
BonMOLière: Small-Sized Libraries of Readily Purchasable Compounds, Optimized to Produce Genuine Hits in Biological Screens across the Protein Space



Abstract
:1. Introduction
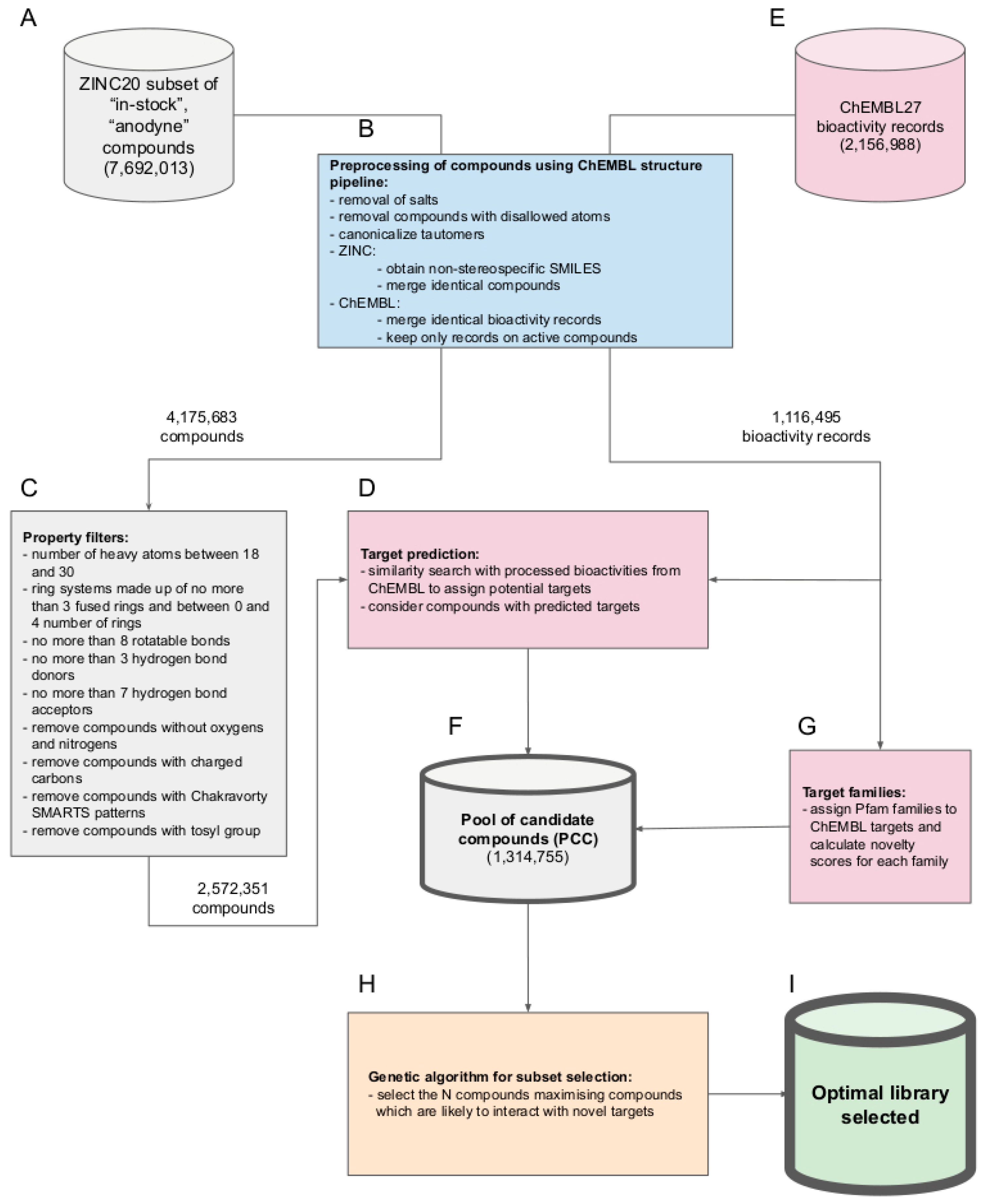
2. Results and Discussion
- The compounds are already made and readily obtainable from the manufacturer (i.e., they are part of the “in-stock” subset of the ZINC20 database).
- The compounds are presumed benign in the context of screening with biological assays (i.e., they are also part of the “anodyne” subset of the ZINC20 database). More specifically, all of these compounds have passed an extensive collection of reactivity filters and PAINS patterns compiled and utilized by the developers of the ZINC database, meaning that they are unlikely reactive or causing pan-assay interference [20].
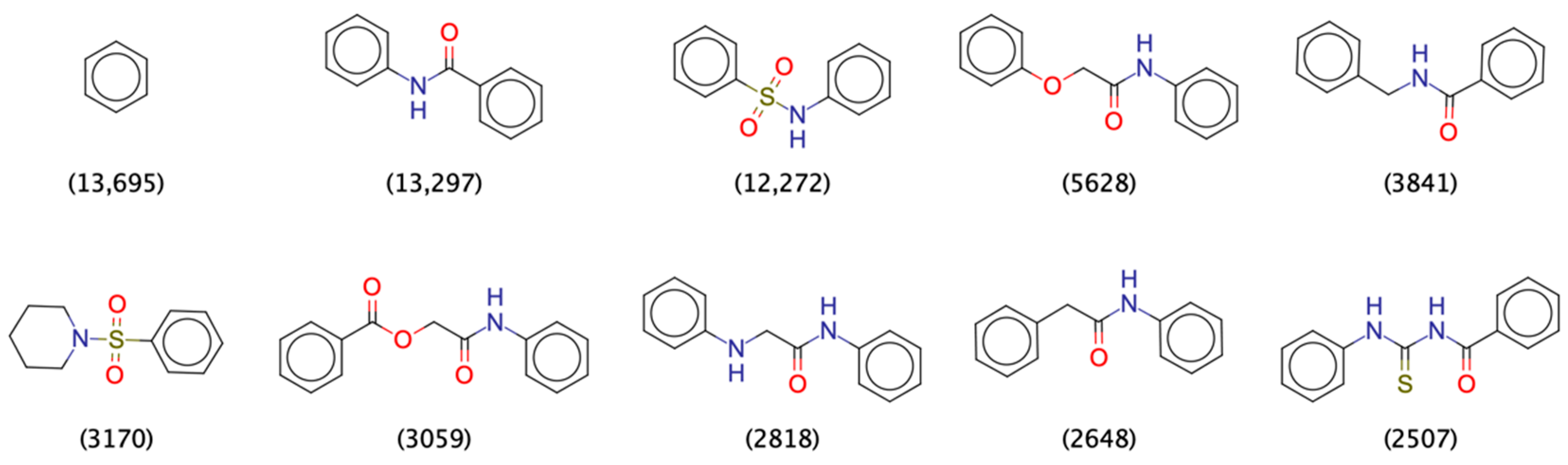
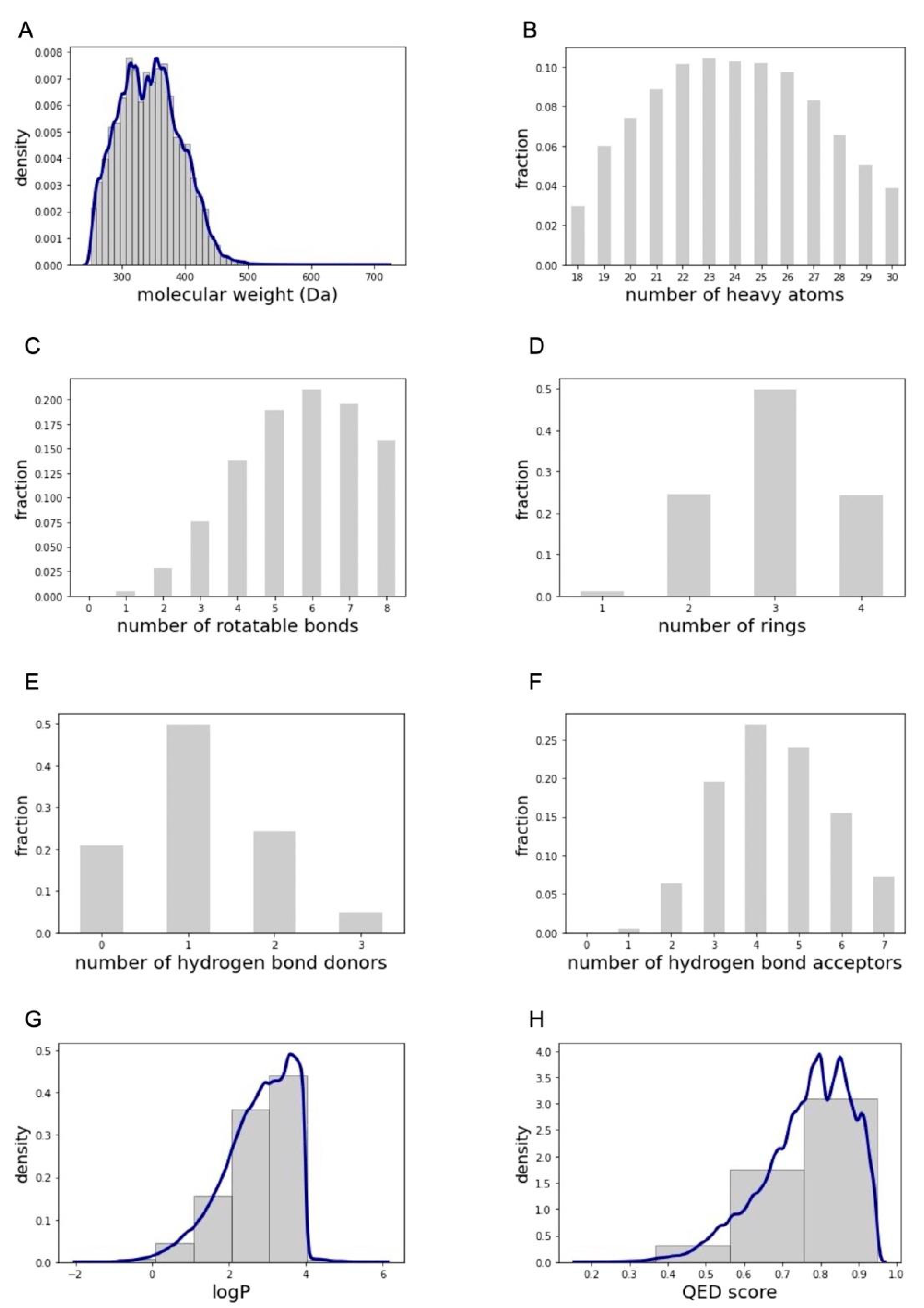
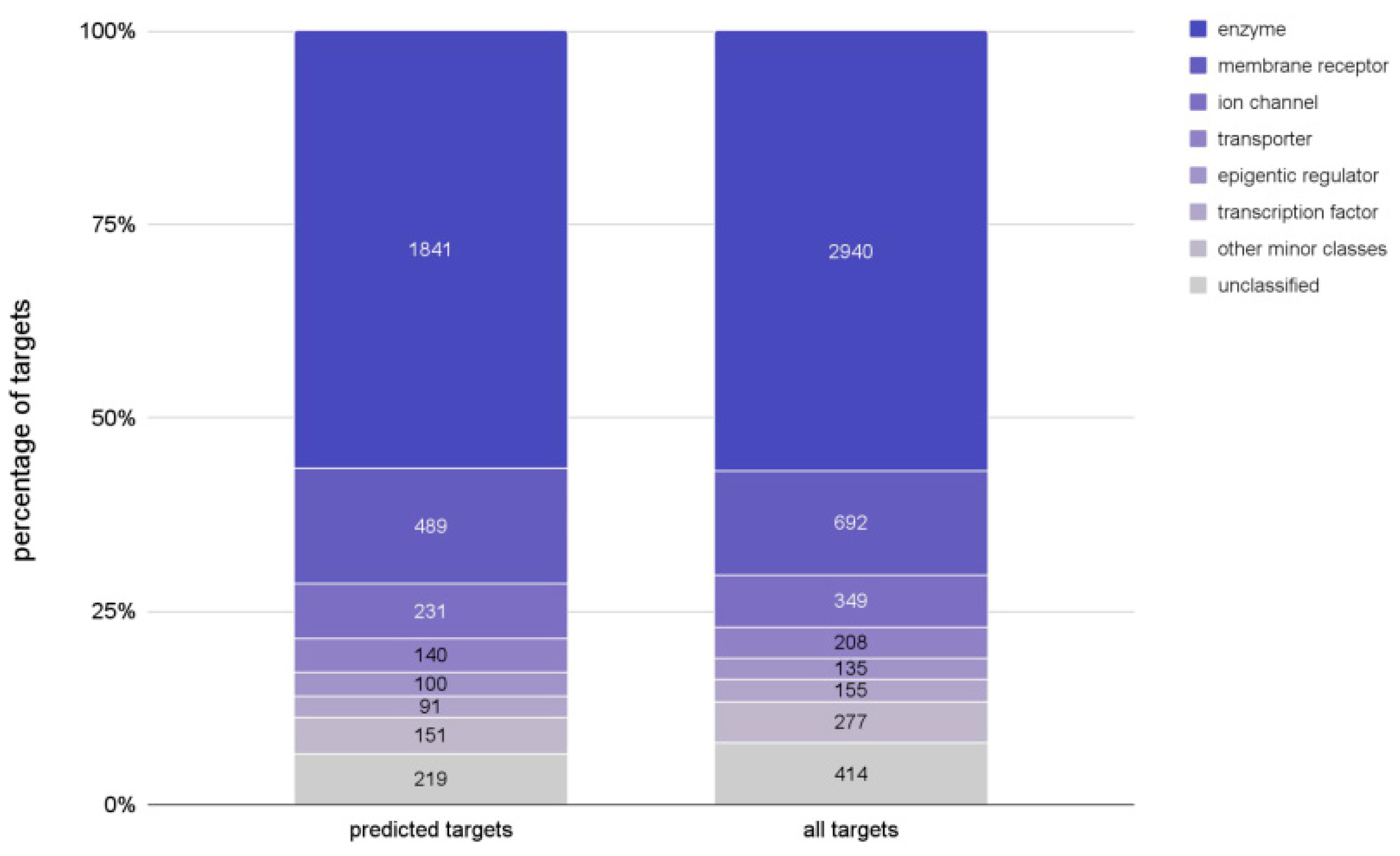
2.1. Characterization of the Pool of Candidate Compounds
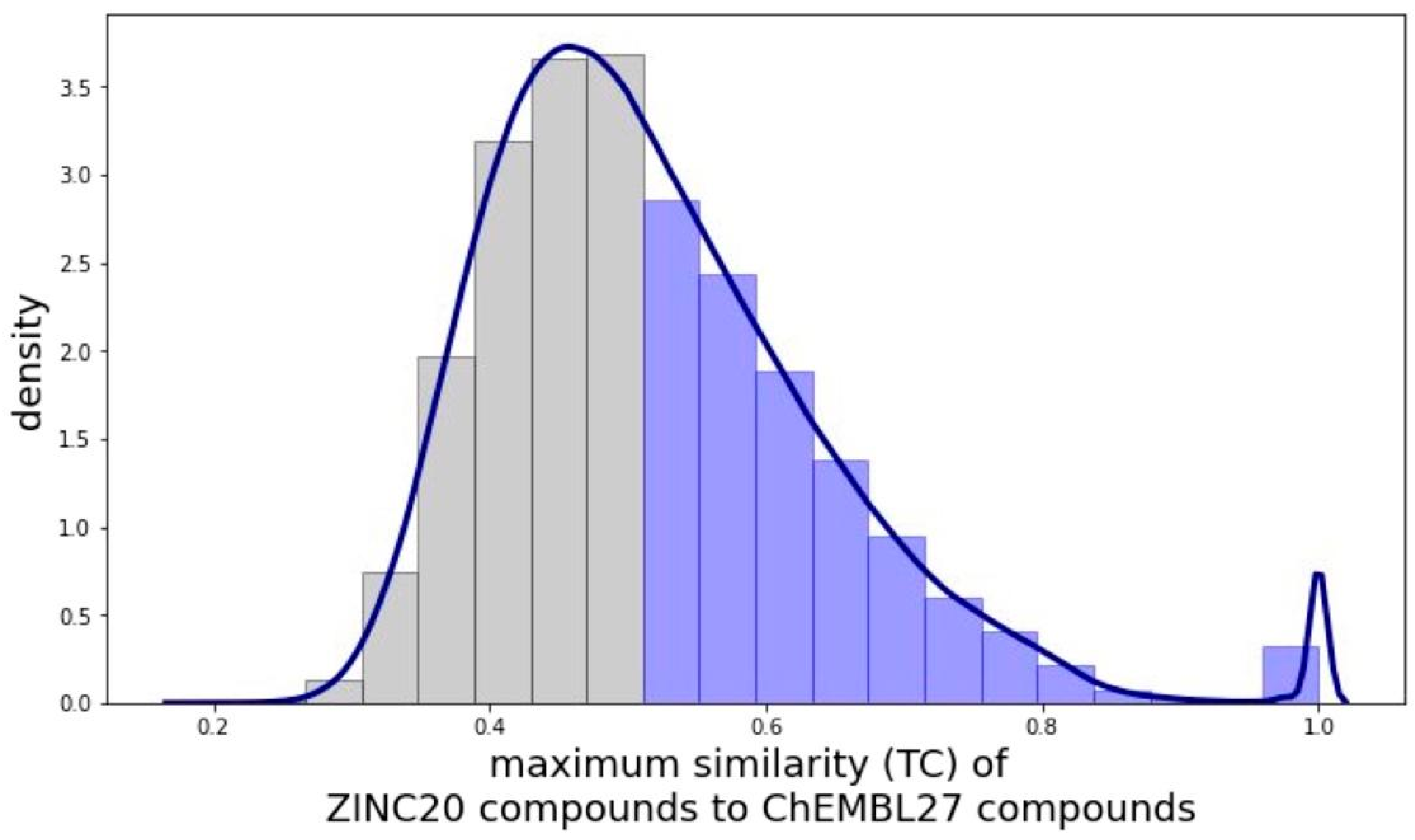
2.1.1. Physicochemical and Structural Characterization of the Pool of Candidate Compounds
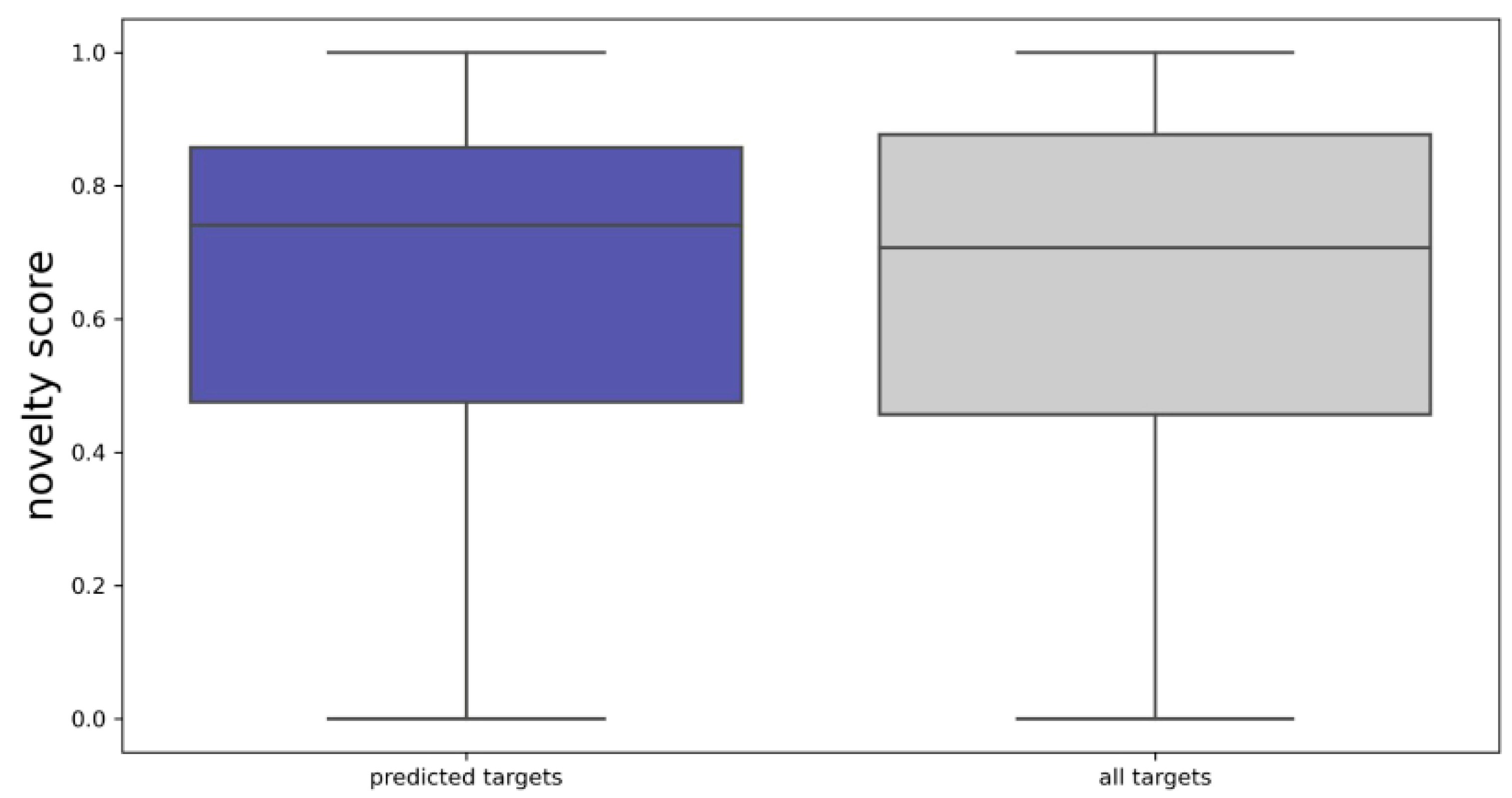
2.1.2. Biological Characterization of the Pool of Candidate Compounds
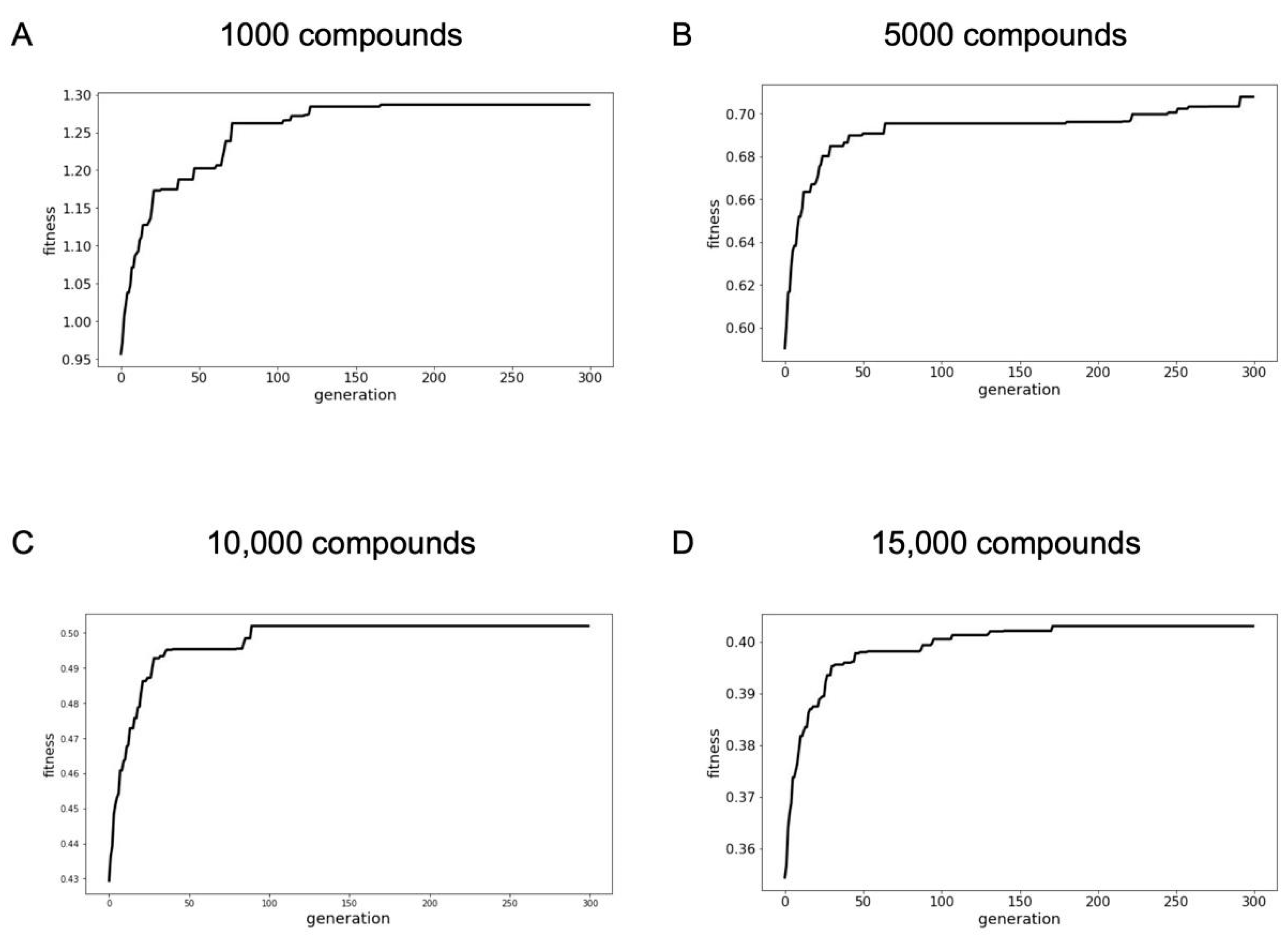
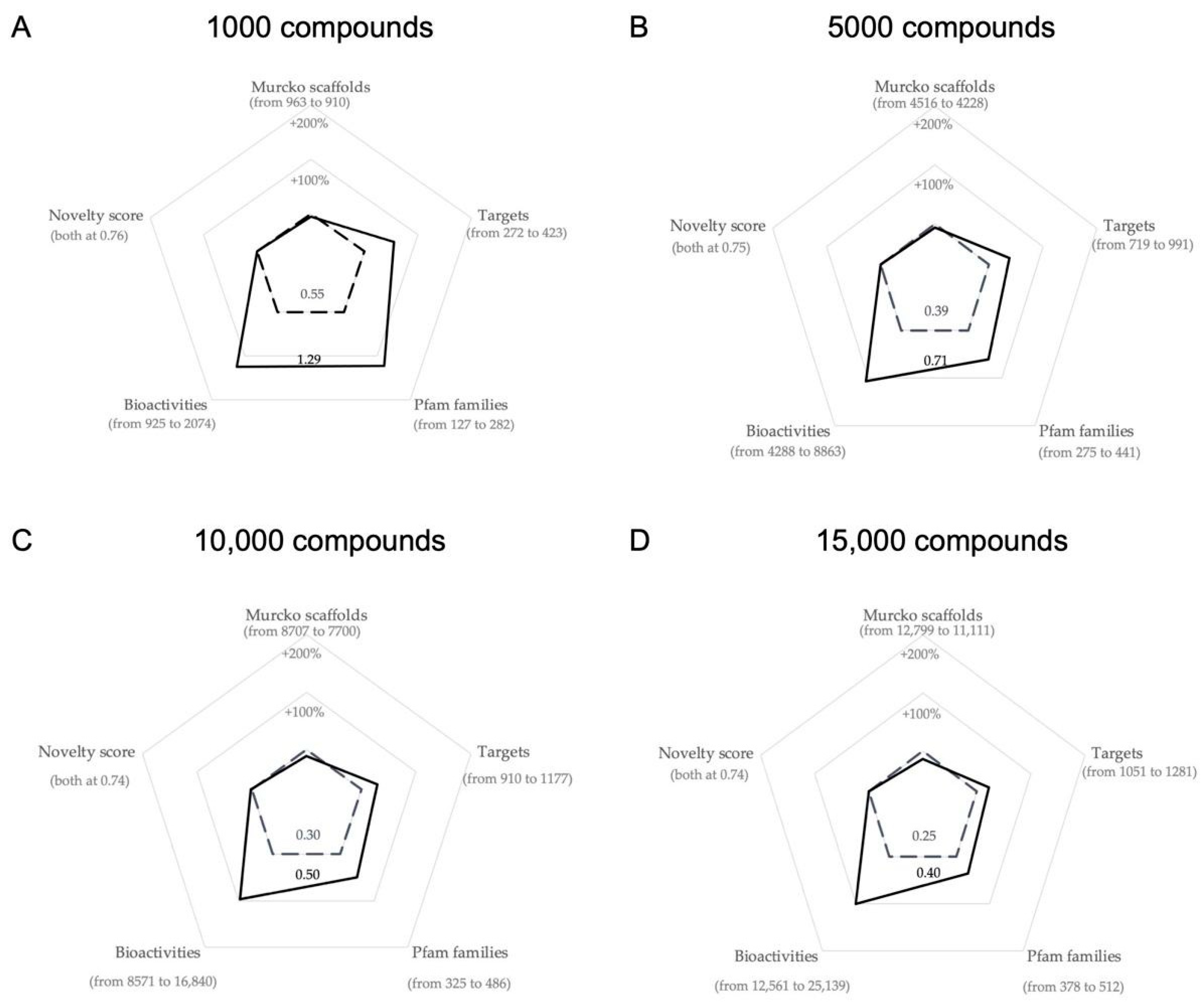
2.2. Characterization of the Optimized Compound Libraries
- more bioactive compounds as a whole
- compounds active on proteins representing more Pfam families (maximizing target diversity)
- compounds active on newer targets (the novelty score is higher for newer targets)
2.2.1. Baseline Compound Libraries
2.2.2. Optimized Compound Libraries
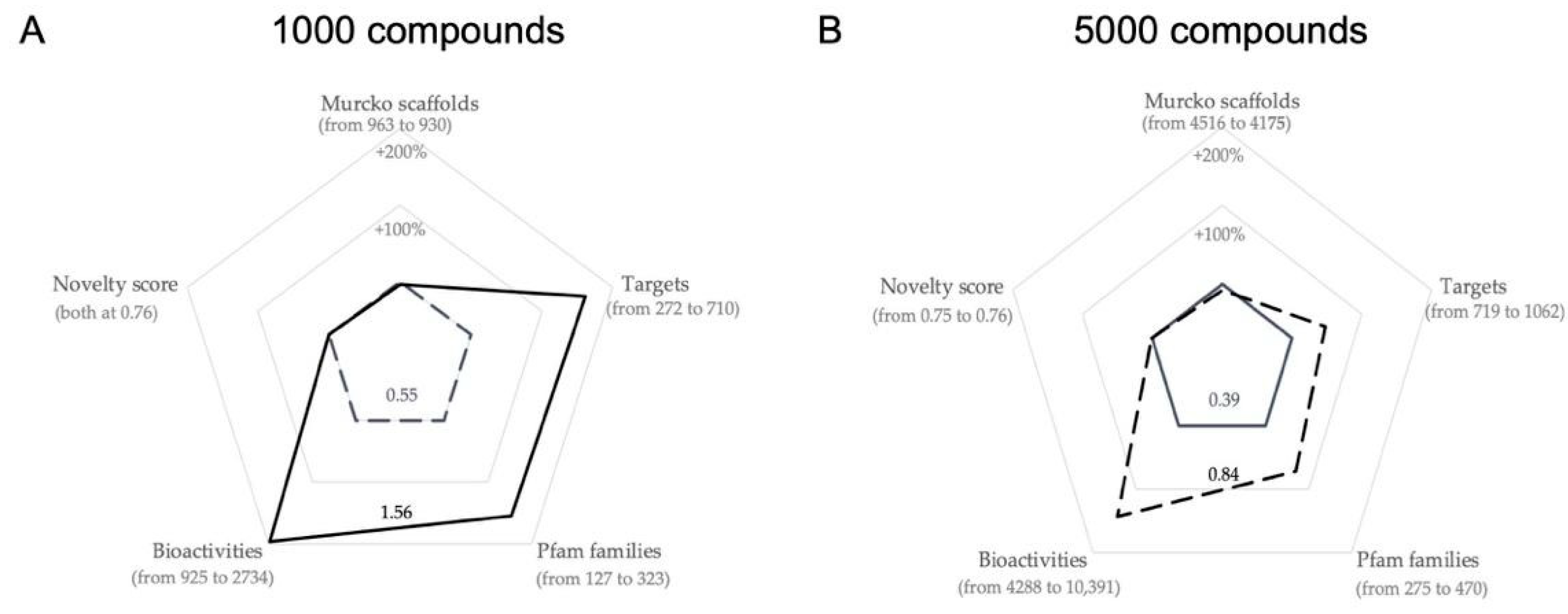
2.2.3. Further Optimization of the Smaller-Sized Compound Libraries
3. Materials and Methods
3.1. Data Sets
3.1.1. ZINC20 Database
3.1.2. ChEMBL27 Database
- Assay covers a single protein or a protein complex (ChEMBL confidence_score is 6, 7, 8, or 9)
- data_validity_comment is null OR “manually validated”
- potential_duplicate is “0”
- standard_type is “Kd”, “Potency”, “AC50”, “IC50”, “Ki”, or “EC50”
- activity_comment is not “Inconclusive”, “inconclusive”, or “unspecified”
- NOT (standard_relation is null AND activity_comment is not “Active” or “active”)
3.2. Chemical Structure Processing and Data Consolidation
3.3. Filtering of the ZINC20 Subset by Molecular Properties
- Less than 18 or more than 30 heavy atoms (calculated using RDKit’s Lipinski.HeavyAtomCount method)
- Less than one or more than four rings (calculated using RDKit’s CalcNumRings method)
- Ring systems with more than three fused rings (calculated using RDKit’s GetRingInfo and AtomRings methods to get the ring systems and number of rings per system present a molecule)
- More than eight rotatable bonds (rdMolDescriptors.CalcNumRotatableBonds)
- More than three hydrogen bond donors (Lipinski.NumHDonors)
- More than seven hydrogen bond acceptors (Lipinski.NumHAcceptors)
- Charged carbon atoms (identified using RDKit atom properties)
- Not at least one oxygen or nitrogen atom (identified using RDKit atom properties)
- Substructures listed in the “remove” and “extreme caution” categories of the SMARTS patterns compiled by Chakravorty et al. [31]. These SMARTS patterns were compiled from a meta-analysis of existing structural filters to identify nuisance compounds and correctly identified 57% of noisy GSK compounds in the study’s validation [31]
- Contain tosyl group (compounds which match the “S(=O)(=O)O” SMARTS pattern).
3.4. Target Prediction
3.5. Additional Descriptions of the Compounds from the ZINC20 Database
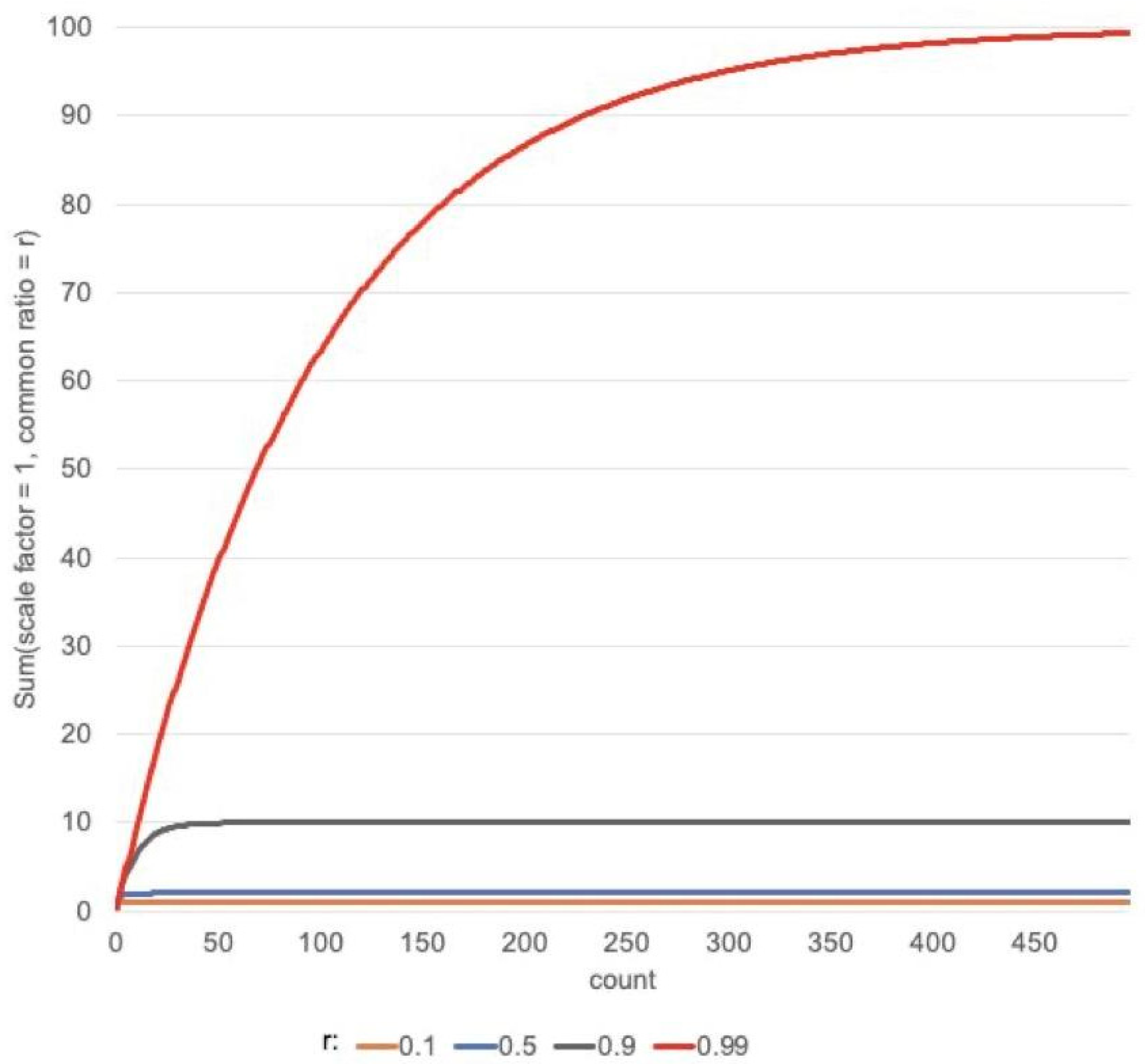
3.6. Calculation of the Novelty of a Target
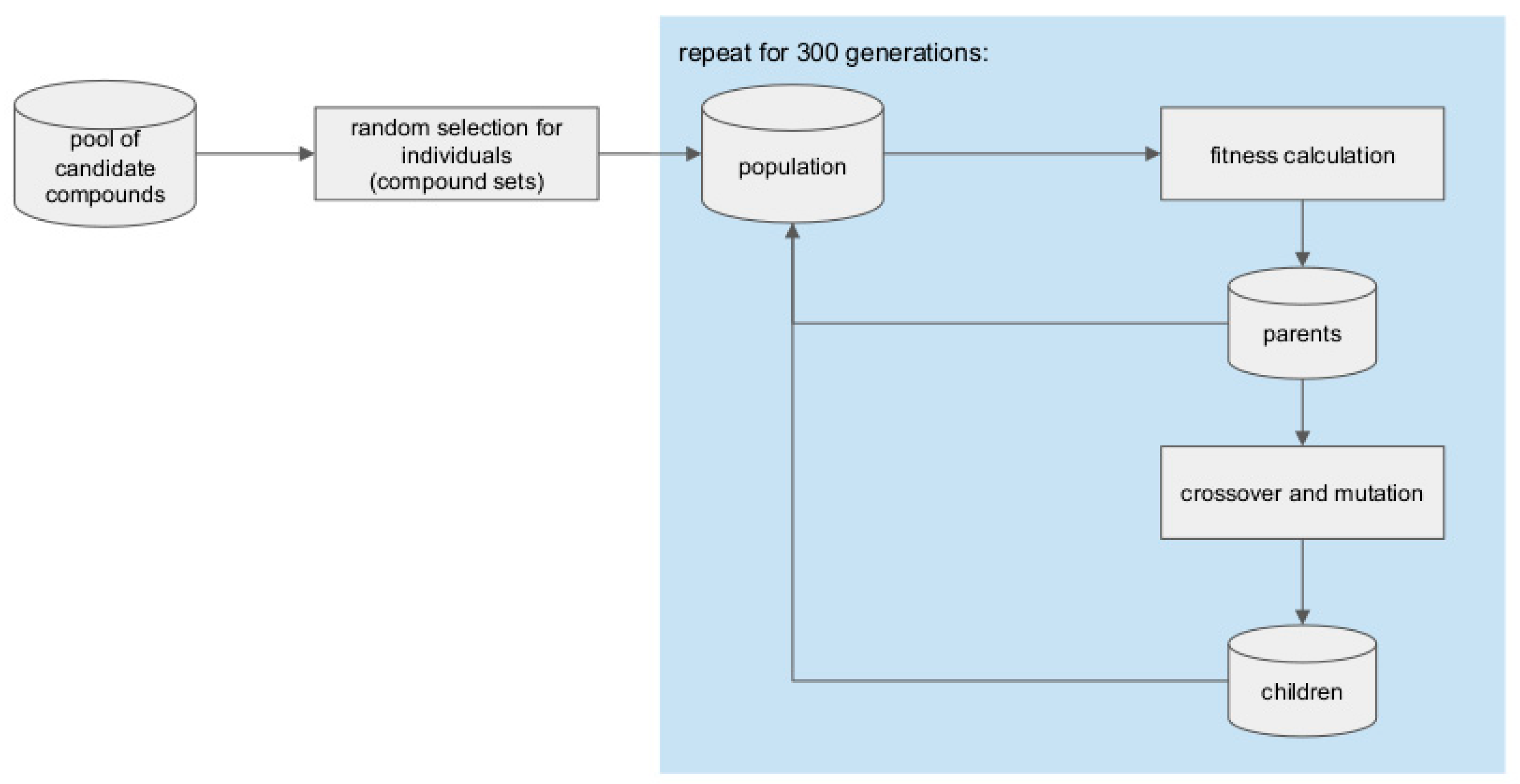
3.7. Genetic Algorithm for Library Generation from the Pool of Candidate Compounds
3.7.1. Calculation of the Fitness Function
3.7.2. Library Optimization Procedure
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Macarron, R.; Banks, M.N.; Bojanic, D.; Burns, D.J.; Cirovic, D.A.; Garyantes, T.; Green, D.V.S.; Hertzberg, R.P.; Janzen, W.P.; Paslay, J.W.; et al. Impact of High-Throughput Screening in Biomedical Research. Nat. Rev. Drug Discov. 2011, 10, 188–195. [Google Scholar] [CrossRef]
- Drewry, D.H.; Macarron, R. Enhancements of Screening Collections to Address Areas of Unmet Medical Need: An Industry Perspective. Curr. Opin. Chem. Biol. 2010, 14, 289–298. [Google Scholar] [CrossRef] [PubMed]
- Baell, J.B. Broad Coverage of Commercially Available Lead-like Screening Space with Fewer than 350,000 Compounds. J. Chem. Inf. Model. 2013, 53, 39–55. [Google Scholar] [CrossRef]
- Paricharak, S.; Méndez-Lucio, O.; Chavan Ravindranath, A.; Bender, A.; IJzerman, A.P.; van Westen, G.J.P. Data-Driven Approaches Used for Compound Library Design, Hit Triage and Bioactivity Modeling in High-Throughput Screening. Brief Bioinform. 2018, 19, 277–285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wassermann, A.M.; Camargo, L.M.; Auld, D.S. Composition and Applications of Focus Libraries to Phenotypic Assays. Front. Pharmacol. 2014, 5, 164. [Google Scholar] [CrossRef] [Green Version]
- Petrone, P.M.; Simms, B.; Nigsch, F.; Lounkine, E.; Kutchukian, P.; Cornett, A.; Deng, Z.; Davies, J.W.; Jenkins, J.L.; Glick, M. Rethinking Molecular Similarity: Comparing Compounds on the Basis of Biological Activity. ACS Chem. Biol. 2012, 7, 1399–1409. [Google Scholar] [CrossRef]
- Janzen, W.P. Screening Technologies for Small Molecule Discovery: The State of the Art. Chem. Biol. 2014, 21, 1162–1170. [Google Scholar] [CrossRef] [Green Version]
- Bakken, G.A.; Bell, A.S.; Boehm, M.; Everett, J.R.; Gonzales, R.; Hepworth, D.; Klug-McLeod, J.L.; Lanfear, J.; Loesel, J.; Mathias, J.; et al. Shaping a Screening File for Maximal Lead Discovery Efficiency and Effectiveness: Elimination of Molecular Redundancy. J. Chem. Inf. Model. 2012, 52, 2937–2949. [Google Scholar] [CrossRef] [PubMed]
- Lahue, B.R.; Glick, M.; Tudor, M.; Johnson, S.A.; Diratsouian, J.; Wildey, M.J.; Burton, M.; Mazzola, R.; Wassermann, A.M. Diversity & Tractability Revisited in Collaborative Small Molecule Phenotypic Screening Library Design. Bioorg. Med. Chem. 2020, 28, 115192. [Google Scholar] [CrossRef]
- Stork, C.; Kirchmair, J. PAIN(S) Relievers for Medicinal Chemists: How Computational Methods Can Assist in Hit Evaluation. Future Med. Chem. 2018, 10, 1533–1535. [Google Scholar] [CrossRef]
- Brenk, R.; Schipani, A.; James, D.; Krasowski, A.; Gilbert, I.H.; Frearson, J.; Wyatt, P.G. Lessons Learnt from Assembling Screening Libraries for Drug Discovery for Neglected Diseases. ChemMedChem 2008, 3, 435–444. [Google Scholar] [CrossRef]
- Spear, K.L.; Brown, S.P. The Evolution of Library Design: Crafting Smart Compound Collections for Phenotypic Screens. Drug Discov. Today Technol. 2017, 61–67. [Google Scholar] [CrossRef]
- Haasen, D.; Schopfer, U.; Antczak, C.; Guy, C.; Fuchs, F.; Selzer, P. How Phenotypic Screening Influenced Drug Discovery: Lessons from Five Years of Practice. Assay Drug Dev. Technol. 2017, 15, 239–246. [Google Scholar] [CrossRef] [PubMed]
- Huggins, D.J.; Venkitaraman, A.R.; Spring, D.R. Rational Methods for the Selection of Diverse Screening Compounds. ACS Chem. Biol. 2011, 6, 208–217. [Google Scholar] [CrossRef] [Green Version]
- Baell, J.B.; Holloway, G.A. New Substructure Filters for Removal of Pan Assay Interference Compounds (PAINS) from Screening Libraries and for Their Exclusion in Bioassays. J. Med. Chem. 2010, 53, 2719–2740. [Google Scholar] [CrossRef] [Green Version]
- Bickerton, G.R.; Paolini, G.V.; Besnard, J.; Muresan, S.; Hopkins, A.L. Quantifying the Chemical Beauty of Drugs. Nat. Chem. 2012, 4, 90–98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schuffenhauer, A.; Schneider, N.; Hintermann, S.; Auld, D.; Blank, J.; Cotesta, S.; Engeloch, C.; Fechner, N.; Gaul, C.; Giovannoni, J.; et al. Evolution of Novartis’ Small Molecule Screening Deck Design. J. Med. Chem. 2020, 63, 14425–14447. [Google Scholar] [CrossRef] [PubMed]
- Irwin, J.J.; Tang, K.G.; Young, J.; Dandarchuluun, C.; Wong, B.R.; Khurelbaatar, M.; Moroz, Y.S.; Mayfield, J.; Sayle, R.A. ZINC20-A Free Ultralarge-Scale Chemical Database for Ligand Discovery. J. Chem. Inf. Model. 2020, 60, 6065–6073. [Google Scholar] [CrossRef]
- ZINC20. Available online: http://zinc20.docking.org (accessed on 26 May 2021).
- Sterling, T.; Irwin, J.J. ZINC 15–Ligand Discovery for Everyone. J. Chem. Inf. Model. 2015, 55, 2324–2337. [Google Scholar] [CrossRef]
- Mathai, N.; Kirchmair, J. Similarity-Based Methods and Machine Learning Approaches for Target Prediction in Early Drug Discovery: Performance and Scope. Int. J. Mol. Sci. 2020, 21, 3585. [Google Scholar] [CrossRef]
- Gaulton, A.; Hersey, A.; Nowotka, M.; Bento, A.P.; Chambers, J.; Mendez, D.; Mutowo, P.; Atkinson, F.; Bellis, L.J.; Cibrián-Uhalte, E.; et al. The ChEMBL Database in 2017. Nucleic Acids Res. 2017, 45, D945–D954. [Google Scholar] [CrossRef] [PubMed]
- Gaulton, A. ChEMBL_27 SARS-CoV-2 Release. Available online: http://chembl.blogspot.com/2020/05/chembl27-sars-cov-2-release.html (accessed on 12 March 2021).
- Mistry, J.; Chuguransky, S.; Williams, L.; Qureshi, M.; Salazar, G.A.; Sonnhammer, E.L.L.; Tosatto, S.C.E.; Paladin, L.; Raj, S.; Richardson, L.J.; et al. Pfam: The Protein Families Database in 2021. Nucleic Acids Res. 2020, 49, D412–D419. [Google Scholar] [CrossRef] [PubMed]
- El-Gebali, S.; Mistry, J.; Bateman, A.; Eddy, S.R.; Luciani, A.; Potter, S.C.; Qureshi, M.; Richardson, L.J.; Salazar, G.A.; Smart, A.; et al. The Pfam Protein Families Database in 2019. Nucleic Acids Res. 2019, 47, D427–D432. [Google Scholar] [CrossRef] [PubMed]
- RDKit: Open-Source Cheminformatics. Available online: http://www.rdkit.org–version2020.09.1.0 (accessed on 8 July 2021).
- Bosc, N.; Atkinson, F.; Felix, E.; Gaulton, A.; Hersey, A.; Leach, A.R. Large Scale Comparison of QSAR and Conformal Prediction Methods and Their Applications in Drug Discovery. J. Cheminform. 2019, 11, 4. [Google Scholar] [CrossRef]
- Patrícia Bento, A.; Hersey, A.; Félix, E.; Landrum, G.; Gaulton, A.; Atkinson, F.; Bellis, L.J.; De Veij, M.; Leach, A.R. An Open Source Chemical Structure Curation Pipeline Using RDKit. J. Cheminform. 2020, 12, 1–16. [Google Scholar] [CrossRef]
- Hann, M.; Hudson, B.; Lewell, X.; Lifely, R.; Miller, L.; Ramsden, N. Strategic Pooling of Compounds for High-Throughput Screening. J. Chem. Inf. Comput. Sci. 1999, 39, 897–902. [Google Scholar] [CrossRef]
- Pearce, B.C.; Sofia, M.J.; Good, A.C.; Drexler, D.M.; Stock, D.A. An Empirical Process for the Design of High-Throughput Screening Deck Filters. J. Chem. Inf. Model. 2006, 46, 1060–1068. [Google Scholar] [CrossRef]
- Chakravorty, S.J.; Chan, J.; Greenwood, M.N.; Popa-Burke, I.; Remlinger, K.S.; Pickett, S.D.; Green, D.V.S.; Fillmore, M.C.; Dean, T.W.; Luengo, J.I.; et al. Nuisance Compounds, PAINS Filters, and Dark Chemical Matter in the GSK HTS Collection. SLAS Discov. 2018, 23, 532–545. [Google Scholar] [CrossRef] [Green Version]
- Dalke, A. The Chemfp Project. J. Cheminformatics 2019, 11, 76. [Google Scholar] [CrossRef] [Green Version]
- Gao, M.; Skolnick, J. A Comprehensive Survey of Small-Molecule Binding Pockets in Proteins. PLoS Comput. Biol. 2013, 9, e1003302. [Google Scholar] [CrossRef] [Green Version]
- Maggiora, G.; Vogt, M.; Stumpfe, D.; Bajorath, J. Molecular Similarity in Medicinal Chemistry. J. Med. Chem. 2014, 57, 3186–3204. [Google Scholar] [CrossRef]
- Wassermann, A.M.; Lounkine, E.; Hoepfner, D.; Le Goff, G.; King, F.J.; Studer, C.; Peltier, J.M.; Grippo, M.L.; Prindle, V.; Tao, J.; et al. Dark Chemical Matter as a Promising Starting Point for Drug Lead Discovery. Nat. Chem. Biol. 2015, 11, 958–966. [Google Scholar] [CrossRef] [PubMed]
- Chemfp Taylor Butina Implementation. Available online: http://dalkescientific.com/writings/taylor_butina.py (accessed on 26 March 2021).
- Taylor, R. Simulation Analysis of Experimental Design Strategies for Screening Random Compounds as Potential New Drugs and Agrochemicals. J. Chem. Inf. Comput. Sci. 1995, 35, 59–67. [Google Scholar] [CrossRef]
- Butina, D. Unsupervised Data Base Clustering Based on Daylight’s Fingerprint and Tanimoto Similarity: A Fast and Automated Way to Cluster Small and Large Data Sets. J. Chem. Inf. Comput. Sci. 1999, 39, 747–750. [Google Scholar] [CrossRef]
- Kim, S.; Chen, J.; Cheng, T.; Gindulyte, A.; He, J.; He, S.; Li, Q.; Shoemaker, B.A.; Thiessen, P.A.; Yu, B.; et al. PubChem in 2021: New Data Content and Improved Web Interfaces. Nucleic Acids Res. 2021, 49, D1388–D1395. [Google Scholar] [CrossRef] [PubMed]
- Mathai, N.; Stork, C.; Kirchmair, J. BonMOLière: Small-Sized Libraries of Readily Purchasable Compounds, Optimized to Produce Genuine Hits in Biological Screens across the Protein Space; CERN: Genève, Switzerland, 2021. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Library Size | Fitness Score | Number of Murcko Scaffolds | Number of Targets | Number of Pfam Families | Number of Bioactivities | Median Novelty Score |
|---|---|---|---|---|---|---|
| 1000 | 0.55 | 963 | 272 | 127 | 925 | 0.76 |
| 5000 | 0.39 | 4516 | 719 | 275 | 4288 | 0.75 |
| 10,000 | 0.30 | 8707 | 910 | 325 | 8571 | 0.74 |
| 15,000 | 0.25 | 12,779 | 1051 | 378 | 12,561 | 0.74 |
| Library Size | % Change in Fitness Score | Δ Number of Murcko Scaffolds | Δ Number of Compound Clusters Represented | Δ Number of Targets | Δ Number of Pfam Families | Δ Number of Bioactivities | % Change in the Median Novelty Score |
|---|---|---|---|---|---|---|---|
| 1000 | +34.48% (0.96→1.29) | −15 (925→910) | 0 (1000 in both) | +59 (364→423) | +102 (180→282) | +395 (1679→2074) | +5.22% (0.72→0.76) |
| 5000 | +19.90% (0.59→0.71) | +17 (4211→4228) | 0 (5000 in both) | +7 (984→991) | +102 (339→441) | +591 (8272→ 8863) | +0% (both at 0.75) |
| 10,000 | +16.89% (0.43→0.50) | −83 (7853→7770) | 0 (10,000 in both) | +40 (1137→1177) | +79 (407→486) | +805 (16,035→16,840) | +6.11% (0.71→0.75) |
| 15,000 | +13.73% (0.35→0.40) | −189 (11,300→11,111) | 0 (15,000 in both) | −81 (1362→1281) | −3 (515→512) | +769 (24,370→25,139) | −0.61% (0.75→0.74) |
| Library Size | Population Size | % Change in Fitness Score | Δ Number of Murcko Scaffolds | Δ Number of Compound Clusters Represented | Δ Number of Targets | Δ Number of Pfam Families | Δ Number of Bioactivities | % Change in the Median Novelty Score |
|---|---|---|---|---|---|---|---|---|
| 1000 | 500 | +46.14% (0.97→1.42) | +6 (912→918) | 0 (1000 in both) | +102 (406→508) | +101 (202→303) | +541 (1657→2198) | +2.26% (0.74→0.76) |
| 1000 | 1000 | +58.16%. (0.99→1.56) | +2 (928→930) | 0 (1000 in both) | +267 (443→710) | +125 (198→323) | +1008 (1735→2743) | +0% (both at 0.76) |
| 5000 | 1000 | +23.12%. (0.59→0.73) | −59 (4252→4193) | 0 (5000 in both) | +58 (859→917) | +15 (406→421) | +959 (8032→8991) | +1.33% (0.74→0.75) |
| 5000 | 5000 | +40.76% (0.60→0.84) | −17 (4192→4175) | 0 (5000 in both) | +230 (832→1062) | +61 (409→ 470) | +2353 (8038→10,391) | +0.92% (0.75→0.76) |
| Library Size/Size of the Individual (N) | Population Size (M) |
|---|---|
| 1000 | 100, 500, 1000 |
| 5000 | 500, 1000, 5000 |
| 10,000 | 1000 |
| 15,000 | 1500 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mathai, N.; Stork, C.; Kirchmair, J. BonMOLière: Small-Sized Libraries of Readily Purchasable Compounds, Optimized to Produce Genuine Hits in Biological Screens across the Protein Space. Int. J. Mol. Sci. 2021, 22, 7773. https://doi.org/10.3390/ijms22157773
Mathai N, Stork C, Kirchmair J. BonMOLière: Small-Sized Libraries of Readily Purchasable Compounds, Optimized to Produce Genuine Hits in Biological Screens across the Protein Space. International Journal of Molecular Sciences. 2021; 22(15):7773. https://doi.org/10.3390/ijms22157773
Chicago/Turabian StyleMathai, Neann, Conrad Stork, and Johannes Kirchmair. 2021. "BonMOLière: Small-Sized Libraries of Readily Purchasable Compounds, Optimized to Produce Genuine Hits in Biological Screens across the Protein Space" International Journal of Molecular Sciences 22, no. 15: 7773. https://doi.org/10.3390/ijms22157773
APA StyleMathai, N., Stork, C., & Kirchmair, J. (2021). BonMOLière: Small-Sized Libraries of Readily Purchasable Compounds, Optimized to Produce Genuine Hits in Biological Screens across the Protein Space. International Journal of Molecular Sciences, 22(15), 7773. https://doi.org/10.3390/ijms22157773