
The Footprint of Kynurenine Pathway in Neurodegeneration: Janus-Faced Role in Parkinson’s Disorder and Therapeutic Implications




 ,
,  , ,
, , 
Abstract
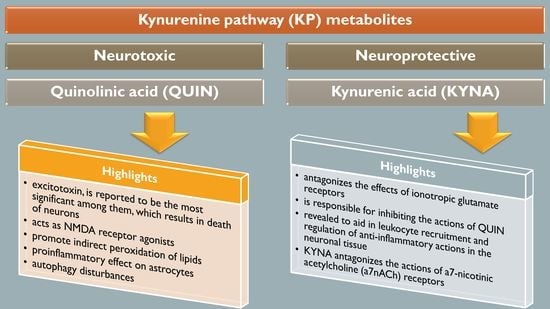
1. Introduction
2. The Kynurenine Pathway
3. The Interaction of Kynurenine Pathway with the Central Nervous System
4. Alteration in KP Metabolites, Parallel to Impairment of Mitochondria Functions, Redox Metals and Oxidative Stress in PD
4.1. Upstream KP Metabolism in PD
4.2. Downstream KP Metabolism in PD
4.3. Microbiota Gut Brain Association with KP in PD
5. Applicability of KP as A Biomarker in PD
6. Exploring the Therapeutic Role of KP in PD
7. Identifying other therapeutic targets of PD
8. Conclusions
9. Future Prospects
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Berg, D.; Postuma, R.B.; Bloem, B.; Chan, P.; Dubois, B.; Gasser, T.; Goetz, C.G.; Halliday, G.M.; Hardy, J.; Lang, A.E. Time to redefine PD? Introductory statement of the MDS Task Force on the definition of Parkinson’s disease. Mov. Disord. 2014, 29, 454–462. [Google Scholar] [CrossRef]
- Pingale, T.; Gupta, G.L. Current and emerging therapeutic targets for Parkinson’s disease. Metab. Brain Dis. 2021, 36, 13–27. [Google Scholar] [CrossRef]
- Tepper, S.; Ashina, M.; Reuter, U.; Brandes, J.L.; Doležil, D.; Silberstein, S.; Winner, P.; Leonardi, D.; Mikol, D.; Lenz, R. Safety and efficacy of erenumab for preventive treatment of chronic migraine: A randomised, double-blind, placebo-controlled phase 2 trial. Lancet Neurol. 2017, 16, 425–434. [Google Scholar] [CrossRef]
- Angot, E.; Brundin, P. Dissecting the potential molecular mechanisms underlying α-synuclein cell-to-cell transfer in Parkinson’s disease. Parkinsonism Relat. Disord. 2009, 15, S143–S147. [Google Scholar] [CrossRef]
- Bartel, W.P.; Van Laar, V.S.; Burton, E.A. Chapter 23—Parkinson’s disease. In Behavioral and Neural Genetics of Zebrafish; Gerlai, R.T., Ed.; Academic Press: Cambridge, MA, USA, 2020; pp. 377–412. [Google Scholar] [CrossRef]
- George, J.; Mok, S.; Moses, D.; Wilkins, S.; Bush, A.I.; Cherny, R.A.; Finkelstein, D.I. Targeting the progression of Parkinson’s disease. Curr. Neuropharmacol. 2009, 7, 9–36. [Google Scholar] [CrossRef]
- Jankovic, J.; Tan, E.K. Parkinson’s disease: Etiopathogenesis and treatment. J. Neurol. Neurosurg. Psychiatry 2020, 91, 795–808. [Google Scholar] [CrossRef]
- Venkatesan, D.; Iyer, M.; Narayanasamy, A.; Siva, K.; Vellingiri, B. Kynurenine pathway in Parkinson’s disease—An update. Eneurologicalsci 2020, 21, 100270. [Google Scholar] [CrossRef]
- Schwarcz, R.; Bruno, J.P.; Muchowski, P.J.; Wu, H.-Q. Kynurenines in the mammalian brain: When physiology meets pathology. Nat. Rev. Neurosci. 2012, 13, 465–477. [Google Scholar] [CrossRef] [PubMed]
- Lim, C.K.; Fernandez-Gomez, F.J.; Braidy, N.; Estrada, C.; Costa, C.; Costa, S.; Bessede, A.; Fernandez-Villalba, E.; Zinger, A.; Herrero, M.T. Involvement of the kynurenine pathway in the pathogenesis of Parkinson’s disease. Prog. Neurobiol. 2017, 155, 76–95. [Google Scholar] [CrossRef]
- Munn, D.H.; Mellor, A.L. Indoleamine 2, 3 dioxygenase and metabolic control of immune responses. Trends Immunol. 2013, 34, 137–143. [Google Scholar] [CrossRef]
- Opitz, C.A.; Heiland, I. Dynamics of NAD-metabolism: Everything but constant. Biochem. Soc. Trans. 2015, 43, 1127–1132. [Google Scholar] [CrossRef] [PubMed]
- Castellano-Gonzalez, G.; Jacobs, K.R.; Don, E.; Cole, N.J.; Adams, S.; Lim, C.K.; Lovejoy, D.B.; Guillemin, G.J. Kynurenine 3-monooxygenase activity in human primary neurons and effect on cellular bioenergetics identifies new neurotoxic mechanisms. Neurotox. Res. 2019, 35, 530–541. [Google Scholar] [CrossRef]
- Erhardt, S.; Schwieler, L.; Imbeault, S.; Engberg, G. The kynurenine pathway in schizophrenia and bipolar disorder. Neuropharmacology 2017, 112, 297–306. [Google Scholar] [CrossRef] [PubMed]
- Stetler, R.A.; Leak, R.K.; Gan, Y.; Li, P.; Zhang, F.; Hu, X.; Jing, Z.; Chen, J.; Zigmond, M.J.; Gao, Y. Preconditioning provides neuroprotection in models of CNS disease: Paradigms and clinical significance. Prog. Neurobiol. 2014, 114, 58–83. [Google Scholar] [CrossRef] [PubMed]
- Török, N.; Török, R.; Szolnoki, Z.; Somogyvári, F.; Klivényi, P.; Vécsei, L. The genetic link between Parkinson’s disease and the kynurenine pathway is still missing. Parkinsons Dis. 2015, 2015, 474135. [Google Scholar] [CrossRef]
- Houser, M.; Tansey, M. The gut-brain axis: Is intestinal inflammation a silent driver of Parkinson’s disease pathogenesis? NPJ Parkinsons Dis. 2017, 3, 3. [Google Scholar] [CrossRef] [PubMed]
- Massudi, H.; Grant, R.; Guillemin, G.J.; Braidy, N. NAD+ metabolism and oxidative stress: The golden nucleotide on a crown of thorns. Redox Rep. 2012, 17, 28–46. [Google Scholar] [CrossRef]
- Castro-Portuguez, R.; Sutphin, G.L. Kynurenine pathway, NAD+ synthesis, and mitochondrial function: Targeting tryptophan metabolism to promote longevity and healthspan. Exp. Gerontol. 2020, 132, 110841. [Google Scholar] [CrossRef] [PubMed]
- Cervenka, I.; Agudelo, L.Z.; Ruas, J.L. Kynurenines: Tryptophan’s metabolites in exercise, inflammation, and mental health. Science 2017, 357, aaf9794. [Google Scholar] [CrossRef]
- Liu, M.; Wang, X.; Wang, L.; Ma, X.; Gong, Z.; Zhang, S.; Li, Y. Targeting the IDO1 pathway in cancer: From bench to bedside. J. Hematol. Oncol. 2018, 11, 100. [Google Scholar] [CrossRef]
- Munn, D.H.; Mellor, A.L. IDO and tolerance to tumors. Trends Mol. Med. 2004, 10, 15–18. [Google Scholar] [CrossRef] [PubMed]
- Dürr, S.; Kindler, V. Implication of indolamine 2,3 dioxygenase in the tolerance toward fetuses, tumors, and allografts. J. Leukoc. Biol. 2013, 93, 681–687. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Mazarei, G.; Leavitt, B.R. Indoleamine 2,3 dioxygenase as a potential therapeutic target in Huntington’s disease. J. Huntingt. Dis. 2015, 4, 109–118. [Google Scholar] [CrossRef] [PubMed]
- Widner, B.; Leblhuber, F.; Fuchs, D. Increased neopterin production and tryptophan degradation in advanced Parkinson’s disease. J. Neural Transm. 2002, 109, 181–189. [Google Scholar] [CrossRef]
- Mellor, A.L.; Baban, B.; Chandler, P.; Marshall, B.; Jhaver, K.; Hansen, A.; Koni, P.A.; Iwashima, M.; Munn, D.H. Cutting edge: Induced indoleamine 2, 3 dioxygenase expression in dendritic cell subsets suppresses T cell clonal expansion. J. Immunol. 2003, 171, 1652–1655. [Google Scholar] [CrossRef]
- Bessede, A.; Gargaro, M.; Pallotta, M.T.; Matino, D.; Servillo, G.; Brunacci, C.; Bicciato, S.; Mazza, E.M.; Macchiarulo, A.; Vacca, C. Aryl hydrocarbon receptor control of a disease tolerance defence pathway. Nature 2014, 511, 184–190. [Google Scholar] [CrossRef]
- Guillemin, G.J.; Smith, D.G.; Smythe, G.A.; Armati, P.J.; Brew, B.J. Expression of the kynurenine pathway enzymes in human microglia and macrophages. Adv. Exp. Med. Biol. 2003, 527, 105–112. [Google Scholar]
- Jones, S.P.; Franco, N.F.; Varney, B.; Sundaram, G.; Brown, D.A.; De Bie, J.; Lim, C.K.; Guillemin, G.J.; Brew, B.J. Expression of the kynurenine pathway in human peripheral blood mononuclear cells: Implications for inflammatory and neurodegenerative disease. PLoS ONE 2015, 10, e0131389. [Google Scholar]
- Guillemin, G.J.; Cullen, K.M.; Lim, C.K.; Smythe, G.A.; Garner, B.; Kapoor, V.; Takikawa, O.; Brew, B.J. Characterization of the kynurenine pathway in human neurons. J. Neurosci. 2007, 27, 12884–12892. [Google Scholar] [CrossRef]
- Vécsei, L.; Szalárdy, L.; Fülöp, F.; Toldi, J. Kynurenines in the CNS: Recent advances and new questions. Nat. Rev. Drug Discov. 2013, 12, 64–82. [Google Scholar] [CrossRef]
- Guillemin, G.J.; Kerr, S.J.; Brew, B.J. Involvement of quinolinic acid in AIDS dementia complex. Neurotox. Res. 2005, 7, 103–123. [Google Scholar] [CrossRef]
- Pierozan, P.; Biasibetti, H.; Schmitz, F.; Ávila, H.; Parisi, M.M.; Barbe-Tuana, F.; Wyse, A.T.S.; Pessoa-Pureur, R. Quinolinic acid neurotoxicity: Differential roles of astrocytes and microglia via FGF-2-mediated signaling in redox-linked cytoskeletal changes. Biochim. Biophys. Acta BBA Mol. Cell Res. 2016, 1863, 3001–3014. [Google Scholar] [CrossRef]
- Chiarugi, A.; Meli, E.; Moroni, F. Similarities and differences in the neuronal death processes activated by 3OH-kynurenine and quinolinic acid. J. Neurochem. 2001, 77, 1310–1318. [Google Scholar] [CrossRef]
- Ramírez-Ortega, D.; Ramiro-Salazar, A.; González-Esquivel, D.; Ríos, C.; Pineda, B.; Pérez de la Cruz, V. 3-Hydroxykynurenine and 3-hydroxyanthranilic acid enhance the toxicity induced by copper in rat astrocyte culture. Oxidative Med. Cell. Longev. 2017, 2017, 2371895. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, L.E.; Leopold, M.C.; Huang, X.; Atwood, C.S.; Saunders, A.J.; Hartshorn, M.; Lim, J.T.; Faget, K.Y.; Muffat, J.A.; Scarpa, R.C. 3-Hydroxykynurenine and 3-hydroxyanthranilic acid generate hydrogen peroxide and promote α-crystallin cross-linking by metal ion reduction. Biochemistry 2000, 39, 7266–7275. [Google Scholar] [CrossRef]
- Grant, R.; Coggan, S.; Smythe, G. The physiological action of picolinic acid in the human brain. Int. J. Tryptophan Res. 2009, 2, IJTR-S2469. [Google Scholar] [CrossRef]
- La Cruz, V.P.-D.; Carrillo-Mora, P.; Santamaría, A. Quinolinic acid, an endogenous molecule combining excitotoxicity, oxidative stress and other toxic mechanisms. Int. J. Tryptophan Res. 2012, 5, IJTR-S8158. [Google Scholar] [CrossRef] [PubMed]
- Rafice, S.A.; Chauhan, N.; Efimov, I.; Basran, J.; Raven, E.L. Oxidation of L-tryptophan in biology: A comparison between tryptophan 2, 3-dioxygenase and indoleamine 2, 3-dioxygenase. Biochem. Soc. Trans. 2009, 37, 408–412. [Google Scholar] [CrossRef] [PubMed]
- Meng, B.; Wu, D.; Gu, J.; Ouyang, S.; Ding, W.; Liu, Z.J. Structural and functional analyses of human tryptophan 2, 3-dioxygenase. Proteins Struct. Funct. Bioinform. 2014, 82, 3210–3216. [Google Scholar] [CrossRef]
- Ren, S.; Correia, M.A. Heme: A regulator of rat hepatic tryptophan 2,3-dioxygenase? Arch. Biochem. Biophys. 2000, 377, 195–203. [Google Scholar] [CrossRef]
- Basile, M.S.; Mazzon, E.; Fagone, P.; Longo, A.; Russo, A.; Fallico, M.; Bonfiglio, V.; Nicoletti, F.; Avitabile, T.; Reibaldi, M. Immunobiology of uveal melanoma: State of the art and therapeutic targets. Front. Oncol. 2019, 9, 1145. [Google Scholar] [CrossRef]
- Li, J.S.; Han, Q.; Fang, J.; Rizzi, M.; James, A.A.; Li, J. Biochemical mechanisms leading to tryptophan 2,3-dioxygenase activation. Arch. Insect Biochem. Physiol. 2007, 64, 74–87. [Google Scholar] [CrossRef]
- Maddison, D.C.; Giorgini, F. The kynurenine pathway and neurodegenerative disease. Semin. Cell Dev. Biol. 2015, 40, 134–141. [Google Scholar] [CrossRef] [PubMed]
- Konan, K.V.; Taylor, M.W. Importance of the two interferon-stimulated response element (ISRE) sequences in the regulation of the human indoleamine 2,3-dioxygenase gene. J. Biol. Chem. 1996, 271, 19140–19145. [Google Scholar] [CrossRef]
- Campbell, B.M.; Charych, E.; Lee, A.W.; Möller, T. Kynurenines in CNS disease: Regulation by inflammatory cytokines. Front. Neurosci. 2014, 8, 12. [Google Scholar] [CrossRef] [PubMed]
- Takikawa, O.; Kuroiwa, T.; Yamazaki, F.; Kido, R. Mechanism of interferon-gamma action. Characterization of indoleamine 2,3-dioxygenase in cultured human cells induced by interferon-gamma and evaluation of the enzyme-mediated tryptophan degradation in its anticellular activity. J. Biol. Chem. 1988, 263, 2041–2048. [Google Scholar] [CrossRef]
- Zunszain, P.A.; Anacker, C.; Cattaneo, A.; Choudhury, S.; Musaelyan, K.; Myint, A.M.; Thuret, S.; Price, J.; Pariante, C.M. Interleukin-1β: A new regulator of the kynurenine pathway affecting human hippocampal neurogenesis. Neuropsychopharmacology 2012, 37, 939–949. [Google Scholar] [CrossRef]
- Connor, T.J.; Starr, N.; O’Sullivan, J.B.; Harkin, A. Induction of indolamine 2,3-dioxygenase and kynurenine 3-monooxygenase in rat brain following a systemic inflammatory challenge: A role for IFN-γ? Neurosci. Lett. 2008, 441, 29–34. [Google Scholar] [CrossRef]
- Molteni, R.; Macchi, F.; Zecchillo, C.; Dell’Agli, M.; Colombo, E.; Calabrese, F.; Guidotti, G.; Racagni, G.; Riva, M.A. Modulation of the inflammatory response in rats chronically treated with the antidepressant agomelatine. Eur. Neuropsychopharmacol. 2013, 23, 1645–1655. [Google Scholar] [CrossRef]
- Giorgini, F.; Möller, T.; Kwan, W.; Zwilling, D.; Wacker, J.L.; Hong, S.; Tsai, L.C.; Cheah, C.S.; Schwarcz, R.; Guidetti, P.; et al. Histone deacetylase inhibition modulates kynurenine pathway activation in yeast, microglia, and mice expressing a mutant huntingtin fragment. J. Biol. Chem. 2008, 283, 7390–7400. [Google Scholar] [CrossRef]
- Kowalska, M.; Fijałkowski, Ł.; Nowaczyk, A. Assessment of Paroxetine Molecular Interactions with Selected Monoamine and γ-Aminobutyric Acid Transporters. Int. J. Mol. Sci. 2021, 22, 6293. [Google Scholar] [CrossRef]
- Zhang, S.; Collier, M.E.W.; Heyes, D.J.; Giorgini, F.; Scrutton, N.S. Advantages of brain penetrating inhibitors of kynurenine-3-monooxygenase for treatment of neurodegenerative diseases. Arch. Biochem. Biophys. 2021, 697, 108702. [Google Scholar] [CrossRef] [PubMed]
- Dang, Y.; Dale, W.E.; Brown, O.R. Comparative effects of oxygen on indoleamine 2,3-dioxygenase and tryptophan 2,3-dioxygenase of the kynurenine pathway. Free Radic. Biol. Med. 2000, 28, 615–624. [Google Scholar] [CrossRef]
- Gál, E.M.; Sherman, A.D. L-kynurenine: Its synthesis and possible regulatory function in brain. Neurochem. Res. 1980, 5, 223–239. [Google Scholar] [CrossRef] [PubMed]
- Speciale, C.; Schwarcz, R. Uptake of kynurenine into rat brain slices. J. Neurochem. 1990, 54, 156–163. [Google Scholar] [CrossRef] [PubMed]
- Guillemin, G.J.; Kerr, S.J.; Smythe, G.A.; Smith, D.G.; Kapoor, V.; Armati, P.J.; Croitoru, J.; Brew, B.J. Kynurenine pathway metabolism in human astrocytes: A paradox for neuronal protection. J. Neurochem. 2001, 78, 842–853. [Google Scholar] [CrossRef]
- Pocivavsek, A.; Notarangelo, F.M.; Wu, H.-Q.; Bruno, J.P.; Schwarcz, R. Astrocytes as pharmacological targets in the treatment of schizophrenia: Focus on kynurenic acid. In Handbook of Behavioral Neuroscience; Elsevier: Amsterdam, The Netherlands, 2016; Volume 23, pp. 423–443. [Google Scholar]
- Gramsbergen, J.B.; Hodgkins, P.S.; Rassoulpour, A.; Turski, W.A.; Guidetti, P.; Schwarcz, R. Brain-specific modulation of kynurenic acid synthesis in the rat. J. Neurochem. 1997, 69, 290–298. [Google Scholar] [CrossRef]
- Rassoulpour, A.; Wu, H.Q.; Poeggeler, B.; Schwarcz, R. Systemic d-amphetamine administration causes a reduction of kynurenic acid levels in rat brain. Brain Res. 1998, 802, 111–118. [Google Scholar] [CrossRef]
- Speciale, C.; Schwarcz, R. On the production and disposition of quinolinic acid in rat brain and liver slices. J. Neurochem. 1993, 60, 212–218. [Google Scholar] [CrossRef]
- Heyes, M.P.; Saito, K.; Major, E.O.; Milstien, S.; Markey, S.P.; Vickers, J.H. A mechanism of quinolinic acid formation by brain in inflammatory neurological disease. Attenuation of synthesis from L-tryptophan by 6-chlorotryptophan and 4-chloro-3-hydroxyanthranilate. Brain 1993, 116, 1425–1450. [Google Scholar] [CrossRef]
- Foster, A.C.; White, R.J.; Schwarcz, R. Synthesis of quinolinic acid by 3-hydroxyanthranilic acid oxygenase in rat brain tissue in vitro. J. Neurochem. 1986, 47, 23–30. [Google Scholar] [CrossRef]
- Fukui, S.; Schwarcz, R.; Rapoport, S.I.; Takada, Y.; Smith, Q.R. Blood-brain barrier transport of kynurenines: Implications for brain synthesis and metabolism. J. Neurochem. 1991, 56, 2007–2017. [Google Scholar] [CrossRef] [PubMed]
- Cardinale, A.; Calabrese, V.; de Iure, A.; Picconi, B. Alpha-Synuclein as a Prominent Actor in the Inflammatory Synaptopathy of Parkinson’s Disease. Int. J. Mol. Sci. 2021, 22, 6517. [Google Scholar] [CrossRef]
- Guidetti, P.; Schwarcz, R. 3-Hydroxykynurenine potentiates quinolinate but not NMDA toxicity in the rat striatum. Eur. J. Neurosci. 1999, 11, 3857–3863. [Google Scholar] [CrossRef] [PubMed]
- Jovanovic, F.; Candido, K.D.; Knezevic, N.N. The Role of the Kynurenine Signaling Pathway in Different Chronic Pain Conditions and Potential Use of Therapeutic Agents. Int. J. Mol. Sci. 2020, 21, 6045. [Google Scholar] [CrossRef]
- Rojewska, E.; Ciapała, K.; Piotrowska, A.; Makuch, W.; Mika, J. Pharmacological inhibition of indoleamine 2, 3-dioxygenase-2 and kynurenine 3-monooxygenase, enzymes of the kynurenine pathway, significantly diminishes neuropathic pain in a rat model. Front. Pharmacol. 2018, 9, 724. [Google Scholar] [CrossRef]
- Török, N.; Maszlag-Török, R.; Molnár, K.; Szolnoki, Z.; Somogyvári, F.; Boda, K.; Tanaka, M.; Klivényi, P.; Vécsei, L. Single Nucleotide Polymorphisms of Indoleamine 2, 3-Dioxygenase 1 Influenced the Age Onset of Parkinson’s Disease. Preprint 2020. [Google Scholar] [CrossRef]
- Jones, S.P.; Guillemin, G.J.; Brew, B.J. The kynurenine pathway in stem cell biology. Int. J. Tryptophan Res. 2013, 6, 57–66. [Google Scholar] [CrossRef]
- Lugo-Huitrón, R.; Ugalde Muñiz, P.; Pineda, B.; Pedraza-Chaverrí, J.; Ríos, C.; Pérez-de la Cruz, V. Quinolinic acid: An endogenous neurotoxin with multiple targets. Oxidative Med. Cell. Longev. 2013, 2013, 104024. [Google Scholar] [CrossRef]
- Perkins, M.N.; Stone, T.W. Pharmacology and regional variations of quinolinic acid-evoked excitations in the rat central nervous system. J. Pharm. Exp. 1983, 226, 551–557. [Google Scholar]
- Vandresen-Filho, S.; Martins, W.C.; Bertoldo, D.B.; Mancini, G.; De Bem, A.F.; Tasca, C.I. Cerebral cortex, hippocampus, striatum and cerebellum show differential susceptibility to quinolinic acid-induced oxidative stress. Neurol. Sci. 2015, 36, 1449–1456. [Google Scholar] [CrossRef]
- Kumar, U. Characterization of striatal cultures with the effect of QUIN and NMDA. Neurosci. Res. 2004, 49, 29–38. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Brew, B.J.; Guillemin, G.J. Characterization of the kynurenine pathway in NSC-34 cell line: Implications for amyotrophic lateral sclerosis. J. Neurochem. 2011, 118, 816–825. [Google Scholar] [CrossRef] [PubMed]
- Ting, K.K.; Brew, B.J.; Guillemin, G.J. Effect of quinolinic acid on human astrocytes morphology and functions: Implications in Alzheimer’s disease. J. Neuroinflamm. 2009, 6, 36. [Google Scholar] [CrossRef]
- Santamaría, A.; Galván-Arzate, S.; Lisý, V.; Ali, S.F.; Duhart, H.M.; Osorio-Rico, L.; Ríos, C.; St’astný, F. Quinolinic acid induces oxidative stress in rat brain synaptosomes. Neuroreport 2001, 12, 871–874. [Google Scholar] [CrossRef] [PubMed]
- Santamaría, A.; Jiménez-Capdeville, M.E.; Camacho, A.; Rodríguez-Martínez, E.; Flores, A.; Galván-Arzate, S. In vivo hydroxyl radical formation after quinolinic acid infusion into rat corpus striatum. Neuroreport 2001, 12, 2693–2696. [Google Scholar] [CrossRef] [PubMed]
- Pláteník, J.; Stopka, P.; Vejrazka, M.; Stípek, S. Quinolinic acid-iron(ii) complexes: Slow autoxidation, but enhanced hydroxyl radical production in the Fenton reaction. Free Radic. Res. 2001, 34, 445–459. [Google Scholar] [CrossRef]
- Steiner, J.; Bogerts, B.; Sarnyai, Z.; Walter, M.; Gos, T.; Bernstein, H.G.; Myint, A.M. Bridging the gap between the immune and glutamate hypotheses of schizophrenia and major depression: Potential role of glial NMDA receptor modulators and impaired blood-brain barrier integrity. World J. Biol. Psychiatry 2012, 13, 482–492. [Google Scholar] [CrossRef]
- St’astný, F.; Skultétyová, I.; Pliss, L.; Jezová, D. Quinolinic acid enhances permeability of rat brain microvessels to plasma albumin. Brain Res. Bull. 2000, 53, 415–420. [Google Scholar] [CrossRef]
- Pierozan, P.; Zamoner, A.; Soska, A.K.; Silvestrin, R.B.; Loureiro, S.O.; Heimfarth, L.; Mello e Souza, T.; Wajner, M.; Pessoa-Pureur, R. Acute intrastriatal administration of quinolinic acid provokes hyperphosphorylation of cytoskeletal intermediate filament proteins in astrocytes and neurons of rats. Exp. Neurol. 2010, 224, 188–196. [Google Scholar] [CrossRef]
- Rahman, A.; Ting, K.; Cullen, K.M.; Braidy, N.; Brew, B.J.; Guillemin, G.J. The excitotoxin quinolinic acid induces tau phosphorylation in human neurons. PLoS ONE 2009, 4, e6344. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.-C.; Ting, K.K.; Adams, S.; Brew, B.J.; Chung, R.; Guillemin, G.J. Characterisation of the Expression of NMDA Receptors in Human Astrocytes. PLoS ONE 2010, 5, e14123. [Google Scholar] [CrossRef] [PubMed]
- Sundaram, G.; Brew, B.J.; Jones, S.P.; Adams, S.; Lim, C.K.; Guillemin, G.J. Quinolinic acid toxicity on oligodendroglial cells: Relevance for multiple sclerosis and therapeutic strategies. J. Neuroinflamm. 2014, 11, 204. [Google Scholar] [CrossRef]
- Guillemin, G.J. Quinolinic acid, the inescapable neurotoxin. FEBS J. 2012, 279, 1356–1365. [Google Scholar] [CrossRef] [PubMed]
- Guillemin, G.J.; Williams, K.R.; Smith, D.G.; Smythe, G.A.; Croitoru-Lamoury, J.; Brew, B.J. Quinolinic Acid In the Pathogenesis of Alzheimer’s Disease. In Developments in Tryptophan and Serotonin Metabolism; Allegri, G., Costa, C.V.L., Ragazzi, E., Steinhart, H., Varesio, L., Eds.; Springer US: Boston, MA, USA, 2003; pp. 167–176. [Google Scholar] [CrossRef]
- Montgomery, E.B.; He, H. Deep Brain Stimulation Frequency—A Divining Rod for New and Novel Concepts of Nervous System Function and Therapy. Brain Sci. 2016, 6, 34. [Google Scholar] [CrossRef]
- Kepplinger, B.; Baran, H.; Sedlnitzky-Semler, B.; Badawi, N.-R.; Erhart, H. Stochastic Resonance Activity Influences Serum Tryptophan Metabolism in Healthy Human Subjects. Int. J. Tryptophan Res. 2011, 4, IJTR-S7986. [Google Scholar] [CrossRef] [PubMed]
- Ji, F.; Wei, J.; Luan, H.; Li, M.; Cai, Z. Study of metabolic disorders associated with BDE-47 exposure in Drosophila model by MS-based metabolomics. Ecotoxicol. Environ. Saf. 2019, 184, 109606. [Google Scholar] [CrossRef]
- Ogawa, T.; Matson, W.R.; Beal, M.F.; Myers, R.H.; Bird, E.D.; Milbury, P.; Saso, S. Kynurenine pathway abnormalities in Parkinson’s disease. Neurology 1992, 42, 1702–1706. [Google Scholar] [CrossRef] [PubMed]
- Beal, M.F.; Matson, W.R.; Storey, E.; Milbury, P.; Ryan, E.A.; Ogawa, T.; Bird, E.D. Kynurenic acid concentrations are reduced in Huntington’s disease cerebral cortex. J. Neurol. Sci. 1992, 108, 80–87. [Google Scholar] [CrossRef]
- Zinger, A.; Barcia, C.; Herrero, M.T.; Guillemin, G.J. The involvement of neuroinflammation and kynurenine pathway in Parkinson’s disease. Parkinsons Dis. 2011, 2011, 716859. [Google Scholar] [CrossRef]
- Widner, B.; Laich, A.; Sperner-Unterweger, B.; Ledochowski, M.; Fuchs, D. Neopterin production, tryptophan degradation, and mental depression—What is the link? Brain Behav. Immun. 2002, 16, 590–595. [Google Scholar] [CrossRef]
- Hartai, Z.; Klivenyi, P.; Janaky, T.; Penke, B.; Dux, L.; Vecsei, L. Kynurenine metabolism in plasma and in red blood cells in Parkinson’s disease. J. Neurol. Sci. 2005, 239, 31–35. [Google Scholar] [CrossRef]
- Barth, M.C.; Ahluwalia, N.; Anderson, T.J.T.; Hardy, G.J.; Sinha, S.; Alvarez-Cardona, J.A.; Pruitt, I.E.; Rhee, E.P.; Colvin, R.A.; Gerszten, R.E. Kynurenic acid triggers firm arrest of leukocytes to vascular endothelium under flow conditions. J. Biol. Chem. 2009, 284, 19189–19195. [Google Scholar] [CrossRef] [PubMed]
- Fujigaki, S.; Saito, K.; Sekikawa, K.; Tone, S.; Takikawa, O.; Fujii, H.; Wada, H.; Noma, A.; Seishima, M. Lipopolysaccharide induction of indoleamine 2,3-dioxygenase is mediated dominantly by an IFN-gamma-independent mechanism. Eur. J. Immunol. 2001, 31, 2313–2318. [Google Scholar] [CrossRef]
- Pinto, J.T.; Krasnikov, B.F.; Alcutt, S.; Jones, M.E.; Dorai, T.; Villar, M.T.; Artigues, A.; Li, J.; Cooper, A.J.L. Kynurenine aminotransferase III and glutamine transaminase L are identical enzymes that have cysteine S-conjugate β-lyase activity and can transaminate L-selenomethionine. J. Biol. Chem. 2014, 289, 30950–30961. [Google Scholar] [CrossRef]
- McNally, L.; Bhagwagar, Z.; Hannestad, J. Inflammation, glutamate, and glia in depression: A literature review. CNS Spectr. 2008, 13, 501–510. [Google Scholar] [CrossRef] [PubMed]
- Marin, C.; Jimenez, A.; Bonastre, M.; Chase, T.N.; Tolosa, E. Non-NMDA receptor-mediated mechanisms are involved in levodopa-induced motor response alterations in Parkinsonian rats. Synapse 2000, 36, 267–274. [Google Scholar] [CrossRef]
- Ceresoli-Borroni, G.; Guidetti, P.; Amori, L.; Pellicciari, R.; Schwarcz, R. Perinatal kynurenine 3-hydroxylase inhibition in rodents: Pathophysiological implications. J. Neurosci. Res. 2007, 85, 845–854. [Google Scholar] [CrossRef] [PubMed]
- Grégoire, L.; Rassoulpour, A.; Guidetti, P.; Samadi, P.; Bédard, P.J.; Izzo, E.; Schwarcz, R.; Di Paolo, T. Prolonged kynurenine 3-hydroxylase inhibition reduces development of levodopa-induced dyskinesias in parkinsonian monkeys. Behav. Brain Res. 2008, 186, 161–167. [Google Scholar] [CrossRef]
- Samadi, P.; Grégoire, L.; Rassoulpour, A.; Guidetti, P.; Izzo, E.; Schwarcz, R.; Bédard, P.J. Effect of kynurenine 3-hydroxylase inhibition on the dyskinetic and antiparkinsonian responses to levodopa in parkinsonian monkeys. Mov. Disord. 2005, 20, 792–802. [Google Scholar] [CrossRef]
- Abdel-Daim, M.M.; Abo-El-Sooud, K.; Aleya, L.; Bungău, S.G.; Najda, A.; Saluja, R. Alleviation of Drugs and Chemicals Toxicity: Biomedical Value of Antioxidants. Oxidative Med. Cell. Longev. 2018, 2018, 6276438. [Google Scholar] [CrossRef]
- Wonodi, I.; Stine, O.C.; Sathyasaikumar, K.V.; Roberts, R.C.; Mitchell, B.D.; Hong, L.E.; Kajii, Y.; Thaker, G.K.; Schwarcz, R. Downregulated kynurenine 3-monooxygenase gene expression and enzyme activity in schizophrenia and genetic association with schizophrenia endophenotypes. Arch. Gen. Psychiatry 2011, 68, 665–674. [Google Scholar] [CrossRef]
- Behl, T.; Kaur, G.; Fratila, O.; Buhas, C.; Judea-Pusta, C.T.; Negrut, N.; Bustea, C.; Bungau, S. Cross-talks among GBA Gene Mutations, GCase, and α-synuclein in GBA Associated Parkinson’s Disease with their Targeted Therapeutic Approaches: A Comprehensive Review. Transl. Neurodegener. 2021, 10, 4. [Google Scholar] [CrossRef]
- Miranda, A.F.; Boegman, R.J.; Beninger, R.J.; Jhamandas, K. Protection against quinolinic acid-mediated excitotoxicity in nigrostriatal dopaminergic neurons by endogenous kynurenic acid. Neuroscience 1997, 78, 967–975. [Google Scholar] [CrossRef]
- Wu, H.Q.; Rassoulpour, A.; Schwarcz, R. Effect of systemic L-DOPA administration on extracellular kynurenate levels in the rat striatum. J. Neural Transm. 2002, 109, 239–249. [Google Scholar] [CrossRef] [PubMed]
- Brotchie, J.M.; Mitchell, I.J.; Sambrook, M.A.; Crossman, A.R. Alleviation of parkinsonism by antagonism of excitatory amino acid transmission in the medial segment of the globus pallidus in rat and primate. Mov. Disord. 1991, 6, 133–138. [Google Scholar] [CrossRef] [PubMed]
- Silva-Adaya, D.; Pérez-De La Cruz, V.; Villeda-Hernández, J.; Carrillo-Mora, P.; González-Herrera, I.G.; García, E.; Colín-Barenque, L.; Pedraza-Chaverrí, J.; Santamaría, A. Protective effect of L-kynurenine and probenecid on 6-hydroxydopamine-induced striatal toxicity in rats: Implications of modulating kynurenate as a protective strategy. Neurotoxicol. Teratol. 2011, 33, 303–312. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.Q.; Rassoulpour, A.; Schwarcz, R. Kynurenic acid leads, dopamine follows: A new case of volume transmission in the brain? J. Neural. Transm. 2007, 114, 33–41. [Google Scholar] [CrossRef]
- Oxenkrug, G.; van der Hart, M.; Roeser, J.; Summergrad, P. Peripheral Tryptophan—Kynurenine Metabolism Associated with Metabolic Syndrome is Different in Parkinson’s and Alzheimer’s Diseases. Endocrinol. Diabetes. Metab. J. 2017, 1. Available online: http://researchopenworld.com/wp-content/uploads/2017/2011/EDMJ-2017-2113-Gregory-F-Oxenkrug-USA.pdf (accessed on 26 April 2021).
- Tanaka, M.; Vécsei, L. Monitoring the redox status in multiple sclerosis. Biomedicines 2020, 8, 406. [Google Scholar] [CrossRef]
- Di Meo, S.; Reed, T.T.; Venditti, P.; Victor, V.M. Role of ROS and RNS sources in physiological and pathological conditions. Oxidative Med. Cell. Longev. 2016, 2016, 1245049. [Google Scholar] [CrossRef]
- Cores, Á.; Piquero, M.; Villacampa, M.; León, R.; Menéndez, J.C. NRF2 regulation processes as a source of potential drug targets against neurodegenerative diseases. Biomolecules 2020, 10, 904. [Google Scholar] [CrossRef]
- Vargas-Mendoza, N.; Morales-González, Á.; Madrigal-Santillán, E.O.; Madrigal-Bujaidar, E.; Álvarez-González, I.; García-Melo, L.F.; Anguiano-Robledo, L.; Fregoso-Aguilar, T.; Morales-Gonzalez, J.A. Antioxidant and adaptative response mediated by Nrf2 during physical exercise. Antioxidants 2019, 8, 196. [Google Scholar] [CrossRef] [PubMed]
- Di Rosa, G.; Brunetti, G.; Scuto, M.; Trovato Salinaro, A.; Calabrese, E.J.; Crea, R.; Schmitz-Linneweber, C.; Calabrese, V.; Saul, N. Healthspan enhancement by olive polyphenols in C. elegans wild type and Parkinson’s models. Int. J. Mol. Sci. 2020, 21, 3893. [Google Scholar] [CrossRef] [PubMed]
- Carrillo-Mora, P.; Pérez-De la Cruz, V.; Estrada-Cortés, B.; Toussaint-González, P.; Martínez-Cortéz, J.A.; Rodríguez-Barragán, M.; Quinzaños-Fresnedo, J.; Rangel-Caballero, F.; Gamboa-Coria, G.; Sánchez-Vázquez, I. Serum Kynurenines Correlate With Depressive Symptoms and Disability in Poststroke Patients: A Cross-sectional Study. Neurorehabil. Neural Repair 2020, 34, 936–944. [Google Scholar] [CrossRef]
- Vazquez, S.; Garner, B.; Sheil, M.M.; Truscott, R.J.W. Characterisation of the major autoxidation products of 3-hydroxykynurenine under physiological conditions. Free Radic. Res. 2000, 32, 11–23. [Google Scholar] [CrossRef]
- Anderson, G.; Maes, M. Neurodegeneration in Parkinson’s disease: Interactions of oxidative stress, tryptophan catabolites and depression with mitochondria and sirtuins. Mol. Neurobiol. 2014, 49, 771–783. [Google Scholar] [CrossRef] [PubMed]
- Anderson, G.; Maes, M. TRYCAT pathways link peripheral inflammation, nicotine, somatization and depression in the etiology and course of Parkinson’s disease. CNS Neurol. Disord. Drug Targets 2014, 13, 137–149. [Google Scholar] [CrossRef] [PubMed]
- Capuron, L.; Miller, A.H. Immune system to brain signaling: Neuropsychopharmacological implications. Pharmacol. Ther. 2011, 130, 226–238. [Google Scholar] [CrossRef]
- Erabi, H.; Okada, G.; Shibasaki, C.; Setoyama, D.; Kang, D.; Takamura, M.; Yoshino, A.; Fuchikami, M.; Kurata, A.; Kato, T.A. Kynurenic acid is a potential overlapped biomarker between diagnosis and treatment response for depression from metabolome analysis. Sci. Rep. 2020, 10, 1–8. [Google Scholar] [CrossRef]
- Hunt, B.S.C.; Cordeiro, T.M.; Robert, S.; de Dios, C.; Leal, V.A.C.; Soares, J.C.; Robert, D.; Antonio, T.; Sudhakar, S.M. Effect of mmune Activation on the Kynurenine Pathway and Depression Symptoms–A Systematic Review and Meta-Analysis. Neurosci. Biobehav. Rev. 2020, 118, 514–523. [Google Scholar] [CrossRef]
- Bay-Richter, C.; Linderholm, K.R.; Lim, C.K.; Samuelsson, M.; Träskman-Bendz, L.; Guillemin, G.J.; Erhardt, S.; Brundin, L. A role for inflammatory metabolites as modulators of the glutamate N-methyl-D-aspartate receptor in depression and suicidality. Brain Behav. Immun. 2015, 43, 110–117. [Google Scholar] [CrossRef] [PubMed]
- Savitz, J.; Drevets, W.C.; Smith, C.M.; Victor, T.A.; Wurfel, B.E.; Bellgowan, P.S.; Bodurka, J.; Teague, T.K.; Dantzer, R. Putative neuroprotective and neurotoxic kynurenine pathway metabolites are associated with hippocampal and amygdalar volumes in subjects with major depressive disorder. Neuropsychopharmacology 2015, 40, 463–471. [Google Scholar] [CrossRef] [PubMed]
- Lindqvist, D.; Kaufman, E.; Brundin, L.; Hall, S.; Surova, Y.; Hansson, O. Non-motor symptoms in patients with Parkinson’s disease-correlations with inflammatory cytokines in serum. PLoS ONE 2012, 7, e47387. [Google Scholar] [CrossRef]
- Khan, M.M.; Kempuraj, D.; Thangavel, R.; Zaheer, A. Protection of MPTP-induced neuroinflammation and neurodegeneration by Pycnogenol. Neurochem. Int. 2013, 62, 379–388. [Google Scholar] [CrossRef]
- Tas, S.W.; Vervoordeldonk, M.J.; Hajji, N.; Schuitemaker, J.H.N.; van der Sluijs, K.F.; May, M.J.; Ghosh, S.; Kapsenberg, M.L.; Tak, P.P.; de Jong, E.C. Noncanonical NF-κB signaling in dendritic cells is required for indoleamine 2,3-dioxygenase (IDO) induction and immune regulation. Blood 2007, 110, 1540–1549. [Google Scholar] [CrossRef] [PubMed]
- Steiner, J.; Walter, M.; Gos, T.; Guillemin, G.J.; Bernstein, H.-G.; Sarnyai, Z.; Mawrin, C.; Brisch, R.; Bielau, H.; zu Schwabedissen, L.M.; et al. Severe depression is associated with increased microglial quinolinic acid in subregions of the anterior cingulate gyrus: Evidence for an immune-modulated glutamatergic neurotransmission? J. Neuroinflamm. 2011, 8, 94. [Google Scholar] [CrossRef]
- De Carvalho, L.P.; Bochet, P.; Rossier, J. The endogenous agonist quinolinic acid and the non endogenous homoquinolinic acid discriminate between NMDAR2 receptor subunits. Neurochem. Int. 1996, 28, 445–452. [Google Scholar] [CrossRef]
- Beal, M.F.; Kowall, N.W.; Ellison, D.W.; Mazurek, M.F.; Swartz, K.J.; Martin, J.B. Replication of the neurochemical characteristics of Huntington’s disease by quinolinic acid. Nature 1986, 321, 168–171. [Google Scholar] [CrossRef] [PubMed]
- Foster, A.C.; Collins, J.F.; Schwarcz, R. On the excitotoxic properties of quinolinic acid, 2,3-piperidine dicarboxylic acids and structurally related compounds. Neuropharmacology 1983, 22, 1331–1342. [Google Scholar] [CrossRef]
- Shear, D.A.; Dong, J.; Haik-Creguer, K.L.; Bazzett, T.J.; Albin, R.L.; Dunbar, G.L. Chronic Administration of Quinolinic Acid in the Rat Striatum Causes Spatial Learning Deficits in a Radial Arm Water Maze Task. Exp. Neurol. 1998, 150, 305–311. [Google Scholar] [CrossRef]
- Vazey, E.M.; Chen, K.; Hughes, S.M.; Connor, B. Transplanted adult neural progenitor cells survive, differentiate and reduce motor function impairment in a rodent model of Huntington’s disease. Exp. Neurol. 2006, 199, 384–396. [Google Scholar] [CrossRef] [PubMed]
- McGeer, E.G.; Singh, E. Neurotoxic effects of endogenous materials: Quinolinic acid, l-pyroglutamic acid, and thyroid releasing hormone (TRH). Exp. Neurol. 1984, 86, 410–413. [Google Scholar] [CrossRef]
- Perkins, M.N.; Stone, T.W. An iontophoretic investigation of the actions of convulsant kynurenines and their interaction with the endogenous excitant quinolinic acid. Brain Res. 1982, 247, 184–187. [Google Scholar] [CrossRef]
- Lee, D.Y.; Lee, K.S.; Lee, H.J.; Noh, Y.H.; Kim, D.H.; Lee, J.Y.; Cho, S.H.; Yoon, O.J.; Lee, W.B.; Kim, K.Y.; et al. Kynurenic acid attenuates MPP(+)-induced dopaminergic neuronal cell death via a Bax-mediated mitochondrial pathway. Eur. J. Cell Biol. 2008, 87, 389–397. [Google Scholar] [CrossRef]
- Butler, E.G.; Bourke, D.W.; Finkelstein, D.I.; Horne, M.K. The effects of reversible inactivation of the subthalamo-pallidal pathway on the behaviour of naive and hemiparkinsonian monkeys. J. Clin. Neurosci. 1997, 4, 218–227. [Google Scholar] [CrossRef]
- Zádori, D.; Klivényi, P.; Plangár, I.; Toldi, J.; Vécsei, L. Endogenous neuroprotection in chronic neurodegenerative disorders: With particular regard to the kynurenines. J. Cell. Mol. Med. 2011, 15, 701–717. [Google Scholar] [CrossRef] [PubMed]
- Johnson, K.A.; Conn, P.J.; Niswender, C.M. Glutamate receptors as therapeutic targets for Parkinson’s disease. CNS Neurol. Disord. Drug Targets 2009, 8, 475–491. [Google Scholar] [CrossRef] [PubMed]
- Kubicova, L.; Hadacek, F.; Bachmann, G.; Weckwerth, W.; Chobot, V. Coordination Complex Formation and Redox Properties of Kynurenic and Xanthurenic Acid Can Affect Brain Tissue Homeodynamics. Antioxidants 2019, 8, 476. [Google Scholar] [CrossRef]
- Del Tredici, K.; Braak, H. A not entirely benign procedure: Progression of Parkinson’s disease. Acta Neuropathol. 2008, 115, 379–384. [Google Scholar] [CrossRef]
- Mulak, A.; Bonaz, B. Brain-gut-microbiota axis in Parkinson’s disease. World J. Gastroenterol. 2015, 21, 10609–10620. [Google Scholar] [CrossRef]
- Böttner, M.; Zorenkov, D.; Hellwig, I.; Barrenschee, M.; Harde, J.; Fricke, T.; Deuschl, G.; Egberts, J.H.; Becker, T.; Fritscher-Ravens, A.; et al. Expression pattern and localization of alpha-synuclein in the human enteric nervous system. Neurobiol. Dis. 2012, 48, 474–480. [Google Scholar] [CrossRef] [PubMed]
- Forsyth, C.B.; Shannon, K.M.; Kordower, J.H.; Voigt, R.M.; Shaikh, M.; Jaglin, J.A.; Estes, J.D.; Dodiya, H.B.; Keshavarzian, A. Increased Intestinal Permeability Correlates with Sigmoid Mucosa alpha-Synuclein Staining and Endotoxin Exposure Markers in Early Parkinson’s Disease. PLoS ONE 2011, 6, e28032. [Google Scholar] [CrossRef] [PubMed]
- Devos, D.; Lebouvier, T.; Lardeux, B.; Biraud, M.; Rouaud, T.; Pouclet, H.; Coron, E.; Bruley des Varannes, S.; Naveilhan, P.; Nguyen, J.M.; et al. Colonic inflammation in Parkinson’s disease. Neurobiol. Dis. 2013, 50, 42–48. [Google Scholar] [CrossRef]
- Westfall, S.; Lomis, N.; Kahouli, I.; Dia, S.Y.; Singh, S.P.; Prakash, S. Microbiome, probiotics and neurodegenerative diseases: Deciphering the gut brain axis. Cell Mol. Life Sci. 2017, 74, 3769–3787. [Google Scholar] [CrossRef] [PubMed]
- Dehhaghi, M.; Kazemi Shariat Panahi, H.; Guillemin, G.J. Microorganisms, Tryptophan Metabolism, and Kynurenine Pathway: A Complex Interconnected Loop Influencing Human Health Status. Int. J. Tryptophan Res. 2019, 12, 1178646919852996. [Google Scholar] [CrossRef]
- Gao, J.; Xu, K.; Liu, H.; Liu, G.; Bai, M.; Peng, C.; Li, T.; Yin, Y. Impact of the Gut Microbiota on Intestinal Immunity Mediated by Tryptophan Metabolism. Front. Cell. Infect. Microbiol. 2018, 8, 13. [Google Scholar] [CrossRef] [PubMed]
- Vujkovic-Cvijin, I.; Dunham, R.M.; Iwai, S.; Maher, M.C.; Albright, R.G.; Broadhurst, M.J.; Hernandez, R.D.; Lederman, M.M.; Huang, Y.; Somsouk, M.; et al. Dysbiosis of the gut microbiota is associated with HIV disease progression and tryptophan catabolism. Sci. Transl. Med. 2013, 5, 193ra191. [Google Scholar] [CrossRef]
- Makkar, R.; Behl, T.; Bungau, S.; Zengin, G.; Mehta, V.; Kumar, A.; Uddin, M.S.; Ashraf, G.M.; Abdel-Daim, M.M.; Arora, S.; et al. Nutraceuticals in Neurological Disorders. Int. J. Mol. Sci. 2020, 21, 4424. [Google Scholar] [CrossRef]
- O’Farrell, K.; Harkin, A. Stress-related regulation of the kynurenine pathway: Relevance to neuropsychiatric and degenerative disorders. Neuropharmacology 2017, 112, 307–323. [Google Scholar] [CrossRef]
- Shoaie, S.; Ghaffari, P.; Kovatcheva-Datchary, P.; Mardinoglu, A.; Sen, P.; Pujos-Guillot, E.; de Wouters, T.; Juste, C.; Rizkalla, S.; Chilloux, J.; et al. Quantifying Diet-Induced Metabolic Changes of the Human Gut Microbiome. Cell Metab. 2015, 22, 320–331. [Google Scholar] [CrossRef] [PubMed]
- El Aidy, S.; Dinan, T.; Cryan, J. Immune modulation of the brain-gut-microbe axis. Front. Microbiol. 2014, 5, 146. [Google Scholar] [CrossRef] [PubMed]
- Clarke, S.F.; Murphy, E.F.; O’Sullivan, O.; Ross, R.P.; O’Toole, P.W.; Shanahan, F.; Cotter, P.D. Targeting the Microbiota to Address Diet-Induced Obesity: A Time Dependent Challenge. PLoS ONE 2013, 8, e65790. [Google Scholar] [CrossRef] [PubMed]
- Wikoff, W.R.; Anfora, A.T.; Liu, J.; Schultz, P.G.; Lesley, S.A.; Peters, E.C.; Siuzdak, G. Metabolomics analysis reveals large effects of gut microflora on mammalian blood metabolites. Proc. Natl. Acad. Sci. USA 2009, 106, 3698–3703. [Google Scholar] [CrossRef]
- Tavassoly, O.; Sade, D.; Bera, S.; Shaham-Niv, S.; Vocadlo, D.J.; Gazit, E. Quinolinic Acid Amyloid-like Fibrillar Assemblies Seed α-Synuclein Aggregation. J. Mol. Biol. 2018, 430, 3847–3862. [Google Scholar] [CrossRef]
- Lewitt, P.A.; Li, J.; Lu, M.; Beach, T.G.; Adler, C.H.; Guo, L. 3-hydroxykynurenine and other Parkinson’s disease biomarkers discovered by metabolomic analysis. Mov. Disord. 2013, 28, 1653–1660. [Google Scholar] [CrossRef]
- Vilas, D.; Fernández-Santiago, R.; Sanchez, E.; Azcona, L.J.; Santos-Montes, M.; Casquero, P.; Argandoña, L.; Tolosa, E.; Paisán-Ruiz, C. A Novel p.Glu298Lys Mutation in the ACMSD Gene in Sporadic Parkinson’s Disease. J. Parkinsons Dis. 2017, 7, 459–463. [Google Scholar] [CrossRef]
- Chahine, L.M.; Stern, M.B.; Chen-Plotkin, A. Blood-based biomarkers for Parkinson’s disease. Parkinsonism Relat. Disord. 2014, 20, S99–S103. [Google Scholar] [CrossRef]
- Baković, J.; López Martínez, D.; Nikolaou, S.; Yu, B.Y.K.; Tossounian, M.-A.; Tsuchiya, Y.; Thrasivoulou, C.; Filonenko, V.; Gout, I. Regulation of the CoA Biosynthetic Complex Assembly in Mammalian Cells. Int. J. Mol. Sci. 2021, 22, 1131. [Google Scholar] [CrossRef]
- Sas, K.; Szabó, E.; Vécsei, L. Mitochondria, Oxidative Stress and the Kynurenine System, with a Focus on Ageing and Neuroprotection. Molecules 2018, 23, 191. [Google Scholar] [CrossRef]
- Burgos, K.; Malenica, I.; Metpally, R.; Courtright, A.; Rakela, B.; Beach, T.; Shill, H.; Adler, C.; Sabbagh, M.; Villa, S.; et al. Profiles of Extracellular miRNA in Cerebrospinal Fluid and Serum from Patients with Alzheimer’s and Parkinson’s Diseases Correlate with Disease Status and Features of Pathology. PLoS ONE 2014, 9, e94839. [Google Scholar] [CrossRef]
- Luan, H.; Liu, L.-F.; Meng, N.; Tang, Z.; Chua, K.-K.; Chen, L.-L.; Song, J.-X.; Mok, V.C.T.; Xie, L.-X.; Li, M.; et al. LC–MS-Based Urinary Metabolite Signatures in Idiopathic Parkinson’s Disease. J. Proteome Res. 2015, 14, 467–478. [Google Scholar] [CrossRef] [PubMed]
- Luan, H.; Liu, L.F.; Tang, Z.; Zhang, M.; Chua, K.K.; Song, J.X.; Mok, V.C.; Li, M.; Cai, Z. Comprehensive urinary metabolomic profiling and identification of potential noninvasive marker for idiopathic Parkinson’s disease. Sci. Rep. 2015, 5, 13888. [Google Scholar] [CrossRef]
- Iwaoka, K.; Otsuka, C.; Maeda, T.; Yamahara, K.; Kato, K.; Takahashi, K.; Takahashi, K.; Terayama, Y. Impaired metabolism of kynurenine and its metabolites in CSF of Parkinson’s disease. Neurosci. Lett. 2020, 714, 134576. [Google Scholar] [CrossRef] [PubMed]
- Demeter, I.; Nagy, K.; Gellért, L.; Vécsei, L.; Fülöp, F.; Toldi, J. A novel kynurenic acid analog (SZR104) inhibits pentylenetetrazole-induced epileptiform seizures. An electrophysiological study: Special issue related to kynurenine. J. Neural Transm. 2012, 119, 151–154. [Google Scholar] [CrossRef] [PubMed]
- Gellért, L.; Fuzik, J.; Göblös, A.; Sárközi, K.; Marosi, M.; Kis, Z.; Farkas, T.; Szatmári, I.; Fülöp, F.; Vécsei, L.; et al. Neuroprotection with a new kynurenic acid analog in the four-vessel occlusion model of ischemia. Eur. J. Pharm. 2011, 667, 182–187. [Google Scholar] [CrossRef]
- Zádori, D.; Klivényi, P.; Toldi, J.; Fülöp, F.; Vécsei, L. Kynurenines in Parkinson’s disease: Therapeutic perspectives. J. Neural Transm. 2012, 119, 275–283. [Google Scholar] [CrossRef]
- Wu, H.Q.; Lee, S.C.; Schwarcz, R. Systemic administration of 4-chlorokynurenine prevents quinolinate neurotoxicity in the rat hippocampus. Eur. J. Pharm. 2000, 390, 267–274. [Google Scholar] [CrossRef]
- Moffett, J.R.; Els, T.; Espey, M.G.; Walter, S.A.; Streit, W.J.; Namboodiri, M.A. Quinolinate immunoreactivity in experimental rat brain tumors is present in macrophages but not in astrocytes. Exp. Neurol. 1997, 144, 287–301. [Google Scholar] [CrossRef]
- Fukuyama, K.; Tanahashi, S.; Hoshikawa, M.; Shinagawa, R.; Okada, M. Zonisamide regulates basal ganglia transmission via astroglial kynurenine pathway. Neuropharmacology 2014, 76, 137–145. [Google Scholar] [CrossRef]
- Copeland, C.S.; Neale, S.A.; Salt, T.E. Actions of Xanthurenic acid, a putative endogenous Group II metabotropic glutamate receptor agonist, on sensory transmission in the thalamus. Neuropharmacology 2013, 66, 133–142. [Google Scholar] [CrossRef]
- Fazio, F.; Lionetto, L.; Molinaro, G.; Bertrand, H.O.; Acher, F.; Ngomba, R.T.; Notartomaso, S.; Curini, M.; Rosati, O.; Scarselli, P.; et al. Cinnabarinic acid, an endogenous metabolite of the kynurenine pathway, activates type 4 metabotropic glutamate receptors. Mol. Pharm. 2012, 81, 643–656. [Google Scholar] [CrossRef]
- Nicoletti, F.; Bockaert, J.; Collingridge, G.L.; Conn, P.J.; Ferraguti, F.; Schoepp, D.D.; Wroblewski, J.T.; Pin, J.P. Metabotropic glutamate receptors: From the workbench to the bedside. Neuropharmacology 2011, 60, 1017–1041. [Google Scholar] [CrossRef]
- Duty, S. Therapeutic potential of targeting group III metabotropic glutamate receptors in the treatment of Parkinson’s disease. Br. J. Pharmacol. 2010, 161, 271–287. [Google Scholar] [CrossRef]
- Hamann, M.; Sander, S.E.; Richter, A. Effects of the kynurenine 3-hydroxylase inhibitor Ro 61-8048 after intrastriatal injections on the severity of dystonia in the dt sz mutant. Eur. J. Pharm. 2008, 586, 156–159. [Google Scholar] [CrossRef] [PubMed]
- Ouattara, B.; Belkhir, S.; Morissette, M.; Dridi, M.; Samadi, P.; Grégoire, L.; Meltzer, L.T.; Di Paolo, T. Implication of NMDA receptors in the antidyskinetic activity of cabergoline, CI-1041, and Ro 61-8048 in MPTP monkeys with levodopa-induced dyskinesias. J. Mol. Neurosci. 2009, 38, 128–142. [Google Scholar] [CrossRef] [PubMed]
- Pomplun, S.; Wang, Y.; Kirschner, A.; Kozany, C.; Bracher, A.; Hausch, F. Rational Design and Asymmetric Synthesis of Potent and Neurotrophic Ligands for FK506-Binding Proteins (FKBPs). Angew. Chem. Int. Ed. 2015, 54, 345–348. [Google Scholar] [CrossRef]
- Van der Perren, A.; Macchi, F.; Toelen, J.; Carlon, M.S.; Maris, M.; de Loor, H.; Kuypers, D.R.; Gijsbers, R.; Van den Haute, C.; Debyser, Z.; et al. FK506 reduces neuroinflammation and dopaminergic neurodegeneration in an α-synuclein-based rat model for Parkinson’s disease. Neurobiol. Aging 2015, 36, 1559–1568. [Google Scholar] [CrossRef] [PubMed]
- Auluck, P.K.; Chan, H.E.; Trojanowski, J.Q.; Lee, V.M.-Y.; Bonini, N.M. Chaperone suppression of α-synuclein toxicity in a Drosophila model for Parkinson’s disease. Science 2002, 295, 865–868. [Google Scholar] [CrossRef]
- Jones, D.R.; Moussaud, S.; McLean, P. Targeting heat shock proteins to modulate α-synuclein toxicity. Ther. Adv. Neurol. Disord. 2014, 7, 33–51. [Google Scholar] [CrossRef]
- Nagel, F.; Falkenburger, B.H.; Tönges, L.; Kowsky, S.; Pöppelmeyer, C.; Schulz, J.B.; Bähr, M.; Dietz, G.P.H. Tat-Hsp70 protects dopaminergic neurons in midbrain cultures and in the substantia nigra in models of Parkinson’s disease. J. Neurochem. 2008, 105, 853–864. [Google Scholar] [CrossRef]
- Tanabe, A.; Yamamura, Y.; Kasahara, J.; Morigaki, R.; Kaji, R.; Goto, S. A novel tyrosine kinase inhibitor AMN107 (nilotinib) normalizes striatal motor behaviors in a mouse model of Parkinson’s disease. Front. Cell. Neurosci. 2014, 8, 50. [Google Scholar] [CrossRef] [PubMed]
- Nuzzo, D. Role of Natural Antioxidants on Neuroprotection and Neuroinflammation. Antioxidants 2021, 10, 608. [Google Scholar] [CrossRef]
- Pizzino, G.; Irrera, N.; Cucinotta, M.; Pallio, G.; Mannino, F.; Arcoraci, V.; Squadrito, F.; Altavilla, D.; Bitto, A. Oxidative stress: Harms and benefits for human health. Oxidative Med. Cell. Longev. 2017, 2017, 8416763. [Google Scholar] [CrossRef]
- Oh, Y.; Do, H.T.T.; Kim, S.; Kim, Y.-M.; Chin, Y.-W.; Cho, J. Memory-Enhancing Effects of Mangosteen Pericarp Water Extract through Antioxidative Neuroprotection and Anti-Apoptotic Action. Antioxidants 2021, 10, 34. [Google Scholar] [CrossRef]
- Burgos, C.; Muñoz-Mingarro, D.; Navarro, I.; Martín-Cordero, C.; Acero, N. Neuroprotective Potential of Verbascoside Isolated from Acanthus mollis L. Leaves through Its Enzymatic Inhibition and Free Radical Scavenging Ability. Antioxidants 2020, 9, 1207. [Google Scholar] [CrossRef]
- Capatina, L.; Todirascu-Ciornea, E.; Napoli, E.M.; Ruberto, G.; Hritcu, L.; Dumitru, G. Thymus vulgaris Essential Oil Protects Zebrafish against Cognitive Dysfunction by Regulating Cholinergic and Antioxidants Systems. Antioxidants 2020, 9, 1083. [Google Scholar] [CrossRef]
- Moisa, C.; Lupitu, A.; Pop, G.; Chambre, D.R.; Copolovici, L.; Cioca, G.; Bungau, S.; Copolovici, D.M. Variation of the Chemical Composition of Thymus Vulgaris Essential Oils by Phenological Stages. Rev. Chim. 2019, 70, 633–637. [Google Scholar] [CrossRef]
- Ramsey, T.L.; Liu, Q.; Massey, B.W.; Brennan, M.D. Genotypic variation in the SV2C gene impacts response to atypical antipsychotics the CATIE study. Schizophr. Res. 2013, 149, 21–25. [Google Scholar] [CrossRef][Green Version]
- Khaliq, Z.M.; Bean, B.P. Pacemaking in dopaminergic ventral tegmental area neurons: Depolarizing drive from background and voltage-dependent sodium conductances. J. Neurosci. 2010, 30, 7401–7413. [Google Scholar] [CrossRef] [PubMed]
- Mosharov, E.V.; Larsen, K.E.; Kanter, E.; Phillips, K.A.; Wilson, K.; Schmitz, Y.; Krantz, D.E.; Kobayashi, K.; Edwards, R.H.; Sulzer, D. Interplay between cytosolic dopamine, calcium, and α-synuclein causes selective death of substantia nigra neurons. Neuron 2009, 62, 218–229. [Google Scholar] [CrossRef]
- Liu, Y.; Harding, M.; Dore, J.; Chen, X. Cav1. 2, but not Cav1. 3, L-type calcium channel subtype mediates nicotine-induced conditioned place preference in mice. Prog. Neuro Psychopharmacol. Biol. Psychiatry 2017, 75, 176–182. [Google Scholar] [CrossRef]
- Wang, X.; Saegusa, H.; Huntula, S.; Tanabe, T. Blockade of microglial Cav1. 2 Ca2+ channel exacerbates the symptoms in a Parkinson’s disease model. Sci. Rep. 2019, 9, 1–13. [Google Scholar]
- Kang, S.; Cooper, G.; Dunne, S.F.; Luan, C.-H.; Surmeier, D.J.; Silverman, R.B. Antagonism of L-type Ca2+ channels CaV1. 3 and CaV1. 2 by 1, 4-dihydropyrimidines and 4H-pyrans as dihydropyridine mimics. Bioorg. Med. Chem. 2013, 21, 4365–4373. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Harding, M.; Pittman, A.; Dore, J.; Striessnig, J.; Rajadhyaksha, A.; Chen, X. Cav1.2 and Cav1.3 L-type calcium channels regulate dopaminergic firing activity in the mouse ventral tegmental area. J. Neurophysiol. 2014, 112, 1119–1130. [Google Scholar] [CrossRef]
- Turrens, J.F. Mitochondrial formation of reactive oxygen species. J. Physiol. 2003, 552, 335–344. [Google Scholar] [CrossRef]
- Behl, T.; Kaur, G.; Sehgal, A.; Bhardwaj, S.; Singh, S.; Buhas, C.; Judea-Pusta, C.; Uivarosan, D.; Munteanu, M.A.; Bungau, S. Multifaceted Role of Matrix Metalloproteinases in Neurodegenerative Diseases: Pathophysiological and Therapeutic Perspectives. Int. J. Mol. Sci. 2021, 22, 1413. [Google Scholar] [CrossRef]
- Elstner, M.; Morris, C.M.; Heim, K.; Bender, A.; Mehta, D.; Jaros, E.; Klopstock, T.; Meitinger, T.; Turnbull, D.M.; Prokisch, H. Expression analysis of dopaminergic neurons in Parkinson’s disease and aging links transcriptional dysregulation of energy metabolism to cell death. Acta Neuropathol. 2011, 122, 75–86. [Google Scholar] [CrossRef] [PubMed]
- Catanesi, M.; d’Angelo, M.; Tupone, M.G.; Benedetti, E.; Giordano, A.; Castelli, V.; Cimini, A. MicroRNAs dysregulation and mitochondrial dysfunction in neurodegenerative diseases. Int. J. Mol. Sci. 2020, 21, 5986. [Google Scholar] [CrossRef] [PubMed]
- López-Gambero, A.J.; Rosell-Valle, C.; Medina-Vera, D.; Navarro, J.A.; Vargas, A.; Rivera, P.; Sanjuan, C.; de Fonseca, F.R.; Suárez, J. A Negative Energy Balance Is Associated with Metabolic Dysfunctions in the Hypothalamus of a Humanized Preclinical Model of Alzheimer’s Disease, the 5XFAD Mouse. Int. J. Mol. Sci. 2021, 22, 5365. [Google Scholar] [CrossRef] [PubMed]
- McNaught, K.S.P.; Jenner, P. Proteasomal function is impaired in substantia nigra in Parkinson’s disease. Neurosci. Lett. 2001, 297, 191–194. [Google Scholar] [CrossRef]
- Kawahata, I.; Fukunaga, K. Degradation of tyrosine hydroxylase by the ubiquitin-proteasome system in the pathogenesis of Parkinson’s disease and dopa-responsive dystonia. Int. J. Mol. Sci. 2020, 21, 3779. [Google Scholar] [CrossRef] [PubMed]
- Ceulemans, A.-G.; Zgavc, T.; Kooijman, R.; Hachimi-Idrissi, S.; Sarre, S.; Michotte, Y. The dual role of the neuroinflammatory response after ischemic stroke: Modulatory effects of hypothermia. J. Neuroinflamm. 2010, 7, 74. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.-Y.; Wang, X.; Liu, C.; Zhang, H.-L. Pharmacological Targeting of Microglial Activation: New Therapeutic Approach. Front. Cell. Neurosci. 2019, 13, 514. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
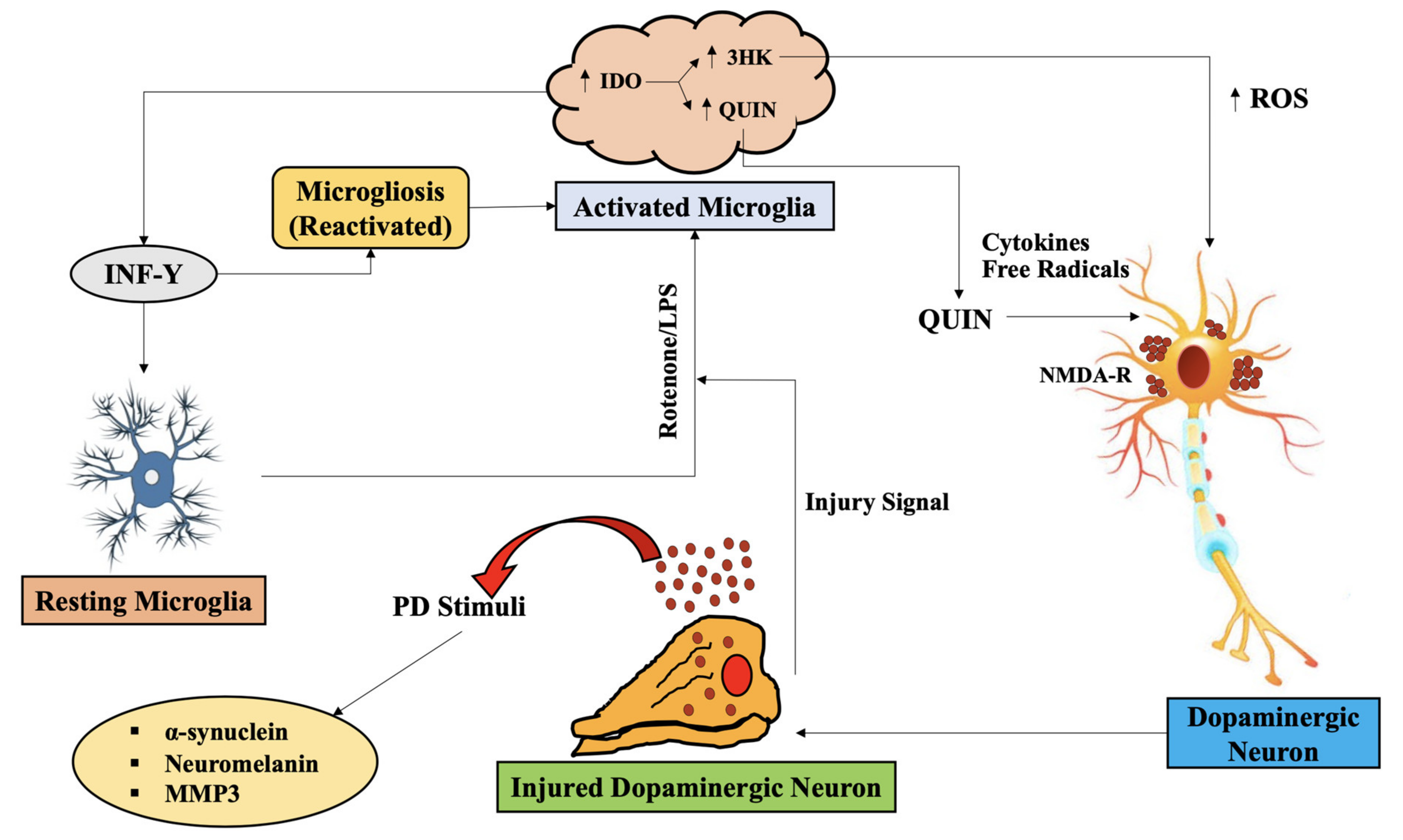{kind=link}
{kind=link}
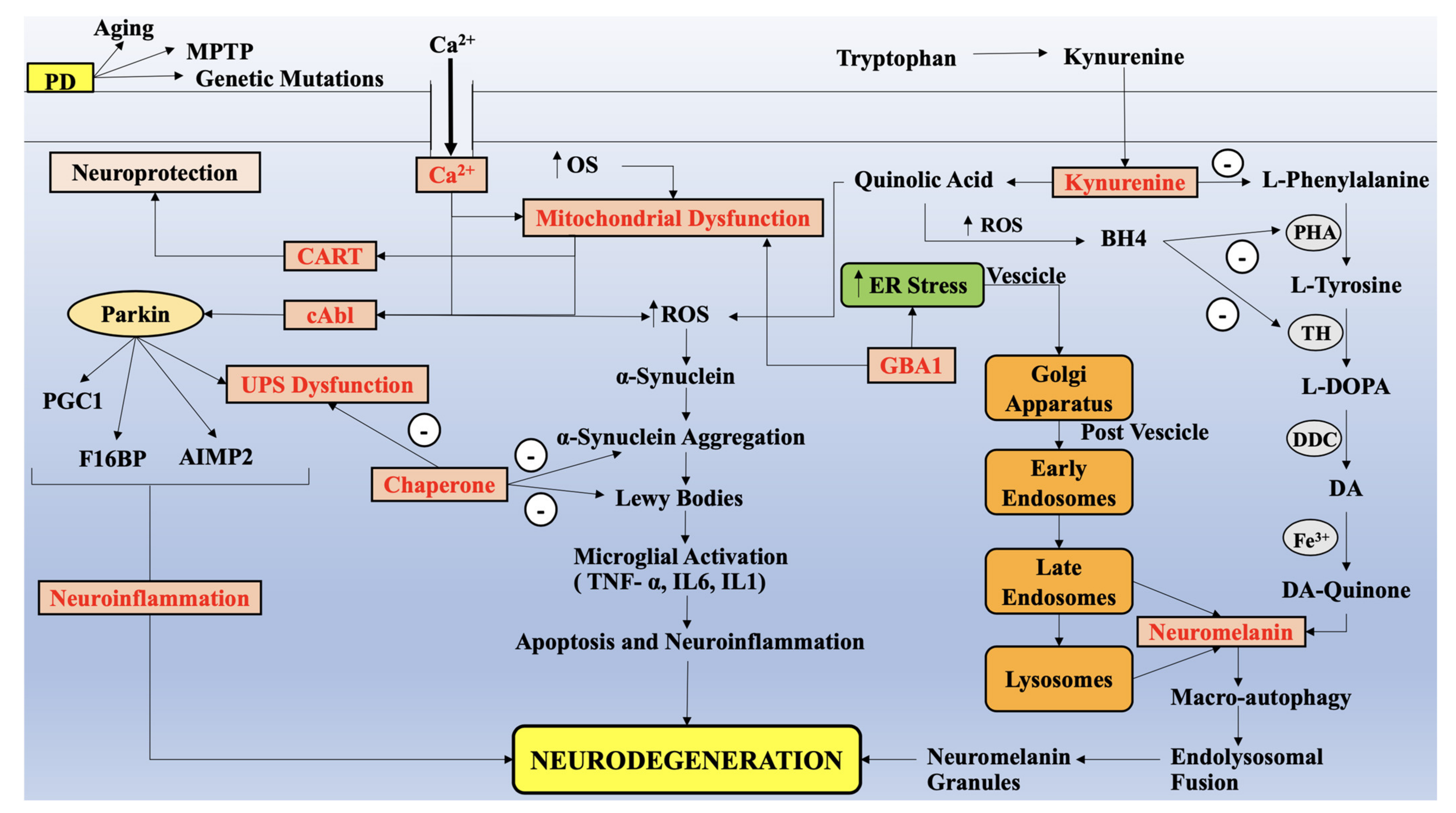{kind=link}
| Biofluid Biomarker | Metabolites | Observation | Ref. |
|---|---|---|---|
| Serum | Kynurenines | Decreased concentration of TRP | [8] |
| Urine | Urinary metabolites | Elevated α-synuclein and modified metabolism of tryptophan | [8] |
| CSF and serum | No metabolites mentioned | Decreased KYNA in CSF; increased KYNA, KYN and QUIN in serum | [8] |
| Plasma | 184 metabolites | Elevated KYN/TRP ratio, KYN, AA, KYN | [108] |
| Increased QUIN/KYNA ratio | [167] | ||
| Increased QUIN | [8] | ||
| CSF | No metabolites mentioned | Increased KYN and 3-HK | [166] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Behl, T.; Kaur, I.; Sehgal, A.; Singh, S.; Bhatia, S.; Al-Harrasi, A.; Zengin, G.; Bumbu, A.G.; Andronie-Cioara, F.L.; Nechifor, A.C.; et al. The Footprint of Kynurenine Pathway in Neurodegeneration: Janus-Faced Role in Parkinson’s Disorder and Therapeutic Implications. Int. J. Mol. Sci. 2021, 22, 6737. https://doi.org/10.3390/ijms22136737
Behl T, Kaur I, Sehgal A, Singh S, Bhatia S, Al-Harrasi A, Zengin G, Bumbu AG, Andronie-Cioara FL, Nechifor AC, et al. The Footprint of Kynurenine Pathway in Neurodegeneration: Janus-Faced Role in Parkinson’s Disorder and Therapeutic Implications. International Journal of Molecular Sciences. 2021; 22(13):6737. https://doi.org/10.3390/ijms22136737
Chicago/Turabian StyleBehl, Tapan, Ishnoor Kaur, Aayush Sehgal, Sukhbir Singh, Saurabh Bhatia, Ahmed Al-Harrasi, Gokhan Zengin, Adrian Gheorghe Bumbu, Felicia Liana Andronie-Cioara, Aurelia Cristina Nechifor, and et al. 2021. "The Footprint of Kynurenine Pathway in Neurodegeneration: Janus-Faced Role in Parkinson’s Disorder and Therapeutic Implications" International Journal of Molecular Sciences 22, no. 13: 6737. https://doi.org/10.3390/ijms22136737
APA StyleBehl, T., Kaur, I., Sehgal, A., Singh, S., Bhatia, S., Al-Harrasi, A., Zengin, G., Bumbu, A. G., Andronie-Cioara, F. L., Nechifor, A. C., Gitea, D., Bungau, A. F., Toma, M. M., & Bungau, S. G. (2021). The Footprint of Kynurenine Pathway in Neurodegeneration: Janus-Faced Role in Parkinson’s Disorder and Therapeutic Implications. International Journal of Molecular Sciences, 22(13), 6737. https://doi.org/10.3390/ijms22136737