
Prediction of Viscoelastic Properties of Enzymatically Crosslinkable Tyramine–Modified Hyaluronic Acid Solutions Using a Dynamic Monte Carlo Kinetic Approach



Abstract
:1. Introduction
2. Enzymatic Crosslinking of Polymer–Phenol Conjugates
2.1. The Postulated Kinetic Mechanism
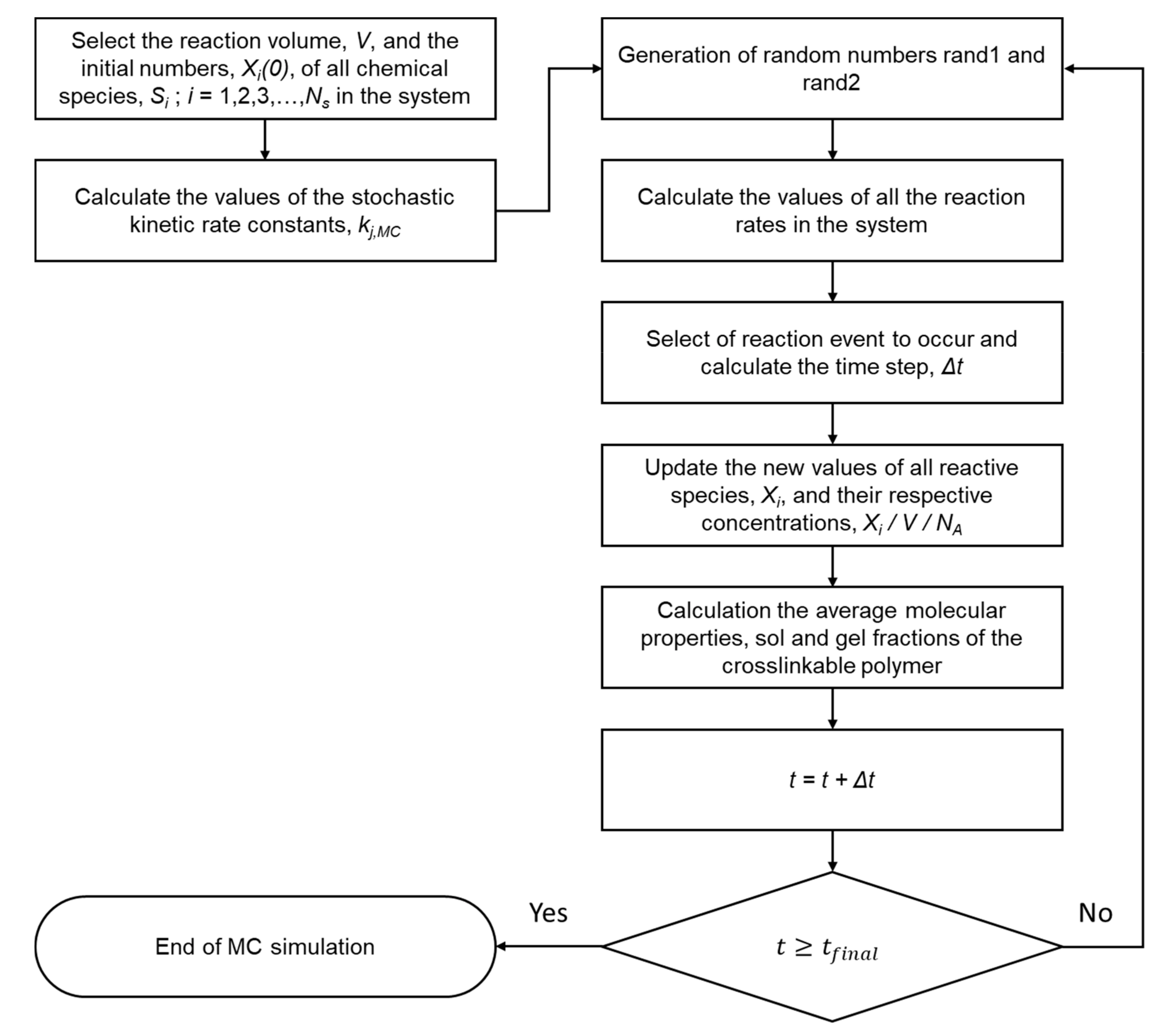
2.2. The Stochastic Monte Carlo Approach
2.3. Development of a 4D MC Kinetic Crosslinking Model
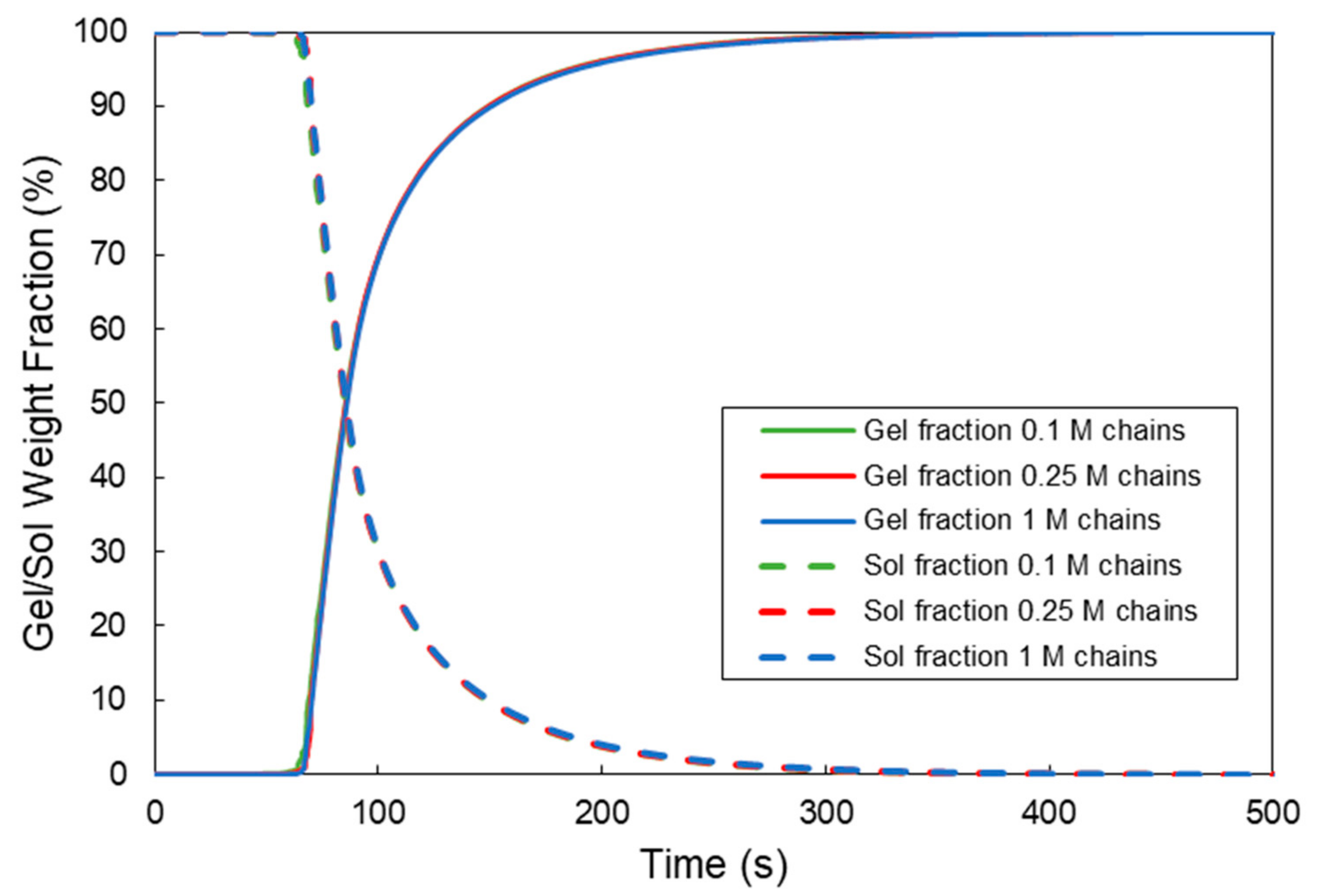
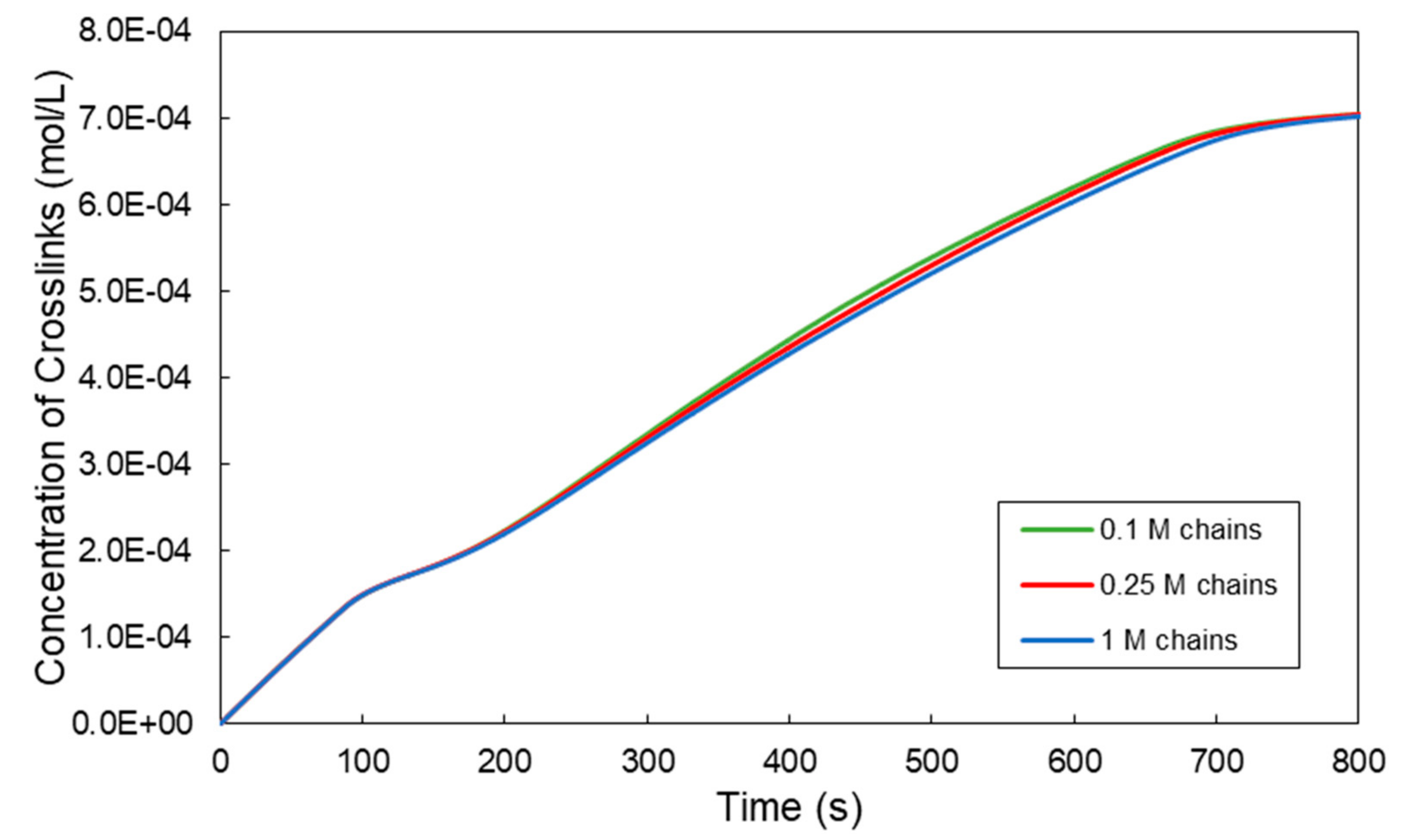
3. Prediction of Viscoelastic Properties of a Crosslinkable Polymer Solution
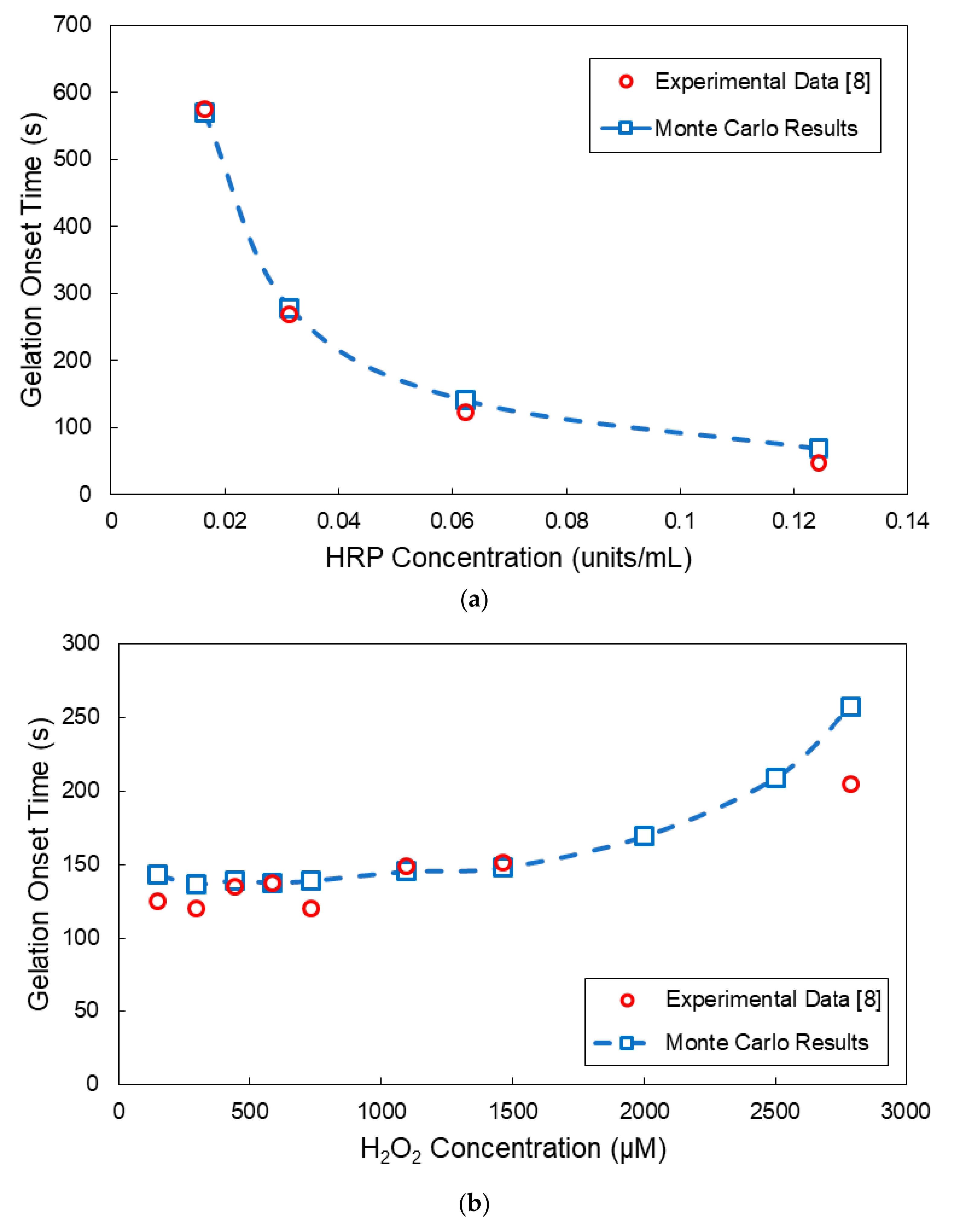
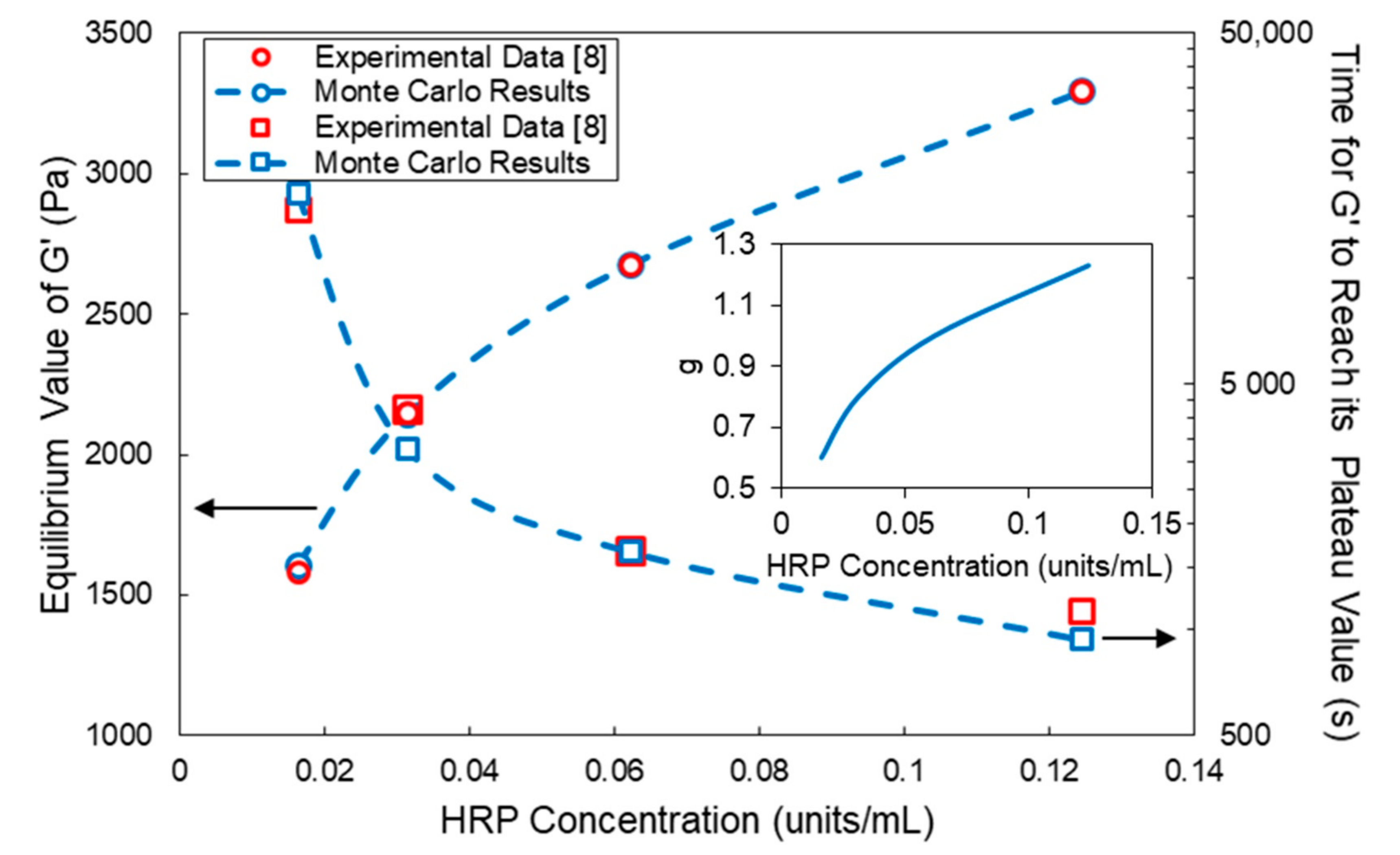
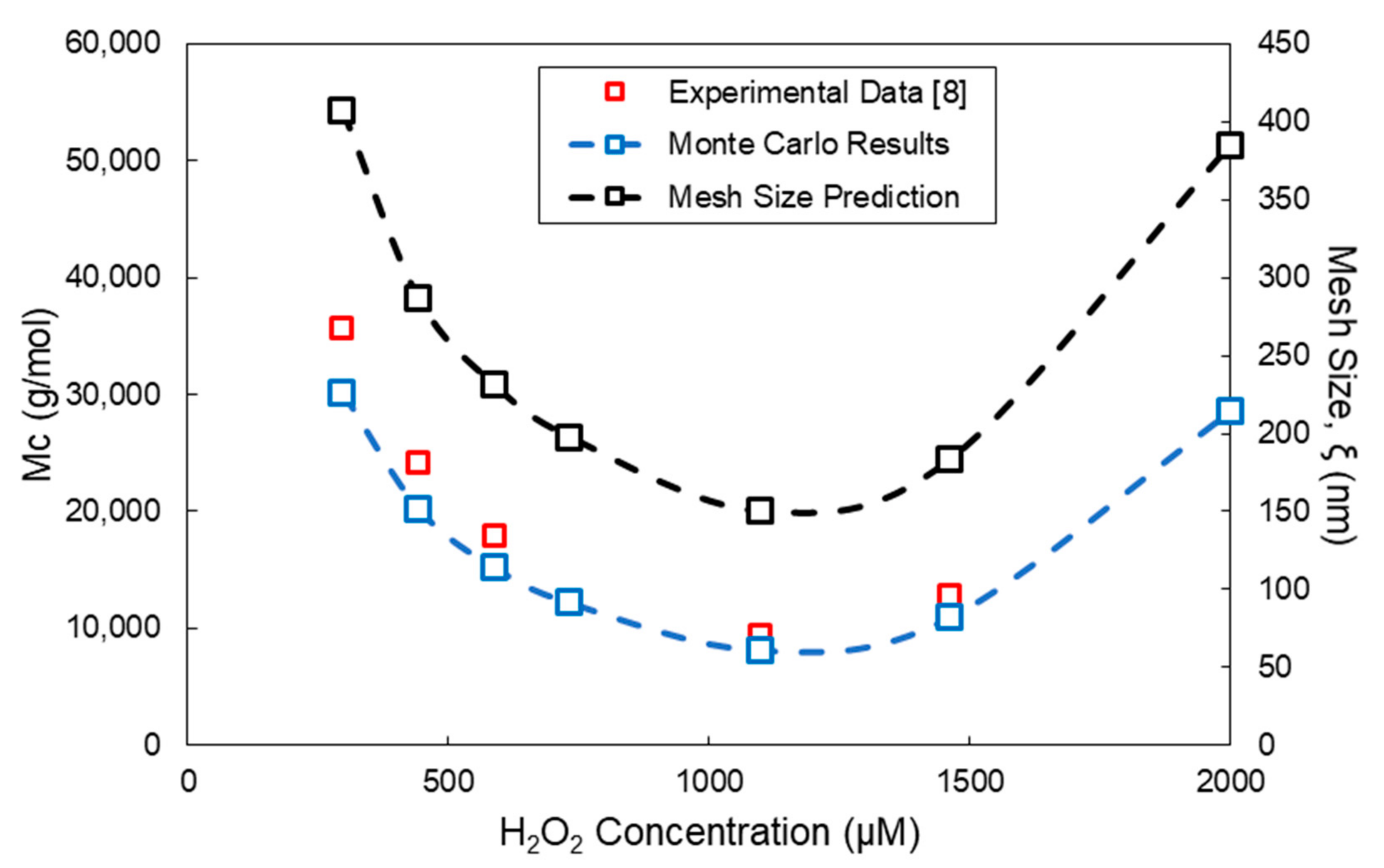
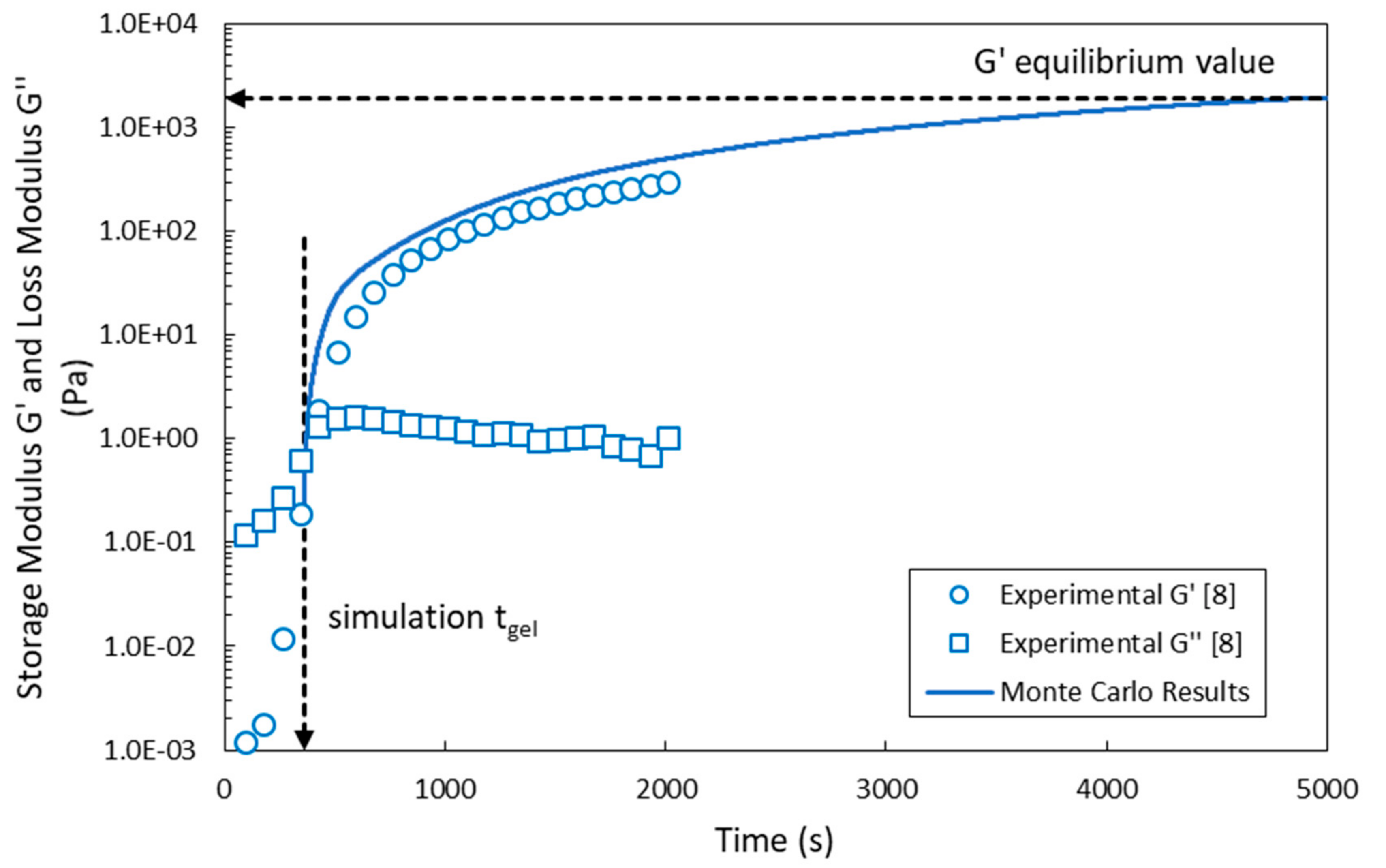
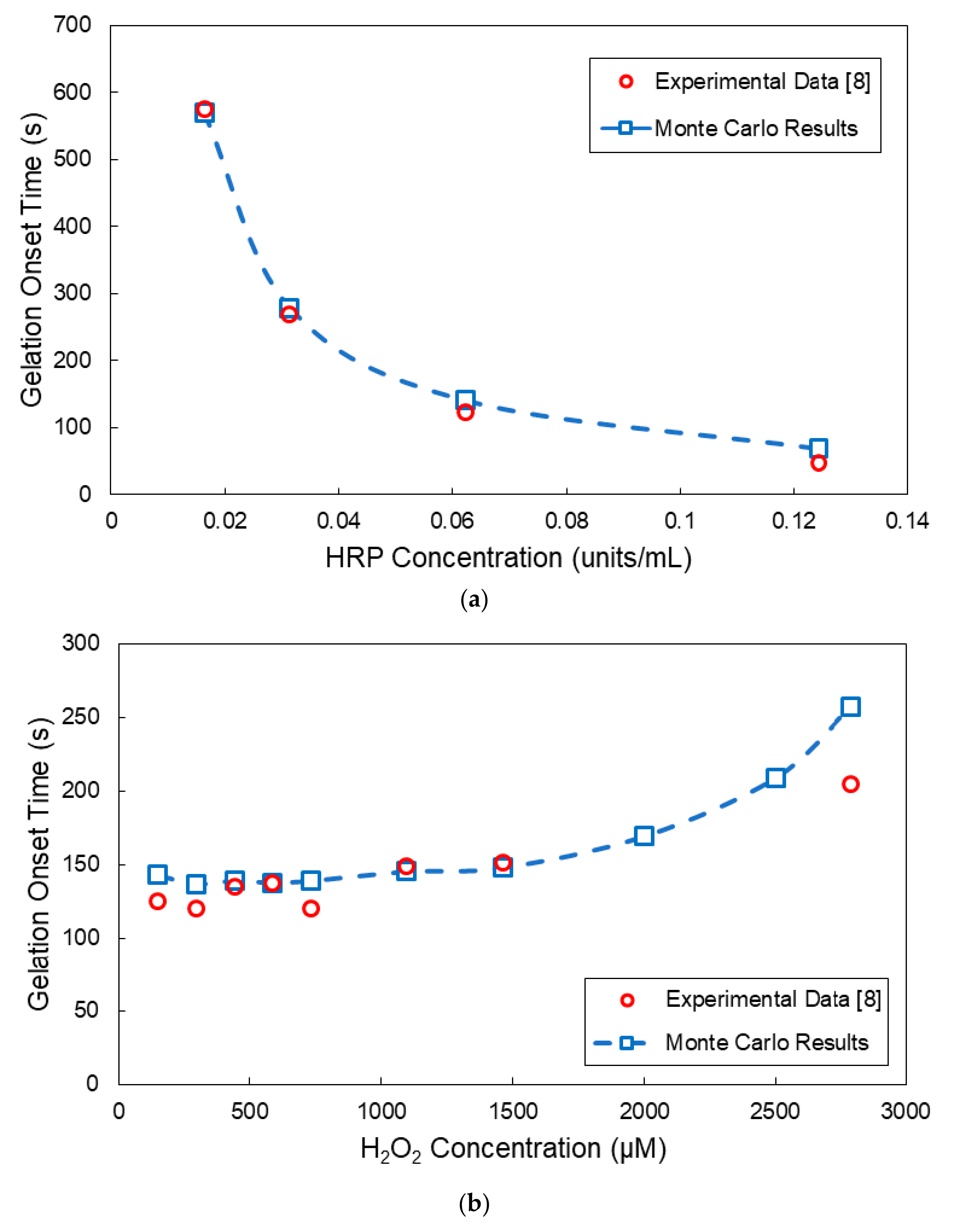
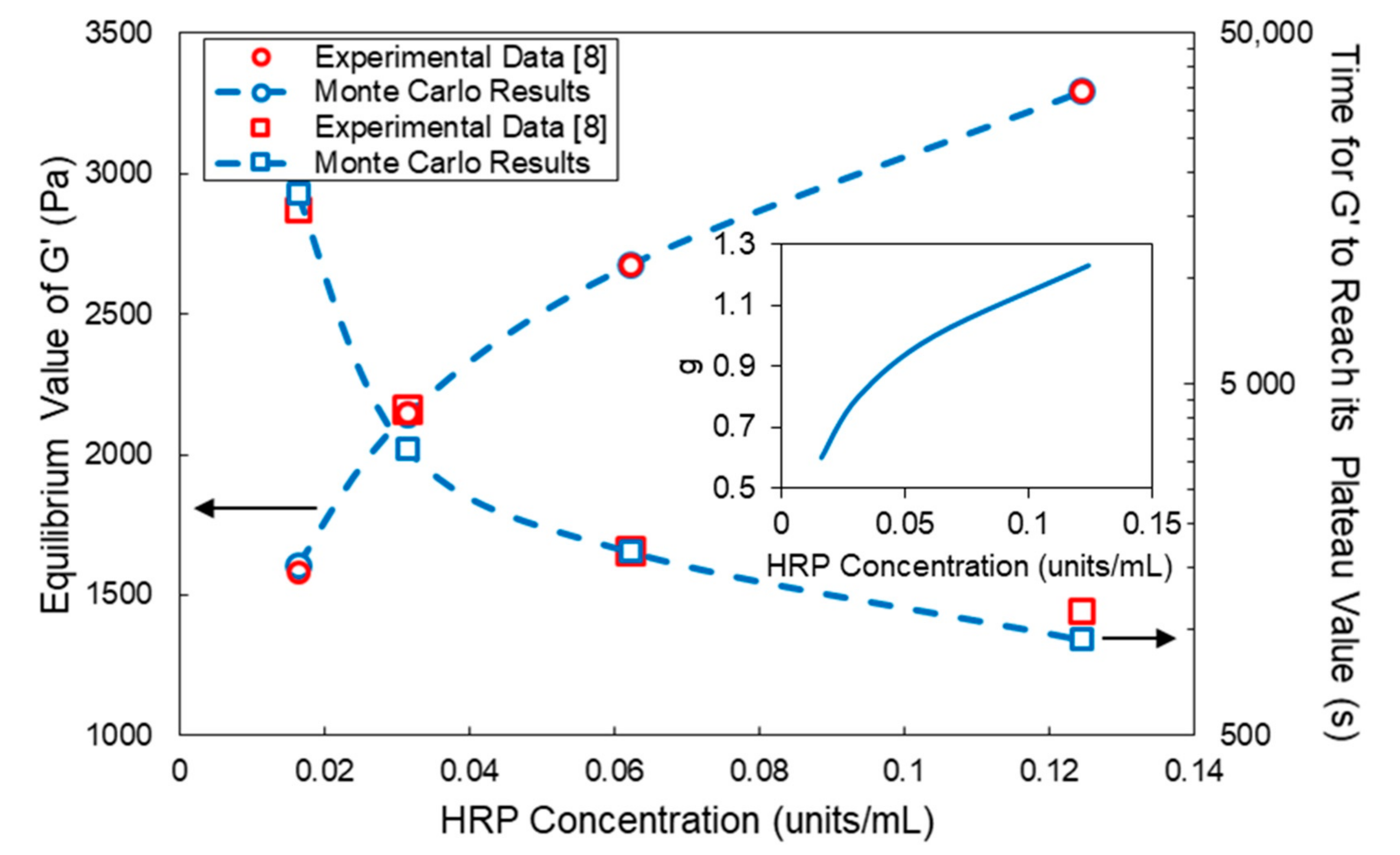
4. Comparison of Model Predictions with Experimental Results
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Nomenclature
| C | concentration of polymer in the solution |
| Cn | Flory’s characteristic ratio |
| E | Horseradish Peroxidase (HRP) |
| g | network correction factor |
| G | relaxation modulus (Pa) |
| G′ | storage modulus (Pa) |
| Ge | equilibrium shear modulus treated by rubberlike elasticity theory (Pa) |
| Gx,m,a,c | largest polymer chain with x monomeric units, m residual phenol groups, a activated phenol groups and c crosslinks |
| k | Boltzmann’s constant (J/K) |
| kj | kinetic rate constant of “j” reaction |
| kj,MC | stochastic kinetic rate constant of “j” reaction |
| Mc | number average molecular weight between two crosslinks |
| Mi | molecular weight of the “ith” polymer chain |
| Mn,sol | number average molecular weight in the solution (g/mol) |
| Mw | weight average molecular weight in the system (g/mol) |
| MWm | molecular weight of a repeating structural unit |
| Mw,sol | weight average molecular weight in the solution (g/mol) |
| NA | Avogadro’s Number |
| NCLD | number chain length distribution |
| NE | the number of enzyme species E |
| NH2O2 | total number of hydrogen peroxide species |
| NR | total number of polymer chains |
| Np | number of primary molecules before crosslinking |
| NRE | total number of reactions |
| rand1 | randomly generated number uniformly distributed in the range of [0, 1] |
| rand2 | randomly generated number uniformly distributed in the range of [0, 1] |
| mean square end-to-end distance of a strand | |
| mean square end-to-end distance of a non-constrained strand | |
| Pi | probability of reaction “i” |
| R | the universal gas constant (J∙mol−1K−1) |
| Rj | the rate of the “j” chemical reaction |
| T | absolute temperature (K) |
| u2,r | polymer volume fraction at the relaxed state |
| u2,s | polymer volume fraction at the equilibrium swollen state |
| V | volume of the mixture |
| V1 | molar volume of the solvent |
| wg | gel mass fraction |
| xi | degree of polymerization of the “ith” polymer chain |
| Xc | total number of possible combinations of reactive species in a reaction |
| Greek Symbols | |
| Δt | the time step between two reactions |
| ν | number of network chains or segments per unit volume (m−3) |
| vc | the number of moles of crosslinks per unit volume (mol∙m−3) |
| ve | number effective network chains or segments per unit volume (m−3) |
| vo | number of crosslinks per unit volume (m−3) |
| λ | backbone bond factor |
| ξ | mesh size of the network |
| ρ | polymer density (kg∙m−3) |
| χ1 | Flory interaction parameter of the polymer-solvent |
References
- Bae, J.W.; Choi, J.H.; Lee, Y.; Park, K.D. Horseradish peroxidase-catalysed in situ-forming hydrogels for tissue-engineering applications. J. Tissue Eng. Regener. Med. 2015, 9, 1225–1232. [Google Scholar] [CrossRef] [PubMed]
- Sakai, S.; Ito, S.; Ogushi, Y.; Hashimoto, I.; Hosoda, N.; Sawae, Y.; Kawakami, K. Enzymatically fabricated and degradable microcapsules for production of multicellular spheroids with well-defined diameters of less than 150 μm. Biomaterials 2009, 30, 5937–5942. [Google Scholar] [CrossRef] [PubMed]
- Sakai, S.; Ogushi, Y.; Kawakami, K. Enzymatically crosslinked carboxymethylcellulose–tyramine conjugate hydrogel: Cellular adhesiveness and feasibility for cell sheet technology. Acta Biomater. 2009, 5, 554–559. [Google Scholar] [CrossRef] [PubMed]
- Pek, Y.S.; Kurisawa, M.; Gao, S.; Chung, J.E.; Ying, J.Y. The development of a nanocrystalline apatite reinforced crosslinked hyaluronic acid–tyramine composite as an injectable bone cement. Biomaterials 2009, 30, 822–828. [Google Scholar] [CrossRef]
- Hu, M.; Kurisawa, M.; Deng, R.; Teo, C.M.; Schumacher, A.; Thong, Y.X.; Wang, L.; Schumacher, K.M.; Ying, J.Y. Cell immobilization in gelatin-hydroxyphenylpropionic acid hydrogel fibers. Biomaterials 2009, 30, 3523–3531. [Google Scholar] [CrossRef]
- Kurisawa, M.; Chung, J.E.; Yang, Y.Y.; Gao, S.J.; Uyama, H. Injectable biodegradable hydrogels composed of hyaluronic ac-id–tyramine conjugates for drug delivery and tissue engineering. Chem. Commun. 2005, 34, 4312–4314. [Google Scholar] [CrossRef] [PubMed]
- Jin, R.; Hiemstra, C.; Zhong, Z.; Feijen, J. Enzyme-mediated fast in situ formation of hydrogels from dextran–tyramine con-jugates. Biomaterials 2007, 28, 2791–2800. [Google Scholar] [CrossRef] [PubMed]
- Lee, F.; Chung, J.E.; Kurisawa, M. An injectable enzymatically crosslinked hyaluronic acid–tyramine hydrogel system with independent tuning of mechanical strength and gelation rate. Soft Matter. 2008, 4, 880–887. [Google Scholar] [CrossRef]
- Jin, R.; Moreira Teixeira, L.S.; Dijkstra, P.J.; Zhong, Z.; van Blitterswijk, C.A.; Karperien, M.; Feijen, J. Enzymatically cross-linked dextran-tyramine hydrogels as injectable scaffolds for cartilage tissue engineering. Tissue Eng. Part A 2010, 16, 2429–2440. [Google Scholar] [CrossRef] [PubMed]
- Ogushi, Y.; Sakai, S.; Kawakami, K. Synthesis of enzymatically-gellable carboxymethylcellulose for biomedical applications. J. Biosci. Bioeng. 2007, 104, 30–33. [Google Scholar] [CrossRef]
- Wang, L.-S.; Chung, J.E.; Chan, P.P.-Y.; Kurisawa, M. Injectable biodegradable hydrogels with tunable mechanical properties for the stimulation of neurogenesic differentiation of human mesenchymal stem cells in 3D culture. Biomaterials 2010, 31, 1148–1157. [Google Scholar] [CrossRef]
- Wennink, J.W.; Niederer, K.; Bochyńska, A.I.; Moreira Teixeira, L.S.; Karperien, M.; Feijen, J.; Dijkstra, P.J. Injectable Hydro-gels by Enzymatic Co-Crosslinking of Dextran and Hyaluronic Acid Tyramine Conjugates. Macromol. Symp. 2011, 309, 213–221. [Google Scholar] [CrossRef]
- Ren, C.D.; Gao, S.; Kurisawa, M.; Ying, J.Y. Cartilage synthesis in hyaluronic acid–tyramine constructs. J. Mater. Chem. B 2015, 3, 1942–1956. [Google Scholar] [CrossRef]
- Bi, B.; Liu, H.; Kang, W.; Zhuo, R.; Jiang, X. An injectable enzymatically crosslinked tyramine-modified carboxymethyl chitin hydrogel for biomedical applications. Colloids Surf. B Biointerfaces 2019, 175, 614–624. [Google Scholar] [CrossRef] [PubMed]
- Flory, P.J. Molecular Size Distribution in Three Dimensional Polymers. II. Trifunctional Branching Units. J. Am. Chem. Soc. 1941, 63, 3091–3096. [Google Scholar] [CrossRef]
- Flory, P.J. Molecular Size Distribution in Three Dimensional Polymers. III. Tetrafunctional Branching Units. J. Am. Chem. Soc. 1941, 63, 3096–3100. [Google Scholar] [CrossRef]
- Ferry, J.D. Protein Gels. Adv. Protein Chem. 1948, 4, 1–78. [Google Scholar] [CrossRef]
- Tobita, H.; Hamielec, A. Crosslinking kinetics in polyacrylamide networks. Polymer 1990, 31, 1546–1552. [Google Scholar] [CrossRef]
- Hamzehlou, S.; Reyes, Y.; Leiza, J.R. A New Insight into the Formation of Polymer Networks: A Kinetic Monte Carlo Simulation of the Cross-Linking Polymerization of S/DVB. Macromolecules 2013, 46, 9064–9073. [Google Scholar] [CrossRef]
- Tripathi, A.K.; Tsavalas, J.G.; Sundberg, D.C. Monte Carlo Simulations of Free Radical Polymerizations with Divinyl Cross-Linker: Pre- and Postgel Simulations of Reaction Kinetics and Molecular Structure. Macromolecules 2014, 48, 184–197. [Google Scholar] [CrossRef]
- Ryu, K.; McEldoon, J.P.; Pokora, A.R.; Cyrus, W.; Dordick, J.S. Numerical and Monte Carlo simulations of phenolic polymerizations catalyzed by peroxidase. Biotechnol. Bioeng. 1993, 42, 807–814. [Google Scholar] [CrossRef]
- Yamagishi, T.A.; Nakatogawa, T.; Ikuji, M.; Nakamoto, Y.; Ishida, S.I. Computational study of network formation of phenolic resins. Angew. Makromol. Chem. 1996, 240, 181–186. [Google Scholar] [CrossRef]
- Kiparissides, C.; Vasileiadou, A.; Karageorgos, F.; Serpetsi, S. A Computational Systems Approach to Rational Design of Nose-to-Brain Delivery of Biopharmaceutics. Ind. Eng. Chem. Res. 2019, 59, 2548–2565. [Google Scholar] [CrossRef]
- Tang, S.; Richardson, B.M.; Anseth, K.S. Dynamic covalent hydrogels as biomaterials to mimic the viscoelasticity of soft tissues. Prog. Mater. Sci. 2020, 120, 100738. [Google Scholar] [CrossRef]
- Huang, D.; Huang, Y.; Xiao, Y.; Yang, X.; Lin, H.; Feng, G.; Zhu, X.; Zhang, X. Viscoelasticity in natural tissues and engineered scaffolds for tissue reconstruction. Acta Biomater. 2019, 97, 74–92. [Google Scholar] [CrossRef] [PubMed]
- Gillespie, D.T. A general method for numerically simulating the stochastic time evolution of coupled chemical reactions. J. Comput. Phys. 1976, 22, 403–434. [Google Scholar] [CrossRef]
- Gillespie, D.T. Exact stochastic simulation of coupled chemical reactions. J. Phys. Chem. 1977, 81, 2340–2361. [Google Scholar] [CrossRef]
- Veitch, N.C. Horseradish peroxidase: A modern view of a classic enzyme. Phytochemistry 2004, 65, 249–259. [Google Scholar] [CrossRef]
- Lee, F.; Bae, K.H.; Kurisawa, M. Injectable hydrogel systems crosslinked by horseradish peroxidase. Biomed. Mater. 2015, 11, 014101. [Google Scholar] [CrossRef]
- Thi, T.T.H.; Lee, Y.; Le Thi, P.; Park, K.D. Engineered horseradish peroxidase-catalyzed hydrogels with high tissue adhesive-ness for biomedical applications. J. Ind. Eng. Chem. 2019, 78, 34–52. [Google Scholar]
- Lopes, G.; Pinto, D.C.G.A.; Silva, A. Horseradish peroxidase (HRP) as a tool in green chemistry. RSC Adv. 2014, 4, 37244–37265. [Google Scholar] [CrossRef]
- Schmidt, A.; Schumacher, J.T.; Reichelt, J.; Hecht, H.J.; Bilitewski, U. Mechanistic and molecular investigations on stabiliza-tion of horseradish peroxidase C. Anal. Chem. 2002, 74, 3037–3045. [Google Scholar] [CrossRef] [PubMed]
- Gross, A.J.; Sizer, I.W. The Oxidation of Tyramine, Tyrosine, and Related Compounds by Peroxidase. J. Biol. Chem. 1959, 234, 1611–1614. [Google Scholar] [CrossRef]
- Somvarsky, J.; Dušek, K. Kinetic Monte-Carlo simulation of network formation. 1. Simulation Method. Polym. Bull. 1994, 33, 369–376. [Google Scholar] [CrossRef]
- Kurdikar, D.L.; Somvarsky, J.; Dusek, K.; Peppas, N.A. Development and evaluation of a Monte Carlo technique for the simulation of multifunctional polymerizations. Macromolecules 1995, 28, 5910–5920. [Google Scholar] [CrossRef]
- Somvarsky, J.; Dušek, K. Kinetic Monte-Carlo simulation of network formation. 2. Effect of System Size. Polym. Bull. 1994, 33, 377–384. [Google Scholar] [CrossRef]
- Brandão, A.L.T.; Soares, J.B.P.; Pinto, J.C.; Alberton, A.L. When Polymer Reaction Engineers Play Dice: Applications of Monte Carlo Models in PRE. Macromol. React. Eng. 2015, 9, 141–185. [Google Scholar] [CrossRef]
- Tobita, H. Molecular weight distribution in free radical polymerization with long-chain branching. J. Polym. Sci. Part. B Polym. Phys. 1993, 31, 1363–1371. [Google Scholar] [CrossRef]
- Meimaroglou, D.; Kiparissides, C. Review of Monte Carlo methods for the prediction of distributed molecular and morpho-logical polymer properties. Ind. Eng. Chem. Res. 2014, 53, 8963–8979. [Google Scholar] [CrossRef]
- Meimaroglou, D.; Krallis, A.; Saliakas, V.; Kiparissides, C. Prediction of the Bivariate Molecular Weight—Long Chain Branching Distribution in Highly Branched Polymerization Systems Using Monte Carlo and Sectional Grid Methods. Macromolecules 2007, 40, 2224–2234. [Google Scholar] [CrossRef]
- Al-Harthi, M.A. MATLAB Programming of Polymerization Processes Using Monte Carlo Techniques; IntechOpen Access Publisher: London, UK, 2011. [Google Scholar]
- Maafa, I.M.; Soares, J.B.; Elkamel, A. Prediction of chain length distribution of polystyrene made in batch reactors with bi-functional free-radical initiators using dynamic Monte Carlo simulation. Macromol. React. Eng. 2007, 1, 364–383. [Google Scholar] [CrossRef]
- Krallis, A.; Meimaroglou, D.; Kiparissides, C. Dynamic prediction of the bivariate molecular weight–copolymer composition distribution using sectional-grid and stochastic numerical methods. Chem. Eng. Sci. 2008, 63, 4342–4360. [Google Scholar] [CrossRef]
- Ferry, J.D. Viscoelastic Properties of Polymers, 3rd ed.; John Wiley & Sons: New York, NY, USA, 1980. [Google Scholar]
- Treloar, L.R.G. The Physics of Rubber Elasticity, 3rd ed.; Oxford University Press: Oxford, UK, 2005. [Google Scholar]
- Flory, P.J. Network Structure and the Elastic Properties of Vulcanized Rubber. Chem. Rev. 1944, 35, 51–75. [Google Scholar] [CrossRef]
- Bueche, A.M. An investigation of the theory of rubber elasticity using irradiated polydimethylsiloxanes. J. Polym. Sci. 1956, 19, 297–306. [Google Scholar] [CrossRef]
- Te Nijenhuis, K. Dynamic Mechanical Studies on Thermo-Reversible Ageing Processes in Gels of Polyvinyl Chloride and of Gelatin. Ph.D. Thesis, Delft University of Technology, Delft, The Netherlands, 1979. [Google Scholar]
- Davidovich-Pinhas, M.; Bianco-Peled, H. A quantitative analysis of alginate swelling. Carbohydr. Polym. 2010, 79, 1020–1027. [Google Scholar] [CrossRef]
- Flory, P.J. Principles of Polymer Chemistry; Cornell University Press: Ithaca, NY, USA, 1953. [Google Scholar]
- Peppas, N.A.; Merrill, E.W. Determination of interaction parameter χ1, for poly(vinyl alcohol) and water in gels crosslinked from solutions. J. Polym. Sci. Polym. Chem. Ed. 1976, 14, 459–464. [Google Scholar] [CrossRef]
- Tobita, H. Crosslinking Kinetics in Free-Radical Copolymerization. Ph.D. Thesis, McMaster University, Hamilton, ON, Canada, 1990. [Google Scholar]
- Canal, T.; Peppas, N.A. Correlation between mesh size and equilibrium degree of swelling of polymeric networks. J. Biomed. Mater. Res. 1989, 23, 1183–1193. [Google Scholar] [CrossRef] [PubMed]
- Richbourg, N.R.; Peppas, N.A. The swollen polymer network hypothesis: Quantitative models of hydrogel swelling, stiffness, and solute transport. Prog. Polym. Sci. 2020, 105, 101243. [Google Scholar] [CrossRef]
- Peppas, N.A.; Hilt, J.Z.; Khademhosseini, A.; Langer, R. Hydrogels in Biology and Medicine: From Molecular Principles to Bionanotechnology. Adv. Mater. 2006, 18, 1345–1360. [Google Scholar] [CrossRef]
- Martini, M.; Hegger, P.S.; Schädel, N.; Minsky, B.B.; Kirchhof, M.; Scholl, S.; Southan, A.; Tovar, G.E.M.; Boehm, H.; Laschat, S. Charged Triazole Cross-Linkers for Hyaluronan-Based Hybrid Hydrogels. Materials 2016, 9, 810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arnao, M.B.; Acosta, M.; Del Rio, J.A.; Varon, R.; Garcia-Canovas, F. A kinetic study on the suicide inactivation of peroxidase by hydrogen peroxide. Biochim. Biophys. Acta Protein Struct. Mol. Enzymol. 1990, 1041, 43–47. [Google Scholar] [CrossRef]
- Vojinović, V.; Carvalho, R.; Lemos, F.; Cabral, J.; Fonseca, L.; Ferreira, B. Kinetics of soluble and immobilized horseradish peroxidase-mediated oxidation of phenolic compounds. Biochem. Eng. J. 2007, 35, 126–135. [Google Scholar] [CrossRef]
- Buchanan, I.D.; Nicell, J.A. Model development for horseradish peroxidase catalyzed removal of aqueous phenol. Biotechnol. Bioeng. 1997, 54, 251–261. [Google Scholar] [CrossRef]
- Buchanan, I. Kinetic Modelling of Horseradish Peroxidase Catalyzed Phenol Removal for Reactor Development. Ph.D. Thesis, McGill University Libraries, Montreal, QC, Canada, 1996. [Google Scholar]
- Vasudevan, P.T.; Li, L.O. Kinetics of phenol oxidation by peroxidase. Appl. Biochem. Biotechnol. 1996, 60, 203–215. [Google Scholar] [CrossRef]
- Karageorgos, F.F.; Kiparissides, C. Modeling of Gelation Kinetics of Enzymatically Cross-linked Polymer-Phenol Conjugates Using Deterministic and Stochastic Methods. 2021; to be submitted. [Google Scholar]
- Mikes, J.; Dušek, K. Simulation of polymer network formation by the Monte Carlo method. Macromolecules 1982, 15, 93–99. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Material | Authors |
|---|---|
| HA-Tyr | Kurisawa, Chung, Yang, Gao and Uyama 2005 [6] |
| Dextran-Tyr | Jin, Hiemstra, Zhong and Feijen 2007 [7] |
| HA-Tyr | Lee, Chung and Kurisawa 2008 [8] |
| Dextran-Tyr | Jin, Moreira Teixeira, Dijkstra, Zhong, Blitterswijk, Karperien and Feijen 2010 [9] |
| Carboxymethylcellulose-tyramine | Ogushi, Sakai and Kawakami 2007 [10] |
| Carboxymethylcellulose-phenolic hydroxyl groups (CMC-Ph) | Sakai, Ogushi and Kawakami 2009 [3] |
| Gelatin-hydroxyphenylpropionic acid (Gtn–HPA) | Wang, Chung, Chan and Kurisawa 2010 [11] |
| Dextran-tyramine (Dex-TA)/Hyaluronic acid-tyramine (HA-TA) conjugates | Wennink, Niederer, Bochynska, Teixeira, Karperien, Feijen and Dijkstra 2011 [12] |
| HA-Tyr | Ren, Gao, Kurisawa and Ying 2015 [13] |
| CMCH-Tyr | Bi, Liu, Kang, Zhuo and Jiang 2019 [14] |
| Equation | Stochastic Reaction Rate | MC Simulation Algorithm |
|---|---|---|
| 1 | ||
| 2 | Selection of | |
| 3 | Selection of | |
| 4 | ||
| 5 | Selection of Selection of Production of Removal of Removal of | |
| 6 | Selection of |
| No of polymer chains | 55,214 | 103,527 | 248,465 | 255,367 | 517,635 | 1,028,368 |
| CPU in sec | 173 | 586 | 4208 | 4371 | 16,657 | 72,839 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Karageorgos, F.F.; Kiparissides, C. Prediction of Viscoelastic Properties of Enzymatically Crosslinkable Tyramine–Modified Hyaluronic Acid Solutions Using a Dynamic Monte Carlo Kinetic Approach. Int. J. Mol. Sci. 2021, 22, 7317. https://doi.org/10.3390/ijms22147317
Karageorgos FF, Kiparissides C. Prediction of Viscoelastic Properties of Enzymatically Crosslinkable Tyramine–Modified Hyaluronic Acid Solutions Using a Dynamic Monte Carlo Kinetic Approach. International Journal of Molecular Sciences. 2021; 22(14):7317. https://doi.org/10.3390/ijms22147317
Chicago/Turabian StyleKarageorgos, Filippos F., and Costas Kiparissides. 2021. "Prediction of Viscoelastic Properties of Enzymatically Crosslinkable Tyramine–Modified Hyaluronic Acid Solutions Using a Dynamic Monte Carlo Kinetic Approach" International Journal of Molecular Sciences 22, no. 14: 7317. https://doi.org/10.3390/ijms22147317
APA StyleKarageorgos, F. F., & Kiparissides, C. (2021). Prediction of Viscoelastic Properties of Enzymatically Crosslinkable Tyramine–Modified Hyaluronic Acid Solutions Using a Dynamic Monte Carlo Kinetic Approach. International Journal of Molecular Sciences, 22(14), 7317. https://doi.org/10.3390/ijms22147317