
Cysteine Proteome Reveals Response to Endogenous Oxidative Stress in Bacillus cereus



Abstract
:1. Introduction
2. Results and Discussion
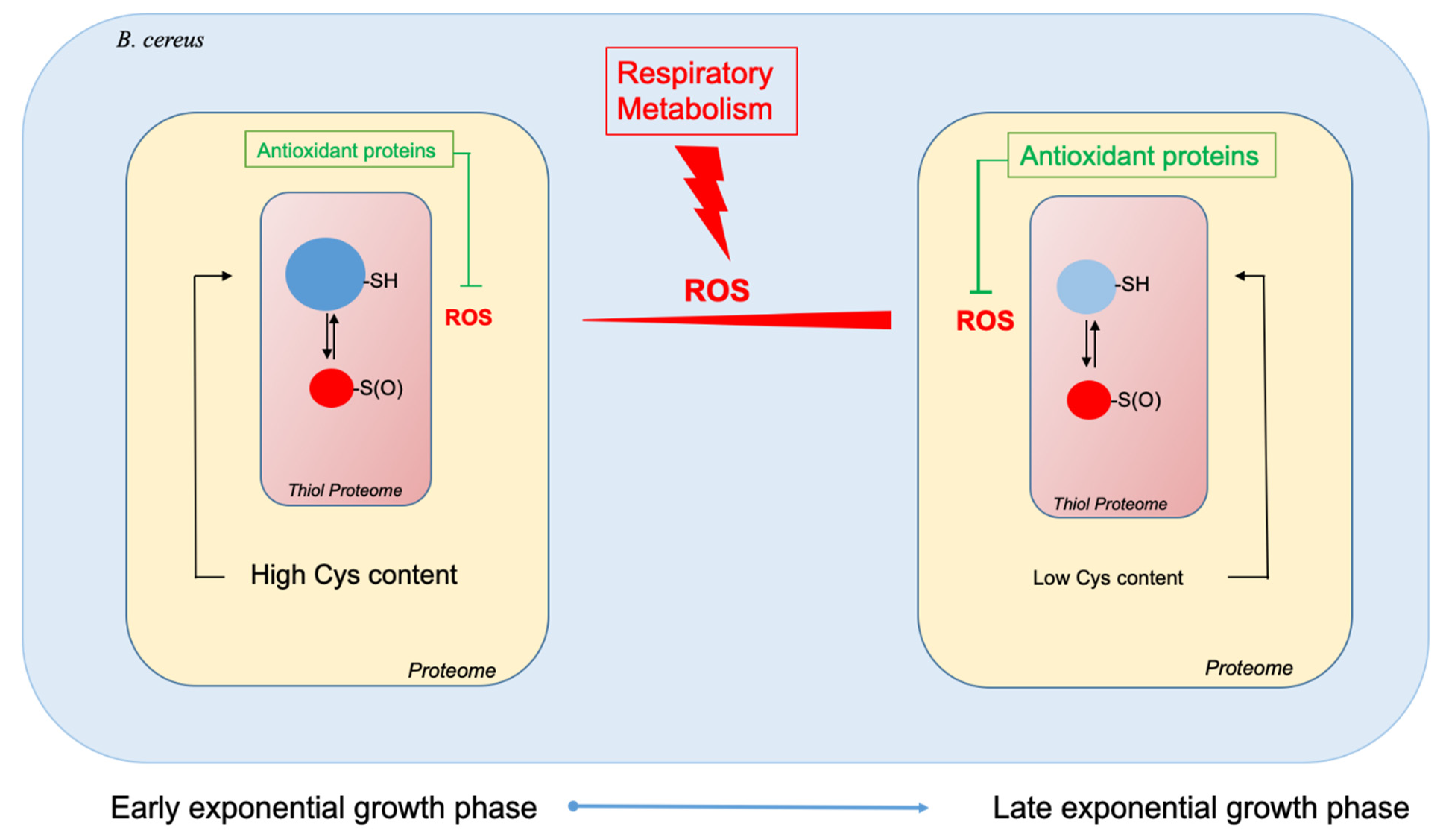
2.1. B. cereus Proteome Is Enriched in Antioxidant Proteins as Aerobic Growth Progresses
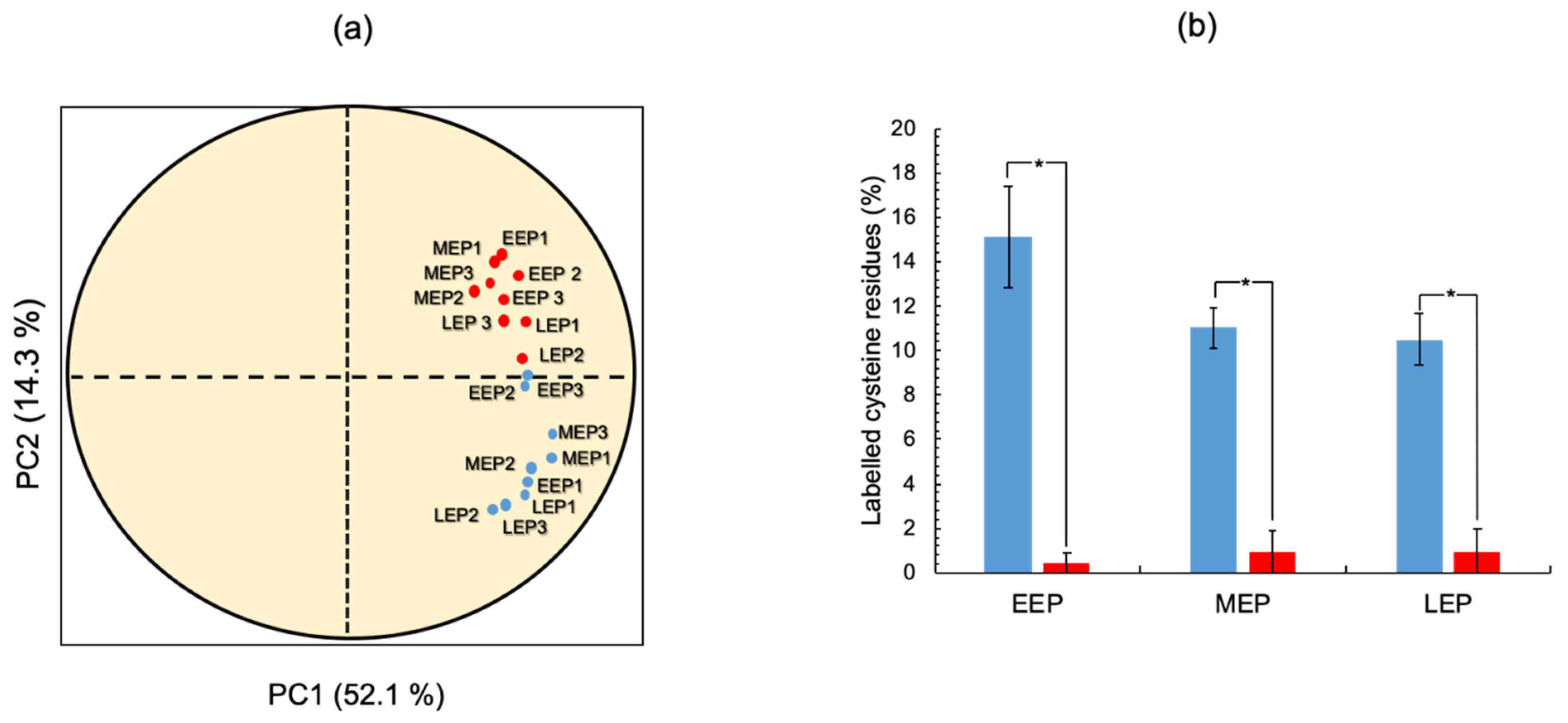
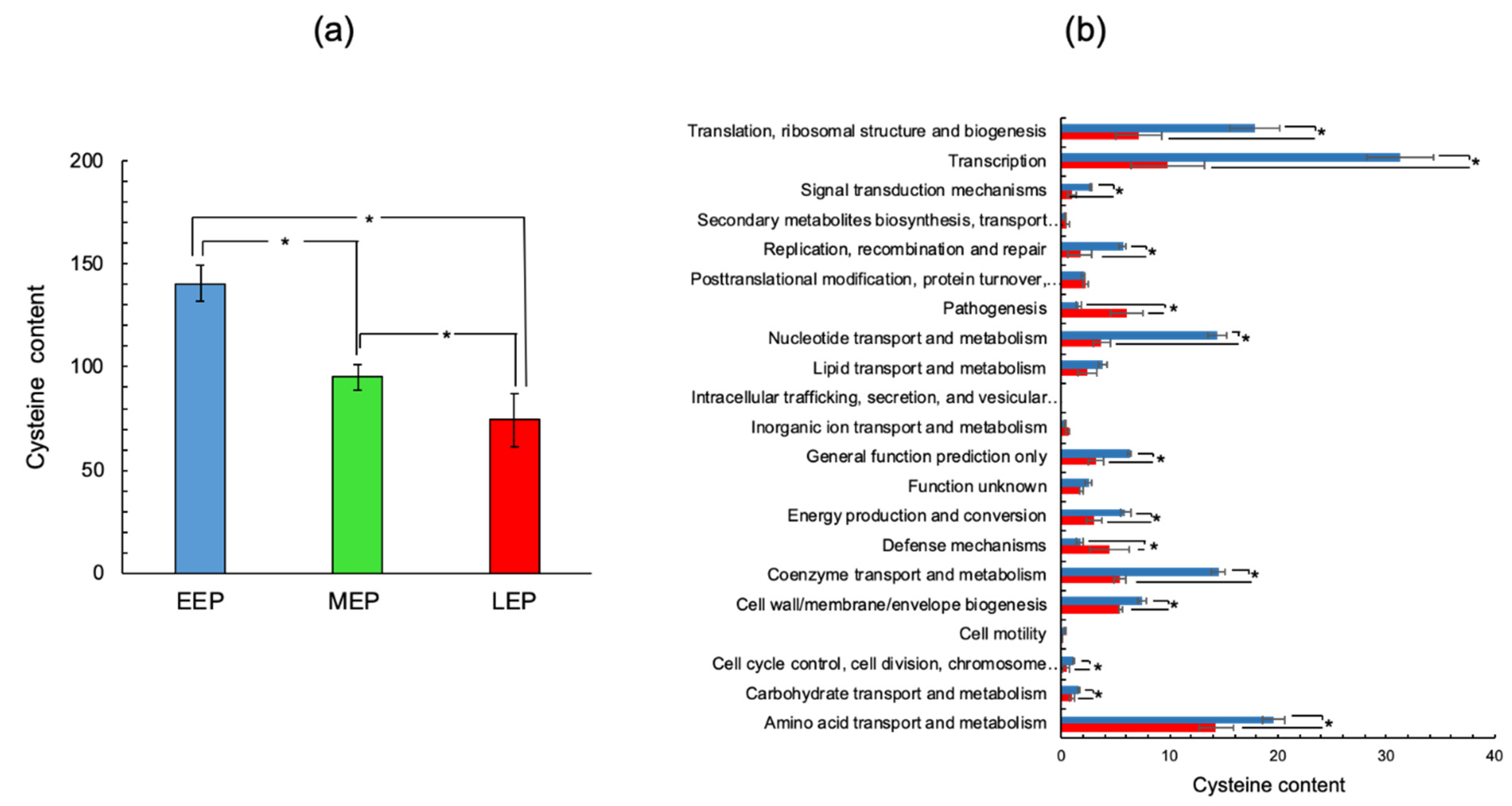
2.2. The Cysteine Content of the B. cereus Proteome Decreases as Growth Progresses
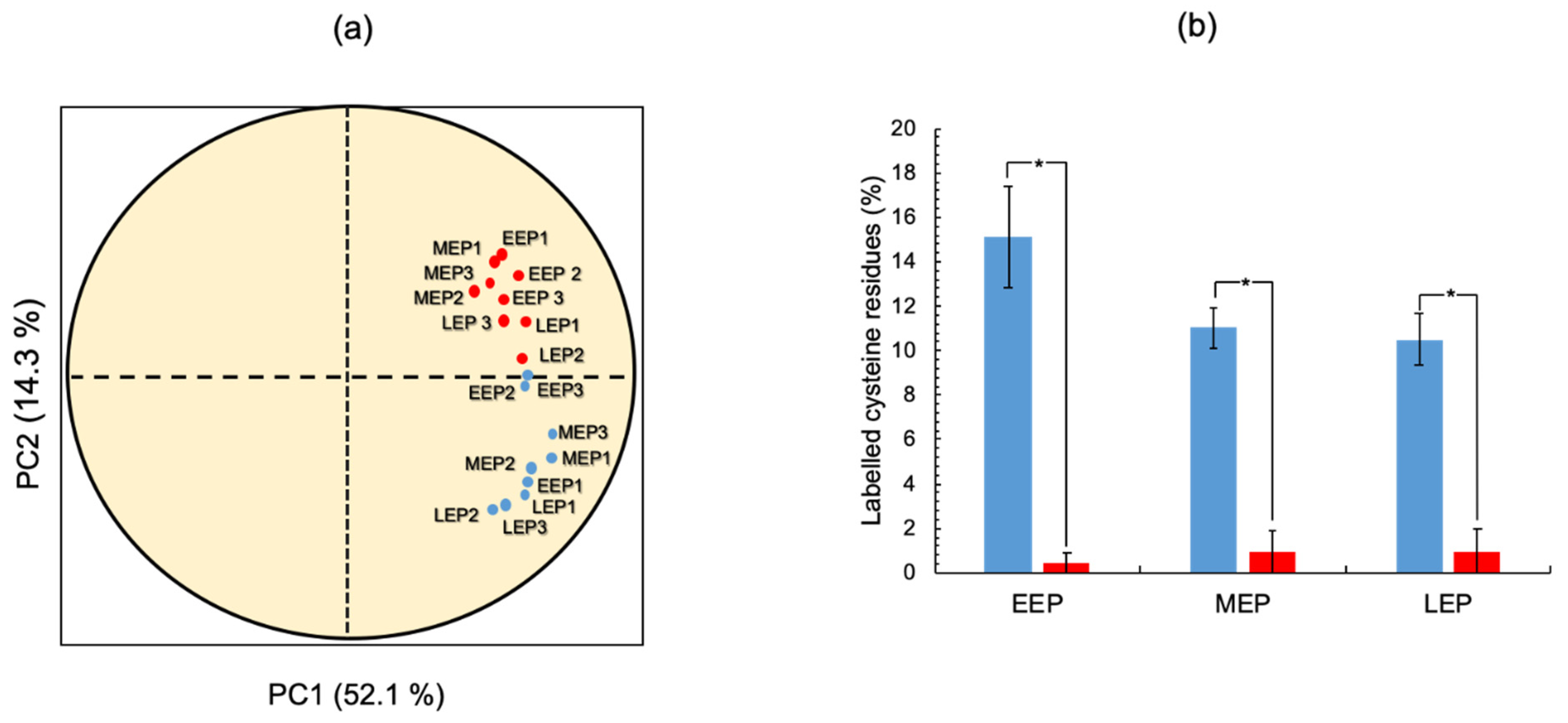
2.3. The Thiol Reduction Status of the B. cereus Proteome Decreases as Aerobic Growth Progresses
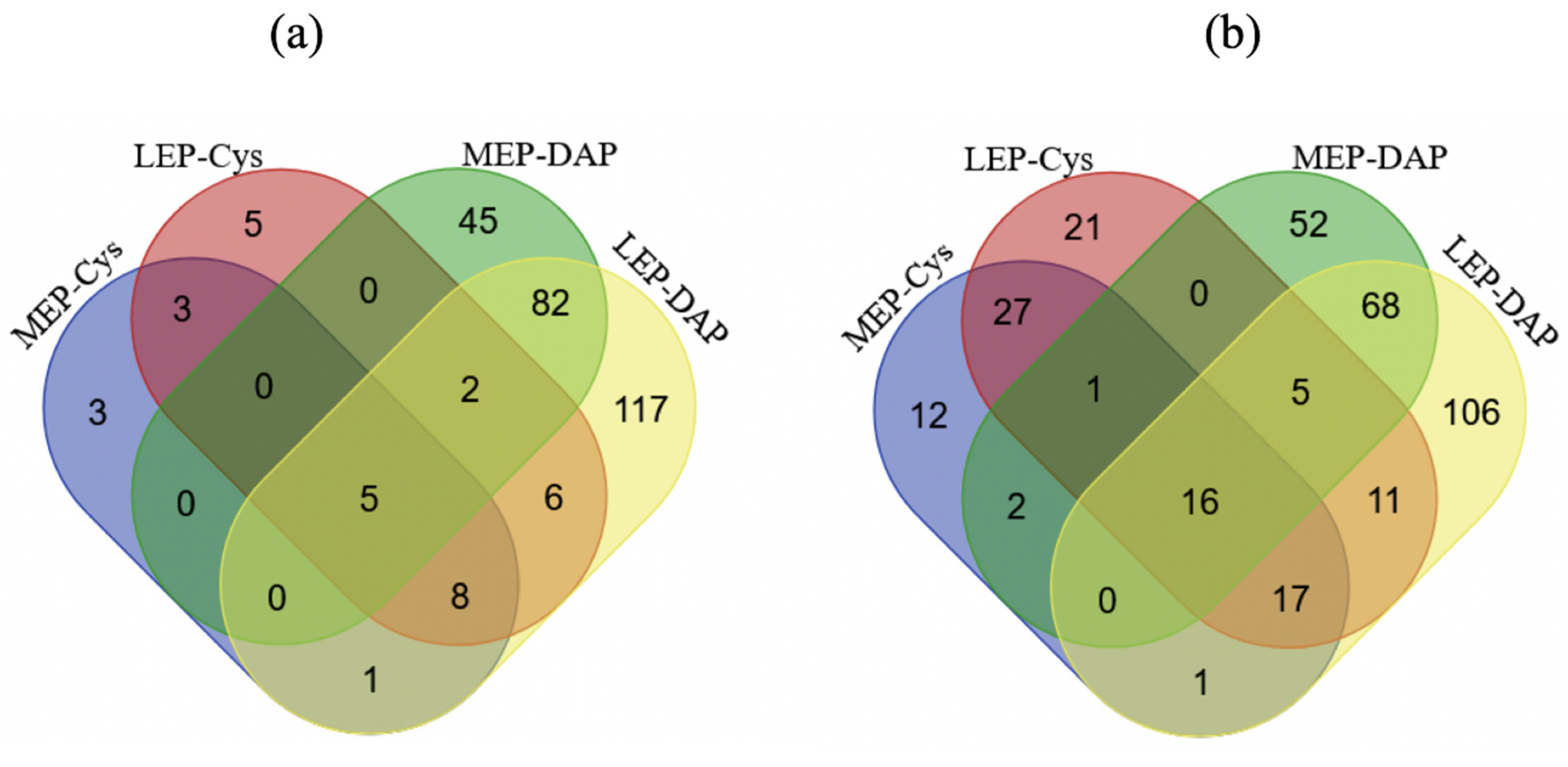
2.3.1. Temporal Dynamics of NEMCys-Labeled Proteins (NEMCys Proteins)
2.3.2. Temporal Dynamics of IAMCys-Labeled Proteins (IAMCys Proteins)
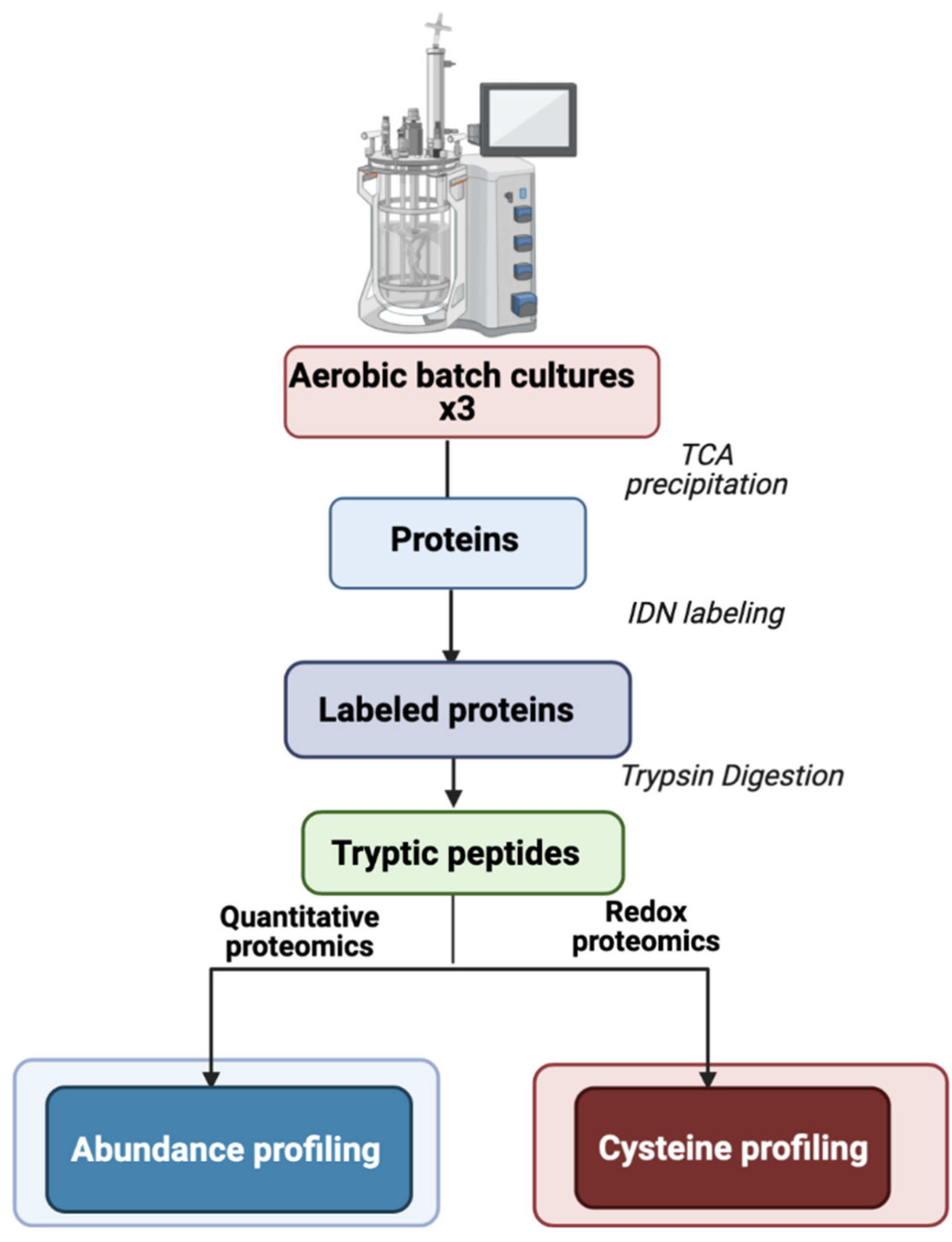
3. Materials and Methods
3.1. B. cereus Growth Conditions
3.2. Protein Extraction for Proteome Analysis
3.3. Protein Identification and Quantification
3.4. Bioinformatics Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Halliwell, B. Reactive Species and Antioxidants. Redox Biology Is a Fundamental Theme of Aerobic Life. Plant Physiol. 2006, 141, 312–322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bueno, E.; Mesa, S.; Bedmar, E.J.; Richardson, D.J.; Delgado, M.J. Bacterial adaptation of respiration from oxic to microoxic and anoxic conditions: Redox control. Antioxid. Redox Signal. 2012, 16, 819–852. [Google Scholar] [CrossRef]
- Lemire, J.; AlHasawi, A.; Appanna, V.; Tharmalingam, S. Metabolic defence against oxidative stress: The road less travelled so far. J. Appl. Microbiol. 2017, 123, 798–809. [Google Scholar] [CrossRef] [Green Version]
- Imlay, J.A. Pathways of Oxidative Damage. Annu. Rev. Microbiol. 2003, 57, 395–418. [Google Scholar] [CrossRef]
- Baatout, S.; De Boever, P.; Mergeay, M. Physiological changes induced in four bacterial strains following oxidative stress. Appl. Biochem. Microbiol. 2006, 42, 369–377. [Google Scholar] [CrossRef]
- Pamplona, R.; Costantini, D. Molecular and structural antioxidant defenses against oxidative stress in animals. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2011, 301, R843–R863. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seaver, L.C.; Imlay, J.A. Alkyl Hydroperoxide Reductase Is the Primary Scavenger of Endogenous Hydrogen Peroxide in Escherichia coli. J. Bacteriol. 2001, 183, 7173–7181. [Google Scholar] [CrossRef] [Green Version]
- Van Laer, K.; Hamilton, C.J.; Messens, J. Low-molecular-weight thiols in thiol–disulfide exchange. Antioxid. Redox Signal. 2013, 18, 1642–1653. [Google Scholar] [CrossRef] [PubMed]
- Messens, J.; Rouhier, N.; Collet, J.-F. Redox Homeostasis. In Oxidative Stress and Redox Regulation; Springer Science and Business Media LLC: Berlin, Germany, 2013; pp. 59–84. [Google Scholar]
- Zhang, J.; Wang, X.; Vikash, V.; Ye, Q.; Wu, D.; Liu, Y.; Dong, W. ROS and ROS-Mediated Cellular Signaling. Oxidative Med. Cell. Longev. 2016, 2016, 4350965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bae, Y.S.; Oh, H.; Rhee, S.G.; Yoo, Y.D. Regulation of reactive oxygen species generation in cell signaling. Mol. Cells 2011, 32, 491–509. [Google Scholar] [CrossRef] [Green Version]
- Rhee, S.G. H2O2, A necessary evil for cell signaling. Science 2006, 312, 1882–1883. [Google Scholar] [CrossRef]
- Poole, L.B. The basics of thiols and cysteines in redox biology and chemistry. Free. Radic. Biol. Med. 2015, 80, 148–157. [Google Scholar] [CrossRef] [Green Version]
- Cross, J.V.; Templeton, D.J. Regulation of Signal Transduction Through Protein Cysteine Oxidation. Antioxid. Redox Signal. 2006, 8, 1819–1827. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Ha, S.; Lee, H.Y.; Lee, K.-J. ROSics: Chemistry and proteomics of cysteine modifications in redox biology. Mass Spectrom. Rev. 2015, 34, 184–208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiappetta, G.; Ndiaye, S.; Igbaria, A.; Kumar, C.; Vinh, J.; Toledano, M.B. Proteome Screens for Cys Residues Oxidation: The Redoxome. Methods. Enzymol. 2010, 473, 199–216. [Google Scholar] [PubMed]
- Duport, C.; Jobin, M.; Schmitt, P. Adaptation in Bacillus cereus: From Stress to Disease. Front. Microbiol. 2016, 7, 1550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mols, M.; Abee, T. Primary and secondary oxidative stress inBacillus. Environ. Microbiol. 2011, 13, 1387–1394. [Google Scholar] [CrossRef]
- Madeira, J.P.; Alpha-Bazin, B.; Armengaud, J.; Duport, C. Time dynamics of the Bacillus cereus exoproteome are shaped by cellular oxidation. Front. Microbiol. 2015, 6, 342. [Google Scholar] [CrossRef] [PubMed]
- Zigha, A.; Rosenfeld, E.; Schmitt, P.; Duport, C. The redox regulator Fnr is required for fermentative growth and enterotoxin synthesis in Bacillus cereus F4430/73. J. Bacteriol. 2007, 189, 2813–2824. [Google Scholar] [CrossRef] [Green Version]
- Clair, G.; Armengaud, J.; Duport, C. Restricting fermentative potential by proteome remodeling: An adaptive strategy evidenced in Bacillus cereus. Mol. Cell. Proteomics 2012, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duport, C.; Zigha, A.; Rosenfeld, E.; Schmitt, P. Control of enterotoxin gene expression in Bacillus cereus F4430/73 involves the redox-sensitive ResDE signal transduction system. J. Bacteriol. 2006, 188, 6640–6651. [Google Scholar] [CrossRef] [Green Version]
- Wojdyla, K.; Rogowska-Wrzesinska, A. Differential alkylation-based redox proteomics—Lessons learnt. Redox Biol. 2015, 6, 240–252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wholey, W.Y.; Jakob, U. Hsp33 confers bleach resistance by protecting elongation factor Tu against oxidative degradation in Vibrio cholerae. Mol. Microbiol. 2012, 83, 981–991. [Google Scholar] [CrossRef] [Green Version]
- Hamitouche, F.; Gaillard, J.-C.; Schmitt, P.; Armengaud, J.; Duport, C.; Dedieu, L. Redox proteomic study of Bacillus cereus thiol proteome during fermentative anaerobic growth. BMC Genom. 2021, in press. [Google Scholar]
- Eymann, C.; Dreisbach, A.; Albrecht, D.; Bernhardt, J.; Becher, D.; Gentner, S.; Thi Tam, L.; Buttner, K.; Buurman, G.; Scharf, C.; et al. A comprehensive proteome map of growing Bacillus subtilis cells. Proteomics 2004, 4, 2849–2876. [Google Scholar] [CrossRef]
- Folio, P.; Chavant, P.; Chafsey, I.; Belkorchia, A.; Chambon, C.; Hébraud, M. Two-dimensional electrophoresis database of Listeria monocytogenes EGDe proteome and proteomic analysis of midlog and stationary growth phase cells. Proteomics 2004, 4, 3187–3201. [Google Scholar] [CrossRef] [PubMed]
- Ceuppens, S.; Rajkovic, A.; Heyndrickx, M.; Tsilia, V.; Van De Wiele, T.; Boon, N.; Uyttendaele, M. Regulation of toxin production by Bacillus cereus and its food safety implications. Crit. Rev. Microbiol. 2011, 37, 188–213. [Google Scholar] [CrossRef]
- Jaishankar, J.; Srivastava, P. Molecular Basis of Stationary Phase Survival and Applications. Front. Microbiol. 2017, 8, 2000. [Google Scholar] [CrossRef] [Green Version]
- Xu, K.; Liu, G.; Fu, C. The Tryptophan Pathway Targeting Antioxidant Capacity in the Placenta. Oxidative Med. Cell. Longev. 2018, 2018, 1054797. [Google Scholar] [CrossRef]
- Nyström, T. Stationary-Phase Physiology. Annu. Rev. Microbiol. 2004, 58, 161–181. [Google Scholar] [CrossRef] [PubMed]
- Cohen, D.P.A.; Renes, J.; Bouwman, F.G.; Zoetendal, E.G.; Mariman, E.; De Vos, W.M.; Vaughan, E.E. Proteomic analysis of log to stationary growth phaseLactobacillus plantarum cells and a 2-DE database. Proteomic 2006, 6, 6485–6493. [Google Scholar] [CrossRef]
- Mols, M.; Abee, T. Bacillus cereus responses to acid stress. Environ. Microbiol. 2011, 13, 2835–2843. [Google Scholar] [CrossRef] [PubMed]
- Anastasiou, D.; Poulogiannis, G.; Asara, J.M.; Boxer, M.B.; Jiang, J.-k.; Shen, M.; Bellinger, G.; Sasaki, A.T.; Locasale, J.W.; Auld, D.S.; et al. Inhibition of pyruvate kinase M2 by reactive oxygen species contributes to cellular antioxidant responses. Science 2011, 334, 1278–1283. [Google Scholar] [CrossRef] [Green Version]
- Clair, G.; Roussi, S.; Armengaud, J.; Duport, C. Expanding the known repertoire of virulence factors pro-duced by Bacillus cereus through early secretome profiling in three redox conditions. Mol. Cell. Proteom. 2010, 9, 1486–1498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daniels, R.; Mellroth, P.; Bernsel, A.; Neiers, F.; Normark, S.; von Heijne, G.; Henriques-Normark, B. Disulfide Bond Formation and Cysteine Exclusion in Gram-positive Bacteria*. J. Biol. Chem. 2010, 285, 3300–3309. [Google Scholar] [CrossRef] [Green Version]
- Billington, S.J.; Jost, B.H.; Songer, J.G. Thiol-activated cytolysins: Structure, function and role in pathogene-sis. FEMS Microbiol. Lett. 2000, 182, 197–205. [Google Scholar] [CrossRef]
- Groitl, B.; Jakob, U. Thiol-based redox switches. Biochim. Biophys. Acta (BBA) Proteins Proteom. 2014, 1844, 1335–1343. [Google Scholar] [CrossRef] [Green Version]
- Van der Reest, J.; Lilla, S.; Zheng, L.; Zanivan, S.; Gottlieb, E. Proteome-wide analysis of cysteine oxidation reveals metabolic sensitivity to redox stress. Nat. Commun. 2018, 9, 1581. [Google Scholar] [CrossRef] [PubMed]
- Peifer, S.; Barduhn, T.; Zimmet, S.; Volmer, D.A.; Heinzle, E.; Schneider, K. Metabolic engineering of the pu-rine biosynthetic pathway in Corynebacterium glutamicum results in increased intracellular pool sizes of IMP and hypoxanthine. Microb. Cell Factories 2012, 11, 138. [Google Scholar] [CrossRef] [Green Version]
- Hochgräfe, F.; Mostertz, J.; Pöther, D.-C.; Becher, D.; Helmann, J.; Hecker, M. S-Cysteinylation Is a General Mechanism for Thiol Protection of Bacillus subtilis Proteins after Oxidative Stress. J. Biol. Chem. 2007, 282, 25981–25985. [Google Scholar] [CrossRef] [Green Version]
- Leichert, L.; Jakob, U. Protein Thiol Modifications Visualized In Vivo. PLoS Biol. 2004, 2, e333. [Google Scholar] [CrossRef]
- Toledano, M.B.; Kumar, C.; Le Moan, N.; Spector, D.; Tacnet, F. The system biology of thiol redox system in Escherichia coli and yeast: Differential functions in oxidative stress, iron metabolism and DNA synthesis. FEBS Lett. 2007, 581, 3598–3607. [Google Scholar] [CrossRef] [Green Version]
- Hwang, Y.-W.; Sanchez, A.; Hwang, M.-C.C.; Miller, D.L. The role of cysteinyl residues in the activity of bac-terial elongation factor Ts, a guanosine nucleotide dissociation protein. Arch. Biochem. Biophys. 1997, 348, 157–162. [Google Scholar] [CrossRef]
- Topf, U.; Suppanz, I.; Samluk, L.; Wrobel, L.; Böser, A.; Sakowska, P.; Knapp, B.; Pietrzyk, M.; Chacinska, A.; Warscheid, B. Quantitative proteomics identifies redox switches for global translation modulation by mitochondrially produced reactive oxygen species. Nat. Commun. 2018, 9, 324. [Google Scholar] [CrossRef]
- Jones-Carson, J.; Yahashiri, A.; Kim, J.-S.; Liu, L.; Fitzsimmons, L.F.; Weiss, D.S.; Vázquez-Torres, A. Nitric oxide disrupts bacterial cytokinesis by poisoning purine metabolism. Sci. Adv. 2020, 6, eaaz0260. [Google Scholar] [CrossRef] [Green Version]
- Rigoulet, M.; Bouchez, C.; Paumard, P.; Ransac, S.; Cuvellier, S.; Duvezin-Caubet, S.; Mazat, J.P.; Devin, A. Cell energy metabolism: An update. Biochim. Biophys. Acta (BBA) Bioenerg. 2020, 1861, 148276. [Google Scholar] [CrossRef] [PubMed]
- Quijano, C.; Trujillo, M.; Castro, L.; Trostchansky, A. Interplay between oxidant species and energy metabolism. Redox Biol. 2016, 8, 28–42. [Google Scholar] [CrossRef] [Green Version]
- Danson, M.J. Dihydrolipoamide dehydrogenase: A ‘new’function for an old enzyme? Biochem. Soc. Trans. 1988, 16, 87–89. [Google Scholar] [CrossRef] [Green Version]
- Vettakkorumakankav, N.N.; Patel, M.S. Dihydrolipoamide dehydrogenase: Structural and mechanistic aspects. Indian J. Biochem. Biophys. 1996, 33, 168–176. [Google Scholar] [PubMed]
- Gutierrez-Correa, J.; Stoppani, A. Inactivation of heart dihydrolipoamide dehydrogenase by copper Fen-ton systems. Effect of thiol compounds and metal chelators. Free Radic. Res. 1995, 22, 239–250. [Google Scholar] [CrossRef] [PubMed]
- Yan, L.-J.; Sumien, N.; Thangthaeng, N.; Forster, M.J. Reversible inactivation of dihydrolipoamide dehydro-genase by mitochondrial hydrogen peroxide. Free Radic. Res. 2013, 47, 123–133. [Google Scholar] [CrossRef] [Green Version]
- Gazaryan, I.G.; Krasnikov, B.F.; Ashby, G.A.; Thorneley, R.N.; Kristal, B.S.; Brown, A.M. Zinc is a potent inhibi-tor of thiol oxidoreductase activity and stimulates reactive oxygen species production by lipoamide de-hydrogenase. J. Biol. Chem. 2002, 277, 10064–10072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, X.; Song, J.; Yan, L.-J. Chronic inhibition of mitochondrial dihydrolipoamide dehydrogenase (DLDH) as an approach to managing diabetic oxidative stress. Antioxidants 2019, 8, 32. [Google Scholar] [CrossRef] [Green Version]
- Grünenfelder, B.; Rummel, G.; Vohradsky, J.; Röder, D.; Langen, H.; Jenal, U. Proteomic analysis of the bac-terial cell cycle. Proc. Natl. Acad. Sci. USA 2001, 98, 4681–4686. [Google Scholar] [CrossRef] [Green Version]
- Varatnitskaya, M.; Degrossoli, A.; Leichert, L.I. Redox regulation in host-pathogen interactions: Thiol switches and beyond. Biol. Chem. 2021, 402, 299–316. [Google Scholar] [CrossRef]
- Dahl, J.-U.; Gray, M.; Jakob, U. Protein Quality Control under Oxidative Stress Conditions. J. Mol. Biol. 2015, 427, 1549–1563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winter, J.; Linke, K.; Jatzek, A.; Jakob, U. Severe oxidative stress causes inactivation of DnaK and activa-tion of the redox-regulated chaperone Hsp33. Mol. Cell 2005, 17, 381–392. [Google Scholar] [CrossRef]
- Imlay, J.A. Transcription Factors That Defend Bacteria Against Reactive Oxygen Species. Annu. Rev. Microbiol. 2015, 69, 93–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perego, M.; Spiegelman, G.B.; Hoch, J.A. Structure of the gene for the transition state regulator, abrB: Regulator synthesis is controlled by the spo0A sporulation gene in Bacillus subtilis. Mol. Microbiol. 1988, 2, 689–699. [Google Scholar] [CrossRef]
- Strauch, M.; Spiegelman, G.B.; Perego, M.; Johnson, W.C.; Burbulys, D.; Hoch, J. The transition state transcription regulator abrB of Bacillus subtilis is a DNA binding protein. EMBO J. 1989, 8, 1615–1621. [Google Scholar] [CrossRef]
- Dietrich, R.; Jessberger, N.; Ehling-Schulz, M.; Märtlbauer, E.; Granum, P.E. The Food Poisoning Toxins of Bacillus cereus. Toxins 2021, 13, 98. [Google Scholar] [CrossRef] [PubMed]
- Ehling-Schulz, M.; Frenzel, E.; Gohar, M. Food–bacteria interplay: Pathometabolism of emetic Bacillus ce-reus. Front. Microbiol. 2015, 6, 704. [Google Scholar] [CrossRef] [Green Version]
- Phillips, Z.E.; Strauch, M. Role of Cys54 in AbrB multimerization and DNA-binding activity. FEMS Microbiol. Lett. 2001, 203, 207–210. [Google Scholar] [CrossRef]
- Rosenfeld, E.; Duport, C.; Zigha, A.; Schmitt, P. Characterization of aerobic and anaerobic vegetative growth of the food-borne pathogen Bacillus cereus F4430/73 strain. Can. J. Microbiol. 2005, 51, 149–158. [Google Scholar] [CrossRef]
- Hartmann, E.M.; Allain, F.; Gaillard, J.-C.; Pible, O.; Armengaud, J. Taking the Shortcut for High-Throughput Shotgun Proteomic Analysis of Bacteria; Springer Science and Business Media LLC: Berlin, Germany, 2014; Volume 1197, pp. 275–285. [Google Scholar]
- Imlay, J.A. Where in the world do bacteria experience oxidative stress? Environ. Microbiol. 2019, 21, 521–530. [Google Scholar] [CrossRef]
- Perez-Gonzalez, A.; Muñoz-Rugeles, L.; Alvarez-Idaboy, J.R. Tryptophan: Antioxidant or target of oxidative stress? A quantum chemistry elucidation. RSC Adv. 2014, 4, 56128–56131. [Google Scholar] [CrossRef]
- Genestet, C.; Le Gouellec, A.; Chaker, H.; Polack, B.; Guery, B.; Toussaint, B.; Stasia, M.J. Scavenging of reactive oxygen species by tryptophan metabolites helps Pseudomonas aeruginosa escape neutrophil killing. Free. Radic. Biol. Med. 2014, 73, 400–410. [Google Scholar] [CrossRef]
- Xu, K.; Liu, H.; Bai, M.; Gao, J.; Wu, X.; Yin, Y. Redox Properties of Tryptophan Metabolism and the Concept of Tryptophan Use in Pregnancy. Int. J. Mol. Sci. 2017, 18, 1595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strauch, M.A.; Hoch, J.A. Transition-state regulators: Sentinels of Bacillus subtilis post-exponential gene expression. Mol. Microbiol. 1993, 7, 337–342. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene ID | Protein | IAM Labeled Cys Peptide | Log2(FC) * | |
|---|---|---|---|---|
| MEP | LEP | |||
| Proteins with increased levels of IAMCys residues | ||||
| Transcription | ||||
| BC4603 | Transcriptional regulator, GntR family | IVC113NLPK | 1.0 | |
| Translation, ribosomal structure, and biogenesis | ||||
| BC0155 | 50S ribosomal protein L36 (RpmJ) | VRPSVKPIC11EK | 2.2 | |
| GKVMVIC27ENPK | ||||
| Signal transduction mechanisms | ||||
| BC0442 | Tellurium resistance protein (TerD) | LSTC79GSIIHSGDNLTGEGAGDDETIFVELHK | 1.6 | |
| LVFVVNIYDC123VNR | ||||
| Secondary metabolite biosynthesis, transport, and catabolism | ||||
| BC2305 | Isochorismatase (DhbB) | C197AVTTSTNLLLK | 1.7 | |
| C67KELGIPVVYTAQPGGQTLEQR | ||||
| Coenzyme transport and metabolism | ||||
| BC4853 | 1,4-dihydroxy-2-naphthoyl-CoA synthase (MenB) | EIWYLC174R | 1.0 | 1.6 |
| BC1086 | Lipoate--protein ligase | AFC71SGGDQKVR | 1.6 | |
| INLAIEEYC274VK | ||||
| Post-translational modification, protein turnover, chaperones | ||||
| BC0517 | Thioredoxin-dependent thiol peroxidase | DMTPGC47TTEAC52DFR | 2.0 | |
| Amino acid transport and metabolism | ||||
| BC1182 | Oligopeptide transport ATP-binding protein (OppD) | VVIAMALAC172NPK | 1.7 | 1.2 |
| Function unknown | ||||
| BC5199 | Xaa-Pro dipeptidase | FIC25YISR | 1.6 | 1.1 |
| BC4341 | GTP pyrophosphokinase | ITC88C89FVEDIYHLK | ||
| BC3302 | Pentapeptide repeat containing protein | SC78NLEEIHIADC88R | 1.8 | 2.1 |
| ASFFDC68DFEFADFR | ||||
| Proteins with decreased levels of IAMCys residues | ||||
| Amino acid transport and metabolism | ||||
| BC1740 | Aspartate ammonia-lyase | AFTDNC393LK | −0.8 | |
| BC1546 | Aminotransferase | DQGIAYDPSEIIVC96NGAK | −1.3 | |
| BC0055 | D-alanyl-D-alanine carboxypeptidase (DacA) | TGSTPEAGDC169FTGTVER | −1.0 | |
| BC4981 | Cysteine desulfurase (SufS) | VDVQDLNC215DFYALSAHK | −1.3 | |
| MC226GPTGIGVLYGK | ||||
| AGHHC361AQPLMK | ||||
| BC3798 | Aspartokinase (DapG) | HLQTVTYNEIC206NMAYQGAK | −2.6 | −2.1 |
| LLQNLGYEPIVTEHC339AK | ||||
| BC4936 | Diaminopimelate epimerase (DapF) | GPAEVIC278R | −1.0 | |
| BC0747 | Glycine oxidase (ThiO) | IENNKVTGVITSEGIVTC201EK | −2.1 | −1.7 |
| BC3799 | Aspartate-semialdehyde dehydrogenase (AsdB) | KIMHMPELEVAATC244VR | −1.3 | −1.8 |
| Energy production and conversion | ||||
| BC3970 | Dihydrolipoyl dehydrogenase | GIIEIDEQC303R | −1.7 | |
| ANLGGVC47LNVGC52IPSK | ||||
| VAVEAISGHASAIDYIGIPAVC353FTDPELASVGYTK | ||||
| BC4157 | Dihydrolipoamide acetyltransferase component of pyruvate dehydrogenase complex | VLDGLIC417GK | −1.1 | |
| DMVNLC404LSLDHR | ||||
| BC2826 | Probable manganese-dependent inorganic pyrophosphatase (PpaC) | NPDTDAIC18SAIAYAELKK | −0.9 | |
| SPTC158TEQDVAAAR | ||||
| C113EPVGC118TATILNK | ||||
| BC4980 | IscU protein | C127ATLAWK | −1.4 | |
| NHGVLEDSVTVNLNNPTC40GDR | ||||
| BC3773 | Pyruvate synthase | AAANVGLNPDELAVISGIGC50SGR | −2.4 | |
| NSVKPNWCPGC16GDFSVQAAIQR | ||||
| BC3616 | Aconitate hydratase | VVEEYC346K | −1.0 | |
| BC3833 | Succinate--CoA ligase subunit alpha (SucD) | LLGPNC124PGVITPDEC133K | −1.2 | |
| IKTMEAC269GIK | ||||
| GLFETC297K | ||||
| BC5387 | Phosphate acetyltransferase | GC306NEEEVYK | −1.7 | −1.9 |
| EEEKYVFADC167AINIAPNSQDLAEIGIESAK | ||||
| BC4517 | Succinate dehydrogenase flavoprotein subunit | EIFDVC295VEQKAMC185EAAPGIIHLMDR | −1.4 | −1.8 |
| AVVDDEGVC170R | ||||
| BC4604 | Malate dehydrogenase | LMEPTFGGVNLEDIAAPNC141FIIEER | −2.1 | −2.5 |
| DLSLAYSPGVAEPC45KEIYDDKSK | ||||
| DIIMC215DR | ||||
| BC4158 | 2-oxoisovalerate dehydrogenase beta subunit | SNNDWTC111PVTIR | −1.1 | −1.1 |
| Carbohydrate transport and metabolism | ||||
| BC4599 | Pyruvate kinase (Pyk) | IVC8TIGPASESIEKLEQLIEAGMNVAR | −2.8 | |
| C265NVLGKPVITATQMLDSMQR | ||||
| BC5135 | Enolase (Eno B) | TGLTSAEMVDFYEELC279K | −2.3 | |
| BC5140 | Glyceraldehyde-3-phosphate dehydrogenase (Gap1) | GILGYSEEPLVSIDYNGC145TASSTIDALSTMVMEGNMVK | −1.6 | −1.5 |
| BC5335 | Fructose-bisphosphate aldolase (Fba) | NVSVEAELGTVGGQEDDVIAEGVIYADPAEC161K | −1.2 | −1.3 |
| C92KEAIDAGFTSVMIDASHHPFEENVETTK | ||||
| HLVEATGIDC172LAPALGSVHGPYK | ||||
| BC4571 | Deblocking aminopeptidase | IGC189AIAIDVLK | −1.1 | |
| BC5318 | Ribose 5-phosphate isomerase (RpiB) | GILVC66GTGIGMSIAANK | −2.0 | −2.1 |
| C84ALVHDTFSAK | ||||
| BC4600 | ATP-dependent 6-phosphofructokinase (PfkA) | C283VGIQDNK | −2.0 | −1.4 |
| Coenzyme transport and metabolism | ||||
| BC5413 | Phosphomethylpyrimidine kinase (ThiD) | GADEALHPETNDC126LR | −1.3 | |
| BC0621 | Putative pyridoxal phosphate-dependent acyltransferase | SRPFLFSTALTPADAAAC284MR | −1.1 | |
| BC4468 | Glutamate-1-semialdehyde 2,1-aminomutase 2 (HemL2) | VAYNC251GQGYYGVTPDLTC265LGK | −1.6 | |
| BC4111 | Riboflavin biosynthesis protein (RibBA) | GLVC56VPITEGYAER | −1.8 | −1.6 |
| VPDLIEC179AK | ||||
| Uncategorized | ||||
| BC3977 | Ribonuclease J (RnjB) | VVILC297TGSQGEPMAALSR | −2.5 | |
| MAEIGKDGVLC191LLSDSTNSEVPNFTMSER | ||||
| BC4425 | Hypothetical transcriptional regulator | VIVC84QHKPAEVR | −0.9 | |
| EKLDAAC169EALDK | ||||
| BC3854 | Predicted kinase related to hydroxyacetone kinase | DTEIDGVAIQKDDFMC280IADGK | −2.3 | |
| YGYC244TEFMVK | ||||
| DUF3797 domain-containing protein | TLYYVQC20PVC23K | −0.9 | ||
| BC0049 | Protein (SspF) | LC133GATPVFVDVR | −1.3 | −1.0 |
| Nucleotide transport and metabolism | ||||
| BC4402 | Adenine phosphoribosyltransferase (Apt B) | GFIIGC64PVSYALEVGFAPVRK | −1.4 | |
| BC0296 | GMP synthase (GuaA) | VLC225ALSGGVDSSVVAVLIHK | −1.1 | |
| AIGDQLTC250IFVDHGLLR | ||||
| GIIFSGGPNSVYGEGALHC72DEK | ||||
| BC5315 | Uracil phosphoribosyltransferase (UppB) | LMC159IVAAPEGVK | −1.3 | |
| BC0331 | Phosphoribosylformylglycinamidine cyclo-ligase (PurM) | GISEGC88R | −1.1 | −1.2 |
| Translation, ribosomal structure, and biogenesis | ||||
| BC0153 | Methionine aminopeptidase (Map) | SLVAQC231EHTVVVTR | −1.9 | |
| LC121QAAVDAFWAAMK | ||||
| BC0144 | 30S ribosomal protein S14 type Z (RpsZ) | C27GRPHSVYR | −2.4 | |
| BC4391 | tRNA-specific 2-thiouridylase (MnmA) | KDSTGIC200FIGER | −1.4 | |
| ILC330DEPIR | ||||
| BC3824 | Elongation factor Ts (EF-Ts) | FFEEIC239LLDQAFVKNPDMK | −1.7 | −1.4 |
| EKTGAGMMDC22KK | ||||
| BC0153 | Methionine--tRNA ligase 2 (MetG2) | VIC506VTNLKPVK | −1.9 | |
| SWESLSTIGC617IPAGTK | ||||
| BC0352 | Aspartyl/glutamyl-tRNA (GatB) | SIIQYTGVSDC182K | −1.2 | −2.8 |
| C191DANISLRPVGQEK | ||||
| AAMALNC68EIATETK | ||||
| BC0108 | Glutamate-tRNA ligase (GltX2) | C108YMTEEELEAEREGQIAR | −1.7 | −1.6 |
| BC3923 | 50S ribosomal protein L32 (RpmF) | VC42KAC45GTYK | −1.5 | −1.9 |
| Cell wall/membrane/envelope biogenesis | ||||
| BC0054 | Bifunctional protein (GlmU) | EINTGTYC175FDNK | −1.4 | |
| BC4444 | Rod shape-determining protein (MreD) | TITVC240SEEITEALKENAAVIVQAAK | −3.2 | −3.3 |
| ILIC101C102PTNITSVEQK | ||||
| BC0257 | D-alanine-D-alanine ligase B (DdlB) | LGYPC180FVKPANLGSSVGINK | −1.1 | −1.7 |
| C239SVVGEIVPK | ||||
| Transcription | ||||
| BC0042 | Transcription state regulatory protein (AbrB) | YKPNMTC54QVTGEVSDGNLSLAEGK | −2.3 | |
| Lipid transport and metabolism | ||||
| BC2302 | 2,3-dihydro−2,3-dihydroxybenzoate dehydrogenase | C182NLVSPGSTETEMQR | −1.6 | |
| BC4276 | 4-hydroxy-3-methylbut-2-en-1-yl diphosphate synthase (IspG) | VAVLGC303AVNGPGEAR | −1.6 | |
| RLEEAGC54QVVR | ||||
| SFGLASNAATLISC268PTCGR | ||||
| BC0353 | DAGKc domain-containing protein | LEQAGYETSC38HATTGPGDATVAAR | 1.0 | |
| SIEEAADIIC116EGK | ||||
| Cell cycle control, cell division, chromosome partitioning | ||||
| BC4442 | Cell division inhibitor (MinD) | QDYDYILIDC120PAGIEQGFK | −1.2 | |
| C68RLPQALIK | ||||
| BC4446 | Cell shape-determining protein (MreB) | MPVLVAEDPLDC319VAIGTGK | −1.0 | −1.4 |
| KPYVMVC108VPSGITAVER | ||||
| Post-translational modification, protein turnover, chaperones | ||||
| BC4312 | Chaperone protein (DnaK) | SKIIGIDLGTTNSC15VAVMEGGEPK | −0.9 | |
| BC4311 | Chaperone protein (DnaJ) | HC192SGSGQVSVEQNTPFGR | −2.5 | −2.2 |
| ELNVEIPVEDPC146DTC149K | ||||
| Intracellular trafficking, secretion, and vesicular transport | ||||
| BC3845 | Signal recognition particle receptor (FtsY) | KVDVLLC208DTAGR | −1.1 | −1.1 |
| Secondary metabolite biosynthesis, transport, and catabolism | ||||
| BC1372 | D-alanine--D-alanyl carrier protein ligase (DltA) | SLPVGYC324K | −2.9 | |
| TFLFC269GEVLPNEVAR | ||||
| AC421SYVEGAVIVPIKK | ||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hamitouche, F.; Armengaud, J.; Dedieu, L.; Duport, C. Cysteine Proteome Reveals Response to Endogenous Oxidative Stress in Bacillus cereus. Int. J. Mol. Sci. 2021, 22, 7550. https://doi.org/10.3390/ijms22147550
Hamitouche F, Armengaud J, Dedieu L, Duport C. Cysteine Proteome Reveals Response to Endogenous Oxidative Stress in Bacillus cereus. International Journal of Molecular Sciences. 2021; 22(14):7550. https://doi.org/10.3390/ijms22147550
Chicago/Turabian StyleHamitouche, Fella, Jean Armengaud, Luc Dedieu, and Catherine Duport. 2021. "Cysteine Proteome Reveals Response to Endogenous Oxidative Stress in Bacillus cereus" International Journal of Molecular Sciences 22, no. 14: 7550. https://doi.org/10.3390/ijms22147550
APA StyleHamitouche, F., Armengaud, J., Dedieu, L., & Duport, C. (2021). Cysteine Proteome Reveals Response to Endogenous Oxidative Stress in Bacillus cereus. International Journal of Molecular Sciences, 22(14), 7550. https://doi.org/10.3390/ijms22147550