole MICs. Med. Mycol. 2017, 55, 453–456. [Google Scholar]
- Daum, G.; Lees, N.D.; Bard, M.; Dickson, R. Biochemistry, cell biology and molecular biology of lipids of Saccharomyces cerevisiae. Yeast 1998, 14, 1471–1510. [Google Scholar] [CrossRef]
- Kelly, S.L.; Lamb, D.C.; Kelly, D.E. Inhibitors of CYP51 as antifungal agents and resistance to azole antifungals. In Molecular and Applied Aspects of Oxidative Drug Metabolizing Enzymes; Springer: Boston, MA, USA, 1999; pp. 157–172. [Google Scholar]
- Lepesheva, G.I.; Friggeri, L.; Waterman, M.R. CYP51 as drug targets for fungi and protozoan parasites: Past, present and future. Parasitology 2018, 145, 1820–1836. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Li, L.; Lv, Q.; Yan, L.; Wang, Y.; Jiang, Y. The fungal CYP51s: Their functions, structures, related drug resistance, and inhibitors. Front. Microbiol. 2019, 10, 691. [Google Scholar] [CrossRef]
- Choi, J.Y.; Podust, L.M.; Roush, W.R. Drug strategies targeting CYP51 in neglected tropical diseases. Chem. Rev. 2014, 114, 11242–11271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burley, S.K.; Berman, H.M.; Bhikadiya, C.; Bi, C.; Chen, L.; Di Costanzo, L.; Christie, C.; Dalenberg, K.; Duarte, J.M.; Dutta, S. RCSB Protein Data Bank: Biological macromolecular structures enabling research and education in fundamental biology, biomedicine, biotechnology and energy. Nucleic Acids Res. 2019, 47, D464–D474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelly, S.L.; Kelly, D.E. Microbial cytochromes P450: Biodiversity and biotechnology. Where do cytochromes P450 come from, what do they do and what can they do for us? Philos. Trans. R. Soc. B Biol. Sci. 2013, 368, 20120476. [Google Scholar] [CrossRef] [Green Version]
- Rodero, L.; Mellado, E.; Rodriguez, A.C.; Salve, A.; Guelfand, L.; Cahn, P.; Cuenca-Estrella, M.; Davel, G.; Rodriguez-Tudela, J.L. G484S amino acid substitution in lanosterol 14-alpha demethylase (ERG11) is related to fluconazole resistance in a recurrent Cryptococcus neoformans clinical isolate. Antimicrob. Agents Chemother. 2003, 47, 3653–3656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sionov, E.; Chang, Y.C.; Garraffo, H.M.; Dolan, M.A.; Ghannoum, M.A.; Kwon-Chung, K.J. Identification of a Cryptococcus neoformans cytochrome P450 lanosterol 14alpha-demethylase (Erg11) residue critical for differential susceptibility between fluconazole/voriconazole and itraconazole/posaconazole. Antimicrob. Agents Chemother. 2012, 56, 1162–1169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kano, R.; Okubo, M.; Hasegawa, A.; Kamata, H. Multi-azole-resistant strains of Cryptococcus neoformans var. grubii isolated from a FLZ-resistant strain by culturing in medium containing voriconazole. Med. Mycol. 2017, 55, 877–882. [Google Scholar]
- Lamb, D.; Corran, A.; Baldwin, B.C.; Kwon-Chung, J.; Kelly, S. Resistant P45051A1 activity in azole antifungal tolerant Cryptococcus neoformans from AIDS patients. FEBS Lett. 1995, 368, 326–330. [Google Scholar] [CrossRef] [Green Version]
- Sheng, C.; Miao, Z.; Ji, H.; Yao, J.; Wang, W.; Che, X.; Dong, G.; Lü, J.; Guo, W.; Zhang, W. Three-dimensional model of lanosterol 14α-demethylase from Cryptococcus neoformans: Active-site characterization and insights into azole binding. Antimicrob. Agents Chemother. 2009, 53, 3487–3495. [Google Scholar] [CrossRef] [Green Version]
- Akapo, O.O.; Padayachee, T.; Chen, W.; Kappo, A.P.; Yu, J.H.; Nelson, D.R.; Syed, K. Distribution and diversity of cytochrome P450 monooxygenases in the fungal class Tremellomycetes. Int. J. Mol. Sci. 2019, 20, 2889. [Google Scholar] [CrossRef] [Green Version]
- Parvez, M.; Qhanya, L.B.; Mthakathi, N.T.; Kgosiemang, I.K.; Bamal, H.D.; Pagadala, N.S.; Xie, T.; Yang, H.; Chen, H.; Theron, C.W.; et al. Molecular evolutionary dynamics of cytochrome P450 monooxygenases across kingdoms: Special focus on mycobacterial P450s. Sci. Rep. 2016, 6, 33099. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ngcobo, N.S.; Chiliza, Z.E.; Chen, W.; Yu, J.-H.; Nelson, D.R.; Tuszynski, J.A.; Preto, J.; Syed, K. Comparative analysis, structural insights, and substrate/drug interaction of CYP128A1 in Mycobacterium tuberculosis. Int. J. Mol. Sci. 2020, 21, 4816. [Google Scholar] [CrossRef]
- Syed, P.R.; Chen, W.; Nelson, D.R.; Kappo, A.P.; Yu, J.H.; Karpoormath, R.; Syed, K. Cytochrome P450 monooxygenase CYP139 family involved in the synthesis of secondary metabolites in 824 mycobacterial species. Int. J. Mol. Sci. 2019, 20, 2690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jawallapersand, P.; Mashele, S.S.; Kovacic, L.; Stojan, J.; Komel, R.; Pakala, S.B.; Krasevec, N.; Syed, K. Cytochrome P450 monooxygenase CYP53 family in fungi: Comparative structural and evolutionary analysis and its role as a common alternative anti-fungal drug target. PLoS ONE 2014, 9, e107209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Syed, K.; Shale, K.; Pagadala, N.S.; Tuszynski, J. Systematic identification and evolutionary analysis of catalytically versatile cytochrome P450 monooxygenase families enriched in model basidiomycete fungi. PLoS ONE 2014, 9, e86683. [Google Scholar] [CrossRef]
- Qhanya, L.B.; Matowane, G.; Chen, W.; Sun, Y.; Letsimo, E.M.; Parvez, M.; Yu, J.H.; Mashele, S.S.; Syed, K. Genome-wide annotation and comparative analysis of cytochrome P450 monooxygenases in Basidiomycete biotrophic plant pathogens. PLoS ONE 2015, 10, e0142100. [Google Scholar] [CrossRef] [Green Version]
- Webb, B.; Sali, A. Comparative Protein Structure Modeling Using MODELLER. Curr. Protoc. Bioinform. 2016, 54, 5.6.1–5.6.37. [Google Scholar] [CrossRef] [Green Version]
- Monk, B.C.; Tomasiak, T.M.; Keniya, M.V.; Huschmann, F.U.; Tyndall, J.D.; O’Connell, J.D.; Cannon, R.D.; McDonald, J.G.; Rodriguez, A.; Finer-Moore, J.S. Architecture of a single membrane spanning cytochrome P450 suggests constraints that orient the catalytic domain relative to a bilayer. Proc. Natl. Acad. Sci. USA 2014, 111, 3865–3870. [Google Scholar] [CrossRef] [Green Version]
- Baker, D.; Sali, A. Protein structure prediction and structural genomics. Science 2001, 294, 93–96. [Google Scholar] [CrossRef] [Green Version]
- Nelson, D.R. The cytochrome p450 homepage. Hum. Genom. 2009, 4, 59–65. [Google Scholar] [CrossRef] [Green Version]
- Podust, L.M.; Stojan, J.; Poulos, T.L.; Waterman, M.R. Substrate recognition sites in 14α-sterol demethylase from comparative analysis of amino acid sequences and X-ray structure of Mycobacterium tuberculosis CYP51. J. Inorg. Biochem. 2001, 87, 227–235. [Google Scholar] [CrossRef]
- Gotoh, O. Substrate recognition sites in cytochrome P450 family 2 (CYP2) proteins inferred from comparative analyses of amino acid and coding nucleotide sequences. J. Biol. Chem. 1992, 267, 83–90. [Google Scholar] [CrossRef]
- Balding, P.R.; Porro, C.S.; McLean, K.J.; Sutcliffe, M.J.; Maréchal, J.-D.; Munro, A.W.; Visser, S.P.d. How do azoles inhibit cytochrome P450 enzymes? A density functional study. J. Phys. Chem. A 2008, 112, 12911–12918. [Google Scholar] [CrossRef] [PubMed]
- Gront, D.; Hansmann, U.H.; Kolinski, A. Exploring protein energy landscapes with hierarchical clustering. Int. J. Quantum Chem. 2005, 105, 826–830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, D.T.; Taylor, W.R.; Thornton, J.M. The rapid generation of mutation data matrices from protein sequences. Bioinformatics 1992, 8, 275–282. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berman, H.M. The protein data bank: A historical perspective. Acta Crystallogr. Sect. A Found. Crystallogr. 2008, 64, 88–95. [Google Scholar] [CrossRef]
- Zimmermann, L.; Stephens, A.; Nam, S.-Z.; Rau, D.; Kübler, J.; Lozajic, M.; Gabler, F.; Söding, J.; Lupas, A.N.; Alva, V. A completely reimplemented MPI bioinformatics toolkit with a new HHpred server at its core. J. Mol. Biol. 2018, 430, 2237–2243. [Google Scholar] [CrossRef]
- Leman, J.K.; Weitzner, B.D.; Lewis, S.M.; Adolf-Bryfogle, J.; Alam, N.; Alford, R.F.; Aprahamian, M.; Baker, D.; Barlow, K.A.; Barth, P. Macromolecular modeling and design in Rosetta: Recent methods and frameworks. Nat. Methods 2020, 17, 665–680. [Google Scholar] [CrossRef] [PubMed]
- Koehler Leman, J.; Weitzner, B.D.; Renfrew, P.D.; Lewis, S.M.; Moretti, R.; Watkins, A.M.; Mulligan, V.K.; Lyskov, S.; Adolf-Bryfogle, J.; Labonte, J.W. Better together: Elements of successful scientific software development in a distributed collaborative community. PLoS Comput. Biol. 2020, 16, e1007507. [Google Scholar] [CrossRef] [PubMed]
- Meiler, J.; Baker, D. ROSETTALIGAND: Protein-small molecule docking with full side-chain flexibility. Proteins Struct. Funct. Bioinform. 2006, 65, 538–548. [Google Scholar] [CrossRef]
- Lemmon, G.; Meiler, J. Rosetta Ligand docking with flexible XML protocols. In Computational Drug Discovery and Design; Springer: New York, NY, USA, 2012; pp. 143–155. [Google Scholar]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An open chemical toolbox. J. Cheminformatics 2011, 3, 33. [Google Scholar] [CrossRef] [Green Version]
- Kothiwale, S.; Mendenhall, J.L.; Meiler, J. BCL:: C onf: Small molecule conformational sampling using a knowledge based rotamer library. J. Cheminformatics 2015, 7, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Chaudhury, S.; Lyskov, S.; Gray, J.J. PyRosetta: A script-based interface for implementing molecular modeling algorithms using Rosetta. Bioinformatics 2010, 26, 689–691. [Google Scholar] [CrossRef]
- Le, K.; Adolf-Bryfogle, J.; Klima, J.; Lyskov, S.; Labonte, J.; Bertolani, S.; Burman, S.R.; Leaver-Fay, A.; Weitzner, B.; Maguire, J. PyRosetta Jupyter Notebooks Teach Biomolecular Structure Prediction and Design. Biophysicist 2021, 2, 108–122. [Google Scholar] [CrossRef]
- Macnar, J.M.; Szulc, N.A.; Kryś, J.D.; Badaczewska-Dawid, A.E.; Gront, D. BioShell 3.0: Library for Processing Structural Biology Data. Biomolecules 2020, 10, 461. [Google Scholar] [CrossRef] [Green Version]
- Gront, D.; Kolinski, A. HCPM—Program for hierarchical clustering of protein models. Bioinformatics 2005, 21, 3179–3180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rose, A.S.; Hildebrand, P.W. NGL Viewer: A web application for molecular visualization. Nucleic Acids Res. 2015, 43, W576–W579. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Wang, S.; Hu, Q.; Gao, S.; Ma, X.; Zhang, W.; Shen, Y.; Chen, F.; Lai, L.; Pei, J. CavityPlus: A web server for protein cavity detection with pharmacophore modelling, allosteric site identification and covalent ligand binding ability prediction. Nucleic Acids Res. 2018, 46, W374–W379. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ligand | No. Clashes | No. Correct | No. Near | No. Pocket |
|---|---|---|---|---|
| Lanosterol | 131 | 212 | 809 | 12,032 |
| Obtusifoliol | 116 | 209 | 720 | 11,670 |
| Eburicol | 116 | 202 | 828 | 12,511 |
| Itraconazole | 185 | 108 | 256 | 9041 |
| average | 76 | 98 | 387 | 7555 |
| VT_1129 | 24 | 76 | 250 | 4552 |
| Clotrimazole | 1 | 25 | 98 | 2825 |
| Voriconazole | 21 | 22 | 195 | 5020 |
| Fluconazole | 42 | 19 | 240 | 5040 |
| Ketoconazole | 55 | 14 | 87 | 5307 |
| CYP51F | Amino Acids Interacting with Ligands (≥5 Ligands) |
|---|---|
| CYP51F CngH99 | F240, Y145, A317, F139, I386, M528, T321, V144, Y131 |
| CYP51F1 TaaCBS2479 | S159, A158, A334, F127, F257, H337, I403, M333, M545, P402, S157, T123, T338, T544 |
| CYP51F1 Tmfri | A307, F126, F228, I376, M521, T311, V131, Y118, Y132 |
| CYP51F Ccur | A307, F128, F230, I376, M518, T124, T311, Y120, Y134 |
| CYP51F1 Nvis | F228, Y120, A305, F128, I374, T309, Y134 |
| CYP51F1 Nen UCDFST68-887 | A210, A214, F137, F35, H217, I283, T218, T31, V40, Y27, Y41 |
| CYP51F1 Kim NRRL Y-17943 | A313, F235, H316, I382, L129, M526, T130, T317, T525, Y126, Y140 |
| CYP51F Cwer | A304, F128, F227, I373, S375, T124, T308, Y120, Y134 |
| CYP51F Cter | A237, F160, F60, H240, L306, L55, M236, M451, T241, T56, Y52, Y66 |
| CYP51F1 Cneo B-3501A | F234, I380, A311, F133, H314, I523, M310, T129, T315, V524, Y125, Y139 |
| CYP51F Cgat EJB2 | F240, A317, F139, I386, M316, M528, T135, T321, Y131, Y145 |
| Species Name | Species Abbreviation | CYP ID | CYP Abbreviation Used in the Study |
|---|---|---|---|
| Cryptococcus neoformans var. grubii H99 | CngH99 | 00040 (NCBI) | CYP51F CngH99 |
| Trichosporon asahii var. asahii CBS 2479 | TaaCBS2479 | 1742 (JGI) | CYP51F1 TaaCBS2479 |
| Tremella mesenterica Fries v1.0 | Tmfri | 38441 (JGI) | CYP51F1 Tmfri |
| Cryptococcus curvatus | Ccur | 367271 (JGI) | CYP51F Ccur |
| Naganishia vishniacii v1.0 | Nvis | 345805 (JGI) | CYP51F1 Nvis |
| Naematella encephela UCDFST 68-887 | Nen UCDFST68-887 | 523016 (JGI) | CYP51F1 Nen UCDFST68-887 |
| Kockovaella imperatae NRRL Y-17943 | Kim NRRL Y-17943 | 627561 (JGI) | CYP51F1 Kim NRRL Y-17943 |
| Cryptococcus werringae | Cwer | 629036 (JGI) | CYP51F Cwer |
| Cryptococcus terricola | Cter | 809380 (JGI) | CYP51F Cter |
| Cryptococcus neoformans var. neoformans B-3501A | Cneo B-3501A | EAL23379 (NCBI) | CYP51F1 Cneo B-3501A |
| Cryptococcus gattii EJB2 | Cgat EJB2 | KIR77383 (NCBI) | CYP51F Cgat EJB2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Akapo, O.O.; Macnar, J.M.; Kryś, J.D.; Syed, P.R.; Syed, K.; Gront, D. In Silico Structural Modeling and Analysis of Interactions of Tremellomycetes Cytochrome P450 Monooxygenases CYP51s with Substrates and Azoles. Int. J. Mol. Sci. 2021, 22, 7811. https://doi.org/10.3390/ijms22157811
Akapo OO, Macnar JM, Kryś JD, Syed PR, Syed K, Gront D. In Silico Structural Modeling and Analysis of Interactions of Tremellomycetes Cytochrome P450 Monooxygenases CYP51s with Substrates and Azoles. International Journal of Molecular Sciences. 2021; 22(15):7811. https://doi.org/10.3390/ijms22157811
Chicago/Turabian StyleAkapo, Olufunmilayo Olukemi, Joanna M. Macnar, Justyna D. Kryś, Puleng Rosinah Syed, Khajamohiddin Syed, and Dominik Gront. 2021. "In Silico Structural Modeling and Analysis of Interactions of Tremellomycetes Cytochrome P450 Monooxygenases CYP51s with Substrates and Azoles" International Journal of Molecular Sciences 22, no. 15: 7811. https://doi.org/10.3390/ijms22157811
APA StyleAkapo, O. O., Macnar, J. M., Kryś, J. D., Syed, P. R., Syed, K., & Gront, D. (2021). In Silico Structural Modeling and Analysis of Interactions of Tremellomycetes Cytochrome P450 Monooxygenases CYP51s with Substrates and Azoles. International Journal of Molecular Sciences, 22(15), 7811. https://doi.org/10.3390/ijms22157811