
A Halomanganates(II) with P,P’-Diprotonated Bis(2-Diphenylphosphinophenyl)ether: Wavelength-Excitation Dependence of the Quantum Yield and Role of the Non-Covalent Interactions


Abstract
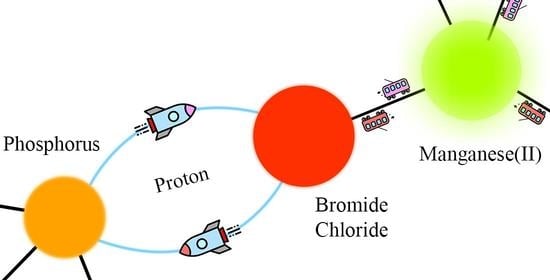
:
1. Introduction
2. Experimental Part
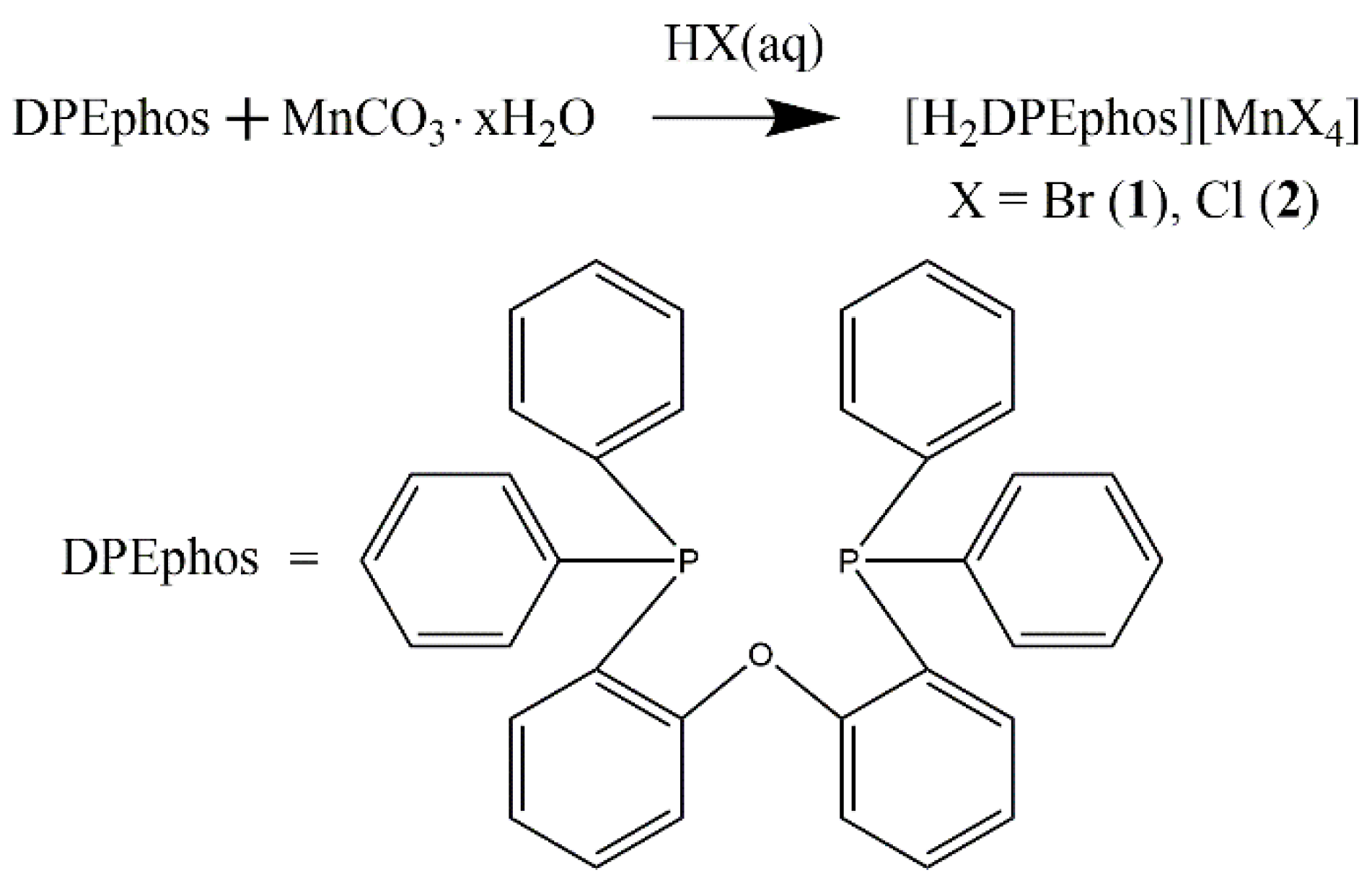
2.1. Synthesis and Characterization Data for 1 and 2
General Procedure for the Synthesis of Complexes 1 and 2
3. Methods
3.1. Structural Descriptions
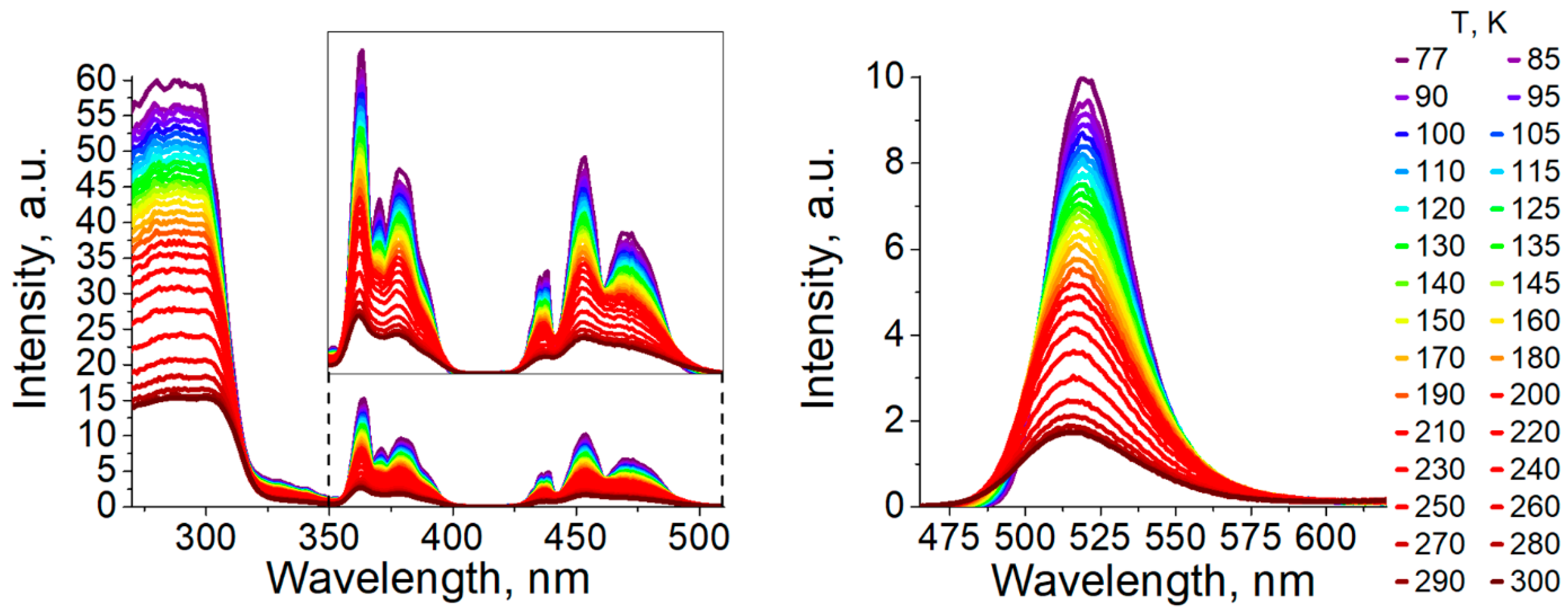
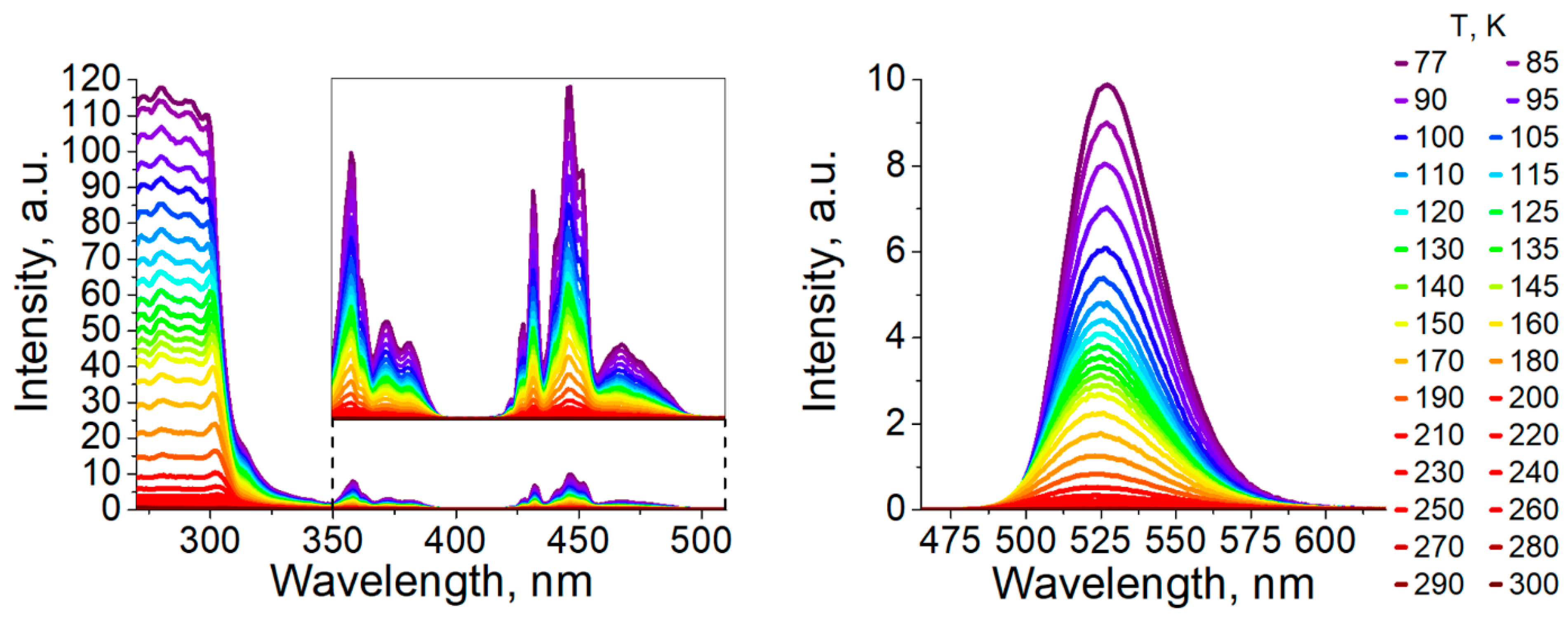
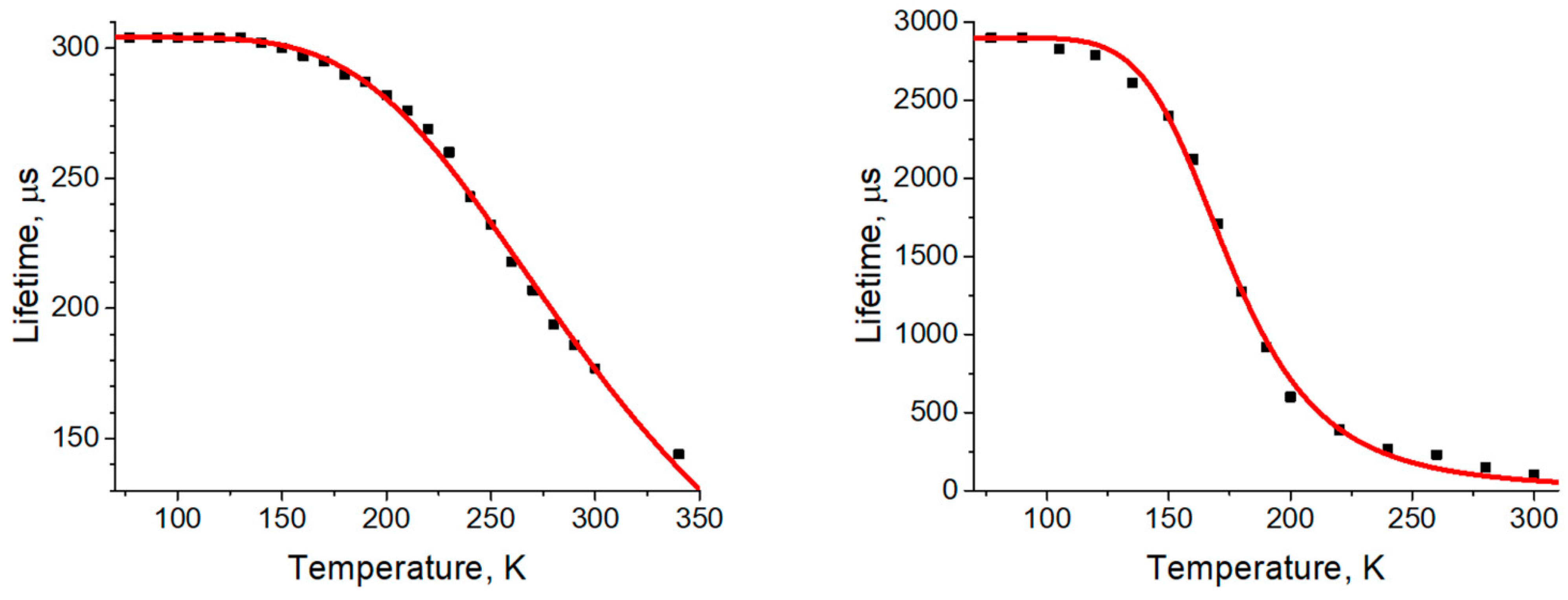
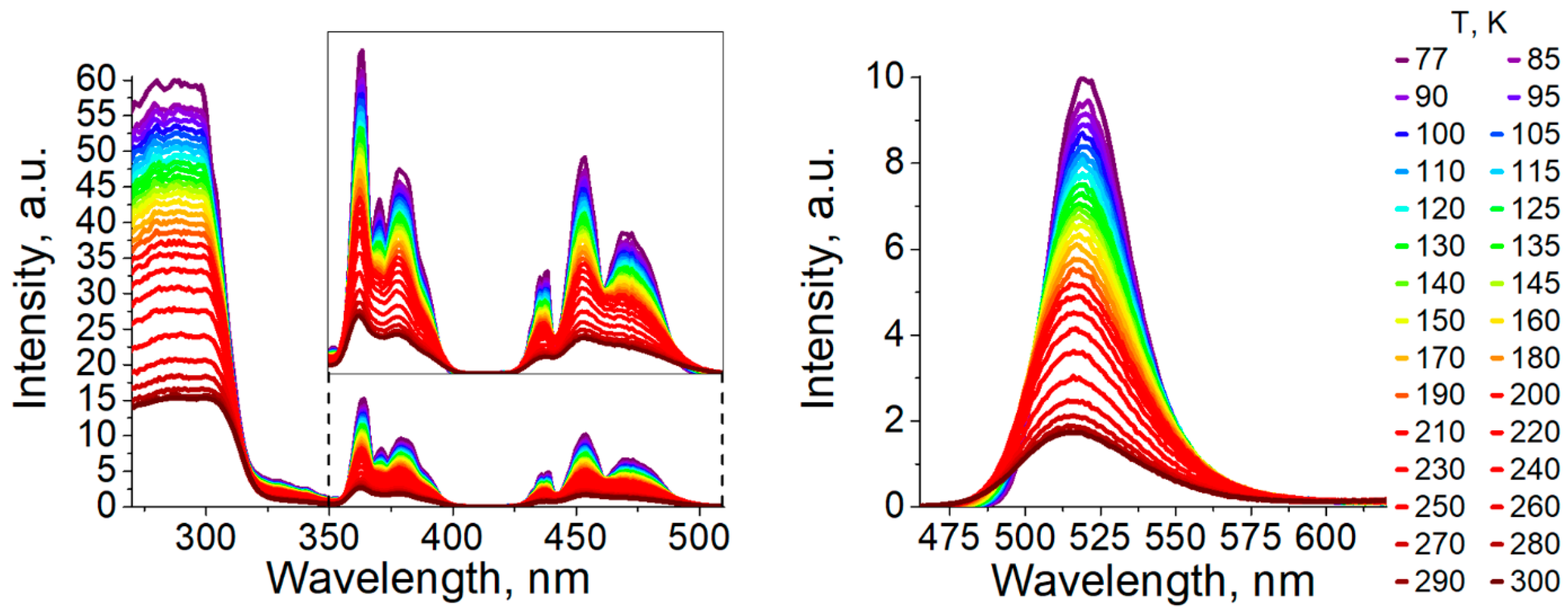
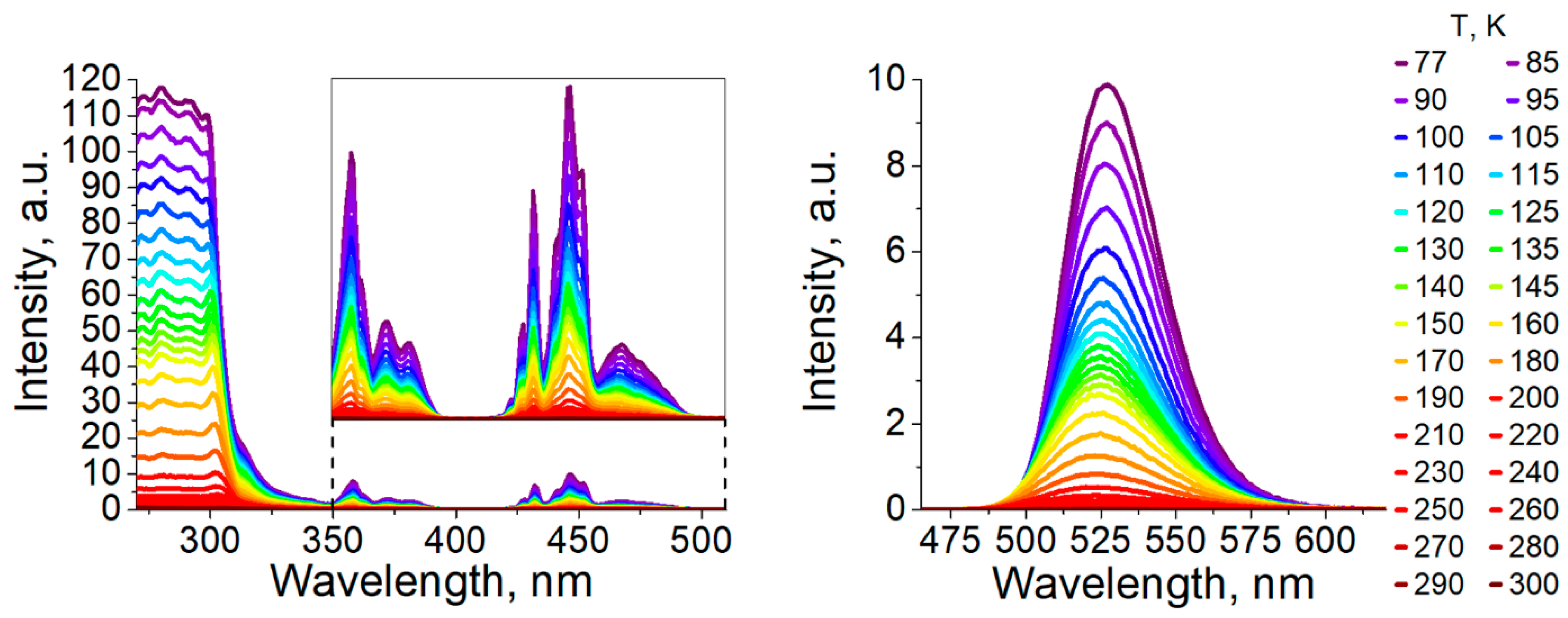
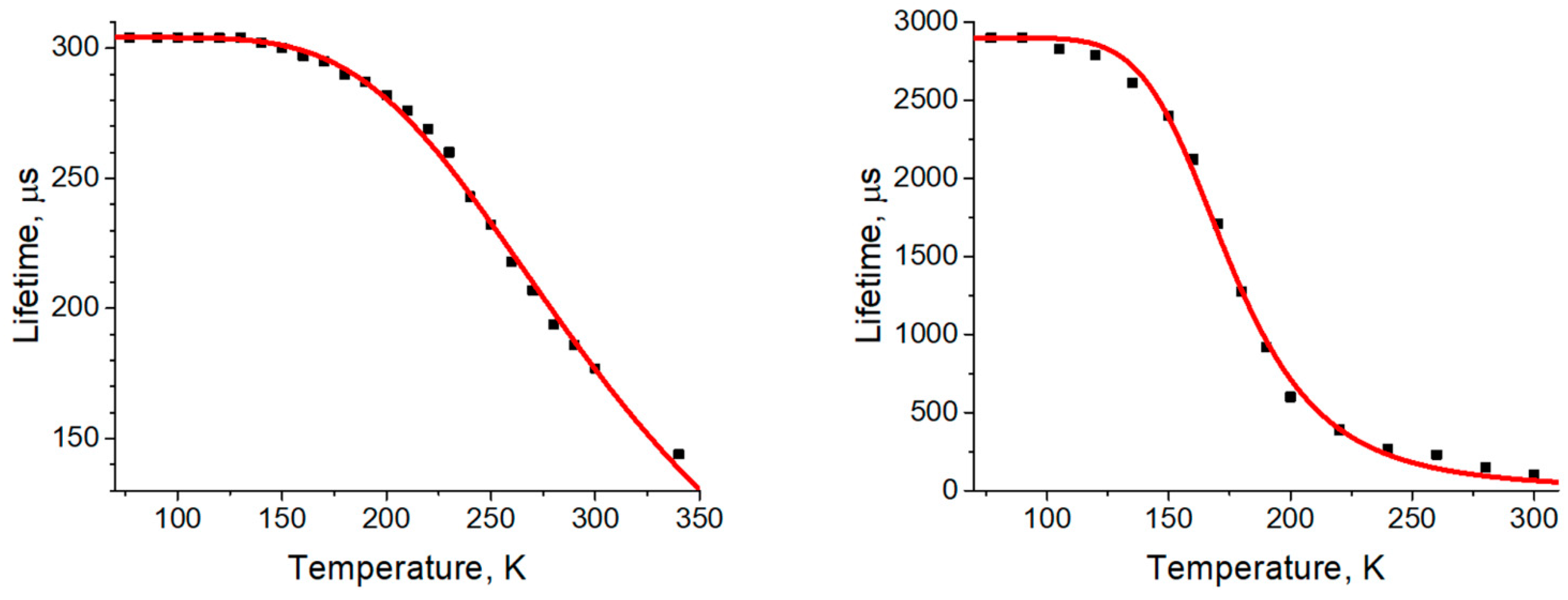
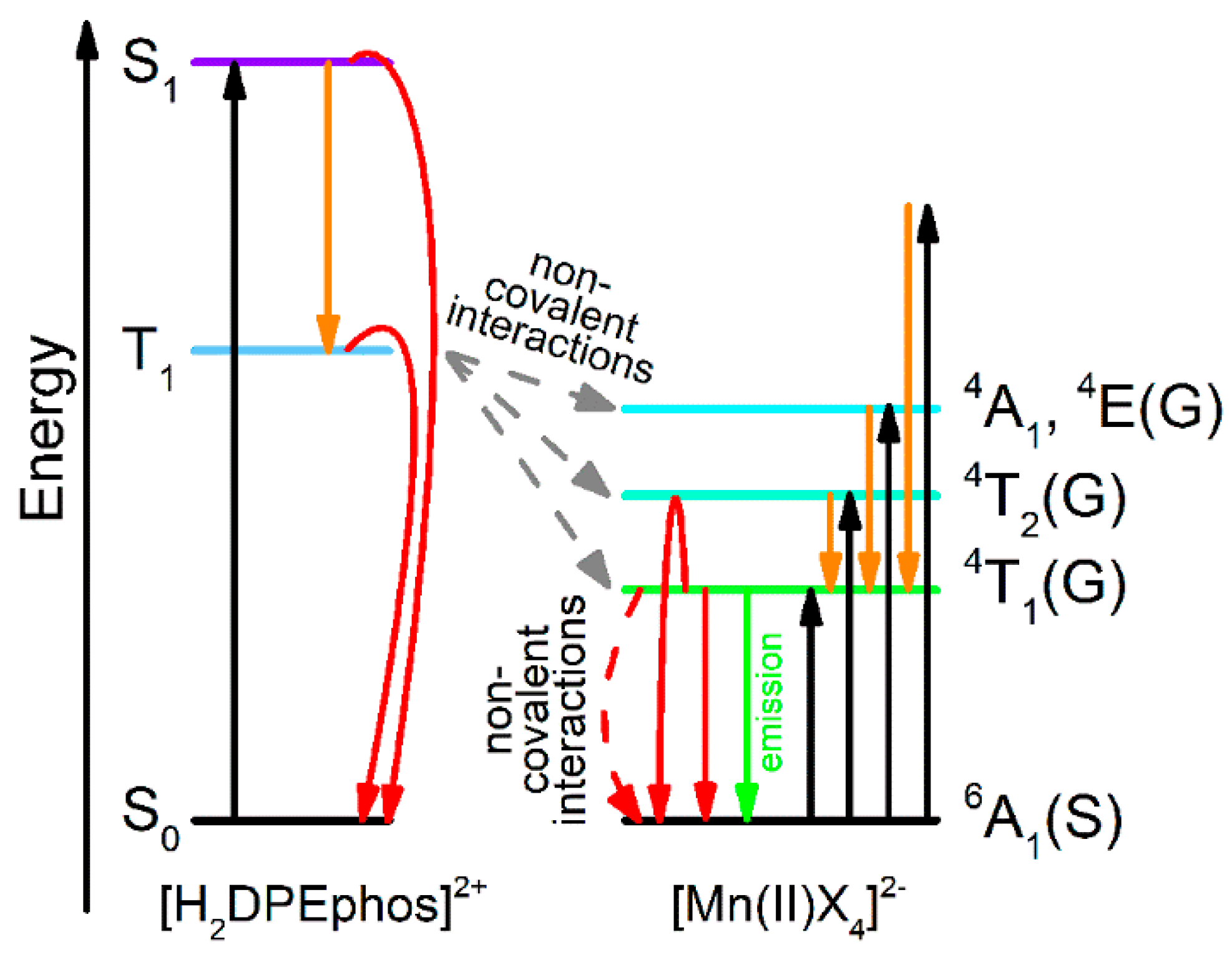
3.2. Photophysical Properties
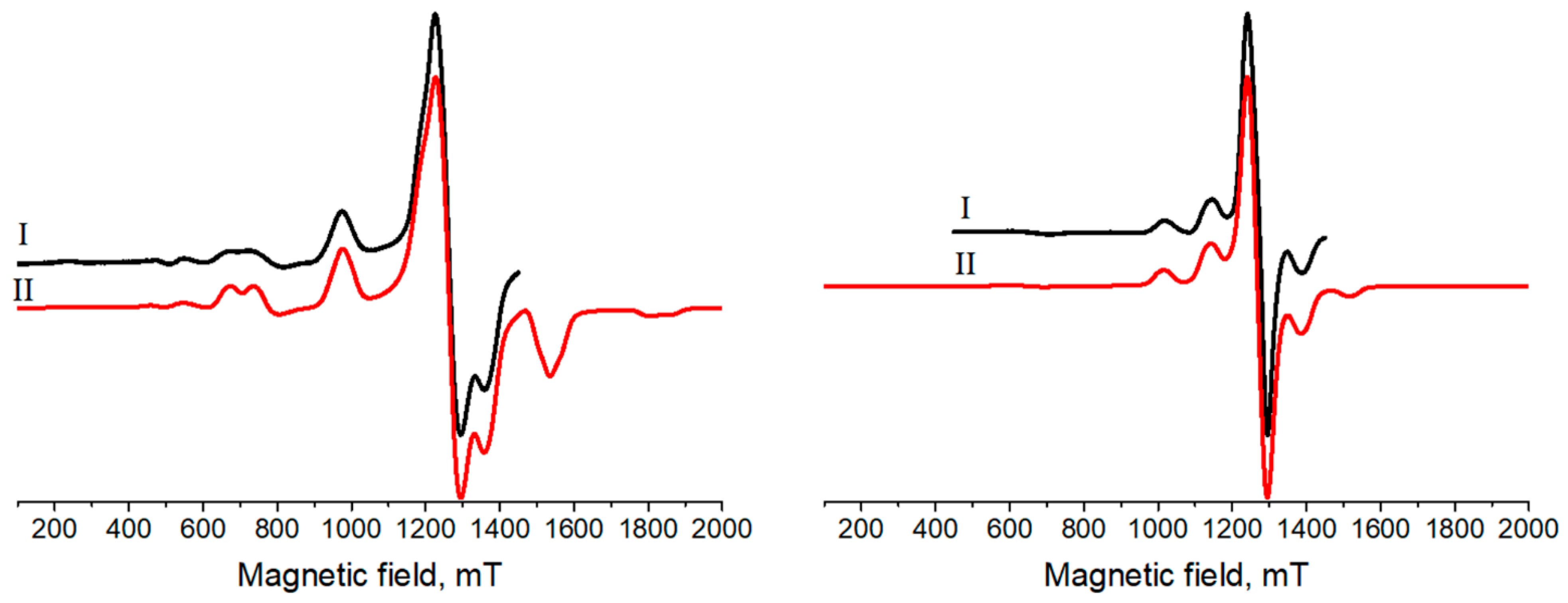
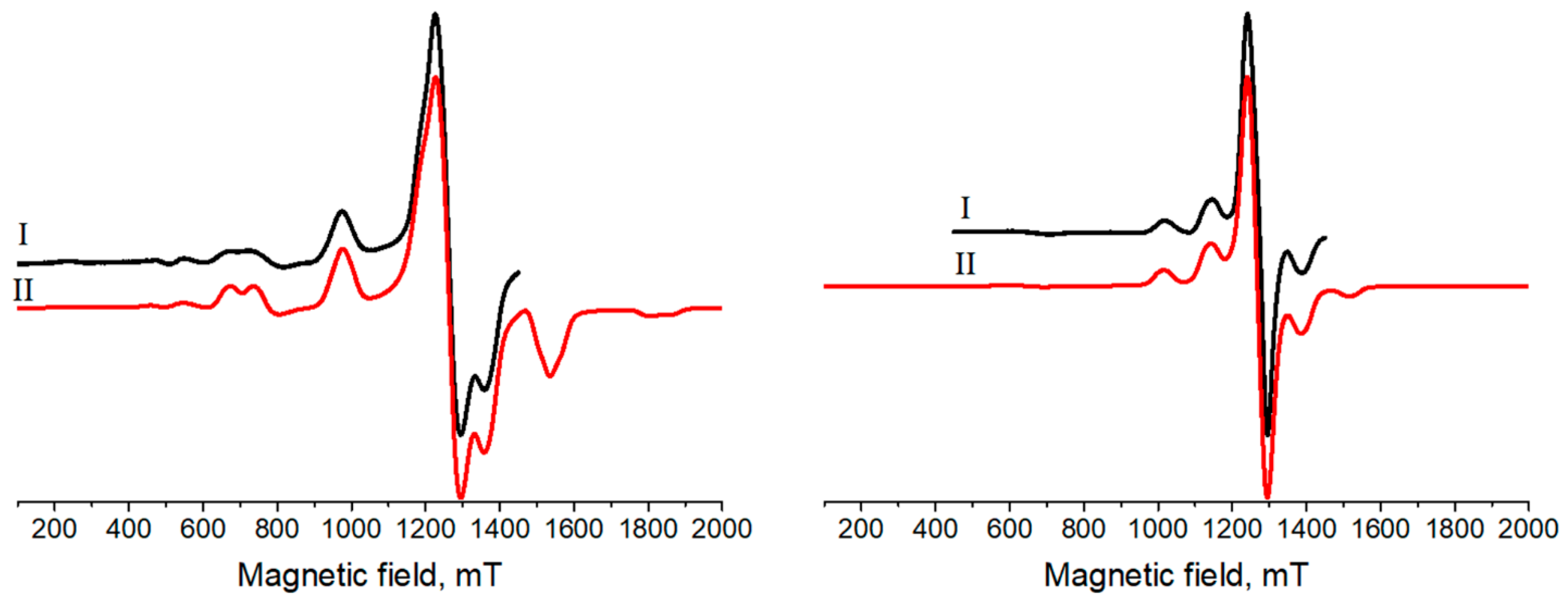
3.3. Electron Paramagnetic Resonance
4. Conclusions
Supplementary Materials
Funding
Acknowledgments
Conflicts of Interest
References
- Hausmann, D.; Kuzmanoski, A.; Feldmann, C. MnBr2/18-crown-6 coordination complexes showing high room temperature luminescence and quantum yield. Dalton Trans. 2016, 45, 6541. [Google Scholar] [CrossRef]
- Morad, V.; Cherninkh, I.; Pottschacher, L.; Shynkarenko, Y.; Yakunin, S.; Kovalenko, M.V. Manganese(II) in tetrahedral halide environment: Factors governing bright green luminescence. Chem. Mater. 2019, 31, 10161. [Google Scholar] [CrossRef] [PubMed]
- Etaiw, S.; Marie, H. Sonochemical nanostructure of Mn(II) supramolecular complex: X-ray structure, sensing and photocatalytic properties. Sens. Actuators B Chem. 2019, 290, 631. [Google Scholar] [CrossRef]
- Godfrey, S.M.; McAuliffe, C.A.; Ndifon, P.T.; Pritchard, R.G. Controlled reaction of molecular oxygen with [MnI2(PPh2Me)2] to form the mixed phosphine–phosphine oxide complex [MnI2(OPPh2Me)(PPh2Me)] and the Bis(phosphine oxide) complex [MnI2(OPPh2Me)2]. J. Chem. Soc. Dalton Trans. 1993, 3373. [Google Scholar] [CrossRef]
- McAuliffe, C.A.; Godfrey, S.M.; Mackie, A.G.; Pritchard, R.G. The reaction of coarse-grain manganese powder with diiodotrimethylphosphorane to form [Mn(PMe3)I2]n, which reacts with trace quantities of molecular oxygen to form the mixed (+2/+3) oxidation state complex [Mn2(PMe3)3I5]PMe3. J. Chem. Soc. Chem. Commun. 1992, 483–485. [Google Scholar] [CrossRef]
- Davydova, M.P.; Bauer, I.A.; Brel, V.K.; Rakhmanova, M.I.; Yu, I. Bagryanskaya and A. V. Artem’ev, Manganese(II) thiocyanate complexes with bis(phosphine oxide) ligands: Synthesis and excitation wavelength-dependent multicolor luminescence. Eur. J. Inorg. Chem. 2020, 8, 695. [Google Scholar] [CrossRef]
- Berezin, A.S.; Vinogradova, K.A.; Nadolinny, V.A.; Sukhikh, T.S.; Krivopalov, V.P.; Nikolaenkova, E.B.; Bushuev, M.B. Temperature- and excitation wavelength-dependent emission in a manganese(II) complex. Dalton Trans. 2018, 47, 1657. [Google Scholar] [CrossRef]
- Stalzer, M.M.; Lohr, T.L.; Marks, T.J. Synthesis, characterization, and thermal properties of N-alkyl beta-diketiminate manganese complexes. Inorg. Chem. 2018, 57, 3017. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.H.; Gao, E.Q.; Hong, M.C.; Gao, S.; Luo, J.H.; Lin, Z.Z.; Han, L.; Cao, R. A three-dimensional manganese(II) complex exhibiting ferrimagnetic and metamagnetic behaviors. Inorg. Chem. 2003, 42, 5486. [Google Scholar] [CrossRef]
- Bouchoucha, A.; Terbouche, A.; Bourouina, A.; Djebbar, S. New complexes of manganese(II), nickel(II) and copper(II) with derived benzoxazole ligands: Synthesis, characterization, DFT, antimicrobial activity, acute and subacute toxicity. Inorg. Chim. Acta 2014, 418, 187. [Google Scholar] [CrossRef]
- Mabad, B.; Cassoux, P.; Tuchagues, J.P.; Hendrickson, D.N. Manganese(II) complexes of polydentate schiff-bases. 1. Synthesis, characterization, magnetic-properties, and molecular-structure. Inorg. Chem. 1986, 25, 1420. [Google Scholar] [CrossRef]
- Eichhofer, A.; Lebedkin, S. 1D and 3D polymeric manganese(II) thiolato complexes: Synthesis, structure, and properties of 3[Mn4(SPh)8] and 1[Mn(SMes)2]. Inorg. Chem. 2018, 57, 602. [Google Scholar] [CrossRef]
- Qin, Y.Y.; She, P.F.; Huang, X.M.; Huang, W.; Zhao, Q. Luminescent manganese(II) complexes: Synthesis, properties and optoelectronic applications. Coord. Chem. Rev. 2020, 416, 213331. [Google Scholar] [CrossRef]
- Berezin, A.S.; Davydova, M.P.; Samsonenko, D.G.; Sukhikh, T.S.; Artem’ev, A.V. A family of brightly emissive homo- and mixed-halomanganates(II): The effect of halide on optical and magnetic properties. J. Lumin. 2021, 236, 118069. [Google Scholar] [CrossRef]
- Chen, J.; Zhang, Q.; Zheng, F.K.; Liu, Z.F.; Wang, S.H.; Wu, A.Q.; Guo, G.C. Intense photo- and tribo-luminescence of three tetrahedral manganese(II) dihalides with chelating bidentate phosphine oxide ligand. Dalton Trans. 2015, 44, 3289. [Google Scholar] [CrossRef]
- Xu, L.-J.; Sun, C.-Z.; Xiao, H.; Wu, Y.; Chen, Z.-N. Green-light-emitting diodes based on tetrabromide manganese(II) complex through solution process. Adv. Mater. 2017, 29, 1605739. [Google Scholar] [CrossRef] [PubMed]
- Ben-Akacha, A.; Zhou, C.; Chaaban, M.; Beery, D.; Lee, S.; Worku, M.; Lin, X.; Westphal, R.; Ma, B. Mechanochemical synthesis of zero dimensional organic-inorganic metal halide hybrids. ChemPhotoChem 2021, 5, 326. [Google Scholar] [CrossRef]
- Xu, L.-J.; Lin, X.; He, Q.; Worku, M.; Ma, B. Highly efficient eco-friendly X-ray scintillators based on an organic manganese halide. Nat. Commun. 2020, 11, 4329. [Google Scholar] [CrossRef]
- Sun, M.-E.; Li, Y.; Dong, X.-Y.; Zang, S.-Q. Thermoinduced structural-transformation and thermochromic luminescence in organic manganese chloride crystals. Chem. Sci. 2019, 10, 3836. [Google Scholar] [CrossRef] [Green Version]
- Gong, L.-K.; Hu, Q.-Q.; Huang, F.-Q.; Zhang, Z.-Z.; Shen, N.-N.; Hu, B.; Song, Y.; Wang, Z.-P.; Du, K.-Z.; Huang, X.-Y. Efficient modulation of photoluminescence by hydrogen bonding interactions between inorganic [MnBr4]2− anions and organic cations. Chem. Commun. 2019, 55, 7303. [Google Scholar] [CrossRef]
- Ratajczak, H. Charge-transfer properties of the hydrogen bond. I. Theory of the enhancement of dipole moment of hydrogen-bonded systems. J. Phys. Chem. 1972, 76, 3000. [Google Scholar] [CrossRef]
- Christodouleas, N.; McGlynn, S.P. Energy transfer in charge-transfer complexes. III. Intersystem crossing. J. Chem. Phys. 1964, 40, 166. [Google Scholar] [CrossRef]
- Trotter, P.J. Internal compression effects. II. Frequency and intensity changes in charge-transfer and hydrogen-bonded complex spectra. J. Chem. Phys. 1968, 48, 2736. [Google Scholar] [CrossRef]
- Rigaku. CrysAlisPro Software System, Version V1.171.41.83a, Rigaku Oxford Diffraction. 2020. Available online: http://www.rigaku.com (accessed on 1 May 2021).
- Sheldrick, G.M. SHELXT-Integrated space-group and crystal-structure determination. Acta Crystallogr. A Found. Adv. 2015, A71, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C Struct. Chem. 2015, C71, 3. [Google Scholar] [CrossRef]
- Artem’ev, A.V.; Davydova, M.P.; Berezin, A.S.; Ryzhikov, M.R.; Samsonenko, D.G. Dicopper(I) paddle-wheel complexes with thermally activated delayed fluorescence adjusted by ancillary ligands. Inorg. Chem. 2020, 59, 10699. [Google Scholar] [CrossRef]
- Stoll, S.; Schweiger, A. EasySpin, a comprehensive software package for spectral simulation and analysis in EPR. J. Magn. Reson. 2006, 178, 42. [Google Scholar] [CrossRef]
- Te Velde, G.T.; Bickelhaupt, F.M.; Baerends, E.J.; Guerra, C.F.; Van Gisbergen, S.J.A.; Snijders, J.G.; Ziegler, T. Chemistry with ADF. J. Comput. Chem. 2001, 22, 931. [Google Scholar] [CrossRef]
- ADF2019, ADF2019.3, SCM, Theoretical Chemistry, Vrije Universiteit, Amsterdam, The Netherlands. 2019. Available online: http://www.scm.com (accessed on 1 May 2021).
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic-behavior. Phys. Rev. A 1988, 38, 3098. [Google Scholar] [CrossRef]
- Perdew, J.P. Density-functional approximation for the correlation-energy of the inhomogeneous electron-gas. Phys. Rev. B 1986, 33, 8822. [Google Scholar] [CrossRef] [PubMed]
- Perdew, J.P. Correction. Phys. Rev. B 1986, 34, 7406. [Google Scholar] [CrossRef]
- van Lenthe, E.; van der Avoird, A.; Wormer, P.E.S. Density functional calculations of molecular g-tensors in the zero-order regular approximation for relativistic effects. J. Chem. Phys. 1997, 107, 2488. [Google Scholar] [CrossRef]
- van Lenthe, E.; Baerends, E.J.; Snijders, J.G. Relativistic regular 2-component Hamiltonians. J. Chem. Phys. 1993, 99, 4597. [Google Scholar] [CrossRef]
- van Lenthe, E.; Baerends, E.J.; Snijders, J.G. Relativistic total-energy using regular approximations. J. Chem. Phys. 1994, 101, 9783. [Google Scholar] [CrossRef]
- van Lenthe, E.; Ehlers, A.E.; Baerends, E.J. Geometry optimizations in the zero order regular approximation for relativistic effects. J. Chem. Phys. 1999, 110, 8943. [Google Scholar] [CrossRef] [Green Version]
- Grimme, S. Accurate description of van der Waals complexes by density functional theory including empirical corrections. J. Comput. Chem. 2004, 25, 1463. [Google Scholar] [CrossRef] [PubMed]
- Ernzerhof, M.; Scuseria, G. Assessment of the Perdew-Burke-Ernzerhof exchange-correlation functional. J. Chem. Phys. 1999, 110, 5029. [Google Scholar] [CrossRef] [Green Version]
- van Wüllen, C. Magnetic anisotropy from density functional calculations. Comparison of different approaches: Mn12O12 acetate as a test case. J. Chem. Phys. 2009, 130, 194109. [Google Scholar] [CrossRef]
- Schmitt, S.; Jost, P.; van Wüllen, C. Zero-field splittings from density functional calculations: Analysis and improvement of known methods. J. Chem. Phys. 2011, 134, 194113. [Google Scholar] [CrossRef] [PubMed]
- van Gisbergen, S.J.A.; Snijders, J.G.; Baerends, E.J. Implementation of time-dependent density functional response equations. Comput. Phys. Commun. 1999, 118, 119. [Google Scholar] [CrossRef]
- Wang, F.; Ziegler, T. A simplified relativistic time-dependent density-functional theory formalism for the calculations of excitation energies including spin-orbit coupling effect. J. Chem. Phys. 2005, 123, 154102. [Google Scholar] [CrossRef] [PubMed]
- Stephens, P.J.; Devlin, F.J.; Chabalowski, C.F.; Frisch, M.J. Ab initio calculation of vibrational absorption and circular dichroism spectra using density functional force fields. J. Phys. Chem. 1994, 98, 11623. [Google Scholar] [CrossRef]
- Yanai, T.; Tew, D.P.; Handy, N.C. A new hybrid exchange–correlation functional using the Coulomb-attenuating method (CAM-B3LYP). Chem. Phys. Lett. 2004, 393, 51. [Google Scholar] [CrossRef] [Green Version]
- Wang, F.; Ziegler, T. Time-dependent density functional theory based on a noncollinear formulation of the exchange-correlation potential. J. Chem. Phys. 2004, 121, 12191. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Ziegler, T. The performance of time-dependent density functional theory based on a noncollinear exchange-correlation potential in the calculations of excitation energies. J. Chem. Phys. 2005, 122, 74109. [Google Scholar] [CrossRef]
- Bader, R.F.W. A quantum theory of molecular structure and its applications. Chem. Rev. 1991, 91, 893. [Google Scholar] [CrossRef]
- Mitoraj, M.; Michalak, A.; Ziegler, T. A combined charge and energy decomposition scheme for bond analysis. J. Chem. Theory Comput. 2009, 5, 962. [Google Scholar] [CrossRef]
- Rodríguez, J.I. An efficient method for computing the QTAIM topology of a scalar field: The electron density case. J. Comput. Chem. 2013, 34, 681. [Google Scholar] [CrossRef]
- Rodríguez, J.I.; Bader, R.F.W.; Ayers, P.W.; Michel, C.; Götz, A.W.; Bo, C. A high performance grid-based algorithm for computing QTAIM properties. Chem. Phys. Lett. 2009, 472, 149. [Google Scholar] [CrossRef]
- Johnson, E.R.; Keinan, S.; Mori-Sánchez, P.; Contreras-García, J.; Cohen, A.J.; Yang, W. Revealing noncovalent interactions. J. Am. Chem. Soc. 2010, 132, 6498. [Google Scholar] [CrossRef] [Green Version]
- Contreras-García, J.; Johnson, E.R.; Keinan, S.; Chaudret, R.; Piquemal, J.-P.; Beratan, D.N.; Yang, W. NCIPLOT: A Program for plotting noncovalent interaction regions. J. Chem. Theory Comput. 2011, 7, 625. [Google Scholar] [CrossRef]
- Aarabi, M.; Gholami, S.; Grabowski, S.J. S−H…O and O−H…O hydrogen bonds-comparison of dimers of thiocarboxylic and carboxylic acids. ChemPhysChem 2020, 21, 1653. [Google Scholar] [CrossRef] [PubMed]
- Poater, J.; Gimferrer, M.; Poater, A. Covalent and ionic capacity of mofs to sorb small gas molecules. Inorg. Chem. 2018, 57, 6981–6990. [Google Scholar] [CrossRef] [PubMed]
- Fluck, E.; Svara, J.; Neumuller, B.; Riffel, H.; Thurn, H. Darstellung und struktur von methylphosphoniumchlorid. Z. Anorg. Allg. Chem. 1986, 536, 129. [Google Scholar] [CrossRef]
- Schroeder, L.W.; Rush, J.J. Neutron diffraction study of the structure and thermal motion of phosphonium bromide. J. Chem. Phys. 1971, 54, 1968. [Google Scholar] [CrossRef]
- Tanabe, Y.; Sugano, S. On the absorption spectra of complex ions. I. J. Phys. Soc. Jpn. 1954, 9, 753. [Google Scholar] [CrossRef] [Green Version]
- Racah, G. Theory of Complex Spectra. III. Phys. Rev. 1943, 63, 367. [Google Scholar] [CrossRef]
- Racah, G. Theory of Complex Spectra. IV. Phys. Rev. 1949, 76, 1352. [Google Scholar] [CrossRef]
- Liu, X.; Dronskowski, R.; Glaum, R.; Tchougréeff, A.L. Experimental and quantum-chemical investigations of the UV/Vis absorption spectrum of manganese carbodiimide, MnNCN. Z. Anorg. Allg. Chem. 2010, 636, 343. [Google Scholar] [CrossRef]
- Gautier, R.; Paris, M.; Massuyeau, F. Hydrogen bonding and broad-band emission in hybrid zinc halide phosphors. Inorg. Chem. 2020, 59, 2626. [Google Scholar] [CrossRef] [PubMed]
- Duboc, C.; Phoeung, T.; Zein, S.; Pécaut, J.; Collomb, M.N.; Neese, F. Origin of the zero-field splitting in mononuclear octahedral dihalide Mn-II complexes: An investigation by multifrequency high-field electron paramagnetic resonance and density functional theory. Inorg. Chem. 2007, 46, 4905. [Google Scholar] [CrossRef] [PubMed]

 —absorption,
—absorption,  —internal conversion and intersystem crossing transition,
—internal conversion and intersystem crossing transition,  —emission,
—emission,  —transfer from [H2DPEphos]2+ to [MnX4]2−,
—transfer from [H2DPEphos]2+ to [MnX4]2−,  —non-radiative relaxation).
—absorption, —internal conversion and intersystem crossing transition, —emission, —transfer from [H2DPEphos]2+ to [MnX4]2−, —non-radiative relaxation).
—non-radiative relaxation).
—absorption, —internal conversion and intersystem crossing transition, —emission, —transfer from [H2DPEphos]2+ to [MnX4]2−, —non-radiative relaxation).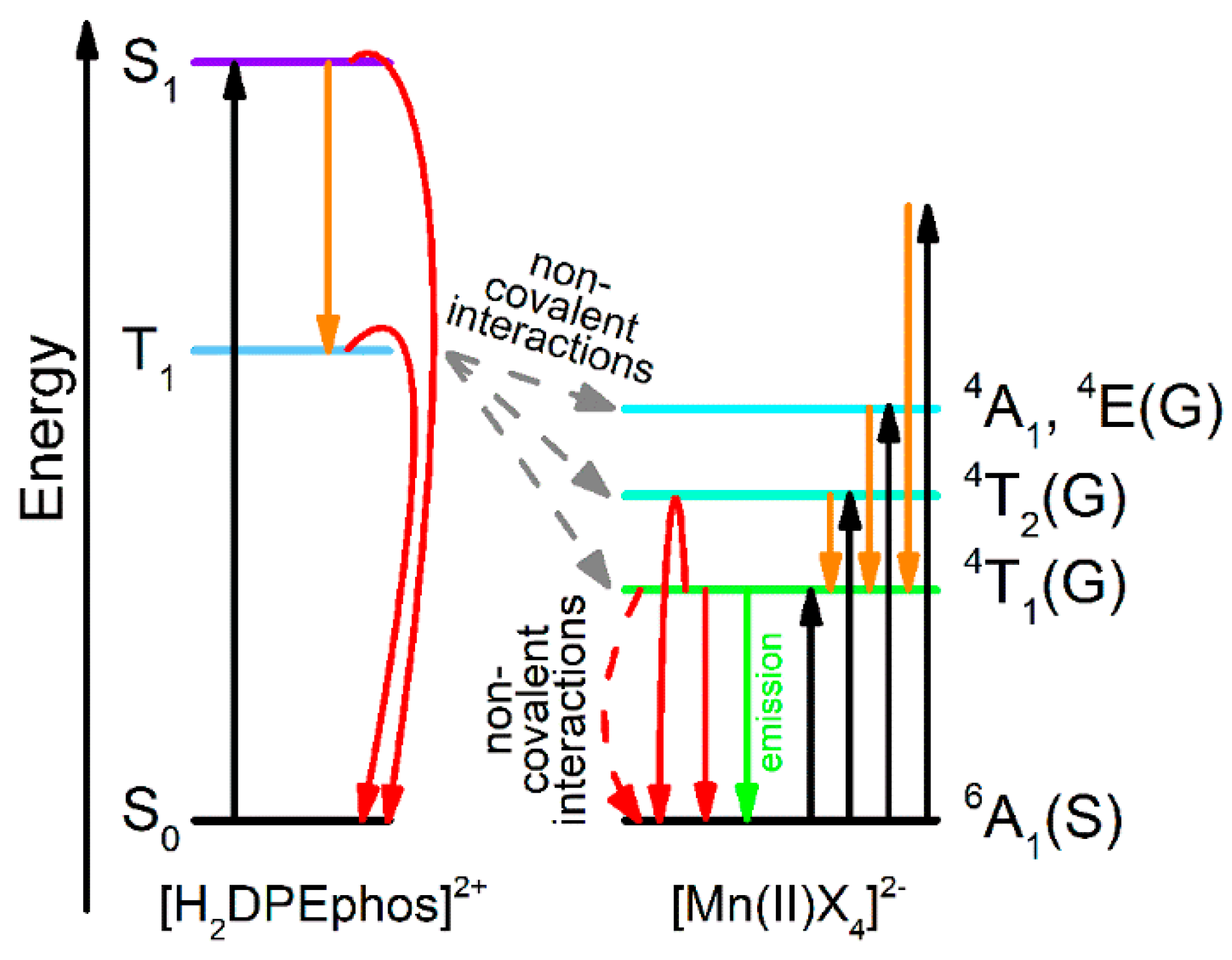

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cation | Halide | Quantum Yield, % | λEm, nm | Ref. |
|---|---|---|---|---|
| Ph3P-Ph | Br | 98 | 516 | [16] |
| Ph3P-Ph | Cl | 40 | 522 | [17] |
| Ph3P-Benzyl | Br | 90 | 510 | [13] |
| Ph3P-Benzyl | Cl | 32 | 509 | [13] |
| Ph3P-Et | Br | 91 | 522 | [13] |
| Ph3P-Et | Cl | 29 | 524 | [13] |
| Ethylenebis- Ph3P | Br | 95 | 517 | [18] |
| Ph(Me)3N | Br | 76 | 520 | [2] |
| Ph(Me)3N | Cl | 89 | 522 | [2] |
| Et4N | Br | 86 | 516 | [2] |
| Et4N | Cl | 75 | 518 | [2] |
| 1 (Br) | 2 (Cl) | |
|---|---|---|
| λmax(300 K) [nm] a | 515 | 523 |
| τ(300 K) [µs] a | 177 | 100 |
| ΦPL(300 K) [%] a | 42 | ≈0.1 |
| kr(300 K) [s–1] a,b | 2.4 × 103 | ≈1.0 × 101 |
| knr(300 K) [s–1] a,c | 3.3 × 103 | ≈1.0 × 104 |
| ΦPL(300 K) [%] e | 60 | 2 |
| kr(300 K) [s–1] e,b | 3.4 × 103 | 2.0 × 102 |
| knr(300 K) [s–1] e,c | 2.3 × 103 | 9.8 × 103 |
| Chromaticity (300 K) [x, y] d | 0.164; 0.750 | 0.259; 0.569 |
| λmax(77 K) [nm] a | 520 | 527 |
| τ(77 K) [µs] a | 304 | 2900 |
| ΦPL(77 K) [%] a | ≈100 | 40 |
| kr(77 K) [s–1] a,b | 3.3 × 103 | 1.4 × 102 |
| knr(77 K) [s−1] a,c | <3.3 × 102 | 2.1 × 102 |
| ΦPL(77 K) [%] e | ≈100 | ≈100 |
| kr(77 K) [s–1] e,b | 3.3 × 103 | 3.4 × 102 |
| knr(77 K) [s−1] e,c | <3.3 × 102 | <3.4 × 101 |
| Chromaticity (77 K) [x, y] d | 0.216; 0.656 | 0.205; 0.744 |
| [cm−1] | 900 | 1100 |
| 10Dqtet [cm−1] | 1730 | 1960 |
| B [cm−1] | 638 | 608 |
| 1 | 2 | |
|---|---|---|
| 5/2 | 5/2 | |
| a | 2.005 | 2.000 |
| a | 2.008 | 2.003 |
| [MHz] b | 4200 | 1760 |
| [MHz] b | 4800 | 2007 |
| [MHz] b | 903 | 380 |
| [MHz] b | 525 | 356 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Berezin, A.S. A Halomanganates(II) with P,P’-Diprotonated Bis(2-Diphenylphosphinophenyl)ether: Wavelength-Excitation Dependence of the Quantum Yield and Role of the Non-Covalent Interactions. Int. J. Mol. Sci. 2021, 22, 6873. https://doi.org/10.3390/ijms22136873
Berezin AS. A Halomanganates(II) with P,P’-Diprotonated Bis(2-Diphenylphosphinophenyl)ether: Wavelength-Excitation Dependence of the Quantum Yield and Role of the Non-Covalent Interactions. International Journal of Molecular Sciences. 2021; 22(13):6873. https://doi.org/10.3390/ijms22136873
Chicago/Turabian StyleBerezin, Alexey S. 2021. "A Halomanganates(II) with P,P’-Diprotonated Bis(2-Diphenylphosphinophenyl)ether: Wavelength-Excitation Dependence of the Quantum Yield and Role of the Non-Covalent Interactions" International Journal of Molecular Sciences 22, no. 13: 6873. https://doi.org/10.3390/ijms22136873
APA StyleBerezin, A. S. (2021). A Halomanganates(II) with P,P’-Diprotonated Bis(2-Diphenylphosphinophenyl)ether: Wavelength-Excitation Dependence of the Quantum Yield and Role of the Non-Covalent Interactions. International Journal of Molecular Sciences, 22(13), 6873. https://doi.org/10.3390/ijms22136873