
Metformin Affects Cardiac Arachidonic Acid Metabolism and Cardiac Lipid Metabolite Storage in a Prediabetic Rat Model

 , , and
, , and
Abstract
:1. Introduction
2. Results
2.1. Characterization of Cardiac Lipid Metabolic Parameters in HHTg Rats
2.2. Effects of Metformin on Basal Metabolic Parameters and Inflammatory Markers
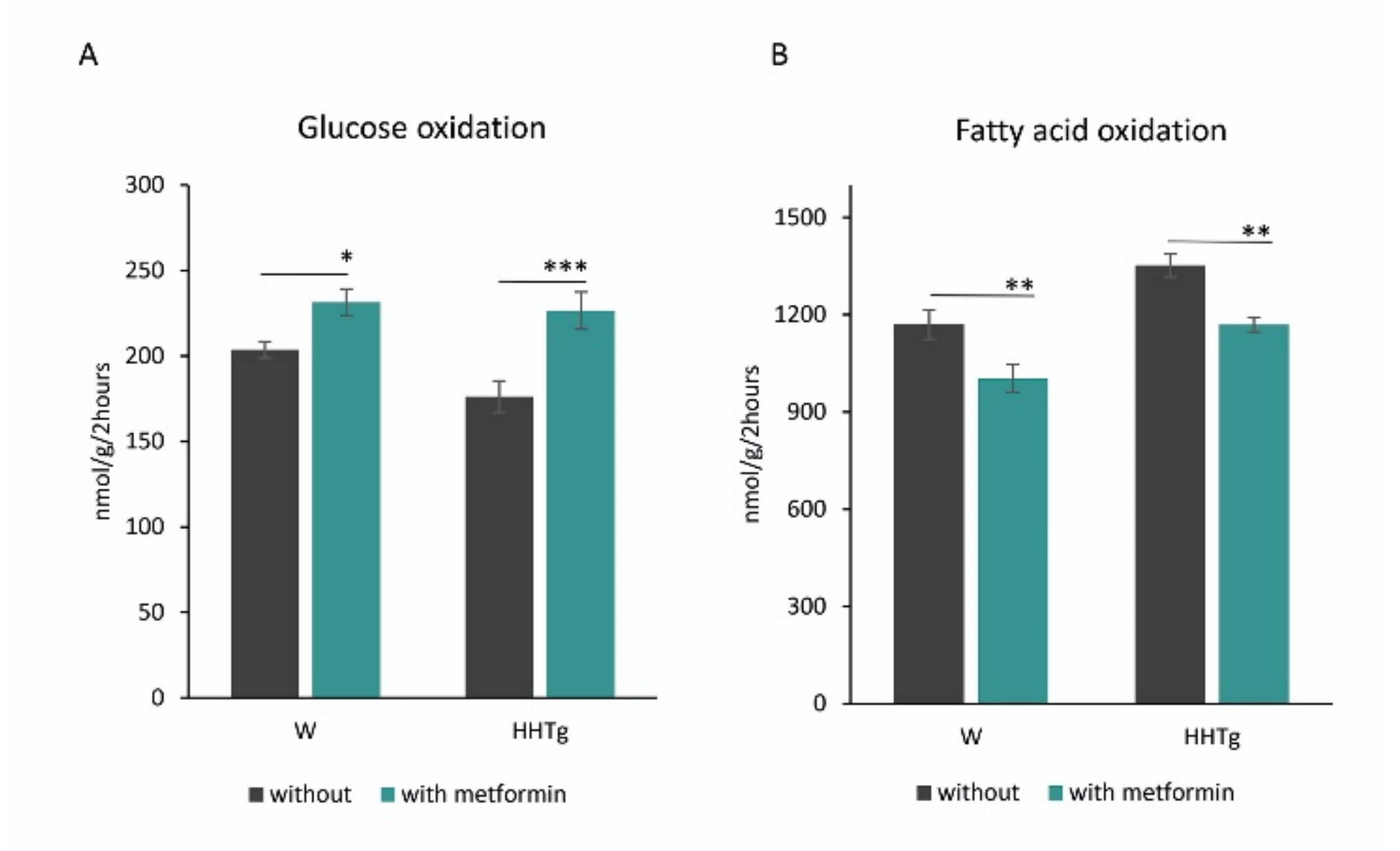
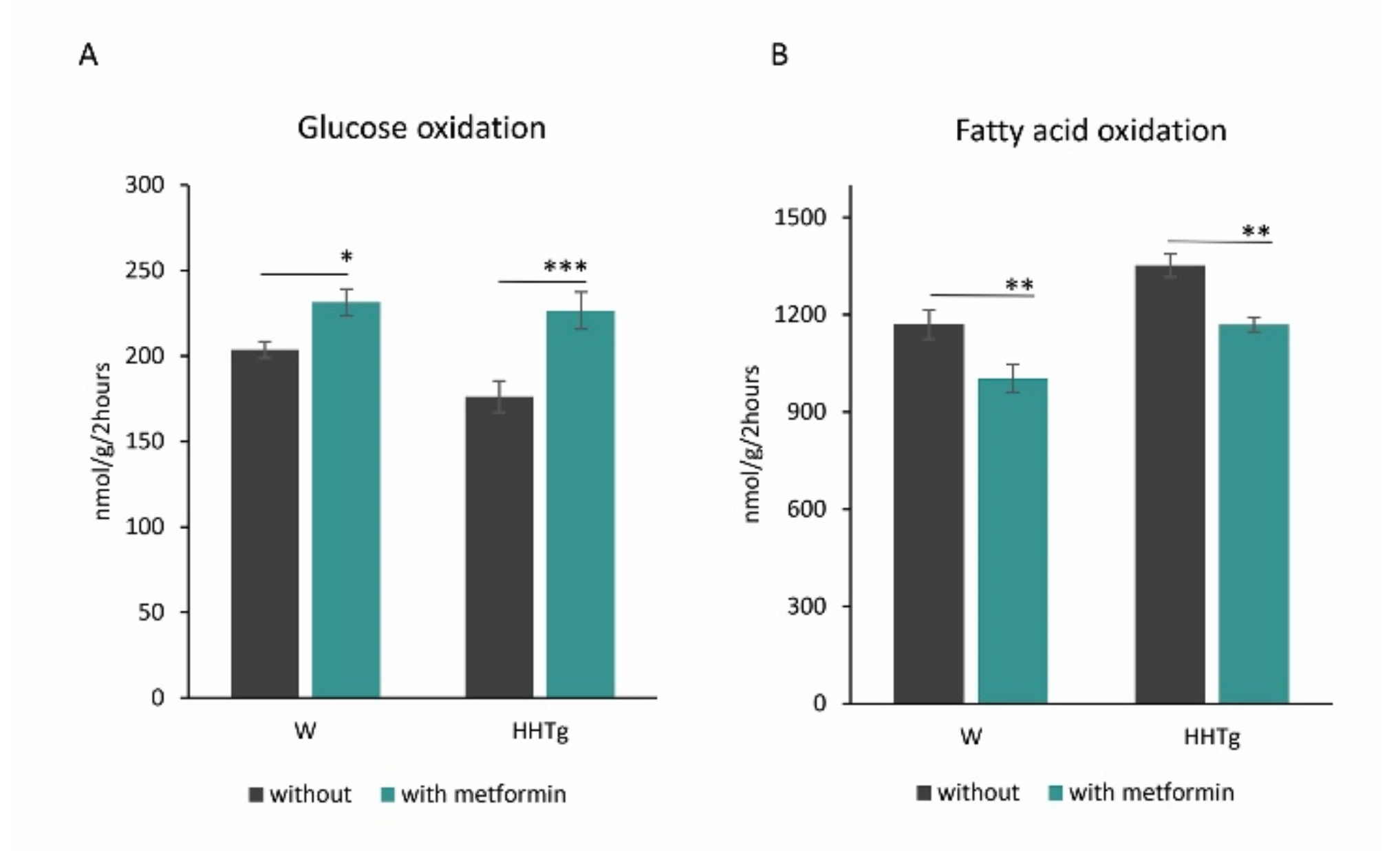
2.3. Effects of Metformin on Myocardial Lipid Storage and Substrate Utilization
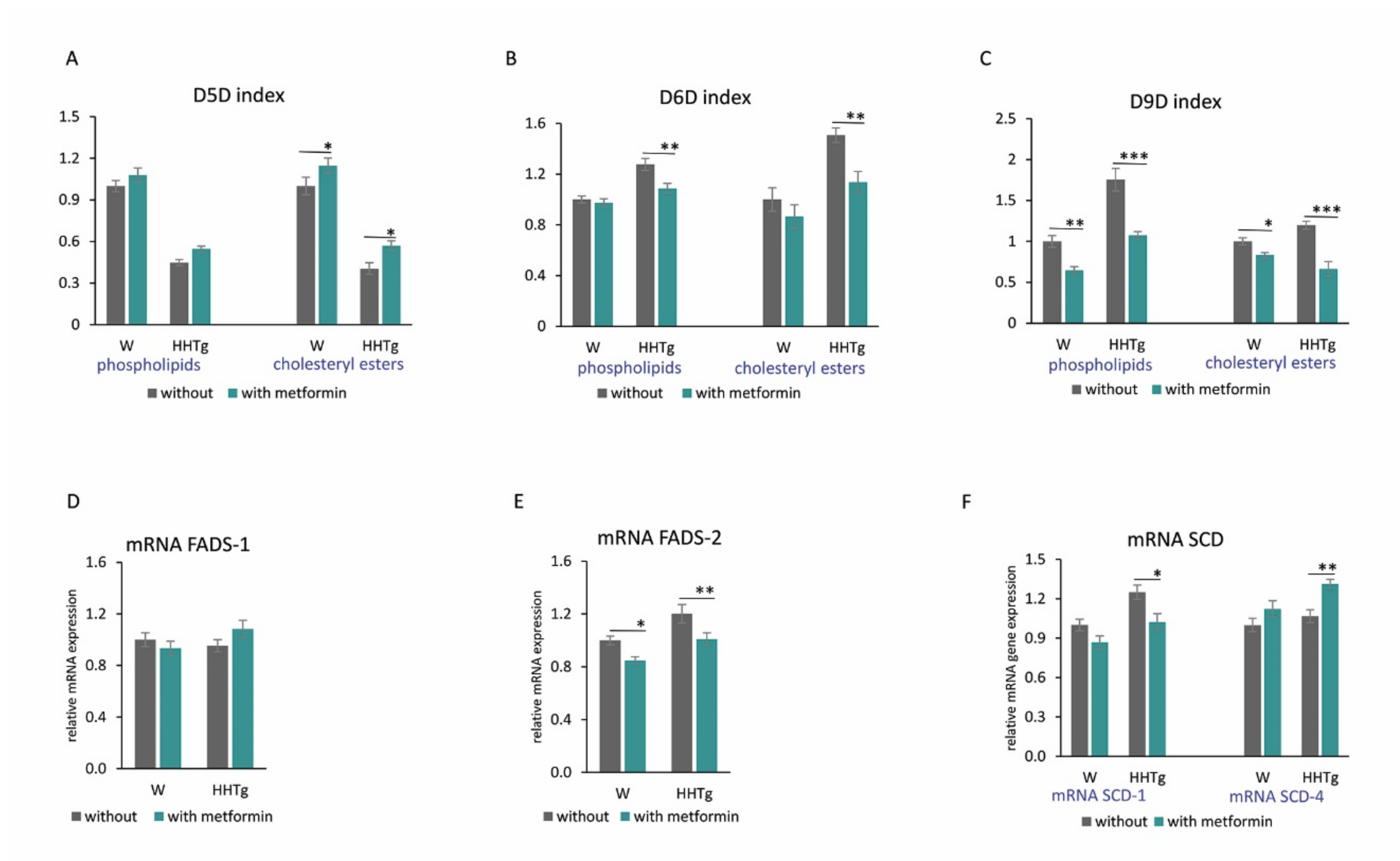
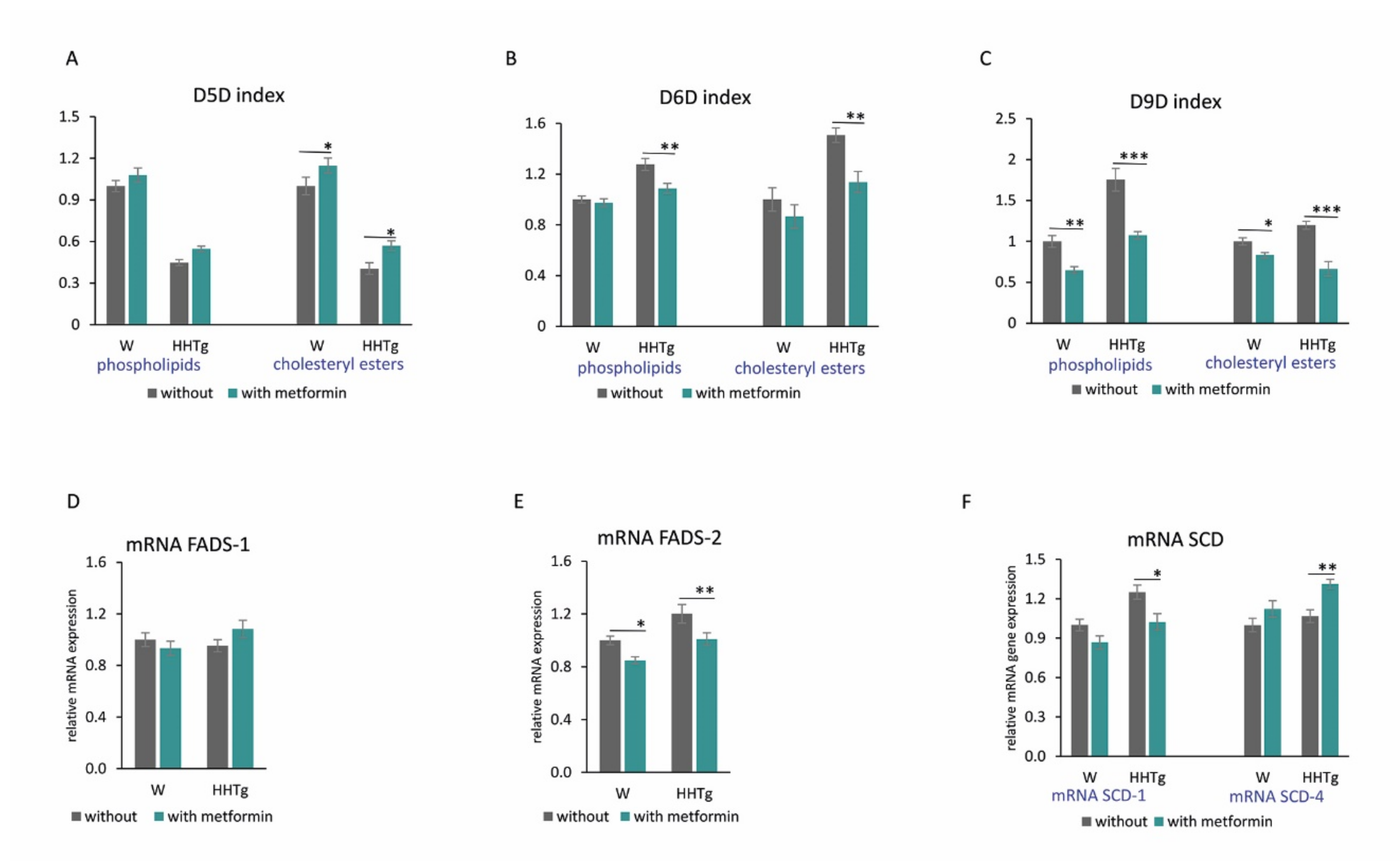
2.4. Effects of Metformin on Fatty Acid Desaturases and Profiles in the Myocardium
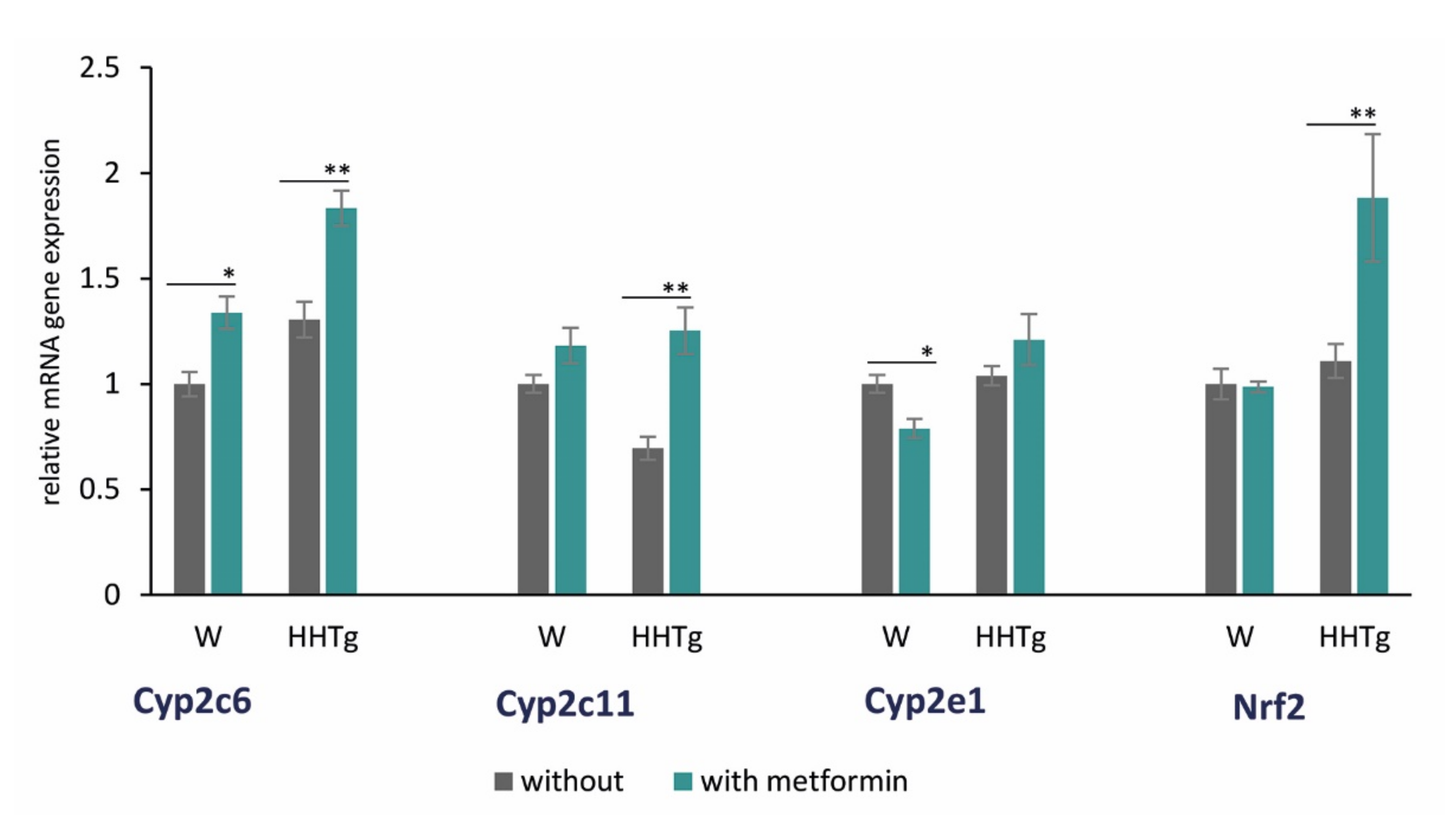
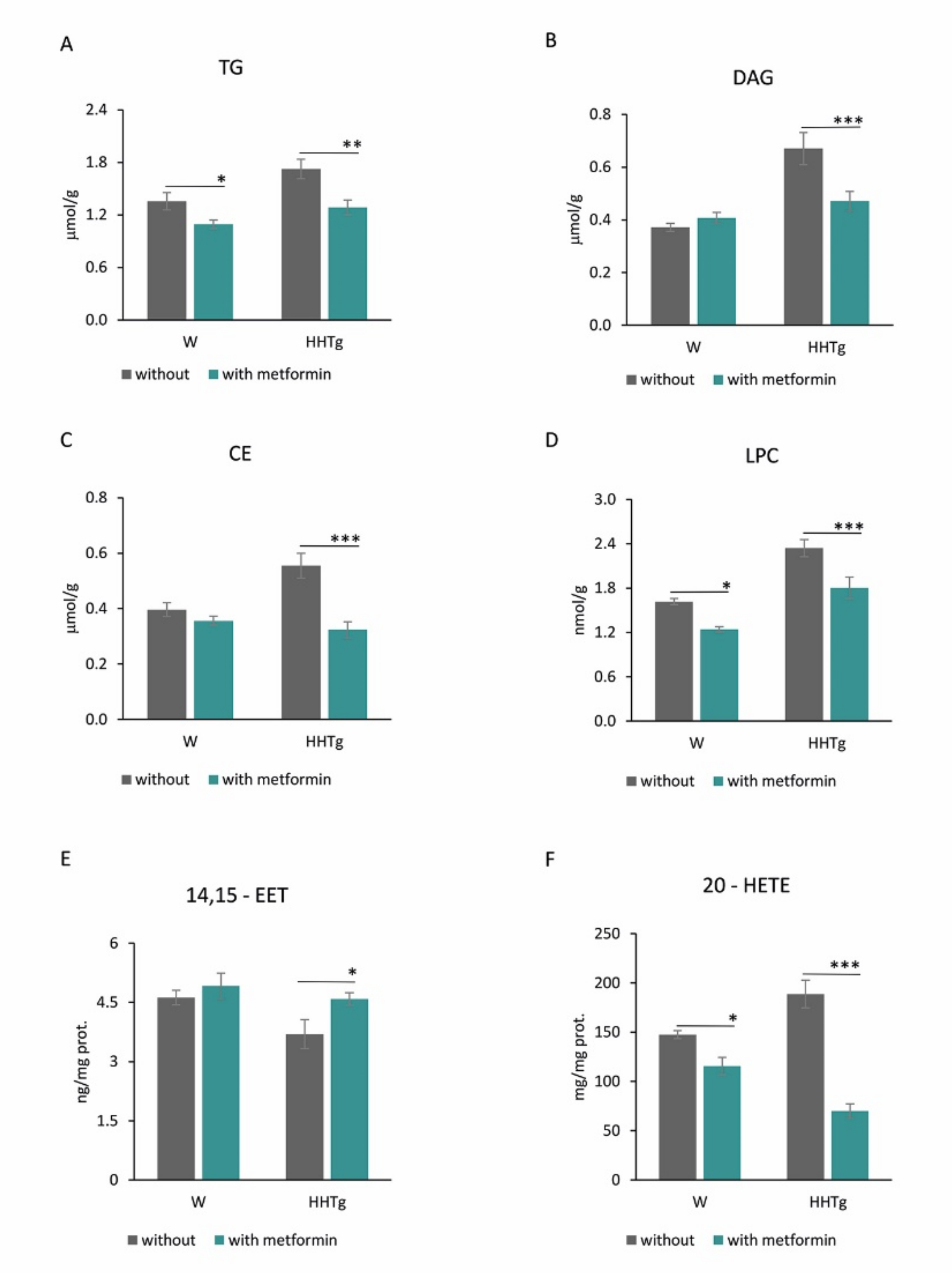
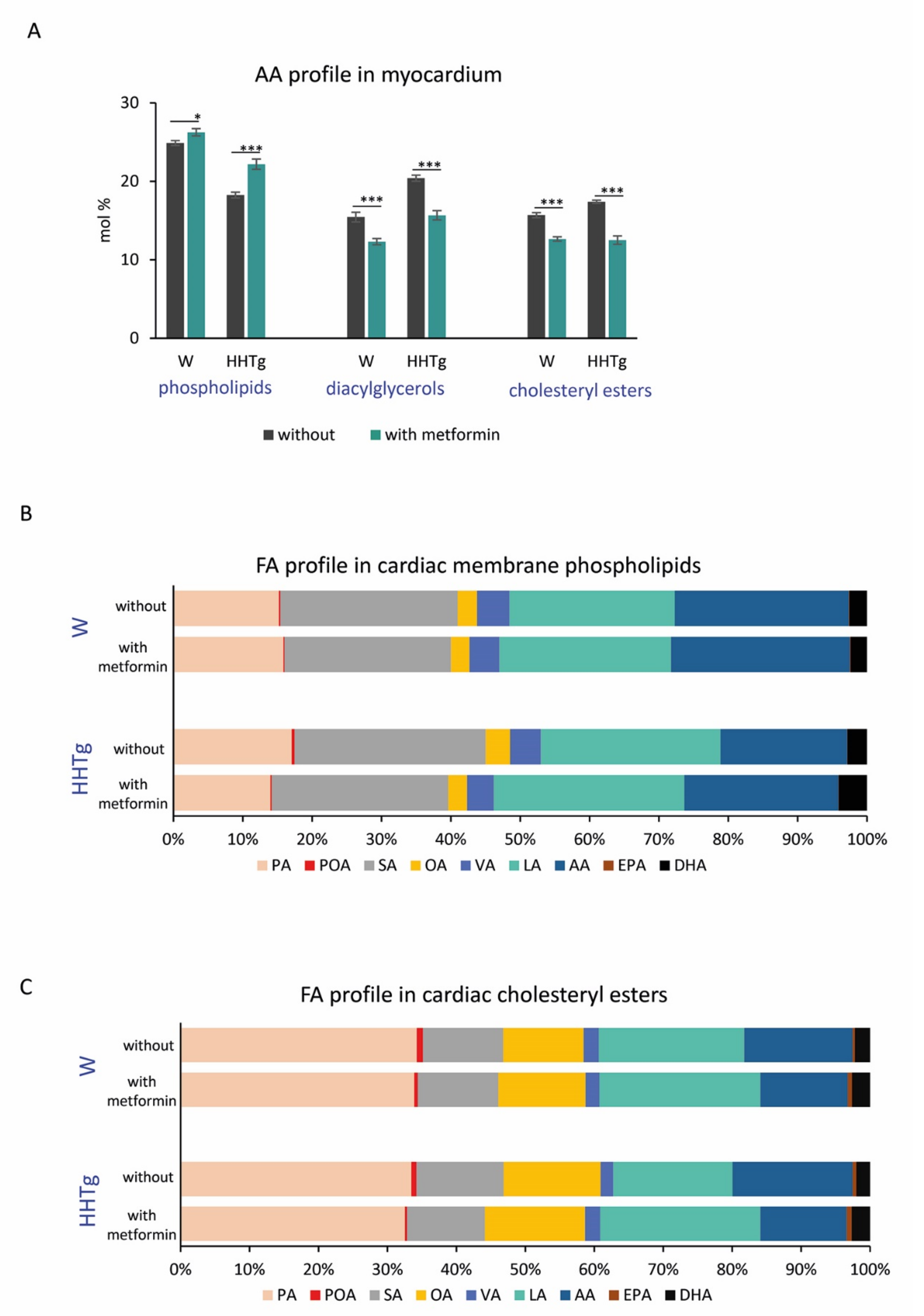
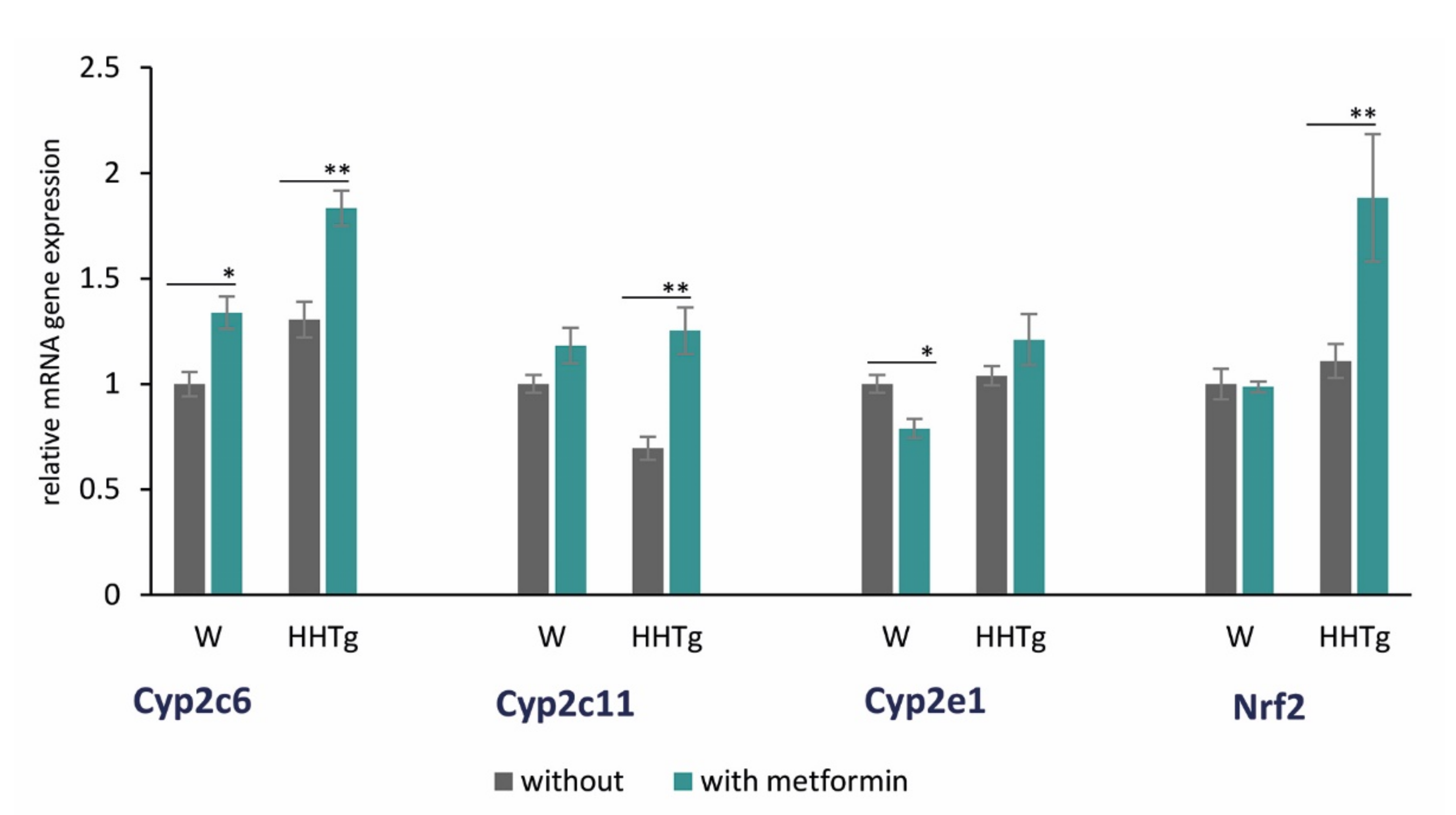
2.5. Effects of Metformin on AA Metabolism and Metabolites in the Myocardium
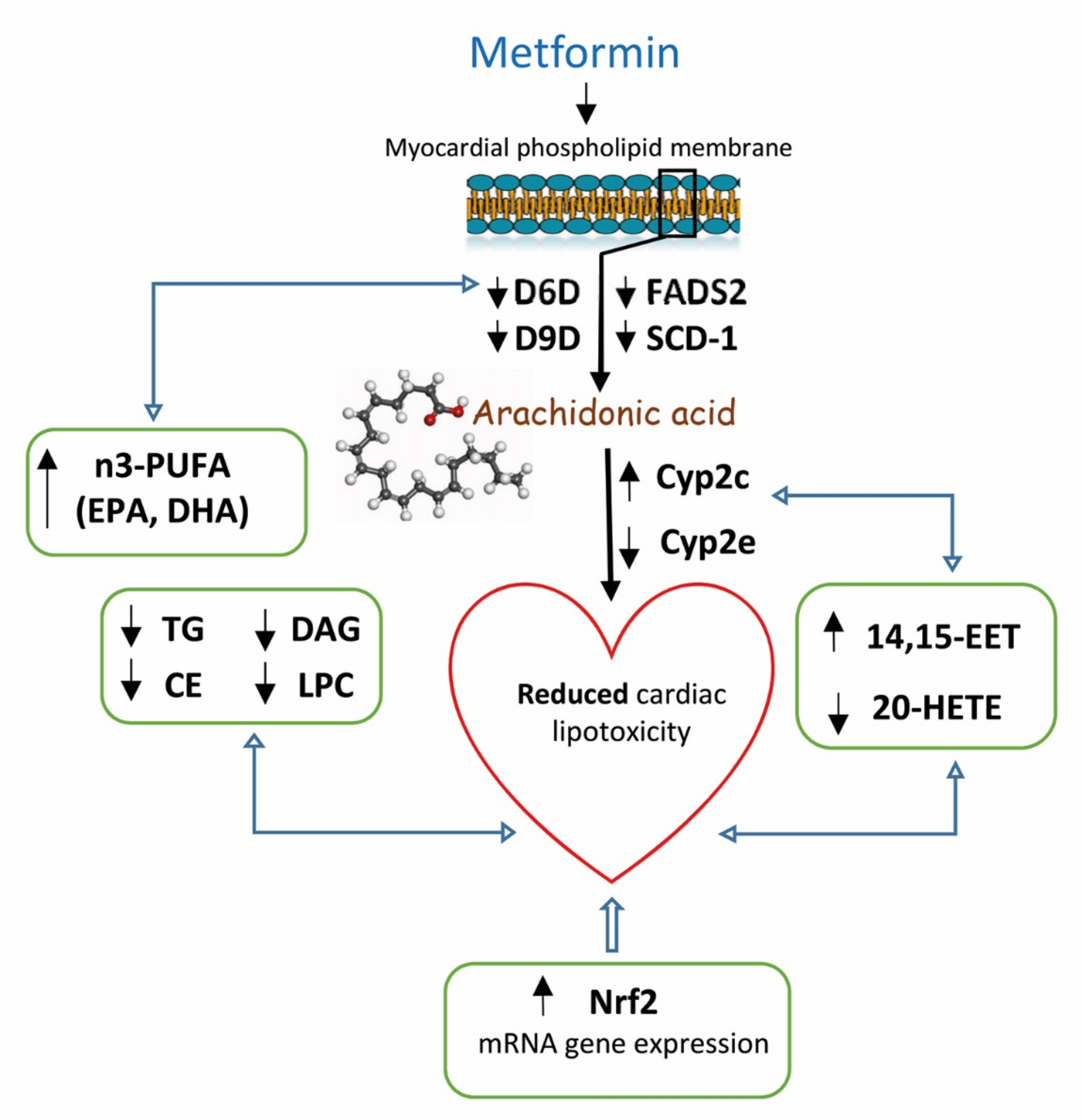
3. Discussion
4. Materials and Methods
4.1. Animals and Diet
4.2. Analytical Methods and Biochemical Analysis
4.3. Fatty Acid Composition and Fatty Acid Desaturases Activity
4.4. Relative mRNA Expression
4.5. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AA | arachidonic acid |
| ACC | acetyl-coenzyme A carboxylase |
| AMPK | adenosine monophosphate-activated protein kinase |
| CE | cholesteryl ester |
| COX | cyclooxygenase |
| CYP450 | cytochrome P450 |
| DAG | diacylglycerol |
| D5D | delta 5-desaturase |
| D6D | delta 6-desaturase |
| D9D | delta 9-desaturase |
| EET | epoxyeicosatrienoic acid |
| FA | fatty acid |
| FADS | fatty acid desaturase |
| FAS | fatty acid synthase |
| HETE | hydroxyeicosatetraenoic acid |
| HMGCR | 3-hydroxy-3-methylglutaryl-coenzyme A reductase |
| hsCRP | high-sensitivity C-reactive protein |
| IL-6 | interleukin 6 |
| LOX | lipoxygenase |
| LPC | lysophosphatidylcholine |
| MAPK | mitogen-activated protein kinase |
| MCP-1 | monocyte chemoattractant protein-1 |
| NEFA | non-esterified fatty acid |
| NF-κB | nuclear factor-κB |
| NRF2 | nuclear factor erythroid-2-related factor 2 |
| PL | phospholipid |
| PPARα | peroxisome proliferator-acivated receptor α |
| SCD | stearoyl-coenzyme A desaturase |
| SREBP1 | sterol regulatory element-binding protein 1 |
| TG | triglyceride |
| TNFα | tumor necrosis factor α |
References
- Grundy, S.M. Metabolic syndrome update. Trends Cardiovasc. Med. 2016, 26, 364–373. [Google Scholar] [CrossRef]
- Roberts, C.K.; Hevener, A.L.; Barnard, R.J. Metabolic syndrome and insulin resistance: Underlying causes and modification by exercise training. Compr. Physiol. 2013, 3, 1–58. [Google Scholar] [PubMed] [Green Version]
- Weijers, R.N. Lipid composition of cell membranes and its relevance in type 2 diabetes mellitus. Curr. Diabetes Rev. 2012, 8, 390–400. [Google Scholar] [CrossRef]
- AM, A.L.; Syed, D.N.; Ntambi, J.M. Insights into Stearoyl-CoA Desaturase-1 Regulation of Systemic Metabolism. Trends Endocrinol. Metab. 2017, 28, 831–842. [Google Scholar]
- Tabaczar, S.; Wolosiewicz, M.; Filip, A.; Olichwier, A.; Dobrzyn, P. The role of stearoyl-CoA desaturase in the regulation of cardiac metabolism. Postepy. Biochem. 2018, 64, 183–189. [Google Scholar]
- Dobrzyn, P.; Bednarski, T.; Dobrzyn, A. Metabolic reprogramming of the heart through stearoyl-CoA desaturase. Prog. Lipid. Res. 2015, 57, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Jamieson, K.L.; Endo, T.; Darwesh, A.M.; Samokhvalov, V.; Seubert, J.M. Cytochrome P450-derived eicosanoids and heart function. Pharmacol. Ther. 2017, 179, 47–83. [Google Scholar] [CrossRef] [PubMed]
- Grammatiki, M.; Sagar, R.; Ajjan, R.A. Metformin: Is it still the first line in type 2 diabetes management algorithm? Curr. Pharm. Des. 2021, 27, 1061–1067. [Google Scholar] [CrossRef]
- Fujita, Y.; Inagaki, N. Metformin: New Preparations and Nonglycemic Benefits. Curr. Diab. Rep. 2017, 17, 5. [Google Scholar] [CrossRef]
- Jenkins, A.J.; Welsh, P.; Petrie, J.R. Metformin, lipids and atherosclerosis prevention. Curr. Opin. Lipidol. 2018, 29, 346–353. [Google Scholar] [CrossRef]
- Woo, S.L.; Xu, H.; Li, H.; Zhao, Y.; Hu, X.; Zhao, J.; Guo, X.; Guo, T.; Botchlett, R.; Qi, T.; et al. Metformin ameliorates hepatic steatosis and inflammation without altering adipose phenotype in diet-induced obesity. PLoS ONE 2014, 9, e91111. [Google Scholar] [CrossRef] [Green Version]
- Marchesini, G.; Brizi, M.; Bianchi, G.; Tomassetti, S.; Zoli, M.; Melchionda, N. Metformin in non-alcoholic steatohepatitis. Lancet 2001, 358, 893–894. [Google Scholar] [CrossRef]
- Geerling, J.J.; Boon, M.R.; Van der Zon, G.C.; Van den Berg, S.A.; Van den Hoek, A.M.; Lombes, M.; Princen, H.M.; Havekes, L.M.; Rensen, P.C.; Guigas, B. Metformin lowers plasma triglycerides by promoting VLDL-triglyceride clearance by brown adipose tissue in mice. Diabetes 2014, 63, 880–891. [Google Scholar] [CrossRef] [Green Version]
- Loomba, R.; Lutchman, G.; Kleiner, D.E.; Ricks, M.; Feld, J.J.; Borg, B.B.; Modi, A.; Nagabhyru, P.; Sumner, A.E.; Liang, T.J.; et al. Clinical trial: Pilot study of metformin for the treatment of non-alcoholic steatohepatitis. Aliment. Pharmacol. Ther. 2009, 29, 172–182. [Google Scholar] [CrossRef] [PubMed]
- Nesti, L.; Natali, A. Metformin effects on the heart and the cardiovascular system: A review of experimental and clinical data. Nutr. Metab. Cardiovasc. Dis. 2017, 27, 657–669. [Google Scholar] [CrossRef] [PubMed]
- Malinska, H.; Skop, V.; Trnovska, J.; Markova, I.; Svoboda, P.; Kazdova, L.; Haluzik, M. Metformin attenuates myocardium dicarbonyl stress induced by chronic hypertriglyceridemia. Physiol. Res. 2018, 67, 181–189. [Google Scholar] [CrossRef] [PubMed]
- Bolivar, S.; Noriega, L.; Ortega, S.; Osorio, E.; Rosales, W.; Mendoza, X.; Mendoza-Torres, E. Novel targets of metformin in cardioprotection: Beyond the effects mediated by AMPK. Curr. Pharm. Des. 2021, 27, 80–90. [Google Scholar] [CrossRef]
- Foretz, M.; Guigas, B.; Bertrand, L.; Pollak, M.; Viollet, B. Metformin: From mechanisms of action to therapies. Cell. Metab. 2014, 20, 953–966. [Google Scholar] [CrossRef] [Green Version]
- Zicha, J.; Pechanova, O.; Cacanyiova, S.; Cebova, M.; Kristek, F.; Torok, J.; Simko, F.; Dobesova, Z.; Kunes, J. Hereditary hypertriglyceridemic rat: A suitable model of cardiovascular disease and metabolic syndrome? Physiol. Res. 2006, 55 (Suppl. 1), S49–S63. [Google Scholar] [PubMed]
- Markova, I.; Miklankova, D.; Huttl, M.; Kacer, P.; Skibova, J.; Kucera, J.; Sedlacek, R.; Kacerova, T.; Kazdova, L.; Malinska, H. The Effect of Lipotoxicity on Renal Dysfunction in a Nonobese Rat Model of Metabolic Syndrome: A Urinary Proteomic Approach. J. Diabetes. Res. 2019, 2019, 8712979. [Google Scholar] [CrossRef]
- Goldberg, I.J.; Trent, C.M.; Schulze, P.C. Lipid metabolism and toxicity in the heart. Cell. Metab. 2012, 15, 805–812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ormazabal, V.; Nair, S.; Elfeky, O.; Aguayo, C.; Salomon, C.; Zuniga, F.A. Association between insulin resistance and the development of cardiovascular disease. Cardiovasc. Diabetol. 2018, 17, 122. [Google Scholar] [CrossRef] [PubMed]
- De Carvalho, L.P.; Tan, S.H.; Ow, G.; Tang, Z.; Ching, J.; Kovalik, J.P.; Poh, S.C.; Chin, C.T.; Richards, A.M.; Martinez, E.C.; et al. Plasma ceramides as prognostic biomarkers and their arterial and myocardial tissue correlates in acute myocardial infarction. JACC Basic. Transl. Sci. 2018, 30, 163–175. [Google Scholar] [CrossRef] [PubMed]
- D’Souza, K.; Nzirorera, C.; Kienesberger, P.C. Lipid metabolism and signaling in cardiac lipotoxicity. Biochim. Biophys. Acta 2016, 1861, 1513–1524. [Google Scholar] [CrossRef] [PubMed]
- Matsui, H.; Yokoyama, T.; Sekiguchi, K.; Iijima, D.; Sunaga, H.; Maniwa, M.; Ueno, M.; Iso, T.; Arai, M.; Kurabayashi, M. Stearoyl-CoA desaturase-1 (SCD1) augments saturated fatty acid-induced lipid accumulation and inhibits apoptosis in cardiac myocytes. PLoS ONE 2012, 7, e33283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rizzo, A.M.; Montorfano, G.; Negroni, M.; Adorni, L.; Berselli, P.; Corsetto, P.; Wahle, K.; Berra, B. A rapid method for determining arachidonic:eicosapentaenoic acid ratios in whole blood lipids: Correlation with erythrocyte membrane ratios and validation in a large Italian population of various ages and pathologies. Lipids. Health Dis. 2010, 9, 7. [Google Scholar] [CrossRef] [Green Version]
- Nelson, J.R.; Raskin, S. The eicosapentaenoic acid:arachidonic acid ratio and its clinical utility in cardiovascular disease. Postgrad. Med. 2019, 131, 268–277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.R.; Jeon, S.Y.; Lee, S.M. The association of cardiovascular risk factors with saturated fatty acids and fatty acid desaturase indices in erythrocyte in middle-aged Korean adults. Lipids Health Dis. 2015, 14, 133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Svendsen, K.; Olsen, T.; Nordstrand Rusvik, T.C.; Ulven, S.M.; Holven, K.B.; Retterstol, K.; Telle-Hansen, V.H. Fatty acid profile and estimated desaturase activities in whole blood are associated with metabolic health. Lipids Health Dis. 2020, 19, 102. [Google Scholar] [CrossRef]
- Warensjo, E.; Sundstrom, J.; Vessby, B.; Cederholm, T.; Riserus, U. Markers of dietary fat quality and fatty acid desaturation as predictors of total and cardiovascular mortality: A population-based prospective study. Am. J. Clin. Nutr. 2008, 88, 203–209. [Google Scholar] [CrossRef]
- Drzewoski, J.; Hanefeld, M. The Current and Potential Therapeutic Use of Metformin-The Good Old Drug. Pharmaceuticals 2021, 14, 122. [Google Scholar] [CrossRef]
- Anabtawi, A.; Miles, J.M. Metformin: Nonglycemic Effects and Potential Novel Indications. Endocr. Pract. 2016, 22, 999–1007. [Google Scholar] [CrossRef] [PubMed]
- Wilson, R.R. In defense of the autopsy. JAMA 1966, 196, 1011–1012. [Google Scholar] [CrossRef]
- Miyazaki, M.; Jacobson, M.J.; Man, W.C.; Cohen, P.; Asilmaz, E.; Friedman, J.M.; Ntambi, J.M. Identification and characterization of murine SCD4, a novel heart-specific stearoyl-CoA desaturase isoform regulated by leptin and dietary factors. J. Biol. Chem. 2003, 278, 33904–33911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dobrzyn, P.; Sampath, H.; Dobrzyn, A.; Miyazaki, M.; Ntambi, J.M. Loss of stearoyl-CoA desaturase 1 inhibits fatty acid oxidation and increases glucose utilization in the heart. Am. J. Physiol. Endocrinol. Metab. 2008, 294, E357–E364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pascual, F.; Coleman, R.A. Fuel availability and fate in cardiac metabolism: A tale of two substrates. Biochim. Biophys. Acta 2016, 1861, 1425–1433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, L.; Shi, X.; Bharadwaj, K.G.; Ikeda, S.; Yamashita, H.; Yagyu, H.; Schaffer, J.E.; Yu, Y.H.; Goldberg, I.J. DGAT1 expression increases heart triglyceride content but ameliorates lipotoxicity. J. Biol. Chem. 2009, 284, 36312–36323. [Google Scholar] [CrossRef] [Green Version]
- Barouch, L.A.; Gao, D.; Chen, L.; Miller, K.L.; Xu, W.; Phan, A.C.; Kittleson, M.M.; Minhas, K.M.; Berkowitz, D.E.; Wei, C.; et al. Cardiac myocyte apoptosis is associated with increased DNA damage and decreased survival in murine models of obesity. Circ. Res. 2006, 98, 119–124. [Google Scholar] [CrossRef] [Green Version]
- DaCosta, R.M.; Rodrigues, D.; Pereira, C.A.; Silva, J.F.; Alves, J.V.; Lobato, N.S.; Tostes, R.C. Nrf2 as a potential mediator of cardiovascular risk in metabolic diseases. Front. Pharmacol. 2019, 10, 382. [Google Scholar] [CrossRef] [Green Version]
- Vashi, R.; Patel, B.M. NRF2 in Cardiovascular Diseases: A Ray of Hope! J. Cardiovasc. Transl. Res. 2020, 14, 573–586. [Google Scholar] [CrossRef]
- Dobrzyn, P.; Pyrkowska, A.; Jazurek, M.; Dobrzyn, A. Increased availability of endogenous and dietary oleic acid contributes to the upregulation of cardiac fatty acid oxidation. Mitochondrion 2012, 12, 132–137. [Google Scholar] [CrossRef]
- Finck, B.N.; Han, X.; Courtois, M.; Aimond, F.; Nerbonne, J.M.; Kovacs, A.; Gross, R.W.; Kelly, D.P. A critical role for PPARalpha-mediated lipotoxicity in the pathogenesis of diabetic cardiomyopathy: Modulation by dietary fat content. Proc. Natl. Acad. Sci. USA 2003, 100, 1226–1231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, Y.; Theken, K.N.; Lee, C.R. Cytochrome P450 epoxygenases, soluble epoxide hydrolase, and the regulation of cardiovascular inflammation. J. Mol. Cell. Cardiol. 2010, 48, 331–341. [Google Scholar] [CrossRef] [Green Version]
- Alsaad, A.M.; Zordoky, B.N.; Tse, M.M.; El-Kadi, A.O. Role of cytochrome P450-mediated arachidonic acid metabolites in the pathogenesis of cardiac hypertrophy. Drug. Metab. Rev. 2013, 45, 173–195. [Google Scholar] [CrossRef] [PubMed]
- Siriwardhana, N.; Kalupahana, N.S.; Moustaid-Moussa, N. Health benefits of n-3 polyunsaturated fatty acids: Eicosapentaenoic acid and docosahexaenoic acid. Adv. Food. Nutr. Res. 2012, 65, 211–222. [Google Scholar]
- Calder, P.C. n-3 polyunsaturated fatty acids, inflammation, and inflammatory diseases. Am. J. Clin. Nutr. 2006, 83, 1505S–1519S. [Google Scholar] [CrossRef]
- Malinska, H.; Huttl, M.; Oliyarnyk, O.; Bratova, M.; Kazdova, L. Conjugated linoleic acid reduces visceral and ectopic lipid accumulation and insulin resistance in chronic severe hypertriacylglycerolemia. Nutrition 2015, 31, 1045–1051. [Google Scholar] [CrossRef] [PubMed]
- Eder, K. Gas chromatographic analysis of fatty acid methyl esters. J. Chromatogr. B Biomed. Appl. 1995, 671, 113–131. [Google Scholar] [CrossRef]
- Pelikanova, T.; Kazdova, L.; Chvojkova, S.; Base, J. Serum phospholipid fatty acid composition and insulin action in type 2 diabetic patients. Metabolism 2001, 50, 1472–1478. [Google Scholar]
- Kahleova, H.; Pelikanova, T. Vegetarian Diets in the Prevention and Treatment of Type 2 Diabetes. J. Am. Coll. Nutr. 2015, 34, 448–458. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| WWITHOUT | WWITH METFORMIN | HHTgWITHOUT | HHTgWITH METFORMIN | PSTRAIN | PTREATMENT | PINTERACTION | |
|---|---|---|---|---|---|---|---|
| Body weight (g) | 428 ± 12 | 432 ± 10 | 411 ± 9 | 395 ± 6 | <0.01 | n.s. | n.s. |
| LV weight (g/100 g b.w.) | 0.131 ± 0.006 | 0.135 ± 0.005 | 0.134 ± 0.003 | 0.143 ± 0.004 | n.s. | n.s. | n.s. |
| EAT weight (g/100 g b.w.) | 1.235 ± 0.034 | 1.196 ± 0.061 | 1.839 ± 0.038 | 1.536 ± 0.048 *** | <0.001 | <0.001 | <0.01 |
| Non-fasting glucose (mmol/L) | 6.6 ± 0.2 | 6.6 ± 0.1 | 8.6 ± 0.2 | 8.1 ± 0.2 * | <0.001 | n.s. | n.s. |
| AUC0-180min (mmol/L) | 1268 ± 22 | 1176 ± 13 ** | 1476 ± 18 | 1417 ± 13 * | <0.001 | <0.001 | n.s. |
| HOMA-IR | 2.1 ± 0.1 | 1.8 ± 0.1 * | 2.7 ± 0.1 | 2.3 ± 0.1 ** | <0.001 | <0.001 | n.s. |
| Insulin (nmol/L) | 0.279 ± 0.022 | 0.240 ± 0.036 | 0.273 ± 0.028 | 0.238 ± 0.041 | n.s. | n.s. | n.s. |
| Glucagon (pg/mL) | 224.61 ± 14.82 | 212.51 ± 12.33 | 191.22 ± 13.38 | 158.84 ± 4.94 | <0.01 | n.s. | n.s. |
| Serum TG (mmol/L) | 1.56 ± 0.11 | 1.03 ± 0.08 * | 3.45 ± 0.26 | 2.65 ± 0.20 ** | <0.001 | <0.001 | n.s. |
| Serum cholesterol (mmol/L) | 1.51 ± 0.07 | 1.37 ± 0.06 | 1.77 ± 0.03 | 1.84 ± 0.03 | <0.001 | n.s. | <0.05 |
| HDL-C (mmol/L) | 1.27 ± 0.08 | 1.15 ± 0.06 | 0.88 ± 0.03 | 0.91 ± 0.03 | <0.001 | n.s. | n.s. |
| NEFA (mmol/L) | 0.496 ± 0.020 | 0.449 ± 0.022 | 0.713 ± 0.015 | 0.646 ± 0.017 * | <0.001 | <0.01 | n.s. |
| WWITHOUT | WWITH METFORMIN | HHTgWITHOUT | HHTgWITH METFORMIN | PSTRAIN | PTREATMENT | PINTERACTION | |
|---|---|---|---|---|---|---|---|
| TNFα (pg/mL) | 2.280 ± 0.259 | 2.738 ± 0.435 | 11.488 ± 0.697 | 8.709 ± 0.280 *** | <0.001 | <0.05 | <0.01 |
| MCP-1 (ng/mL) | 4.794 ± 0.242 | 4.873 ± 0.236 | 10.552 ± 1.492 | 6.744 ± 0.525 ** | <0.001 | <0.05 | <0.05 |
| hsCRP (mg/mL) | 1.253 ± 0.069 | 1.457 ± 0.086 | 1.304 ± 0.049 | 1.329 ± 0.058 | n.s. | n.s. | n.s. |
| IL-6 (pg/mL) | 126.5 ± 12.5 | 120.9 ± 8.9 | 171.0 ± 8.3 | 144.0 ± 3.6 * | <0.001 | n.s. | n.s. |
| Leptin (ng/mL) | 5.51 ± 0.64 | 5.45 ± 0.55 | 7.51 ± 0.22 | 6.59 ± 0.16 | <0.01 | n.s. | n.s. |
| HMW Adiponectin (μg/mL) | 1.015 ± 0.034 | 1.358 ± 0.068 *** | 0.893 ± 0.030 | 1.245 ± 0.072 *** | <0.05 | <0.001 | n.s. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Miklankova, D.; Markova, I.; Hüttl, M.; Zapletalova, I.; Poruba, M.; Malinska, H. Metformin Affects Cardiac Arachidonic Acid Metabolism and Cardiac Lipid Metabolite Storage in a Prediabetic Rat Model. Int. J. Mol. Sci. 2021, 22, 7680. https://doi.org/10.3390/ijms22147680
Miklankova D, Markova I, Hüttl M, Zapletalova I, Poruba M, Malinska H. Metformin Affects Cardiac Arachidonic Acid Metabolism and Cardiac Lipid Metabolite Storage in a Prediabetic Rat Model. International Journal of Molecular Sciences. 2021; 22(14):7680. https://doi.org/10.3390/ijms22147680
Chicago/Turabian StyleMiklankova, Denisa, Irena Markova, Martina Hüttl, Iveta Zapletalova, Martin Poruba, and Hana Malinska. 2021. "Metformin Affects Cardiac Arachidonic Acid Metabolism and Cardiac Lipid Metabolite Storage in a Prediabetic Rat Model" International Journal of Molecular Sciences 22, no. 14: 7680. https://doi.org/10.3390/ijms22147680
APA StyleMiklankova, D., Markova, I., Hüttl, M., Zapletalova, I., Poruba, M., & Malinska, H. (2021). Metformin Affects Cardiac Arachidonic Acid Metabolism and Cardiac Lipid Metabolite Storage in a Prediabetic Rat Model. International Journal of Molecular Sciences, 22(14), 7680. https://doi.org/10.3390/ijms22147680