
Epithelial Cell Transformation and Senescence as Indicators of Genome Aging: Current Advances and Unanswered Questions

 , , ,
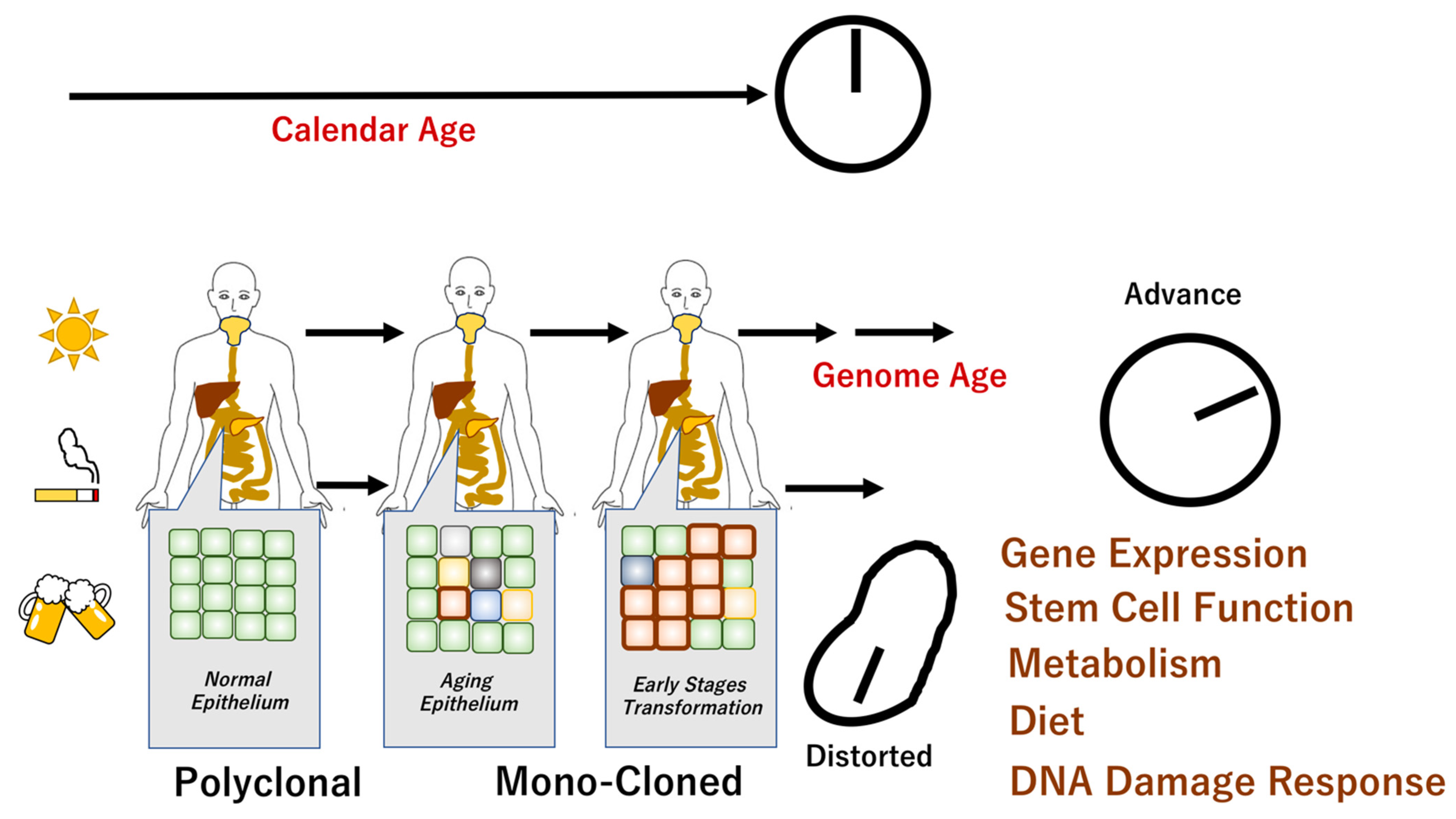
, , , 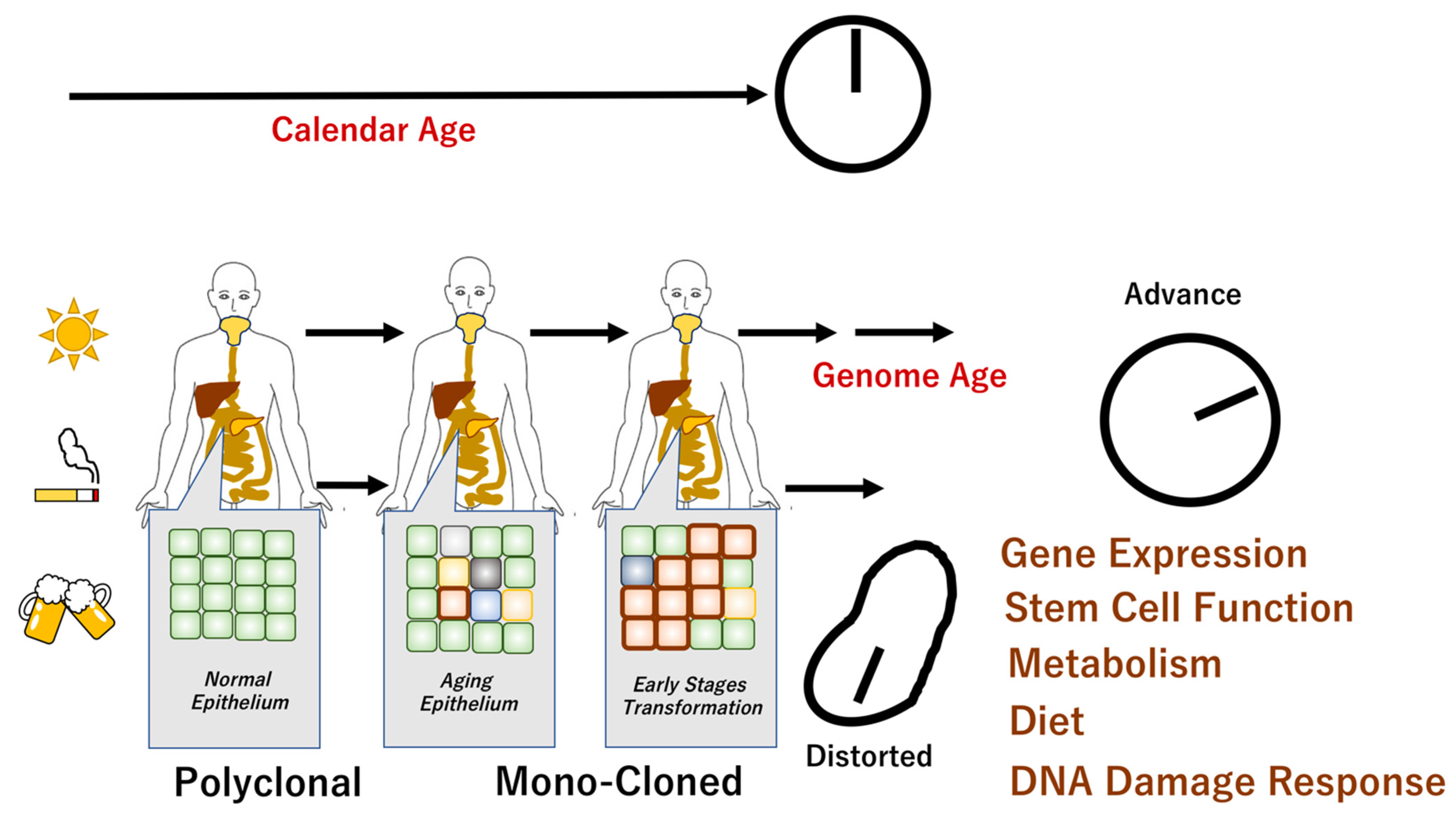{kind=link}
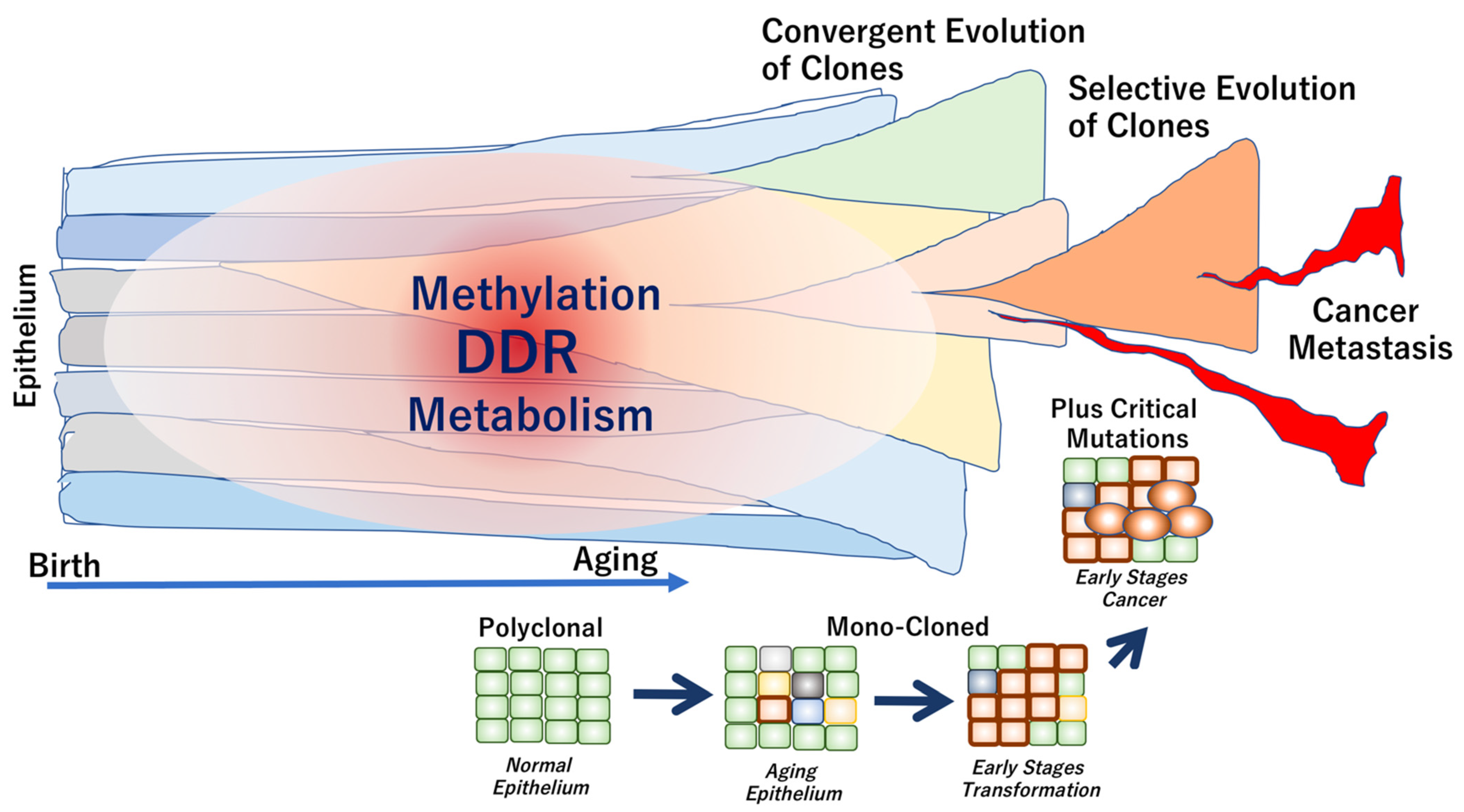
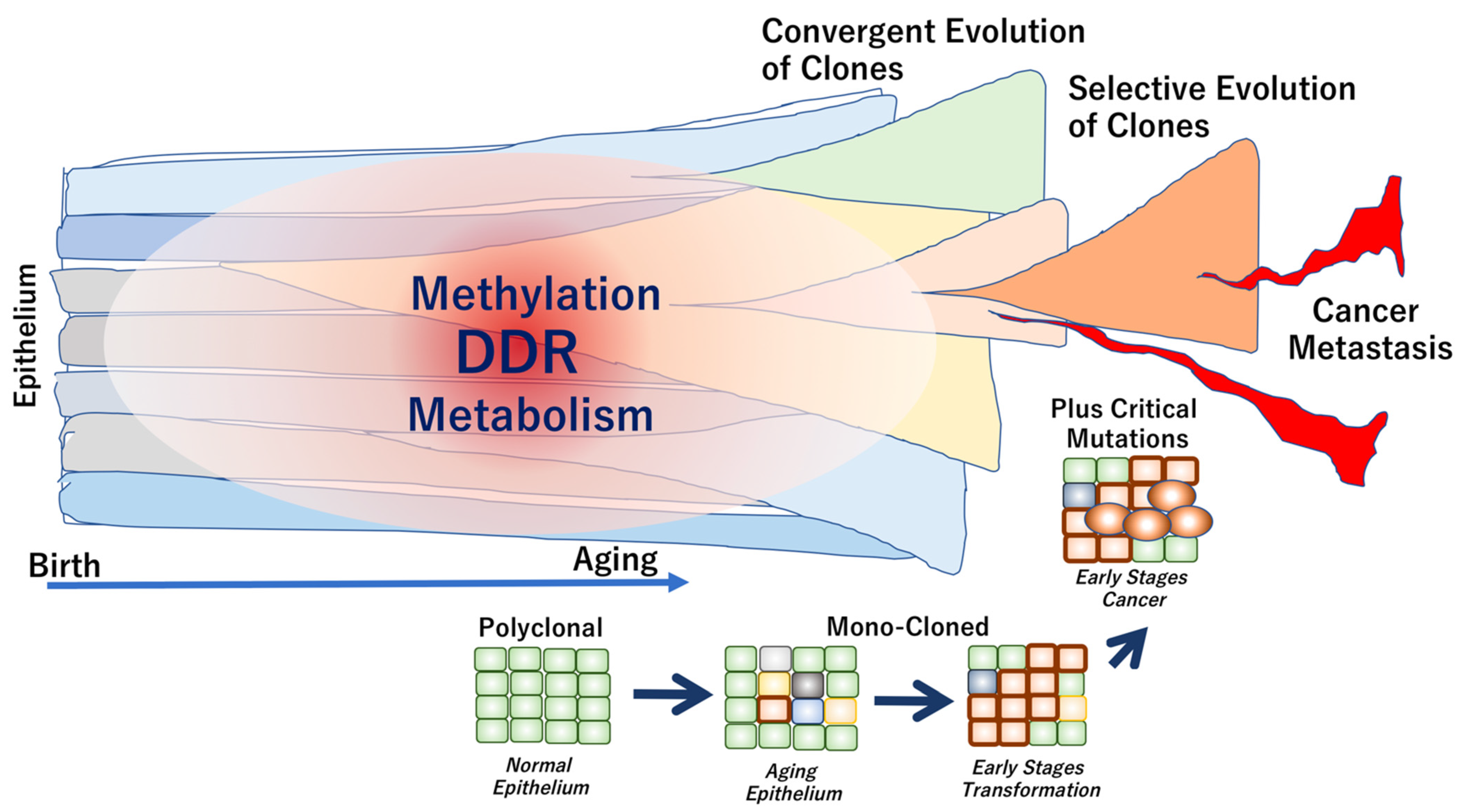{kind=link}
Abstract
:1. Introduction
2. Age-Related Genetic Alterations in Normal Tissues
3. Systematization of Disease Concept According to Genome Aging Theory
4. Cells as Structural and Functional Units of Living Organisms
5. Measurement of Genome Aging
6. Age-Related Clonal Hematopoiesis
7. Age-Related Remodeling of Epithelium
8. The Mechanisms That Accelerate or Decelerate Genome Aging
8.1. DNA Damage and Cellular Senescence
8.2. Metabolism
8.3. Diet
9. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Di Micco, R.; Krizhanovsky, V.; Baker, D.; d’Adda di Fagagna, F. Cellular senescence in ageing: From mechanisms to therapeutic opportunities. Nat. Rev. Mol. Cell. Biol. 2021, 22, 75–95. [Google Scholar] [CrossRef] [PubMed]
- Chan, A.S.; Mowla, S.N.; Arora, P.; Jat, P.S. Tumour suppressors and cellular senescence. IUBMB Life 2014, 66, 812–822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Svensson, E.I.; Arnold, S.J.; Bürger, R.; Csilléry, K.; Draghi, J.; Henshaw, J.M.; Jones, A.G.; de Lisle, S.; Marques, D.A.; McGuigan, K. Correlational selection in the age of genomics. Nat. Ecol. Evol. 2021, 5, 562–573. [Google Scholar] [CrossRef]
- Yokoyama, A.; Kakiuchi, N.; Yoshizato, T.; Nannya, Y.; Suzuki, H.; Takeuchi, Y.; Shiozawa, Y.; Sato, Y.; Aoki, K.; Kim, S.K.; et al. Age-related remodelling of oesophageal epithelia by mutated cancer drivers. Nature 2019, 565, 312–317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turajlic, S.; Sottoriva, A.; Graham, T.; Swanton, C. Resolving genetic heterogeneity in cancer. Nat. Rev. Genet. 2019, 20, 404–416. [Google Scholar] [CrossRef]
- Berben, L.; Floris, G.; Wildiers, H.; Hatse, S. Cancer and Aging: Two tightly interconnected biological processes. Cancers 2021, 13, 1400. [Google Scholar] [CrossRef] [PubMed]
- Feng, E.; Balint, E.; Poznanski, S.M.; Ashkar, A.A.; Loeb, M. Aging and interferons: Impacts on inflammation and viral disease outcomes. Cells 2021, 10, 708. [Google Scholar] [CrossRef]
- Nidadavolu, L.S.; Walston, J.D. Underlying vulnerabilities to the cytokine storm and adverse COVID-19 outcomes in the aging immune system. J. Gerontol. A Biol. Sci. Med. Sci. 2021, 76, e13–e18. [Google Scholar] [CrossRef]
- Süβ, P.; Lana, A.J.; Schlachetzki, J.C.M. Chronic peripheral inflammation: A possible contributor to neurodegenerative diseases. Neural Regen. Res. 2021, 16, 1711–1714. [Google Scholar]
- Unnikrishnan, A.; Freeman, W.M.; Jackson, J.; Wren, J.D.; Porter, H.; Richardson, A. The role of DNA methylation in epigenetics of aging. Pharm. Ther. 2019, 195, 172–185. [Google Scholar] [CrossRef] [PubMed]
- Vijg, J.; Suh, Y. Genome instability and aging. Annu. Rev. Physiol. 2013, 75, 645–668. [Google Scholar] [CrossRef] [PubMed]
- Ahadi, S.; Zhou, W.; Rose, S.M.S.F.; Sailani, M.R.; Contrepois, K.; Avina, M.; Ashland, M.; Brunet, A.; Snyder, M. Personal aging markers and ageotypes revealed by deep longitudinal profiling. Nat. Med. 2020, 26, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Sailani, M.R.; Contrepois, K.; Zhou, Y.; Ahadi, S.; Leopold, S.R.; Zhang, M.J.; Rao, V.; Avina, M.; Mishra, T.; et al. Longitudinal multi-omics of host-microbe dynamics in prediabetes. Nature 2019, 569, 663–671. [Google Scholar] [CrossRef]
- Jones, M.J.; Goodman, S.J.; Kobor, M.S. DNA methylation and healthy human aging. Aging Cell 2015, 14, 924–932. [Google Scholar] [CrossRef]
- Grün, D.; van Oudenaarden, A. Design and analysis of single-cell sequencing experiments. Cell 2015, 163, 799–810. [Google Scholar] [CrossRef] [Green Version]
- Baslan, T.; Hicks, J. Unravelling biology and shifting paradigms in cancer with single-cell sequencing. Nat. Rev. Cancer 2017, 17, 557–569. [Google Scholar] [CrossRef]
- Lappalainen, T.; Scott, A.J.; Brandt, M.; Hall, I.M. Genomic analysis in the age of human genome sequencing. Cell 2019, 177, 70–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goss, C.M. The historical background of Schwann′s cell theory. Yale J. Biol. Med. 1937, 10, 125–144. [Google Scholar] [PubMed]
- Schmidt, A.; Weber, O.F. In memoriam of Rudolf virchow: A historical retrospective including aspects of inflammation, infection and neoplasia. Contrib. Microbiol. 2006, 13, 1–15. [Google Scholar]
- Lambert, T.J.; Waters, J.C. Navigating challenges in the application of superresolution microscopy. J. Cell. Biol. 2017, 216, 53–63. [Google Scholar] [CrossRef]
- Strzelecka, P.M.; Damm, F. Haematopoietic ageing through the lens of single-cell technologies. Dis. Model. Mech. 2021, 14, dmm047340. [Google Scholar] [CrossRef]
- Hwang, B.; Lee, J.H.; Bang, D. Single-cell RNA sequencing technologies and bioinformatics pipelines. Exp. Mol. Med. 2018, 50, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Gao, S. Data analysis in single-cell transcriptome sequencing. Methods Mol. Biol. 2018, 1754, 311–326. [Google Scholar]
- Angelidis, I.; Simon, L.M.; Fernandez, I.E.; Strunz, M.; Mayr, C.H.; Greiffo, F.R.; Tsitsiridis, G.; Ansari, M.; Graf, E.; Strom, T.; et al. An atlas of the aging lung mapped by single cell transcriptomics and deep tissue proteomics. Nat. Commun. 2019, 10, 963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, W.; Zhang, S.; Yan, P.; Ren, J.; Song, M.; Li, J.; Lei, J.; Pan, H.; Wang, S.; Ma, X.; et al. A single-cell transcriptomic landscape of primate arterial aging. Nat. Commun. 2020, 11, 2202. [Google Scholar] [CrossRef]
- He, D.; Wu, H.; Xiang, J.; Ruan, X.; Peng, P.; Ruan, Y.; Chen, Y.; Wang, Y.; Yu, Q.; Zhang, H.; et al. Gut stem cell aging is driven by mTORC1 via a p38 MAPK-p53 pathway. Nat. Commun. 2020, 11, 37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zou, Z.; Long, X.; Zhao, Q.; Zheng, Y.; Song, M.; Ma, S.; Jing, Y.; Wang, S.; He, Y.; Esteban, C.R.; et al. A single-cell transcriptomic atlas of human skin aging. Dev. Cell 2021, 56, 383–397.e8. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.; Studer, L. Aging in iPS cells. Aging 2014, 6, 246–247. [Google Scholar] [CrossRef] [PubMed]
- Salama, R.; Sadaie, M.; Hoare, M.; Narita, M. Cellular senescence and its effector programs. Gene. Dev. 2014, 28, 99–114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Battram, A.M.; Bachiller, M.; Martín-Antonio, B. Senescence in the development and response to cancer with immunotherapy: A double-edged sword. Int. J. Mol. Sci. 2020, 21, 4346. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, L.; Wen, X.; Hao, D.; Zhang, N.; He, G.; Jiang, X. NF-kappaB signaling in skin aging. Mech Ageing Dev. 2019, 184, 111160. [Google Scholar] [CrossRef]
- Wang, Z.N.; Su, R.N.; Yang, B.Y.; Yang, K.X.; Yang, L.F.; Yan, Y.; Chen, Z.-G. Potential role of cellular senescence in asthma. Front. Cell. Dev. Biol. 2020, 8, 59. [Google Scholar] [CrossRef] [PubMed]
- Lasry, A.; Ben-Neriah, Y. Senescence-associated inflammatory responses: Aging and cancer perspectives. Trend. Immunol. 2015, 36, 217–228. [Google Scholar] [CrossRef]
- Prata, L.G.P.L.; Ovsyannikova, I.G.; Tchkonia, T.; Kirkland, J.L. Senescent cell clearance by the immune system: Emerging therapeutic opportunities. Semin. Immunol. 2018, 40, 101275. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Blas, D.; Gorostieta-Salas, E.; Pommer-Alba, A.; Muciño-Hernández, G.; Gerónimo-Olvera, C.; Maciel-Barón, L.A.; Konigsberg, M.; Massieu, L.; Castro-Obregón, S. Cortical neurons develop a senescence-like phenotype promoted by dysfunctional autophagy. Aging 2019, 11, 6175–6198. [Google Scholar] [CrossRef]
- Cianflone, E.; Torella, M.; Biamonte, F.; De Angelis, A.; Urbanek, K.; Costanzo, F.S.; Rota, M.; Ellison-Hughes, G.M.; Torella, D. Targeting cardiac stem cell senescence to treat cardiac aging and disease. Cells 2020, 9, 1558. [Google Scholar] [CrossRef] [PubMed]
- Mu, X.; Lin, C.Y.; Hambright, W.S.; Tang, Y.; Ravuri, S.; Lu, A.; Matre, P.; Chen, W.; Gao, X.; Cui, Y.; et al. Aberrant RhoA activation in macrophages increases senescence-associated secretory phenotypes and ectopic calcification in muscular dystrophic mice. Aging 2020, 12, 24853–24871. [Google Scholar] [CrossRef] [PubMed]
- Coppé, J.-P.; Patil, C.K.; Rodier, F.; Sun, Y.; Muñoz, D.P.; Goldstein, J.; Nelson, P.S.; Desprez, P.Y.; Campisi, J. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. 2008, 6, 2853–2868. [Google Scholar] [CrossRef]
- Zhang, X.; Wang, Y.; Zhao, R.; Hu, X.; Zhang, B.; Lv, X.; Guo, Z.; Zhang, Z.; Yuan, J.; Chu, X.; et al. Folic acid supplementation suppresses sleep deprivation-induced telomere dysfunction and senescence-associated secretory phenotype (SASP). Oxid. Med. Cell. Longev. 2019, 2019, 4569614. [Google Scholar] [CrossRef]
- Yamanaka, S. Elite and stochastic models for induced pluripotent stem cell generation. Nature 2009, 460, 49–52. [Google Scholar] [CrossRef]
- Ji, G.; Ruan, W.; Liu, K.; Wang, F.; Sakellariou, D.; Chen, J.; Yang, Y.; Okuka, M.; Han, J.; Liu, Z.; et al. Telomere reprogramming and maintenance in porcine iPS cells. PLoS ONE 2013, 8, e74202. [Google Scholar] [CrossRef]
- Shlush, L.I. Age-related clonal hematopoiesis. Blood 2018, 131, 496–504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hitzeroth, H.W.; Bender, K.; Ropers, H.H.; Geerthsen, J.M. Tentative evidence for 3–4 haematopoetic stem cells in man. Hum. Genet. 1977, 35, 175–183. [Google Scholar] [CrossRef] [PubMed]
- Hitzeroth, H.W.; Bender, K. Age-dependency of somatic selection in South African Negro G-6-PD heterozygotes. Hum. Genet. 1981, 58, 338–343. [Google Scholar] [CrossRef] [PubMed]
- Laurie, C.C.; Laurie, C.A.; Rice, K.; Doheny, K.F.; Zelnick, L.R.; McHugh, C.P.; Ling, H.; Hetrick, K.N.; Pugh, E.W.; Amos, C.; et al. Detectable clonal mosaicism from birth to old age and its relationship to cancer. Nat. Genet. 2012, 44, 642–650. [Google Scholar] [CrossRef] [Green Version]
- Jacobs, K.B.; Yeager, M.; Zhou, W.; Wacholder, S.; Wang, Z.; Rodriguez-Santiago, B.; Hutchinson, A.; Deng, X.; Liu, C.; Horner, M.; et al. Detectable clonal mosaicism and its relationship to aging and cancer. Nat. Genet. 2012, 44, 651–658. [Google Scholar] [CrossRef] [PubMed]
- Abkowitz, J.L.; Taboada, M.; Shelton, G.H.; Catlin, S.N.; Guttorp, P.; Kiklevich, J.V. An X chromosome gene regulates hematopoietic stem cell kinetics. Proc. Natl. Acad. Sci. USA 1998, 95, 3862–3866. [Google Scholar] [CrossRef] [Green Version]
- Gealy, W.J.; Dwyer, J.M.; Harley, J.B. Allelic exclusion of glucose-6-phosphate dehydrogenase in platelets and T lymphocytes from a Wiskott-Aldrich syndrome carrier. Lancet 1980, 1, 63–65. [Google Scholar] [CrossRef]
- Conley, M.E.; Brown, P.; Pickard, A.R.; Buckley, R.H.; Miller, D.S.; Raskind, W.H.; Singer, J.W.; Fialkow, P.J. Expression of the gene defect in X-linked agammaglobulinemia. N. Engl. J. Med. 1986, 315, 564–567. [Google Scholar] [CrossRef]
- Conley, M.E.; Lavoie, A.; Briggs, C.; Brown, P.; Guerra, C.; Puck, J.M. Nonrandom X chromosome inactivation in B cells from carriers of X chromosome-linked severe combined immunodeficiency. Proc. Natl. Acad. Sci. USA 1988, 85, 3090–3094. [Google Scholar] [CrossRef] [Green Version]
- Nyhan, W.L.; Bakay, B.; Connor, J.D.; Marks, J.F.; Keele, D.K. Hemizygous expression of glucose-6-phosphate dehydrogenase in erythrocytes of heterozygotes for the Lesch-Nyhan syndrome. Proc. Natl. Acad. Sci. USA 1970, 65, 214–218. [Google Scholar] [CrossRef] [Green Version]
- Mimori, K.; Saito, T.; Niida, A.; Miyano, S. Cancer evolution and heterogeneity. Ann. Gastroenterol. Surg. 2018, 2, 332–338. [Google Scholar] [CrossRef]
- Hayflick, L.; Moorhead, P.S. The serial cultivation of human diploid cell strains. Exp. Cell. Res. 1961, 25, 585–621. [Google Scholar] [CrossRef]
- Goldstein, S. Replicative senescence: The human fibroblast comes of age. Science 1990, 249, 1129–1133. [Google Scholar] [CrossRef] [PubMed]
- Kim, N.W.; Piatyszek, M.A.; Prowse, K.R.; Harley, C.B.; West, M.D.; Ho, P.L.; Coviello, G.M.; Wright, W.E.; Weinrich, S.L.; Shay, J.W. Specific association of human telomerase activity with immortal cells and cancer. Science 1994, 266, 2011–2015. [Google Scholar] [CrossRef] [PubMed]
- Herbig, U.; Jobling, W.A.; Chen, B.P.; Chen, D.J.; Sedivy, J.M. Telomere shortening triggers senescence of human cells through a pathway involving ATM, p53, and p21(CIP1), but not p16(INK4a). Mol. Cell 2004, 14, 501–513. [Google Scholar] [CrossRef]
- de Lange, T. Protection of mammalian telomeres. Oncogene 2002, 21, 532–540. [Google Scholar] [CrossRef] [Green Version]
- Seeman, P.; Gebertová, K.; Paderová, K.; Sperling, K.; Seemanová, E. Nijmegen breakage syndrome in 13% of age-matched Czech children with primary microcephaly. Pediatr. Neurol. 2004, 30, 195–200. [Google Scholar] [CrossRef]
- Shiloh, Y.; Lederman, H.M. Ataxia-telangiectasia (A-T): An emerging dimension of premature ageing. Ageing Res. Rev. 2017, 33, 76–88. [Google Scholar] [CrossRef]
- d’Adda di Fagagna, F.; Reaper, P.M.; Clay-Farrace, L.; Fiegler, H.; Carr, P.; Von Zglinicki, T.; Saretzki, G.; Carter, N.P.; Jackson, S.P. A DNA damage checkpoint response in telomere-initiated senescence. Nature 2003, 426, 194–198. [Google Scholar] [CrossRef]
- Chen, J.H.; Stoeber, K.; Kingsbury, S.; Ozanne, S.E.; Williams, G.H.; Hales, C.N. Loss of proliferative capacity and induction of senescence in oxidatively stressed human fibroblasts. J. Biol. Chem. 2004, 279, 49439–49446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takai, H.; Smogorzewska, A.; de Lange, T. DNA damage foci at dysfunctional telomeres. Curr. Biol. 2003, 13, 1549–1556. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.-H.; Hales, C.N.; Ozanne, S.E. DNA damage, cellular senescence and organismal ageing: Causal or correlative? Nucleic Acids Res. 2007, 35, 7417–7428. [Google Scholar] [CrossRef] [PubMed]
- Donato, A.J.; Morgan, R.G.; Walker, A.E.; Lesniewski, L.A. Cellular and molecular biology of aging endothelial cells. J. Mol. Cell Cardiol. 2015, 89, 122–135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- May-Panloup, P.; Boucret, L.; Chao de la Barca, J.M.; Desquiret-Dumas, V.; Ferré-L’Hotellier, V.; Morinière, C.; Descamps, P.; Procaccio, V.; Reynier, P. Ovarian ageing: The role of mitochondria in oocytes and follicles. Hum. Reprod. Update 2016, 22, 725–743. [Google Scholar] [CrossRef] [Green Version]
- Mora, A.L.; Bueno, M.; Rojas, M. Mitochondria in the spotlight of aging and idiopathic pulmonary fibrosis. J. Clin. Invest. 2017, 127, 405–414. [Google Scholar] [CrossRef]
- Wang, H.; Lautrup, S.; Caponio, D.; Zhang, J.; Fang, E.F. DNA damage-induced neurodegeneration in accelerated ageing and Alzheimer’s disease. Int. J. Mol. Sci. 2021, 22, 6748. [Google Scholar] [CrossRef]
- Zhang, L.; Wang, W.; Zhu, B.; Wang, X. Epithelial mitochondrial dysfunction in lung disease. Adv. Exp. Med. Biol. 2017, 1038, 201–217. [Google Scholar]
- Fukunaga, H. Mitochondrial DNA copy number and developmental origins of health and disease (DOHaD). Int. J. Mol. Sci. 2021, 22, 6634. [Google Scholar] [CrossRef]
- Covarrubias, A.J.; Perrone, R.; Grozio, A.; Verdin, E. NAD+ metabolism and its roles in cellular processes during ageing. Nat. Rev. Mol. Cell Biol. 2021, 22, 119–141. [Google Scholar] [CrossRef]
- Minhas, P.S.; Liu, L.; Moon, P.K.; Joshi, A.U.; Dove, C.; Mhatre, S.; Contrepois, K.; Wang, Q.; Lee, B.A.; Coronado, M.; et al. Macrophage de novo NAD(+) synthesis specifies immune function in aging and inflammation. Nat. Immunol. 2019, 20, 50–63. [Google Scholar] [CrossRef] [PubMed]
- Essuman, K.; Summers, D.W.; Sasaki, Y.; Mao, X.; DiAntonio, A.; Milbrandt, J. The SARM1 Toll/Interleukin-1 receptor domain possesses intrinsic NAD(+) cleavage activity that promotes pathological axonal degeneration. Neuron 2017, 93, 1334–1343.e5. [Google Scholar] [CrossRef] [Green Version]
- Hu, X.; Lu, Z.; Yu, S.; Reilly, J.; Liu, F.; Jia, D.; Qin, Y.; Han, S.; Liu, X.; Qu, Z.; et al. CERKL regulates autophagy via the NAD-dependent deacetylase SIRT1. Autophagy 2019, 15, 453–465. [Google Scholar] [CrossRef]
- Hannun, Y.A.; Obeid, L.M. Sphingolipids and their metabolism in physiology and disease. Nat. Rev. Mol. Cell Biol. 2018, 19, 175–191. [Google Scholar] [CrossRef] [PubMed]
- Shamseddine, A.A.; Clarke, C.J.; Carroll, B.; Airola, M.V.; Mohammed, S.; Rella, A.; Obeid, L.M.; Hannun, Y.A. P53-dependent upregulation of neutral sphingomyelinase-2: Role in doxorubicin-induced growth arrest. Cell Death Dis. 2015, 6, e1947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Filosto, S.; Ashfaq, M.; Chung, S.; Fry, W.; Goldkorn, T. Neutral sphingomyelinase 2 activity and protein stability are modulated by phosphorylation of five conserved serines. J. Biol. Chem. 2012, 287, 514–522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fontana, L.; Partridge, L. Promoting health and longevity through diet: From model organisms to humans. Cell 2015, 161, 106–118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anisimov, V.N.; Bartke, A. The key role of growth hormone-insulin-IGF-1 signaling in aging and cancer. Crit. Rev. Oncol. Hematol. 2013, 87, 201–223. [Google Scholar] [CrossRef] [Green Version]
- Weichhart, T. mTOR as regulator of lifespan, aging, and cellular senescence: A mini-review. Gerontology 2018, 64, 127–134. [Google Scholar] [CrossRef]
- Soukas, A.A.; Hao, H.; Wu, L. Metformin as anti-aging therapy: Is it for everyone? Trend. Endocrinol. Metab. 2019, 30, 745–755. [Google Scholar] [CrossRef]
- Canfield, C.-A.; Bradshaw, P.C. Amino acids in the regulation of aging and aging-related diseases. Transl. Med. Aging 2019, 3, 70–89. [Google Scholar]
- Yang, Z.; Roth, K.; Agarwal, M.; Liu, W.; Petriello, M.C. The transcription factors CREBH, PPARa, and FOXO1 as critical hepatic mediators of diet-induced metabolic dysregulation. J. Nutr. Biochem. 2021, 95, 108633. [Google Scholar] [CrossRef] [PubMed]
- See, P.; Lum, J.; Chen, J.; Ginhoux, F. A single-cell sequencing guide for immunologists. Front. Immunol. 2018, 9, 2425. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kitakaze, M.; Chijimatsu, R.; Vecchione, A.; Kitagawa, T.; Doki, Y.; Eguchi, H.; Ishii, H. Epithelial Cell Transformation and Senescence as Indicators of Genome Aging: Current Advances and Unanswered Questions. Int. J. Mol. Sci. 2021, 22, 7544. https://doi.org/10.3390/ijms22147544
Kitakaze M, Chijimatsu R, Vecchione A, Kitagawa T, Doki Y, Eguchi H, Ishii H. Epithelial Cell Transformation and Senescence as Indicators of Genome Aging: Current Advances and Unanswered Questions. International Journal of Molecular Sciences. 2021; 22(14):7544. https://doi.org/10.3390/ijms22147544
Chicago/Turabian StyleKitakaze, Masatoshi, Ryota Chijimatsu, Andrea Vecchione, Toru Kitagawa, Yuichiro Doki, Hidetoshi Eguchi, and Hideshi Ishii. 2021. "Epithelial Cell Transformation and Senescence as Indicators of Genome Aging: Current Advances and Unanswered Questions" International Journal of Molecular Sciences 22, no. 14: 7544. https://doi.org/10.3390/ijms22147544
APA StyleKitakaze, M., Chijimatsu, R., Vecchione, A., Kitagawa, T., Doki, Y., Eguchi, H., & Ishii, H. (2021). Epithelial Cell Transformation and Senescence as Indicators of Genome Aging: Current Advances and Unanswered Questions. International Journal of Molecular Sciences, 22(14), 7544. https://doi.org/10.3390/ijms22147544