
The Diverse Roles of TNNI3K in Cardiac Disease and Potential for Treatment


Abstract

1. Introduction
2. TNNI3K Upstream Regulators and Downstream Targets
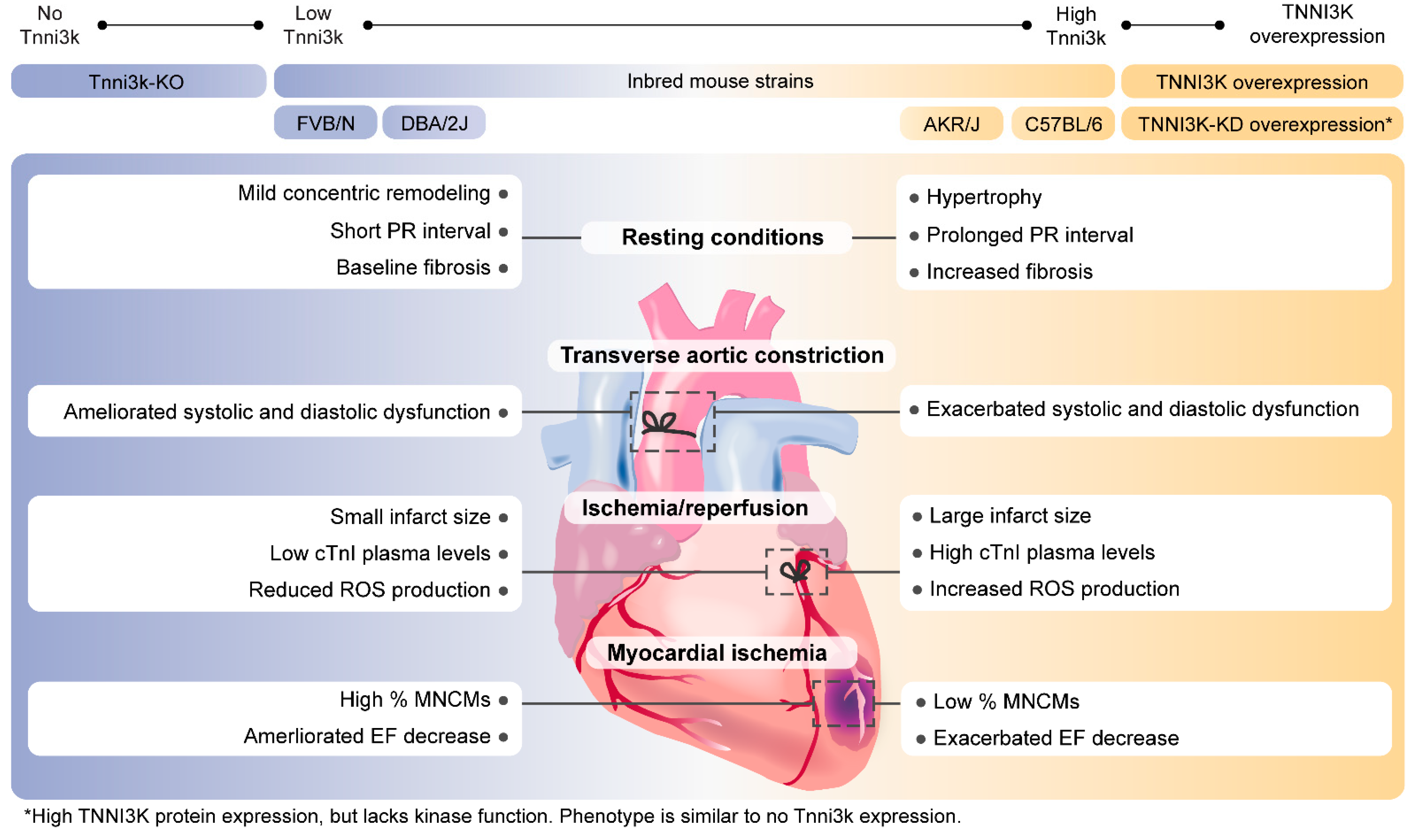
3. TNNI3K and Cardiac Hypertrophy
4. TNNI3K in Myocardial Infarction and Heart Failure
5. TNNI3K in Cardiac Regeneration
6. TNNI3K in Cardiac Conduction
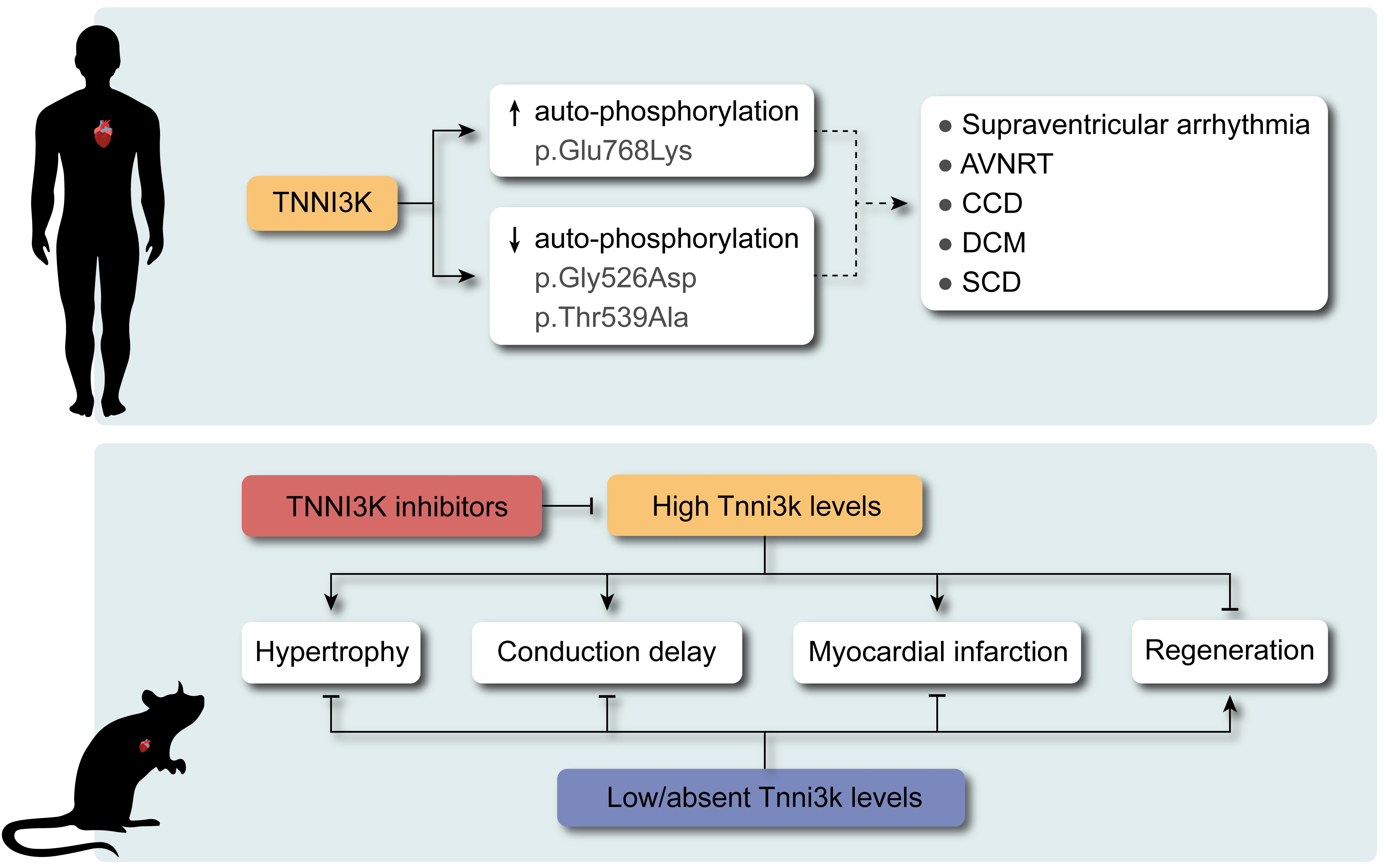
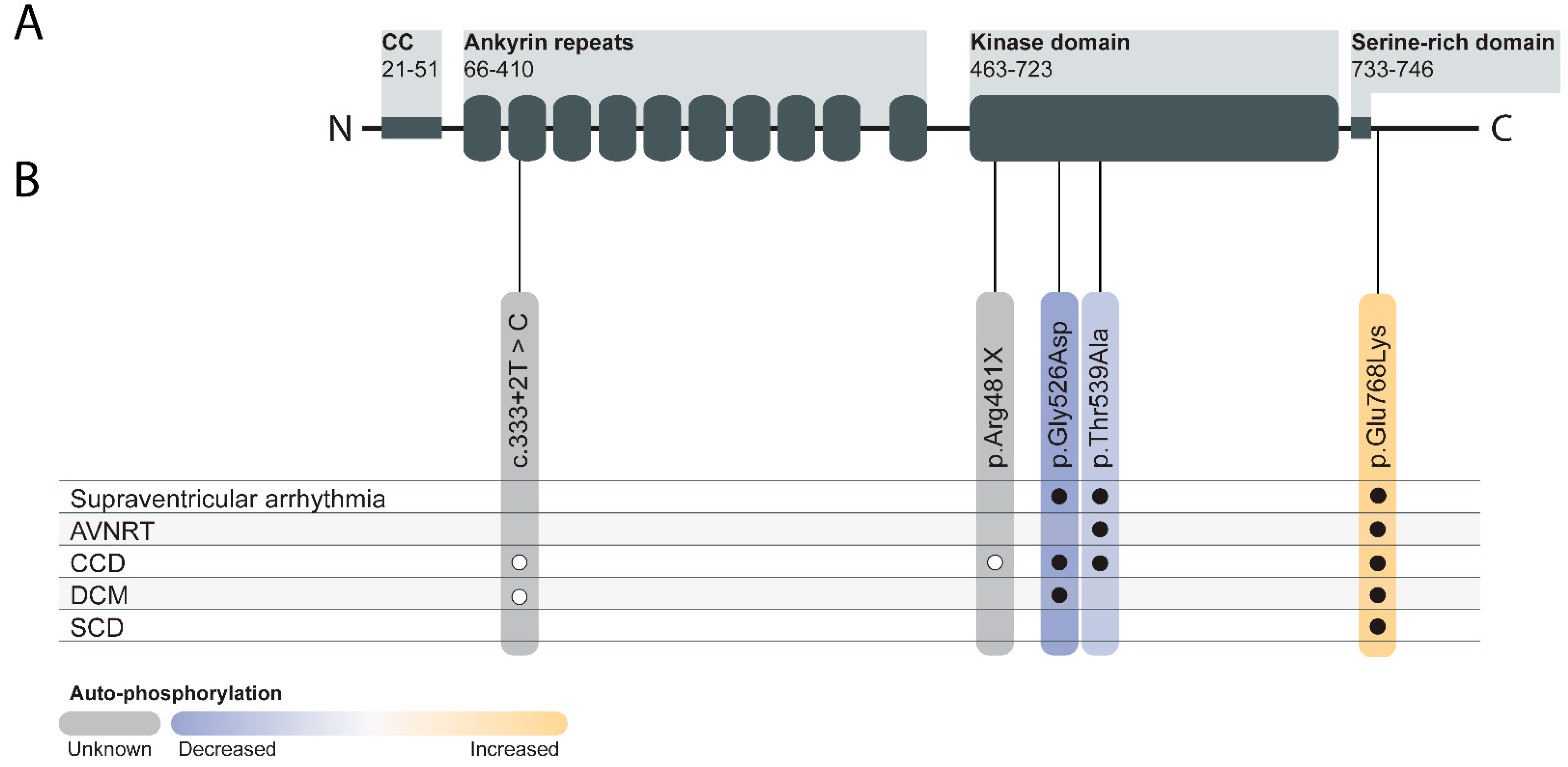
7. TNNI3K in Human Genetics
8. TNNI3K as a Therapeutic Target for Cardiac Diseases
9. Other Potential Roles of TNNI3K
10. Discussion
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Abbreviations
| AF | Atrial fibrillation |
| AOP-1 | Antioxidant protein 1 |
| AV | Atrioventricular |
| AVNRT | Atrioventricular nodal re-entrant tachycardia |
| CCD | Cardiac conduction disease |
| CM | Cardiomyocyte |
| Csq | Calsequestrin |
| cTnI | Troponin-I |
| DCM | Dilated cardiomyopathy |
| EF | Ejection fraction |
| ECG | Electrocardiogram |
| FPGT | Fucose-1-phosphate guanylyltransferase |
| ICM | Ischemic cardiomyopathy |
| ILK | Integrin-linked kinase |
| I/R | Ischemia/reperfusion |
| IC50 | Half maximal inhibitory concentration |
| KD | Kinase dead |
| KO | Knockout |
| LAD | Left anterior descending artery |
| LV | Left ventricle/ventricular |
| MNCM | Mononuclear cardiomyocytes |
| NMD | Nonsense mediated decay |
| MI | Myocardial infarction |
| QTL | Quantitative trait locus |
| ROS | Reactive oxygen species |
| SCD | Sudden cardiac death |
| SNPs | Single nucleotide polymorphisms |
| TAC | Transverse aortic constriction |
| TNNI3K | Troponin I interacting kinase |
| TNNI3Ktg | Transgenic mouse model overexpressing TNNI3K |
| TNNI3K-KDtg | Transgenic mouse model overexpressing kinase-dead TNNI3K |
| Vms1 | Viral myocarditis susceptibility 1 |
| VUS | Variants of unknown significance |
| WT | Wild-type |
References
- Zhao, Y.; Meng, X.-M.; Wei, Y.-J.; Zhao, X.-W.; Liu, D.-Q.; Cao, H.-Q.; Liew, C.-C.; Ding, J.-F. Cloning and characterization of a novel cardiac-specific kinase that interacts specifically with cardiac troponin I. J. Mol. Med. 2003, 81, 297–304. [Google Scholar] [CrossRef] [PubMed]
- Vagnozzi, R.J.; Gatto, G.J., Jr.; Kallander, L.S.; Hoffman, N.E.; Mallilankaraman, K.; Ballard, V.L.; Lawhorn, B.G.; Stoy, P.; Philp, J.; Graves, A.P.; et al. Inhibition of the cardiomyocyte-specific kinase TNNI3K limits oxidative stress, injury, and adverse re-modeling in the ischemic heart. Sci. Transl. Med. 2013, 16, 207ra141. [Google Scholar] [CrossRef]
- Wheeler, F.C.; Tang, H.; Marks, O.A.; Hadnott, T.N.; Chu, P.-L.; Mao, L.; Rockman, H.A.; Marchuk, U.A. Tnni3k Modifies Disease Progression in Murine Models of Cardiomyopathy. PLoS Genet. 2009, 5, e1000647. [Google Scholar] [CrossRef] [PubMed]
- Lodder, E.M.; Scicluna, B.; Milano, A.; Sun, A.Y.; Tang, H.; Remme, C.A.; Moerland, P.D.; Tanck, M.; Pitt, G.; Marchuk, D.A.; et al. Dissection of a Quantitative Trait Locus for PR Interval Duration Identifies Tnni3k as a Novel Modulator of Cardiac Conduction. PLoS Genet. 2012, 8, e1003113. [Google Scholar] [CrossRef]
- Feng, Y.; Liu, D.-Q.; Wang, Z.; Liu, Z.; Cao, H.-Q.; Wang, L.-Y.; Shi, N.; Meng, X.-M. AOP-1 interacts with cardiac-specific protein kinase TNNI3K and down-regulates its kinase activity. Biochemistry (Moscow) 2007, 72, 1199–1204. [Google Scholar] [CrossRef]
- Mosavi, L.K.; Cammett, T.J.; Desrosiers, D.C.; Peng, Z.Y. The ankyrin repeat as molecular architecture for protein recognition. Protein Sci. 2004, 13, 1435–1448. [Google Scholar] [CrossRef]
- Prakash, T.; Sharma, V.K.; Adati, N.; Ozawa, R.; Kumar, N.; Nishida, Y.; Fujikake, T.; Takeda, T.; Taylor, T.D. Expression of Conjoined Genes: Another Mechanism for Gene Regulation in Eukaryotes. PLoS ONE 2010, 5, e13284. [Google Scholar] [CrossRef]
- Wang, H.; Chen, C.; Song, X.; Chen, J.; Zhen, Y.; Sun, K.; Hui, R. Mef2c is an essential regulatory element required for unique expression of the cardiac-specific CARK gene. J. Cell. Mol. Med. 2007, 12, 304–315. [Google Scholar] [CrossRef]
- Luft, F.C. Hearts of this ILK rely on TNNI3K, a MAPKKK that regulated TNNI3. J. Mol. Med. 2003, 81, 279–280. [Google Scholar] [CrossRef]
- Wang, X.; Wang, J.; Su, M.; Wang, C.; Chen, J.; Wang, H.; Song, L.; Zou, Y.; Zhang, L.; Zhang, Y.; et al. TNNI3K, a Cardiac-Specific Kinase, Promotes Physiological Cardiac Hypertrophy in Transgenic Mice. PLoS ONE 2013, 8, e58570. [Google Scholar] [CrossRef]
- Gan, P.; Patterson, M.; Velasquez, A.; Wang, K.; Tian, D.; Windle, J.J.; Tao, G.; Judge, D.; Makita, T.; Park, T.J.; et al. Tnni3k alleles influence ventricular mononuclear diploid cardiomyocyte frequency. PLoS Genet. 2019, 15, e1008354. [Google Scholar] [CrossRef]
- Nakamura, M.; Sadoshima, J. Mechanisms of physiological and pathological cardiac hypertrophy. Nat. Rev. Cardiol. 2018, 15, 387–407. [Google Scholar] [CrossRef]
- Wang, L.; Wang, H.; Ye, J.; Xu, R.X.; Song, L.; Shi, N.; Zhang, Y.W.; Chen, X.; Meng, X.M. Adenovirus-mediated overexpression of cardiac troponin I-interacting kinase promotes cardiomyocyte hypertrophy. Clin. Exp. Pharmacol. Physiol. 2011, 38, 278–284. [Google Scholar] [CrossRef]
- Wang, Y.-S.; Zhou, J.; Hong, K.; Cheng, X.-S.; Li, Y.-G. MicroRNA-223 Displays a Protective Role Against Cardiomyocyte Hypertrophy by Targeting Cardiac Troponin I-Interacting Kinase. Cell. Physiol. Biochem. 2015, 35, 1546–1556. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.; Xiao, K.; Mao, L.; Rockman, H.A.; Marchuk, D.A. Overexpression of TNNI3K, a cardiac-specific MAPKKK, promotes cardiac dysfunction. J. Mol. Cell. Cardiol. 2013, 54, 101–111. [Google Scholar] [CrossRef] [PubMed]
- Patterson, M.; Barske, L.; Van Handel, B.; Rau, C.D.; Gan, P.; Sharma, A.; Parikh, S.; Denholtz, M.; Huang, Y.; Yamaguchi, Y.; et al. Frequency of mononuclear diploid cardiomyocytes underlies natural variation in heart regeneration. Nat. Genet. 2017, 49, 1346–1353. [Google Scholar] [CrossRef] [PubMed]
- Gan, P.; Baicu, C.; Watanabe, H.; Wang, K.; Tao, G.; Judge, D.P.; Zile, M.R.; Makita, T.; Mukherjee, R.; Sucov, H.M. The prevalent I686T human variant and loss-of-function mutations in the cardiomyocyte-specific kinase gene TNNI3K cause adverse contractility and concentric remodeling in mice. Hum. Mol. Genet. 2021, 29, 3504–3515. [Google Scholar] [CrossRef]
- Roth, G.A.; Johnson, C.; Abajobir, A.; Abd-Allah, F.; Abera, S.F.; Abyu, G.; Ahmed, M.; Aksut, B.; Alam, T.; Alam, K.; et al. Global, Regional, and National Burden of Cardiovascular Diseases for 10 Causes, 1990 to 2015. J. Am. Coll. Cardiol. 2017, 70, 1–25. [Google Scholar] [CrossRef]
- Lai, Z.-F.; Chen, Y.-Z.; Feng, L.-P.; Meng, X.-M.; Ding, J.-F.; Wang, L.-Y.; Ye, J.; Li, P.; Cheng, X.-S.; Kitamoto, Y.; et al. Overexpression of TNNI3K, a cardiac-specific MAP kinase, promotes P19CL6-derived cardiac myogenesis and prevents myocardial infarction-induced injury. Am. J. Physiol. Circ. Physiol. 2008, 295, H708–H716. [Google Scholar] [CrossRef] [PubMed]
- E Adams, J.; Bodor, G.S.; Dávila-Román, V.G.; A Delmez, J.; Apple, F.S.; Ladenson, J.H.; Jaffe, A.S. Cardiac troponin I. A marker with high specificity for cardiac injury. Circulation 1993, 88, 101–106. [Google Scholar] [CrossRef]
- Soares, R.O.S.; Losada, D.M.; Jordani, M.C.; Évora, P.; Castro-E-Silva, O. Ischemia/Reperfusion Injury Revisited: An Overview of the Latest Pharmacological Strategies. Int. J. Mol. Sci. 2019, 20, 5034. [Google Scholar] [CrossRef]
- Porrello, E.R.; Mahmoud, A.I.; Simpson, E.; Hill, J.A.; Richardson, J.A.; Olson, E.N.; Sadek, H. Transient Regenerative Potential of the Neonatal Mouse Heart. Science 2011, 331, 1078–1080. [Google Scholar] [CrossRef]
- Piquereau, J.; Ventura-Clapier, R. Maturation of Cardiac Energy Metabolism During Perinatal Development. Front. Physiol. 2018, 9, 959. [Google Scholar] [CrossRef] [PubMed]
- Bergmann, O.; Zdunek, S.; Felker, A.; Salehpour, M.; Alkass, K.; Bernard, S.; Sjostrom, S.L.; Szewczykowska, M.; Jackowska, T.; Dos Remedios, C.; et al. Dynamics of Cell Generation and Turnover in the Human Heart. Cell 2015, 161, 1566–1575. [Google Scholar] [CrossRef] [PubMed]
- González-Rosa, J.M.; Sharpe, M.; Field, D.; Soonpaa, M.H.; Field, L.J.; Burns, C.E. Myocardial Polyploidization Creates a Barrier to Heart Regeneration in Zebrafish. Dev. Cell 2018, 44, 433–446.e7. [Google Scholar] [CrossRef]
- Fuller, S.J.; Osborne, S.A.; Leonard, S.J.; Hardyman, M.A.; Vaniotis, G.; Allen, B.G.; Sugden, P.H.; Clerk, A. Cardiac protein kinases: The cardiomyocyte kinome and differential kinase expression in human failing hearts. Cardiovasc. Res. 2015, 108, 87–98. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wang, S.-Q.; Wang, L.-P.; Yao, Y.-H.; Ma, C.-Y.; Ding, J.-F.; Ye, J.; Meng, X.-M.; Xu, R.-X.; Li, J.-J. Overexpression of Cardiac-Specific Kinase TNNI3K Promotes Mouse Embryonic Stem Cells Differentiation into Cardiomyocytes. Cell. Physiol. Biochem. 2017, 41, 381–398. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S. Long-term Outcomes in Individuals With Prolonged PR Interval or First-Degree Atrioventricular Block. JAMA 2009, 301, 2571–2577. [Google Scholar] [CrossRef]
- Li, Y.-Q.; Zhao, S.; Chen, K.-P.; Su, Y.-G.; Hua, W.; Chen, S.-L.; Liang, Z.-G.; Xu, W.; Dai, Y.; Fan, X.-H.; et al. Heart rate-adjusted PR as a prognostic marker of long-term ventricular arrhythmias and cardiac death in ICD/CRT-D recipients. J. Geriatr. Cardiol. 2019, 16, 259–264. [Google Scholar] [CrossRef]
- Theis, J.L.; Zimmermann, M.T.; Larsen, B.T.; Rybakova, I.N.; Long, P.A.; Evans, J.M.; Middha, S.; De Andrade, M.; Moss, R.L.; Wieben, E.D.; et al. TNNI3K mutation in familial syndrome of conduction system disease, atrial tachyarrhythmia and dilated cardiomyopathy. Hum. Mol. Genet. 2014, 23, 5793–5804. [Google Scholar] [CrossRef]
- Xi, Y.; Honeywell, C.; Zhang, D.; Schwartzentruber, J.; Beaulieu, C.L.; Tetreault, M.; Hartley, T.; Marton, J.; Vidal, S.M.; Majewski, J.; et al. Whole exome sequencing identifies the TNNI3K gene as a cause of familial conduction system disease and congenital junctional ectopic tachycardia. Int. J. Cardiol. 2015, 185, 114–116. [Google Scholar] [CrossRef]
- Podliesna, S.; Delanne, J.; Miller, L.; Tester, D.J.; Uzunyan, M.; Yano, S.; Klerk, M.; Cannon, B.C.; Khongphatthanayothin, A.; Laurent, G.; et al. Supraventricular tachycardias, conduction disease, and cardiomyopathy in 3 families with the same rare variant in TNNI3K (p.Glu768Lys). Hear. Rhythm. 2019, 16, 98–105. [Google Scholar] [CrossRef]
- Fan, L.-L.; Huang, H.; Jin, J.-Y.; Li, J.-J.; Chen, Y.-Q.; Zhao, S.-P.; Xiang, R. Whole exome sequencing identifies a novel mutation (c.333 + 2T > C) of TNNI3K in a Chinese family with dilated cardiomyopathy and cardiac conduction disease. Gene 2018, 648, 63–67. [Google Scholar] [CrossRef]
- Liu, J.; Liu, D.; Li, M.; Wu, K.; Liu, N.; Zhao, C.; Shi, X.; Liu, Q. Identification of a nonsense mutation in TNNI3K associated with cardiac conduction disease. J. Clin. Lab. Anal. 2020, 34, e23418. [Google Scholar] [CrossRef] [PubMed]
- Karczewski, K.J.; Francioli, L.C.; MacArthur, D.G. The mutational constraint spectrum quantified from variation in 141,456 humans. Yearb. Paediatr. Endocrinol. 2020, 581, 434–443. [Google Scholar] [CrossRef]
- Liebson, P.R.; Grandits, G.; Prineas, R.; Dianzumba, S.; Flack, J.M.; A Cutler, J.; Grimm, R.; Stamler, J. Echocardiographic correlates of left ventricular structure among 844 mildly hypertensive men and women in the Treatment of Mild Hypertension Study (TOMHS). Circulation 1993, 87, 476–486. [Google Scholar] [CrossRef] [PubMed]
- Lawhorn, B.G.; Philp, J.; Zhao, Y.; Louer, C.; Hammond, M.; Cheung, M.; Fries, H.; Graves, A.P.; Shewchuk, L.; Wang, L.; et al. Identification of Purines and 7-Deazapurines as Potent and Selective Type I Inhibitors of Troponin I-Interacting Kinase (TNNI3K). J. Med. Chem. 2015, 58, 7431–7448. [Google Scholar] [CrossRef]
- Philp, J.; Lawhorn, B.G.; Graves, A.P.; Shewchuk, L.; Rivera, K.L.; Jolivette, L.J.; Holt, D.A.; Gatto, G.J.; Kallander, L.S. 4,6-Diaminopyrimidines as Highly Preferred Troponin I-Interacting Kinase (TNNI3K) Inhibitors. J. Med. Chem. 2018, 61, 3076–3088. [Google Scholar] [CrossRef]
- I Davis, M.; Hunt, J.P.; Herrgard, S.; Ciceri, P.; Wodicka, L.M.; Pallares, G.; Hocker, M.; Treiber, D.K.; Zarrinkar, P.P. Comprehensive analysis of kinase inhibitor selectivity. Nat. Biotechnol. 2011, 29, 1046–1051. [Google Scholar] [CrossRef]
- Lawhorn, B.G.; Philp, J.; Graves, A.P.; Shewchuk, L.; Holt, D.A.; Gatto, G.J., Jr.; Kallander, L.S. GSK114: A selective inhibitor for elucidating the biological role of TNNI3K. Bioorganic Med. Chem. Lett. 2016, 26, 3355–3358. [Google Scholar] [CrossRef] [PubMed]
- Asquith, C.R.M.; Laitinen, T.; Wells, C.I.; Tizzard, G.J.; Zuercher, W.J. New Insights into 4-Anilinoquinazolines as Inhibitors of Cardiac Troponin I-Interacting Kinase (TNNi3K). Molecules 2020, 25, 1697. [Google Scholar] [CrossRef]
- Pang, H.; Wang, N.; Chai, J.; Wang, X.; Zhang, Y.; Bi, Z.; Wu, W.; He, G. Discovery of novel TNNI3K inhibitor suppresses pyroptosis and apoptosis in murine myocardial infarction injury. Eur. J. Med. Chem. 2020, 197, 112314. [Google Scholar] [CrossRef]
- Wiltshire, S.A.; Leiva-Torres, G.A.; Vidal, S.M. Quantitative Trait Locus Analysis, Pathway Analysis, and Consomic Mapping Show Genetic Variants of Tnni3k, Fpgt, orH28Control Susceptibility to Viral Myocarditis. J. Immunol. 2011, 186, 6398–6405. [Google Scholar] [CrossRef]
- Yeh, C.-N.; Chen, M.-H.; Chang, Y.-C.; Wu, R.-C.; Tsao, L.-C.; Wang, S.-Y.; Cheng, C.-T.; Chiang, K.-C.; Chen, T.-W.; Hsiao, M.; et al. Over-expression of TNNI3K is associated with early-stage carcinogenesis of cholangiocarcinoma. Mol. Carcinog. 2019, 58, 270–278. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Bradfield, J.P.; Zhang, H.; Sleiman, P.M.; Kim, C.E.; Glessner, J.T.; Deliard, S.; Thomas, K.A.; Frackelton, E.C.; Li, M.; et al. Role of BMI-Associated Loci Identified in GWAS Meta-Analyses in the Context of Common Childhood Obesity in European Americans. Obesity 2011, 19, 2436–2439. [Google Scholar] [CrossRef] [PubMed]
- Renkonen, R. Fucose-1-Phosphate Guanylyltransferase (FPGT). In Handbook of Glycosyltransferases and Related Genes; Springer Science and Business Media LLC: Berlin, Germany, 2014; Volume 2, pp. 1631–1635. [Google Scholar]
- Miyoshi, E. Fucosylation and Cancer. In Experimental Glycoscience; Springer Science and Business Media LLC: Berlin, Germany, 2009; pp. 235–237. [Google Scholar]
- Graff, M.; Ngwa, J.S.; Workalemahu, T.; Homuth, G.; Schipf, S.; Teumer, A.; Völzke, H.; Wallaschofski, H.; Abecasis, G.R.; Edward, L.; et al. Genome-wide analysis of BMI in adolescents and young adults reveals additional insight into the effects of genetic loci over the life course. Hum. Mol. Genet. 2013, 22, 3597–3607. [Google Scholar] [CrossRef] [PubMed]
- Cornelis, M.C.; Rimm, E.B.; Curhan, G.C.; Kraft, P.; Hunter, D.J.; Hu, F.B.; Van Dam, R.M. Obesity susceptibility loci and uncontrolled eating, emotional eating and cognitive restraint behaviors in men and women. Obesity 2014, 22, E135–E141. [Google Scholar] [CrossRef]
- Felix, J.F.; Bradfield, J.P.; Monnereau, C.; Van Der Valk, R.J.; Stergiakouli, E.; Chesi, A.; Gaillard, R.; Feenstra, B.; Thiering, E.; Kreiner-Møller, E.; et al. Genome-wide association analysis identifies three new susceptibility loci for childhood body mass index. Hum. Mol. Genet. 2016, 25, 389–403. [Google Scholar] [CrossRef]
- Liu, H.Y.; Alyass, A.; Abadi, A.; Peralta-Romero, J.; Suarez, F.; Gomez-Zamudio, J.H.; Audirac, A.; Parra, E.J.; Cruz, M.; Meyre, D. Fine-mapping of 98 obesity loci in Mexican children. Int. J. Obes. 2018, 43, 23–32. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Model Name | Genetic Model | Genetic Background | Intervention | Results | Reference |
|---|---|---|---|---|---|
| Congenic | Physiological Tnni3k expression (DBA.AKR.hrtfm2) | DBA/2J | N/A | PR interval prolongation compared to DBA/2J WT | [4] |
| TNNI3Ktg | Transgenic human TNNI3K overexpression | DBA/2J | TAC | Greater cardiac dysfunction | [3,15] |
| N/A | PR interval prolongation | [4] | |||
| C57BL/6J | N/A | Concentric hypertrophy | [10] | ||
| FVB/N | I/R | Increased infarct size, mitochondrial dysfunction, increased cell death, and ROS production | [2] | ||
| TNNI3K-KDtg | Transgenic human TNNI3K-p.K489R (kinase-dead) overexpression | DBA/2J | TAC | Similar cardiac dysfunction compared to WT | [15] |
| FVB/N | I/R | Smaller infarct size, reduced cTnI levels | [2] | ||
| C57BL/6J | N/A | Increased MNCMs %, shorter and wider CM dimensions | [11,17] | ||
| TNNI3Ktg/Csqtg | Transgenic human TNNI3K overexpression with transgenic overexpression of Csq | DBA/2J | N/A | Reduced survival rate, cardiac function, and cardiac dilatation | [3] |
| Tnni3k-KO | Complete Tnni3k knockout | C57BL/6J | N/A | Mild concentric remodeling, increased MNCMs %, shorter and wider CM dimensions | [11,17] |
| MI | Increased CM proliferation in the border zone | [16] | |||
| I/R | Smaller infarct size, reduced cTnI levels, and less cell death | [2] | |||
| I685T/I685T | Knock-in allele resulting in Tnni3k-p.Ile685Thr | C57BL/6J | N/A | Increased MNCMs %, shorter and wider CM dimensions | [17] |
| Compound | TNNI3K IC50 (nM) | Other Target(s) | Chemical Structure | Reference |
|---|---|---|---|---|
| GSK854 | <10 | ZAK/MLTK (tested at 100 nM) |  | [2] |
| GSK329 | 10 | Axl, DDR2, Flt1, Flt3, Flt4, KDR, Mer, MuSK, PTK5, TAO2, TAO3 (tested at 100 nM) |  | [2] |
| GSK114 | 25 | ACK1, B-Raf, GAK, MEK5, PDGFRB, STK36, ZAK (tested at 1 µM) |  | [40,41] |
| 6O | 410 | TAK1 (tested at 10 µM) |  | [42] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pham, C.; Muñoz-Martín, N.; Lodder, E.M. The Diverse Roles of TNNI3K in Cardiac Disease and Potential for Treatment. Int. J. Mol. Sci. 2021, 22, 6422. https://doi.org/10.3390/ijms22126422
Pham C, Muñoz-Martín N, Lodder EM. The Diverse Roles of TNNI3K in Cardiac Disease and Potential for Treatment. International Journal of Molecular Sciences. 2021; 22(12):6422. https://doi.org/10.3390/ijms22126422
Chicago/Turabian StylePham, Caroline, Noelia Muñoz-Martín, and Elisabeth M. Lodder. 2021. "The Diverse Roles of TNNI3K in Cardiac Disease and Potential for Treatment" International Journal of Molecular Sciences 22, no. 12: 6422. https://doi.org/10.3390/ijms22126422
APA StylePham, C., Muñoz-Martín, N., & Lodder, E. M. (2021). The Diverse Roles of TNNI3K in Cardiac Disease and Potential for Treatment. International Journal of Molecular Sciences, 22(12), 6422. https://doi.org/10.3390/ijms22126422