
Coregulation Analysis of Mechanistic Biomarkers in Autosomal Dominant Polycystic Kidney Disease

 , , , ,
, , , ,
Abstract
1. Introduction
2. Results
2.1. Baseline Characteristics of the Patient Cohorts
2.2. Biomarker Selection Based on a Generated ADPKD Molecular Model
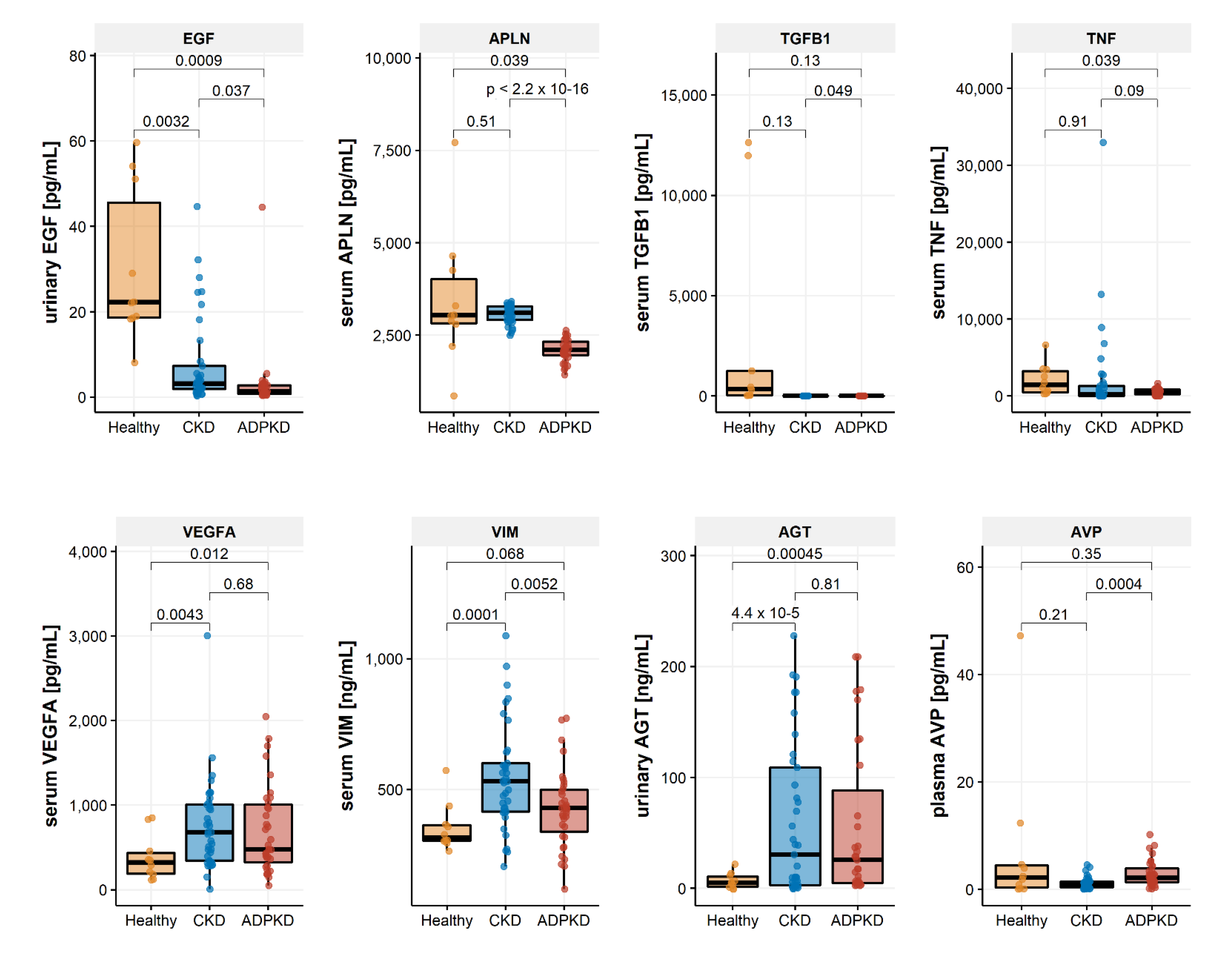
2.3. Biomarker Regulation in ADPKD
2.4. Biomarker Association with eGFR and Age
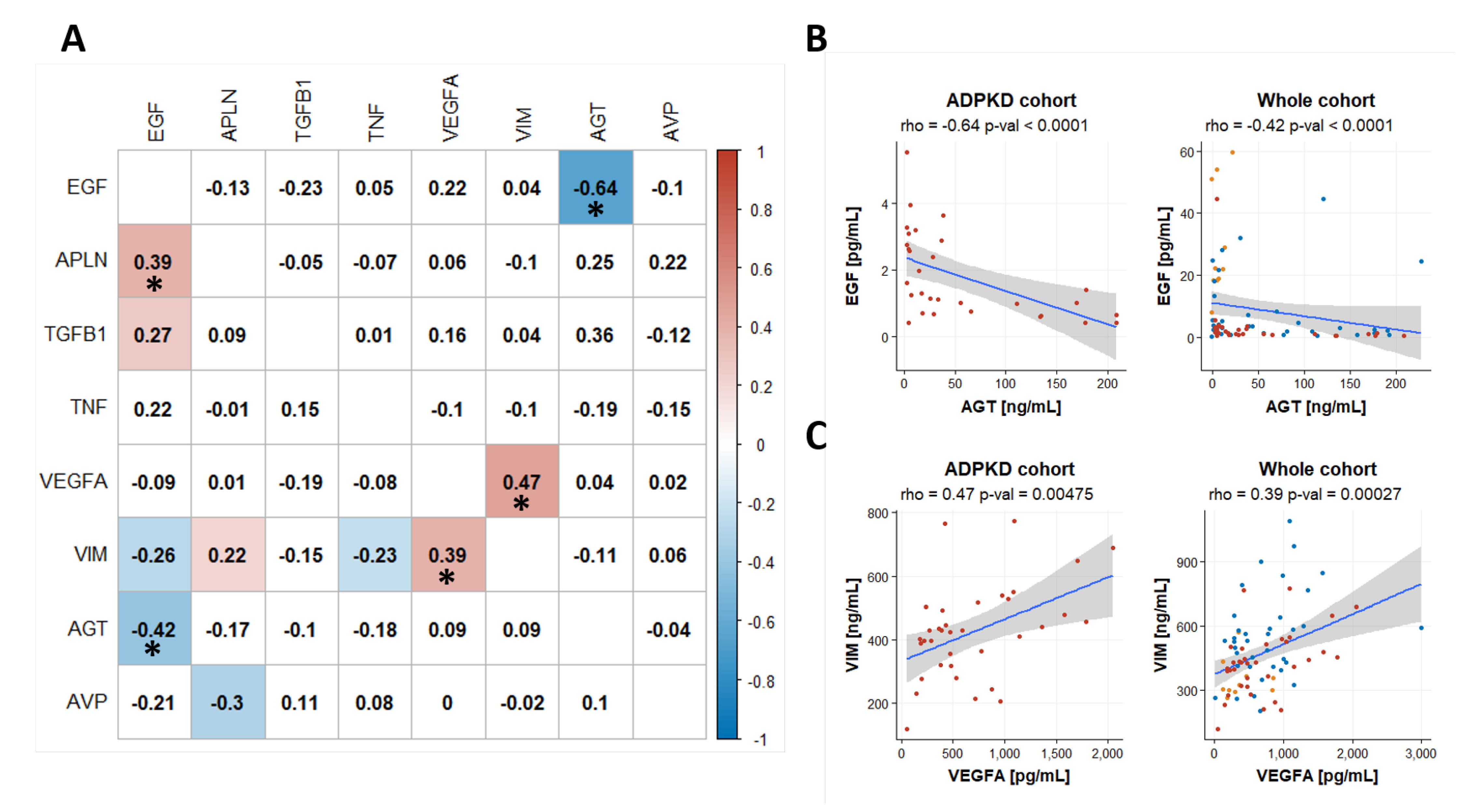
2.5. Coregulation of Biomakers
3. Discussion
4. Methods
4.1. Study Design and Populations
4.2. ADPKD Molecular Model Construction
4.3. Biomarker Panel Selection
4.4. Biomarker Measurements
4.5. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Menezes, L.F.; Germino, G.G. Systems Biology of Polycystic Kidney Disease: A Critical Review. WIREs Syst. Biol. Med. 2015, 7, 39–52. [Google Scholar] [CrossRef]
- Pei, Y.; Watnick, T. Diagnosis and Screening of Autosomal Dominant Polycystic Kidney Disease. Adv. Chronic Kidney Dis. 2010, 17, 140–152. [Google Scholar] [CrossRef]
- Chebib, F.T.; Torres, V.E. Recent Advances in the Management of Autosomal Dominant Polycystic Kidney Disease. Clin. J. Am. Soc. Nephrol. 2018, 13, 1765–1776. [Google Scholar] [CrossRef] [PubMed]
- Pei, Y.; Watnick, T.; He, N.; Wang, K.; Liang, Y.; Parfrey, P.; Germino, G.; George-Hyslop, P.S. Somatic PKD2 Mutations in Individual Kidney and Liver Cysts Support a “Two-Hit” Model of Cystogenesis in Type 2 Autosomal Dominant Polycystic Kidney Disease. J. Am. Soc. Nephrol. 1999, 10, 1524–1529. [Google Scholar] [CrossRef] [PubMed]
- Lanktree, M.B.; Haghighi, A.; di Bari, I.; Song, X.; Pei, Y. Insights into Autosomal Dominant Polycystic Kidney Disease from Genetic Studies. Clin. J. Am. Soc. Nephrol. 2021, 16, 790–799. [Google Scholar] [CrossRef]
- Ong, A.C.M.; Ward, C.J.; Butler, R.J.; Biddolph, S.; Bowker, C.; Torra, R.; Pei, Y.; Harris, P.C. Coordinate Expression of the Autosomal Dominant Polycystic Kidney Disease Proteins, Polycystin-2 And Polycystin-1, in Normal and Cystic Tissue. Am. J. Pathol. 1999, 154, 1721–1729. [Google Scholar] [CrossRef]
- Torres, V.E.; Chapman, A.B.; Devuyst, O.; Gansevoort, R.T.; Grantham, J.J.; Higashihara, E.; Perrone, R.D.; Krasa, H.B.; Ouyang, J.; Czerwiec, F.S. Tolvaptan in Patients with Autosomal Dominant Polycystic Kidney Disease. N. Engl. J. Med. 2012, 367, 2407–2418. [Google Scholar] [CrossRef]
- Gansevoort, R.T.; Meijer, E.; Chapman, A.B.; Czerwiec, F.S.; Devuyst, O.; Grantham, J.J.; Higashihara, E.; Krasa, H.B.; Ouyang, J.; Perrone, R.D.; et al. Albuminuria and Tolvaptan in Autosomal-Dominant Polycystic Kidney Disease: Results of the TEMPO 3:4 Trial. Nephrol. Dial. Transplant. 2016, 31, 1887–1894. [Google Scholar] [CrossRef]
- Amberger, J.S.; Bocchini, C.A.; Schiettecatte, F.; Scott, A.F.; Hamosh, A. OMIM.Org: Online Mendelian Inheritance in Man (OMIM®), an Online Catalog of Human Genes and Genetic Disorders. Nucleic Acids Res. 2015, 43, D789–D798. [Google Scholar] [CrossRef] [PubMed]
- Law, V.; Knox, C.; Djoumbou, Y.; Jewison, T.; Guo, A.C.; Liu, Y.; Maciejewski, A.; Arndt, D.; Wilson, M.; Neveu, V.; et al. DrugBank 4.0: Shedding New Light on Drug Metabolism. Nucleic Acids Res. 2014, 42, D1091–D1097. [Google Scholar] [CrossRef] [PubMed]
- Malas, T.B.; Formica, C.; Leonhard, W.N.; Rao, P.; Granchi, Z.; Roos, M.; Peters, D.J.M.; ’t Hoen, P.A.C. Meta-Analysis of Polycystic Kidney Disease Expression Profiles Defines Strong Involvement of Injury Repair Processes. Am. J. Physiol. Ren. Physiol. 2017, 312, F806–F817. [Google Scholar] [CrossRef]
- Fechete, R.; Heinzel, A.; Söllner, J.; Perco, P.; Lukas, A.; Mayer, B. Using Information Content for Expanding Human Protein Coding Gene Interaction Networks. J. Comput. Sci. Syst. Biol. 2013, 6, 073–082. [Google Scholar]
- Shillingford, J.M.; Murcia, N.S.; Larson, C.H.; Low, S.H.; Hedgepeth, R.; Brown, N.; Flask, C.A.; Novick, A.C.; Goldfarb, D.A.; Kramer-Zucker, A.; et al. The MTOR Pathway Is Regulated by Polycystin-1, and Its Inhibition Reverses Renal Cystogenesis in Polycystic Kidney Disease. Proc. Natl. Acad. Sci. USA 2006, 103, 5466–5471. [Google Scholar] [CrossRef]
- Bhunia, A.K.; Piontek, K.; Boletta, A.; Liu, L.; Qian, F.; Xu, P.-N.; Germino, F.J.; Germino, G.G. PKD1 Induces P21waf1 and Regulation of the Cell Cycle via Direct Activation of the JAK-STAT Signaling Pathway in a Process Requiring PKD2. Cell 2002, 109, 157–168. [Google Scholar] [CrossRef]
- Kim, D.Y.; Park, J.H. Genetic mechanisms of ADPKD. In Cystogenesis; Park, J.H., Ahn, C., Eds.; Advances in Experimental Medicine and Biology; Springer: Singapore, 2016; pp. 13–22. ISBN 978-981-10-2041-4. [Google Scholar]
- O’Dowd, B.F.; Heiber, M.; Chan, A.; Heng, H.H.Q.; Tsui, L.-C.; Kennedy, J.L.; Shi, X.; Petronis, A.; George, S.R.; Nguyen, T. A Human Gene That Shows Identity with the Gene Encoding the Angiotensin Receptor Is Located on Chromosome 11. Gene 1993, 136, 355–360. [Google Scholar] [CrossRef]
- Tatemoto, K.; Hosoya, M.; Habata, Y.; Fujii, R.; Kakegawa, T.; Zou, M.-X.; Kawamata, Y.; Fukusumi, S.; Hinuma, S.; Kitada, C.; et al. Isolation and Characterization of a Novel Endogenous Peptide Ligand for the Human APJ Receptor. Biochem. Biophys. Res. Commun. 1998, 251, 471–476. [Google Scholar] [CrossRef]
- Huang, Z.; Wu, L.; Chen, L. Apelin/APJ System: A Novel Potential Therapy Target for Kidney Disease. J. Cell. Physiol. 2018, 233, 3892–3900. [Google Scholar] [CrossRef]
- Zhang, B.; Wang, W.; Wang, H.; Yin, J.; Zeng, X. Promoting Effects of the Adipokine, Apelin, on Diabetic Nephropathy. PLoS ONE 2013, 8, e60457. [Google Scholar] [CrossRef]
- Guo, C.; Liu, Y.; Zhao, W.; Wei, S.; Zhang, X.; Wang, W.; Zeng, X. Apelin Promotes Diabetic Nephropathy by Inducing Podocyte Dysfunction via Inhibiting Proteasome Activities. J. Cell. Mol. Med. 2015, 19, 2273–2285. [Google Scholar] [CrossRef]
- Liu, Y.; Zhang, J.; Wang, Y.; Zeng, X. Apelin Involved in Progression of Diabetic Nephropathy by Inhibiting Autophagy in Podocytes. Cell Death Dis. 2017, 8, e3006. [Google Scholar] [CrossRef]
- Kocer, D.; Karakukcu, C.; Ozturk, F.; Eroglu, E.; Kocyigit, I. Evaluation of Fibrosis Markers: Apelin and Transforming Growth Factor-Β1 in Autosomal Dominant Polycystic Kidney Disease Patients. Ther. Apher. Dial. Off. Peer-Rev. J. Int. Soc. Apher. Jpn. Soc. Apher. Jpn. Soc. Dial. Ther. 2016, 20, 517–522. [Google Scholar] [CrossRef]
- Lacquaniti, A.; Chirico, V.; Lupica, R.; Buemi, A.; Loddo, S.; Caccamo, C.; Salis, P.; Bertani, T.; Buemi, M. Apelin and Copeptin: Two Opposite Biomarkers Associated with Kidney Function Decline and Cyst Growth in Autosomal Dominant Polycystic Kidney Disease. Peptides 2013, 49, 1–8. [Google Scholar] [CrossRef]
- Melenhorst, W.B.W.H.; Mulder, G.M.; Xi, Q.; Hoenderop, J.G.J.; Kimura, K.; Eguchi, S.; van Goor, H. Epidermal Growth Factor Receptor Signaling in the Kidney. Hypertension 2008, 52, 987–993. [Google Scholar] [CrossRef] [PubMed]
- Torres, V.E.; Harris, P.C. Mechanisms of Disease: Autosomal Dominant and Recessive Polycystic Kidney Diseases. Nat. Rev. Nephrol. 2006, 2, 40–55. [Google Scholar] [CrossRef]
- Park, H.C.; Kang, A.-Y.; Jang, J.Y.; Kim, H.; Han, M.; Oh, K.-H.; Kim, S.H.; Noh, J.W.; Cheong, H.I.; Hwang, Y.-H.; et al. Increased Urinary Angiotensinogen/Creatinine (AGT/Cr) Ratio May Be Associated with Reduced Renal Function in Autosomal Dominant Polycystic Kidney Disease Patients. BMC Nephrol. 2015, 16, 86. [Google Scholar] [CrossRef]
- Harskamp, L.R.; Gansevoort, R.T.; Boertien, W.E.; van Oeveren, W.; Engels, G.E.; van Goor, H.; Meijer, E. Urinary EGF Receptor Ligand Excretion in Patients with Autosomal Dominant Polycystic Kidney Disease and Response to Tolvaptan. Clin. J. Am. Soc. Nephrol. 2015, 10, 1749–1756. [Google Scholar] [CrossRef]
- Koppelstaetter, C.; Leierer, J.; Rudnicki, M.; Kerschbaum, J.; Kronbichler, A.; Melk, A.; Mayer, G.; Perco, P. Computational Drug Screening Identifies Compounds Targeting Renal Age-Associated Molecular Profiles. Comput. Struct. Biotechnol. J. 2019, 17, 843–853. [Google Scholar] [CrossRef]
- Rudnicki, M.; Perco, P.; Enrich, J.; Eder, S.; Heininger, D.; Bernthaler, A.; Wiesinger, M.; Sarközi, R.; Noppert, S.-J.; Schramek, H.; et al. Hypoxia Response and VEGF-A Expression in Human Proximal Tubular Epithelial Cells in Stable and Progressive Renal Disease. Lab. Investig. 2009, 89, 337–346. [Google Scholar] [CrossRef] [PubMed]
- Ju, W.; Nair, V.; Smith, S.; Zhu, L.; Shedden, K.; Song, P.X.K.; Mariani, L.H.; Eichinger, F.H.; Berthier, C.C.; Randolph, A.; et al. Tissue Transcriptome-Driven Identification of Epidermal Growth Factor as a Chronic Kidney Disease Biomarker. Sci. Transl. Med. 2015, 7, 316ra193. [Google Scholar] [CrossRef] [PubMed]
- Kobori, H.; Ohashi, N.; Katsurada, A.; Miyata, K.; Satou, R.; Saito, T.; Yamamoto, T. Urinary Angiotensinogen as a Potential Biomarker of Severity of Chronic Kidney Diseases. J. Am. Soc. Hypertens. 2008, 2, 349–354. [Google Scholar] [CrossRef]
- Loghman-Adham, M.; Soto, C.E.; Inagami, T.; Cassis, L. The Intrarenal Renin-Angiotensin System in Autosomal Dominant Polycystic Kidney Disease. Am. J. Physiol. Ren. Physiol. 2004, 287, F775–F788. [Google Scholar] [CrossRef] [PubMed]
- Hian, C.K.; Lee, C.L.; Thomas, W. Renin-Angiotensin-Aldosterone System Antagonism and Polycystic Kidney Disease Progression. Nephron 2016, 134, 59–63. [Google Scholar] [CrossRef]
- Kocyigit, I.; Yilmaz, M.I.; Unal, A.; Ozturk, F.; Eroglu, E.; Yazici, C.; Orscelik, O.; Sipahioglu, M.H.; Tokgoz, B.; Oymak, O. A Link between the Intrarenal Renin Angiotensin System and Hypertension in Autosomal Dominant Polycystic Kidney Disease. Am. J. Nephrol. 2013, 38, 218–225. [Google Scholar] [CrossRef] [PubMed]
- Kocyigit, I.; Eroglu, E. Urinary Angiotensinogen Can Be Useful for Screening Patients with Polycystic Kidney Disease. Kidney Int. 2014, 85, 714. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Zheleznova, N.N.; Wilson, P.D.; Staruschenko, A. Epidermal Growth Factor-Mediated Proliferation and Sodium Transport in Normal and PKD Epithelial Cells. Biochim. Biophys. Acta 2011, 1812, 1301–1313. [Google Scholar] [CrossRef]
- Yang, C.W.; Ahn, H.J.; Kim, W.Y.; Shin, M.J.; Kim, S.K.; Park, J.H.; Kim, Y.O.; Kim, Y.S.; Kim, J.; Bang, B.K. Influence of the Renin-Angiotensin System on Epidermal Growth Factor Expression in Normal and Cyclosporine-Treated Rat Kidney. Kidney Int. 2001, 60, 847–857. [Google Scholar] [CrossRef][Green Version]
- Zeng, F.; Harris, R.C. Epidermal Growth Factor, from Gene Organization to Bedside. Semin. Cell Dev. Biol. 2014, 28, 2–11. [Google Scholar] [CrossRef]
- Boertien, W.E.; Meijer, E.; Li, J.; Bost, J.E.; Struck, J.; Flessner, M.F.; Gansevoort, R.T.; Torres, V.E. Consortium for Radiologic Imaging Studies of Polycystic Kidney Disease CRISP Relationship of Copeptin, a Surrogate Marker for Arginine Vasopressin, with Change in Total Kidney Volume and GFR Decline in Autosomal Dominant Polycystic Kidney Disease: Results from the CRISP Cohort. Am. J. Kidney Dis. Off. J. Natl. Kidney Found. 2013, 61, 420–429. [Google Scholar] [CrossRef]
- Bello-Reuss, E.; Holubec, K.; Rajaraman, S. Angiogenesis in Autosomal-Dominant Polycystic Kidney Disease. Kidney Int. 2001, 60, 37–45. [Google Scholar] [CrossRef]
- Kraus, A.; Peters, D.J.M.; Klanke, B.; Weidemann, A.; Willam, C.; Schley, G.; Kunzelmann, K.; Eckardt, K.-U.; Buchholz, B. HIF-1α Promotes Cyst Progression in a Mouse Model of Autosomal Dominant Polycystic Kidney Disease. Kidney Int. 2018, 94, 887–899. [Google Scholar] [CrossRef]
- Bhattarai, D.; Xu, X.; Lee, K. Hypoxia-Inducible Factor-1 (HIF-1) Inhibitors from the Last Decade (2007 to 2016): A “Structure–Activity Relationship” Perspective. Med. Res. Rev. 2018, 38, 1404–1442. [Google Scholar] [CrossRef]
- Roskoski, R. Vascular Endothelial Growth Factor (VEGF) and VEGF Receptor Inhibitors in the Treatment of Renal Cell Carcinomas. Pharmacol. Res. 2017, 120, 116–132. [Google Scholar] [CrossRef]
- Chea, S.W.; Lee, K.-B. TGF-β Mediated Epithelial-Mesenchymal Transition in Autosomal Dominant Polycystic Kidney Disease. Yonsei Med. J. 2009, 50, 105–111. [Google Scholar] [CrossRef][Green Version]
- Bauer, A.; Mylroie, H.; Thornton, C.C.; Calay, D.; Birdsey, G.M.; Kiprianos, A.P.; Wilson, G.K.; Soares, M.P.; Yin, X.; Mayr, M.; et al. Identification of Cyclins A1, E1 and Vimentin as Downstream Targets of Heme Oxygenase-1 in Vascular Endothelial Growth Factor-Mediated Angiogenesis. Sci. Rep. 2016, 6, 29417. [Google Scholar] [CrossRef]
- Raman, A.; Parnell, S.C.; Zhang, Y.; Reif, G.A.; Dai, Y.; Khanna, A.; Daniel, E.; White, C.; Vivian, J.L.; Wallace, D.P. Periostin Overexpression in Collecting Ducts Accelerates Renal Cyst Growth and Fibrosis in Polycystic Kidney Disease. Am. J. Physiol. Ren. Physiol. 2018, 315, F1695–F1707. [Google Scholar] [CrossRef]
- Ma, M.; Yung, S.; Chan, T. MTOR Inhibition and Kidney Diseases. Transplantation 2018, 102, S32–S40. [Google Scholar] [CrossRef]
- Xue, C.; Dai, B.; Mei, C. Long-Term Treatment with Mammalian Target of Rapamycin Inhibitor Does Not Benefit Patients with Autosomal Dominant Polycystic Kidney Disease: A Meta-Analysis. Nephron Clin. Pract. 2013, 124, 10–16. [Google Scholar] [CrossRef] [PubMed]
- Kocyigit, I.; Kaya, M.G.; Orscelik, O.; Kaya, C.; Akpek, M.; Zengin, H.; Sipahioglu, M.H.; Unal, A.; Yilmaz, M.I.; Tokgoz, B.; et al. Early Arterial Stiffness and Inflammatory Bio-Markers in Normotensive Polycystic Kidney Disease Patients. Am. J. Nephrol. 2012, 36, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Raptis, V.; Bakogiannis, C.; Loutradis, C.; Boutou, A.K.; Sioulis, A.; Balaskas, E.; Zebekakis, P.; Sarafidis, P.A. Serum Fas Ligand, Serum Myostatin and Urine TGF-Β1 Are Elevated in Autosomal Dominant Polycystic Kidney Disease Patients with Impaired and Preserved Renal Function. Kidney Blood Press. Res. 2018, 43, 744–754. [Google Scholar] [CrossRef]
- Bankir, L.; Bichet, D.G.; Morgenthaler, N.G. Vasopressin: Physiology, Assessment and Osmosensation. J. Intern. Med. 2017, 282, 284–297. [Google Scholar] [CrossRef]
- Song, X.; Di Giovanni, V.; He, N.; Wang, K.; Ingram, A.; Rosenblum, N.D.; Pei, Y. Systems Biology of Autosomal Dominant Polycystic Kidney Disease (ADPKD): Computational Identification of Gene Expression Pathways and Integrated Regulatory Networks. Hum. Mol. Genet. 2009, 18, 2328–2343. [Google Scholar] [CrossRef]
- Bastos, A.P.; Onuchic, L.F. Molecular and Cellular Pathogenesis of Autosomal Dominant Polycystic Kidney Disease. Braz. J. Med. Biol. Res. 2011, 44, 606–617. [Google Scholar] [CrossRef] [PubMed]
- Torres, V.E.; Harris, P.C. Strategies Targeting CAMP Signaling in the Treatment of Polycystic Kidney Disease. J. Am. Soc. Nephrol. 2014, 25, 18–32. [Google Scholar] [CrossRef]
- Harris, P.C.; Torres, V.E. Genetic Mechanisms and Signaling Pathways in Autosomal Dominant Polycystic Kidney Disease. J. Clin. Investig. 2014, 124, 2315–2324. [Google Scholar] [CrossRef]
- Seeger-Nukpezah, T.; Geynisman, D.M.; Nikonova, A.S.; Benzing, T.; Golemis, E.A. The Hallmarks of Cancer: Relevance to the Pathogenesis of Polycystic Kidney Disease. Nat. Rev. Nephrol. 2015, 11, 515–534. [Google Scholar] [CrossRef]
- Mangolini, A. Role of Calcium in Polycystic Kidney Disease: From Signaling to Pathology. World J. Nephrol. 2016, 5, 76. [Google Scholar] [CrossRef]
- Hama, T.; Park, F. Heterotrimeric G Protein Signaling in Polycystic Kidney Disease. Physiol. Genom. 2016, 48, 429–445. [Google Scholar] [CrossRef]
- Bader, G.D.; Hogue, C.W.V. An Automated Method for Finding Molecular Complexes in Large Protein Interaction Networks. BMC Bioinform. 2003, 4, 2. [Google Scholar] [CrossRef] [PubMed]
- Mayer, B.; Heinzel, A.; Lukas, A.; Perco, P. Predictive Biomarkers for Linking Disease Pathology and Drug Effect. Curr. Pharm. Des. 2017, 23, 29–54. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| ADPKD [n = 37] | CKD [n = 37] | Controls [n = 10] | |
|---|---|---|---|
| Age (years) | |||
| Min | 26 | 24 | 26 |
| Max | 82 | 82 | 62 |
| Mean (SD) | 54 ± 13 | 54 ± 13 | 47 ± 11 |
| Gender, n (%) | |||
| Female | 19 (51.4%) | 19 (51.4%) | 6 (60.0%) |
| eGFR (mL/min/1.73 m2) | |||
| Min | 3.8 | 6.6 | 71.8 |
| Max | 119.5 | 175.9 | 129.1 |
| Mean (SD) | 30.4 ± 24.9 | 36.1 ± 32.8 | 90.1 ± 16.7 |
| CKD stage, n (%) | |||
| G1 (≥90 mL/min/1.73 m2) | 1 (2.7%) | 2 (5.4%) | 3 (30%) |
| G2 (60–89 mL/min/1.73 m2) | 5 (13.5%) | 4 (10.8%) | 7 (70%) |
| G3 (30–59 mL/min/1.73 m2) | 6 (16.2%) | 7 (18.9%) | 0 (0%) |
| G4 (15–29 mL/min/1.73 m2) | 14 (37.8%) | 17 (45.9%) | 0 (0%) |
| G5 (<15 mL/min/1.73 m2) | 11 (29.7%) | 7 (18.9%) | 0 (0%) |
| Albuminuria stage, n (%) | |||
| A1 (<30 mg/g) | 8 (21.6%) | 7 (18.9%) | 10 (100%) |
| A2 (30–300 mg/g) | 19 (51.4%) | 10 (27.0%) | 0 (0%) |
| A3 (>300 mg/g) | 6 (16.2%) | 15 (40.5%) | 0 (0%) |
| NA | 4 (10.8%) | 5 (13.5%) | 0 (0%) |
| RAAS inhibitor use, n (%) | 25 (67.6%) | 19 (51.3%) | NA |
| Symbol | Gene Name | Molecular Function/Pathway Membership | Sample Matrix | Dilution | ELISA (Company/Cat No.) |
|---|---|---|---|---|---|
| AGT | angiotensinogen | precursor of angiotensin II; renin angiotensin aldosterone signaling | urine | undil. | Cloud-Clone Corp./SEA797Hu |
| APLN | apelin | endogenous ligand for the G-protein apelin receptor; angiogenesis | serum | 1:5 | Cloud-Clone Corp./CED065Hu |
| AVP | arginine vasopressin | hormonal growth factor; anti-diuretic activity | plasma | undil | Alpco/ 74-VSPHU-E01.1 |
| EGF | epidermal growth factor | growth factor; cell growth, proliferation, and differentiation | urine | 1:20 | R&D Systems/ DEG00 |
| TGFB1 | transforming growth factor beta 1 | growth factor; cell growth, proliferation, and differentiation | serum | 1:3 | Promocell/ PromoKine/ PK-EL-63506 |
| TNF | tumor necrosis factor | proinflammatory cytokine; inflammation, cell differentiation, apoptosis | serum | undil. | Promocell/ PromoKine/ PK-EL-63707 |
| VEGFA | vascular endothelial growth factor A | growth factor; angiogenesis | serum | 1:2 | R&D Systems/ DVE00 |
| VIM | vimentin | type III filament protein; maintenance of cell shape and integrity of the cytoplasm | serum | 1:500 | Cusabio Biotech Co.LTD/ CSB-E08982h |
| ADPKD [n = 37] | CKD [n = 37] | Healthy [n = 10] | p-Value (ANOVA) | |
|---|---|---|---|---|
| EGF [pg/mL] | 3.10 (7.66) | 7.80 (10.66) | 30.20 (17.94) | <0.001 |
| APLN [ng/mL] | 2088.18 (298.81) | 3074.12 (246.03) | 3472.47 (1813.75) | <0.001 |
| TGFB1 [pg/mL] | 0.54 (1.42) | 0.03 (0.09) | 2971.93 (5303.70) | <0.001 |
| TNF [ng/mL] | 509.73 (389.43) | 2204.97 (5904.43) | 2067.02 (2031.68) | 0.189 |
| VEGFA [pg/mL] | 701.48 (516.17) | 753.39 (533.95) | 376.85 (267.43) | 0.113 |
| VIM [ng/mL] | 426.19 (148.68) | 546.84 (202.85) | 353.27 (90.72) | 0.001 |
| AGT [ng/mL] | 56.21 (69.50) | 60.22 (69.47) | 6.58 (7.14) | 0.068 |
| AVP [pg/mL] | 2.85 (2.41) | 1.16 (1.09) | 7.34 (14.48) | 0.005 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Leierer, J.; Perco, P.; Hofer, B.; Eder, S.; Dzien, A.; Kerschbaum, J.; Rudnicki, M.; Mayer, G. Coregulation Analysis of Mechanistic Biomarkers in Autosomal Dominant Polycystic Kidney Disease. Int. J. Mol. Sci. 2021, 22, 6885. https://doi.org/10.3390/ijms22136885
Leierer J, Perco P, Hofer B, Eder S, Dzien A, Kerschbaum J, Rudnicki M, Mayer G. Coregulation Analysis of Mechanistic Biomarkers in Autosomal Dominant Polycystic Kidney Disease. International Journal of Molecular Sciences. 2021; 22(13):6885. https://doi.org/10.3390/ijms22136885
Chicago/Turabian StyleLeierer, Johannes, Paul Perco, Benedikt Hofer, Susanne Eder, Alexander Dzien, Julia Kerschbaum, Michael Rudnicki, and Gert Mayer. 2021. "Coregulation Analysis of Mechanistic Biomarkers in Autosomal Dominant Polycystic Kidney Disease" International Journal of Molecular Sciences 22, no. 13: 6885. https://doi.org/10.3390/ijms22136885
APA StyleLeierer, J., Perco, P., Hofer, B., Eder, S., Dzien, A., Kerschbaum, J., Rudnicki, M., & Mayer, G. (2021). Coregulation Analysis of Mechanistic Biomarkers in Autosomal Dominant Polycystic Kidney Disease. International Journal of Molecular Sciences, 22(13), 6885. https://doi.org/10.3390/ijms22136885