
Improvement of Biophysical Properties and Affinity of a Human Anti-L1CAM Therapeutic Antibody through Antibody Engineering Based on Computational Methods

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
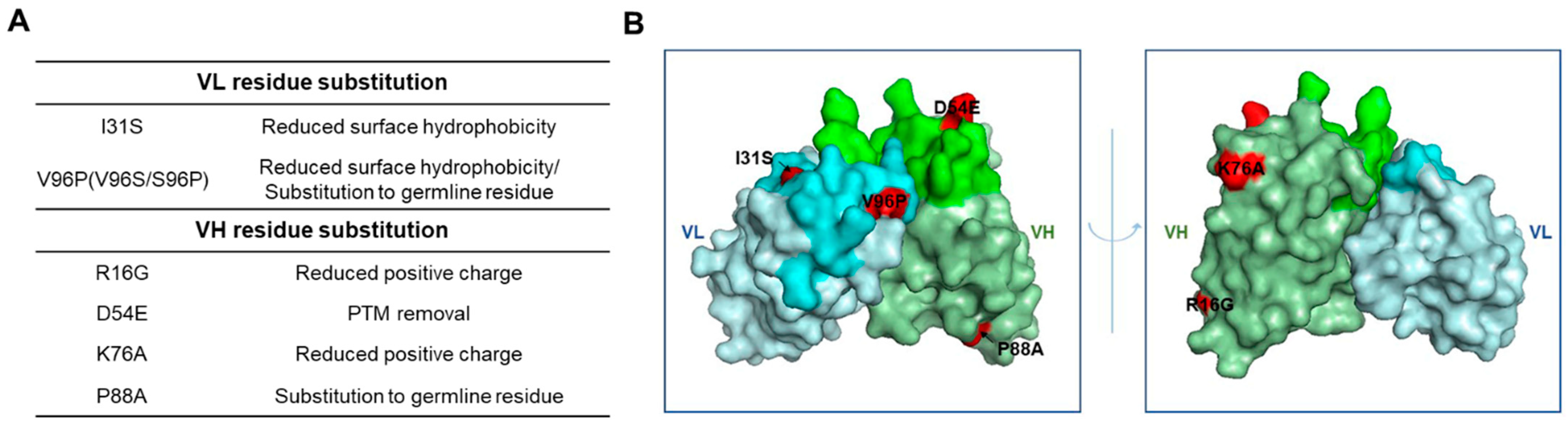
2.1. Design and Selection of an Ab417 Variant with Improved Biophysical Properties
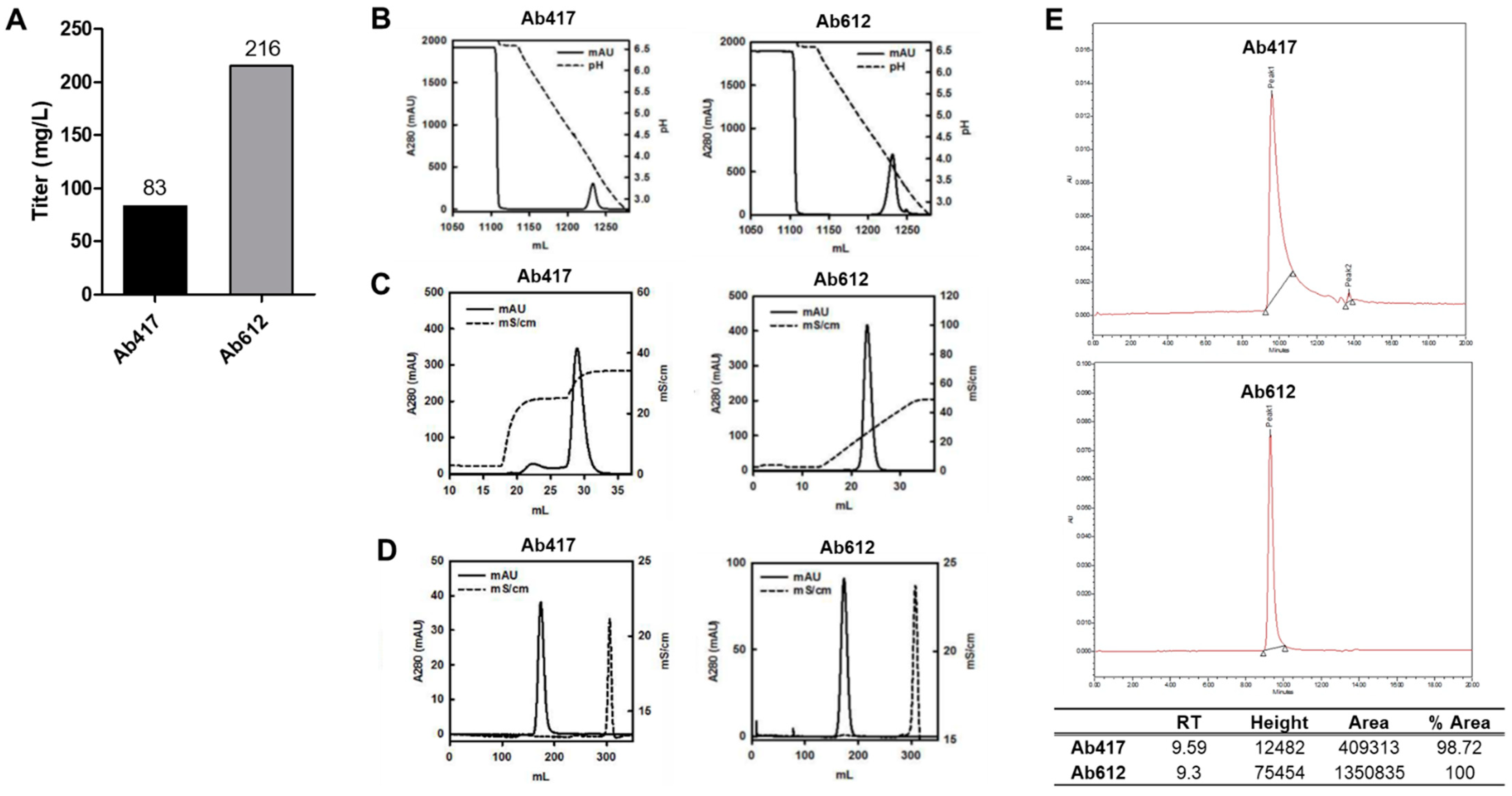
2.2. Expression Analysis and Optimization of Purification Process of Ab417 and Ab612
2.3. Thermal Stability of Ab417 and Ab612
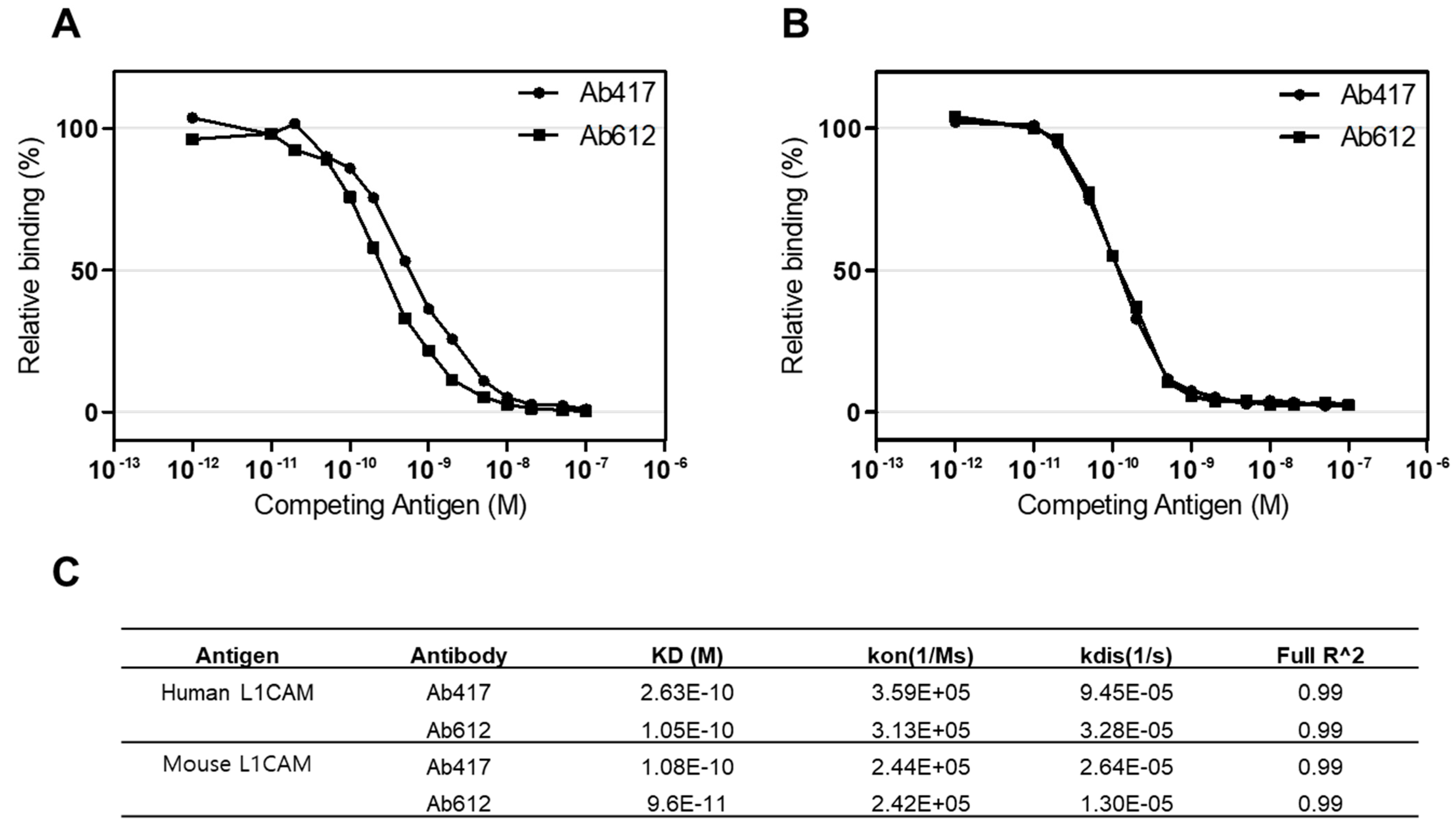
2.4. Affinity and pI Values of Ab417 and Ab612
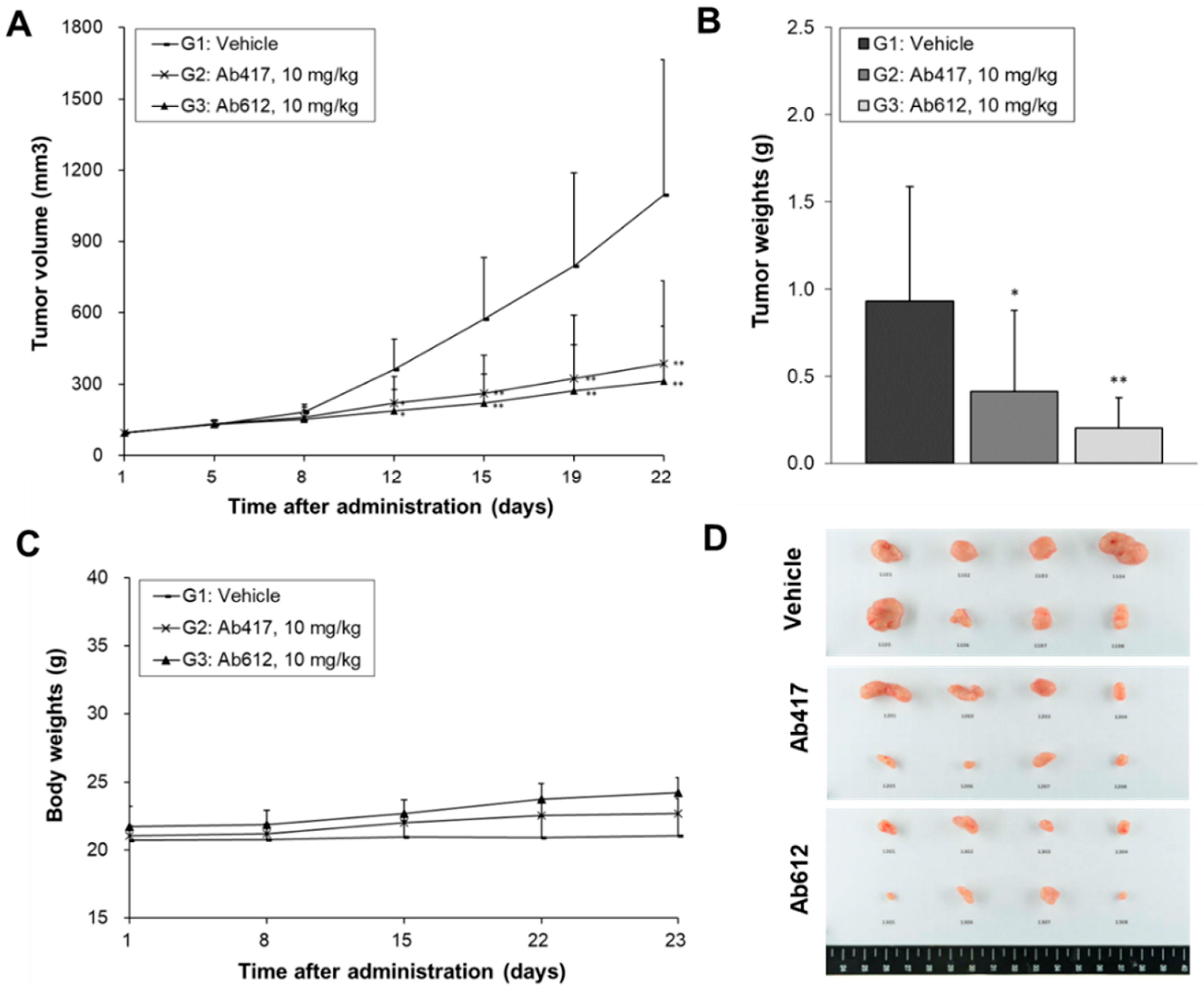
2.5. In Vivo Anti-Tumor Activities of Ab417 and Ab612
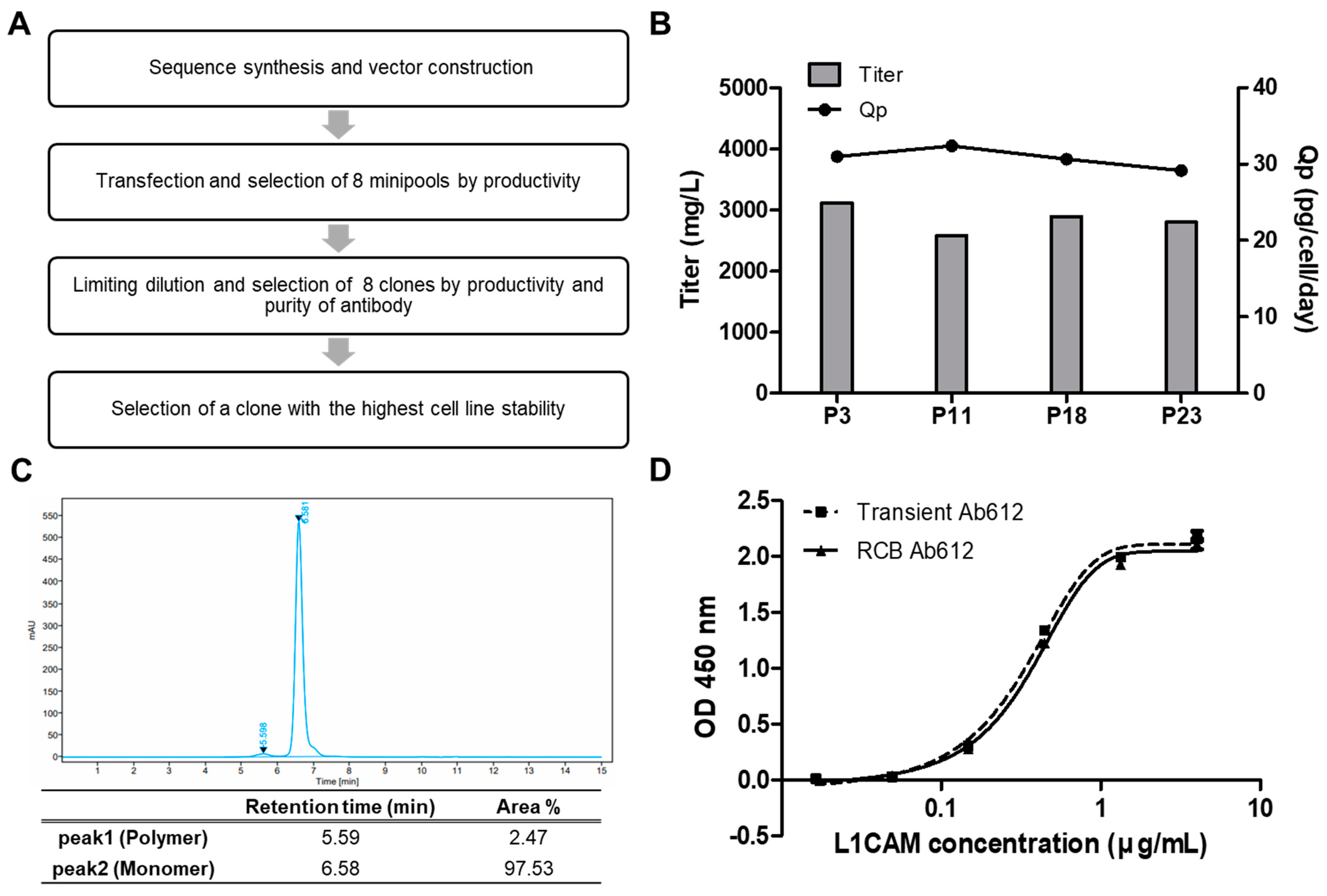
2.6. Generation of an RCB for Preclinical Development of Ab612
3. Discussion
4. Materials and Methods
4.1. Design of Ab417 Variants with Improved Biophysical Properties
4.1.1. Sequence Analysis for Potential PTMs
4.1.2. Construction and Comparison of 3D Models of Ab417 and Its Variants
4.1.3. Calculation of Aggregation Propensity and Assessment of Potential Substitutions
4.1.4. Analysis of Th Epitopes
4.2. Construction of Expression Plasmids and Transient Expression of Antibodies
4.3. ELISAs
4.4. Purification of Antibodies
4.5. Dynamic Light Scattering (DLS)
4.6. Affinity Determination of Antibodies Using Octet red384 System
4.7. Capillary Isoelectric Focusing (cIEF) Analysis
4.8. In Vivo Antitumor Activities of Ab417 or Ab612
4.9. Production of Ab612 from Research Cell Bank (RCB)
4.9.1. Construction of RCB
4.9.2. Stability Study of Research Cell Bank (RCB)
4.9.3. Production and Purification of Antibodies
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Lu, R.-M.; Hwang, Y.-C.; Liu, I.-J.; Lee, C.-C.; Tsai, H.-Z.; Li, H.-J.; Wu, H.-C. Development of therapeutic antibodies for the treatment of diseases. J. Biomed. Sci. 2020, 27, 1–30. [Google Scholar] [CrossRef] [PubMed]
- Morrison, S.L.; Johnson, M.J.; Herzenberg, L.A.; Oi, V.T. Chimeric human antibody molecules: Mouse antigen-binding domains with human constant region domains. Proc. Natl. Acad. Sci. USA 1984, 81, 6851–6855. [Google Scholar] [CrossRef] [Green Version]
- Jones, P.T.; Dear, P.H.; Foote, J.; Neuberger, M.S.; Winter, G. Replacing the complementarity-determining regions in a human antibody with those from a mouse. Nature 1986, 321, 522–525. [Google Scholar] [CrossRef] [PubMed]
- McCafferty, J.; Griffiths, A.D.; Winter, G.; Chiswell, D.J. Phage antibodies: Filamentous phage displaying antibody variable domains. Nature 1990, 348, 552–554. [Google Scholar] [CrossRef]
- Lonberg, N.; Taylor, L.D.; Harding, F.A.; Trounstine, M.; Higgins, K.M.; Schramm, S.R.; Kuo, C.-C.; Mashayekh, R.; Wymore, K.; McCabe, J.G. Antigen-specific human antibodies from mice comprising four distinct genetic modifications. Nature 1994, 368, 856–859. [Google Scholar] [CrossRef]
- Green, L.; Hardy, M.; Maynard-Currie, C.; Tsuda, H.; Louie, D.; Mendez, M.; Abderrahim, H.; Noguchi, M.; Smith, D.; Zeng, Y. Antigen–specific human monoclonal antibodies from mice engineered with human Ig heavy and light chain YACs. Nat. Genet. 1994, 7, 13–21. [Google Scholar] [CrossRef]
- Derer, S.; Kellner, C.; Berger, S.; Valerius, T.; Peipp, M. Fc engineering: Design, expression, and functional characterization of antibody variants with improved effector function. Methods Mol. Biol. 2012, 907, 519–536. [Google Scholar] [PubMed]
- Drake, P.M.; Rabuka, D. An emerging playbook for antibody–drug conjugates: Lessons from the laboratory and clinic suggest a strategy for improving efficacy and safety. Curr. Opin. Chem. Biol. 2015, 28, 174–180. [Google Scholar] [CrossRef]
- Labrijn, A.F.; Janmaat, M.L.; Reichert, J.M.; Parren, P.W. Bispecific antibodies: A mechanistic review of the pipeline. Nat. Rev. Drug Discov. 2019, 18, 585–608. [Google Scholar] [CrossRef]
- Jones, S.D.; Seymour, P.; Levine, H.L. CMC activities for development of mAbs. Contract Pharm. Apr. 2010, 60–64. Available online: https://www.contractpharma.com/issues/2010-04/view_features/cmc-activities-for-development-of-mabs/ (accessed on 10 June 2021).
- Chartrain, M.; Chu, L. Development and production of commercial therapeutic monoclonal antibodies in Mammalian cell expression systems: An overview of the current upstream technologies. Curr. Pharm. Biotechnol. 2008, 9, 447–467. [Google Scholar] [CrossRef]
- Liu, H.F.; Ma, J.; Winter, C.; Bayer, R. Recovery and purification process development for monoclonal antibody production. MAbs 2010, 2, 480–499. [Google Scholar] [CrossRef] [Green Version]
- Elgundi, Z.; Reslan, M.; Cruz, E.; Sifniotis, V.; Kayser, V. The state-of-play and future of antibody therapeutics. Adv. Drug Deliv. Rev. 2017, 122, 2–19. [Google Scholar] [CrossRef]
- Joubert, M.K.; Hokom, M.; Eakin, C.; Zhou, L.; Deshpande, M.; Baker, M.P.; Goletz, T.J.; Kerwin, B.A.; Chirmule, N.; Narhi, L.O. Highly aggregated antibody therapeutics can enhance the in vitro innate and late-stage T-cell immune responses. J. Biol. Chem. 2012, 287, 25266–25279. [Google Scholar] [CrossRef] [Green Version]
- Rehder, D.S.; Chelius, D.; McAuley, A.; Dillon, T.M.; Xiao, G.; Crouse-Zeineddini, J.; Vardanyan, L.; Perico, N.; Mukku, V.; Brems, D.N. Isomerization of a single aspartyl residue of anti-epidermal growth factor receptor immunoglobulin γ2 antibody highlights the role avidity plays in antibody activity. Biochemistry 2008, 47, 2518–2530. [Google Scholar] [CrossRef]
- Yan, Y.; Wei, H.; Fu, Y.; Jusuf, S.; Zeng, M.; Ludwig, R.; Krystek, S.R., Jr.; Chen, G.; Tao, L.; Das, T.K. Isomerization and oxidation in the complementarity-determining regions of a monoclonal antibody: A study of the modification–structure–function correlations by hydrogen–deuterium exchange mass spectrometry. Anal. Chem. 2016, 88, 2041–2050. [Google Scholar] [CrossRef]
- Ahmadi, M.; Bryson, C.J.; Cloake, E.A.; Welch, K.; Filipe, V.; Romeijn, S.; Hawe, A.; Jiskoot, W.; Baker, M.P.; Fogg, M.H. Small amounts of sub-visible aggregates enhance the immunogenic potential of monoclonal antibody therapeutics. Pharm. Res. 2015, 32, 1383–1394. [Google Scholar] [CrossRef]
- Bessa, J.; Boeckle, S.; Beck, H.; Buckel, T.; Schlicht, S.; Ebeling, M.; Kiialainen, A.; Koulov, A.; Boll, B.; Weiser, T. The immunogenicity of antibody aggregates in a novel transgenic mouse model. Pharm. Res. 2015, 32, 2344–2359. [Google Scholar] [CrossRef]
- Manning, M.C.; Chou, D.K.; Murphy, B.M.; Payne, R.W.; Katayama, D.S. Stability of protein pharmaceuticals: An update. Pharm. Res. 2010, 27, 544–575. [Google Scholar] [CrossRef]
- Sahin, E.; Grillo, A.O.; Perkins, M.D.; Roberts, C.J. Comparative effects of pH and ionic strength on protein–protein interactions, unfolding, and aggregation for IgG1 antibodies. J. Pharm. Sci. 2010, 99, 4830–4848. [Google Scholar] [CrossRef]
- Arosio, P.; Rima, S.; Morbidelli, M. Aggregation mechanism of an IgG2 and two IgG1 monoclonal antibodies at low pH: From oligomers to larger aggregates. Pharm. Res. 2013, 30, 641–654. [Google Scholar] [CrossRef]
- Telikepalli, S.N.; Kumru, O.S.; Kalonia, C.; Esfandiary, R.; Joshi, S.B.; Middaugh, C.R.; Volkin, D.B. Structural characterization of IgG1 mAb aggregates and particles generated under various stress conditions. J. Pharm. Sci. 2014, 103, 796–809. [Google Scholar] [CrossRef] [Green Version]
- Harris, R.J.; Kabakoff, B.; Macchi, F.D.; Shen, F.J.; Kwong, M.; Andya, J.D.; Shire, S.J.; Bjork, N.; Totpal, K.; Chen, A.B. Identification of multiple sources of charge heterogeneity in a recombinant antibody. J. Chromatogr. B Biomed. Sci. Appl. 2001, 752, 233–245. [Google Scholar] [CrossRef]
- Pace, A.L.; Wong, R.L.; Zhang, Y.T.; Kao, Y.-H.; Wang, Y.J. Asparagine deamidation dependence on buffer type, pH, and temperature. J. Pharm. Sci. 2013, 102, 1712–1723. [Google Scholar] [CrossRef]
- Zhang, Y.T.; Hu, J.; Pace, A.L.; Wong, R.; Wang, Y.J.; Kao, Y.-H. Characterization of asparagine 330 deamidation in an Fc-fragment of IgG1 using cation exchange chromatography and peptide mapping. J. Chromatogr. B 2014, 965, 65–71. [Google Scholar] [CrossRef] [PubMed]
- Yi, L.; Beckley, N.; Gikanga, B.; Zhang, J.; Wang, Y.J.; Chih, H.-W.; Sharma, V.K. Isomerization of Asp–Asp motif in model peptides and a monoclonal antibody Fab fragment. J. Pharm. Sci. 2013, 102, 947–959. [Google Scholar] [CrossRef]
- Vlasak, J.; Ionescu, R. Fragmentation of monoclonal antibodies. MAbs 2011, 3, 253–263. [Google Scholar] [CrossRef] [Green Version]
- Chennamsetty, N.; Voynov, V.; Kayser, V.; Helk, B.; Trout, B.L. Design of therapeutic proteins with enhanced stability. Proc. Natl. Acad. Sci. USA 2009, 106, 11937–11942. [Google Scholar] [CrossRef] [Green Version]
- Courtois, F.; Schneider, C.P.; Agrawal, N.J.; Trout, B.L. Rational design of biobetters with enhanced stability. J. Pharm. Sci. 2015, 104, 2433–2440. [Google Scholar] [CrossRef]
- Angarica, V.E.; Sancho, J. Protein dynamics governed by interfaces of high polarity and low packing density. PLoS ONE 2012, 7, e48212. [Google Scholar] [CrossRef]
- Cho, S.; Park, I.; Kim, H.; Jeong, M.S.; Lim, M.; Lee, E.S.; Kim, J.H.; Kim, S.; Hong, H.J. Generation, characterization and preclinical studies of a human anti-L1CAM monoclonal antibody that cross-reacts with rodent L1CAM. MAbs 2016, 8, 414–425. [Google Scholar] [CrossRef] [Green Version]
- Cho, S.; Lee, T.S.; Song, I.H.; Kim, A.-R.; Lee, Y.-J.; Kim, H.; Hwang, H.; Jeong, M.S.; Kang, S.G.; Hong, H.J. Combination of anti-L1 cell adhesion molecule antibody and gemcitabine or cisplatin improves the therapeutic response of intrahepatic cholangiocarcinoma. PLoS ONE 2017, 12, e0170078. [Google Scholar] [CrossRef]
- Jain, T.; Sun, T.; Durand, S.; Hall, A.; Houston, N.R.; Nett, J.H.; Sharkey, B.; Bobrowicz, B.; Caffry, I.; Yu, Y. Biophysical properties of the clinical-stage antibody landscape. Proc. Natl. Acad. Sci. USA 2017, 114, 944–949. [Google Scholar] [CrossRef] [Green Version]
- Katoh, K.; Misawa, K.; Kuma, K.; Miyata, T. MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pawar, A.P.; Dubay, K.F.; Zurdo, J.; Chiti, F.; Vendruscolo, M.; Dobson, C.M. Prediction of “aggregation-prone” and “aggregation-susceptible” regions in proteins associated with neurodegenerative diseases. J. Mol. Biol. 2005, 350, 379–392. [Google Scholar] [CrossRef]
- Laupeze, B.; Fardel, O.; Onno, M.; Bertho, N.; Drenou, B.; Fauchet, R.; Amiot, L. Differential expression of major histocompatibility complex class Ia, Ib, and II molecules on monocytes-derived dendritic and macrophagic cells. Hum. Immunol. 1999, 60, 591–597. [Google Scholar] [CrossRef]
- Ryu, C.J.; Kim, Y.K.; Hur, H.; Kim, H.S.; Oh, J.M.; Kang, Y.J.; Hong, H.J. Mouse monoclonal antibodies to hepatitis B virus preS1 produced after immunization with recombinant preS1 peptide. Hybridoma 2000, 19, 185–189. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chae, H.; Cho, S.; Jeong, M.; Kwon, K.; Choi, D.; Lee, J.; Nam, W.; Hong, J.; Lee, J.; Yoon, S.; et al. Improvement of Biophysical Properties and Affinity of a Human Anti-L1CAM Therapeutic Antibody through Antibody Engineering Based on Computational Methods. Int. J. Mol. Sci. 2021, 22, 6696. https://doi.org/10.3390/ijms22136696
Chae H, Cho S, Jeong M, Kwon K, Choi D, Lee J, Nam W, Hong J, Lee J, Yoon S, et al. Improvement of Biophysical Properties and Affinity of a Human Anti-L1CAM Therapeutic Antibody through Antibody Engineering Based on Computational Methods. International Journal of Molecular Sciences. 2021; 22(13):6696. https://doi.org/10.3390/ijms22136696
Chicago/Turabian StyleChae, Heesu, Seulki Cho, Munsik Jeong, Kiyoung Kwon, Dongwook Choi, Jaeyoung Lee, Woosuk Nam, Jisu Hong, Jiwoo Lee, Seonjoo Yoon, and et al. 2021. "Improvement of Biophysical Properties and Affinity of a Human Anti-L1CAM Therapeutic Antibody through Antibody Engineering Based on Computational Methods" International Journal of Molecular Sciences 22, no. 13: 6696. https://doi.org/10.3390/ijms22136696
APA StyleChae, H., Cho, S., Jeong, M., Kwon, K., Choi, D., Lee, J., Nam, W., Hong, J., Lee, J., Yoon, S., & Hong, H. (2021). Improvement of Biophysical Properties and Affinity of a Human Anti-L1CAM Therapeutic Antibody through Antibody Engineering Based on Computational Methods. International Journal of Molecular Sciences, 22(13), 6696. https://doi.org/10.3390/ijms22136696