
Combined Naïve Bayesian, Chemical Fingerprints and Molecular Docking Classifiers to Model and Predict Androgen Receptor Binding Data for Environmentally- and Health-Sensitive Substances



Abstract
:1. Introduction
2. Methods
2.1. Data Sets and Molecular Structures
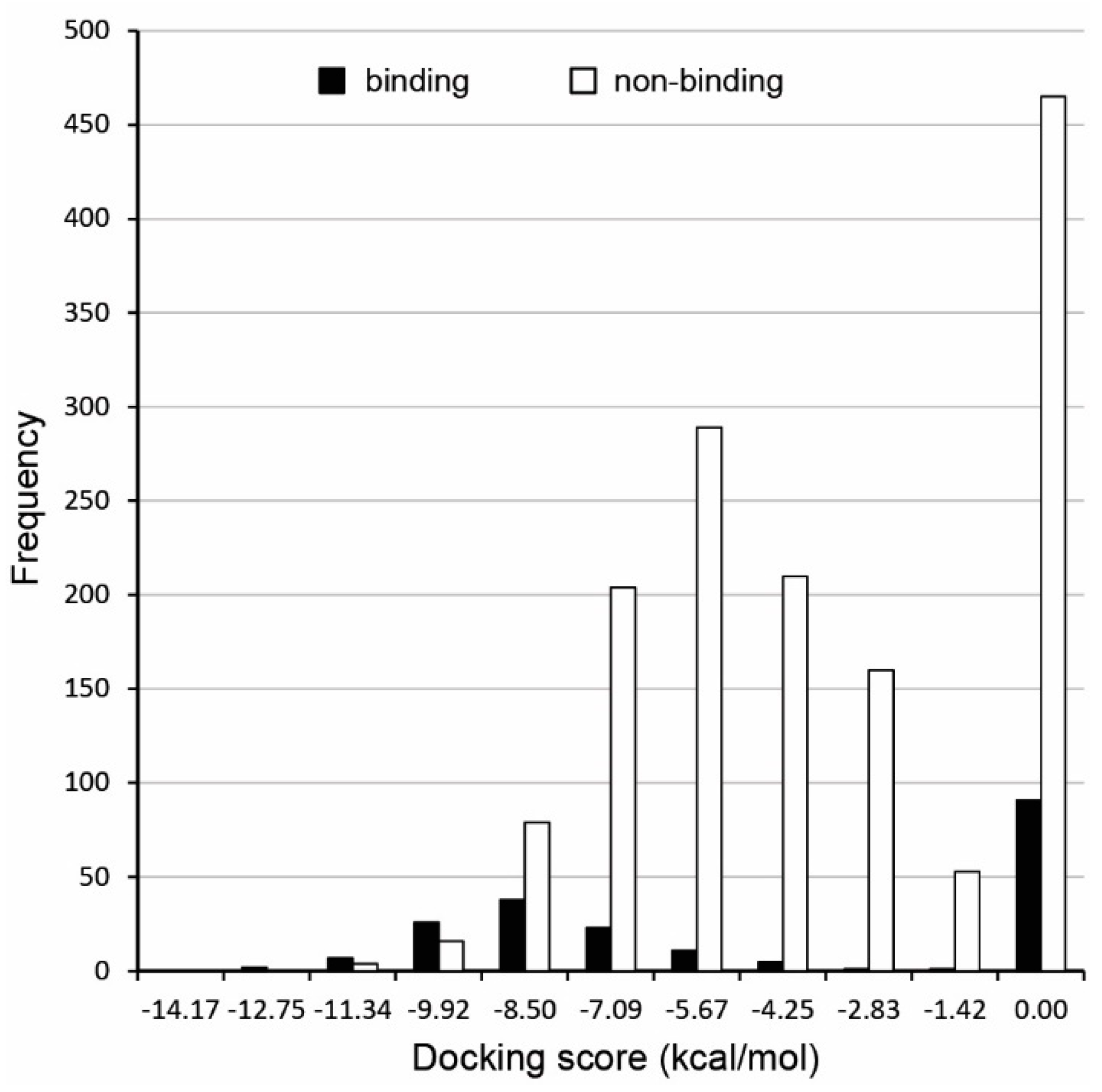
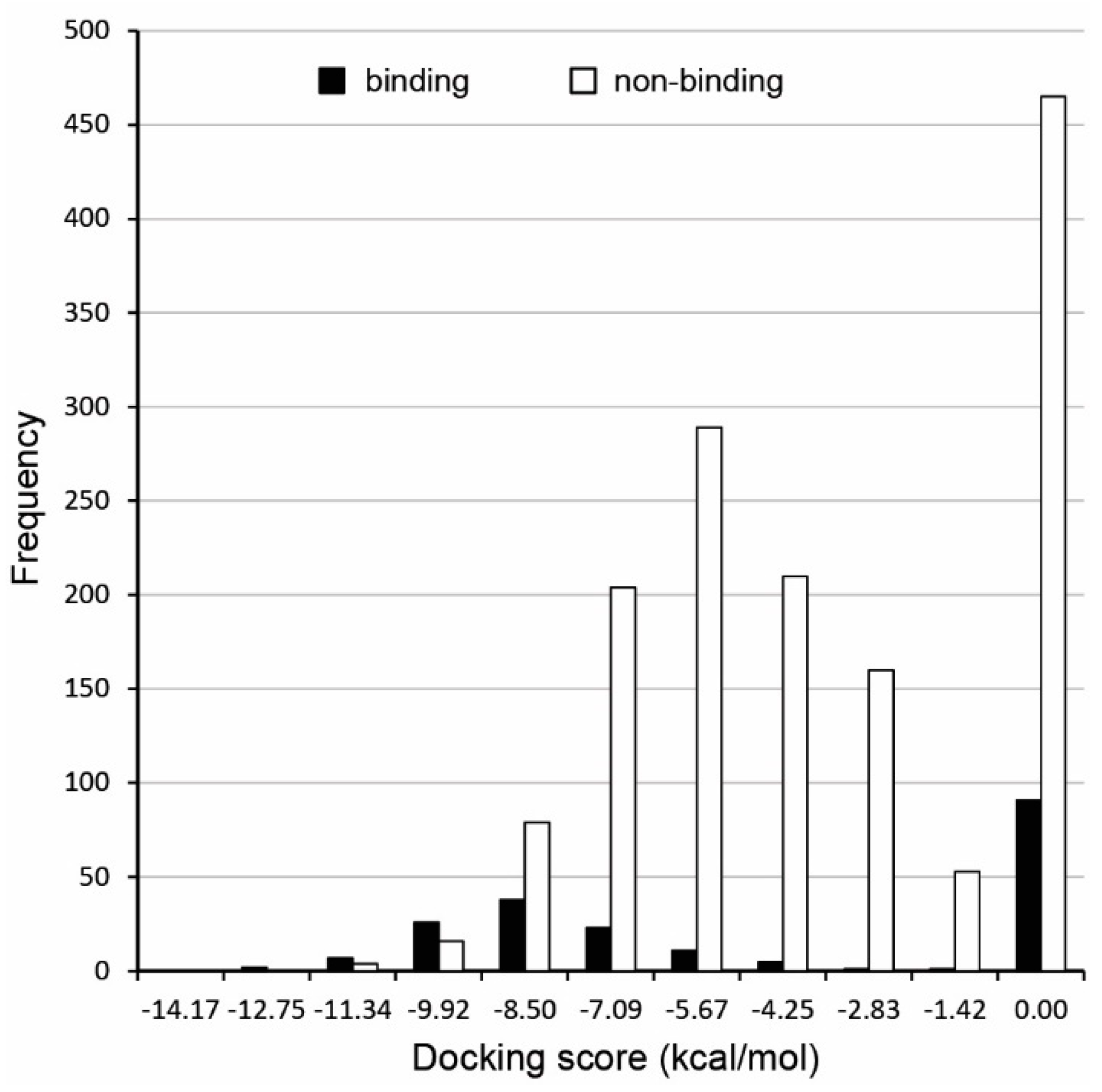
2.2. Molecular Docking
2.3. Protein Structures
2.4. Characterization and Comparison of Ligands
2.5. Naïve Bayesians
2.6. Multivariate Logistic Regression
2.7. Performance Analysis
2.8. Availability of Best Model
3. Results
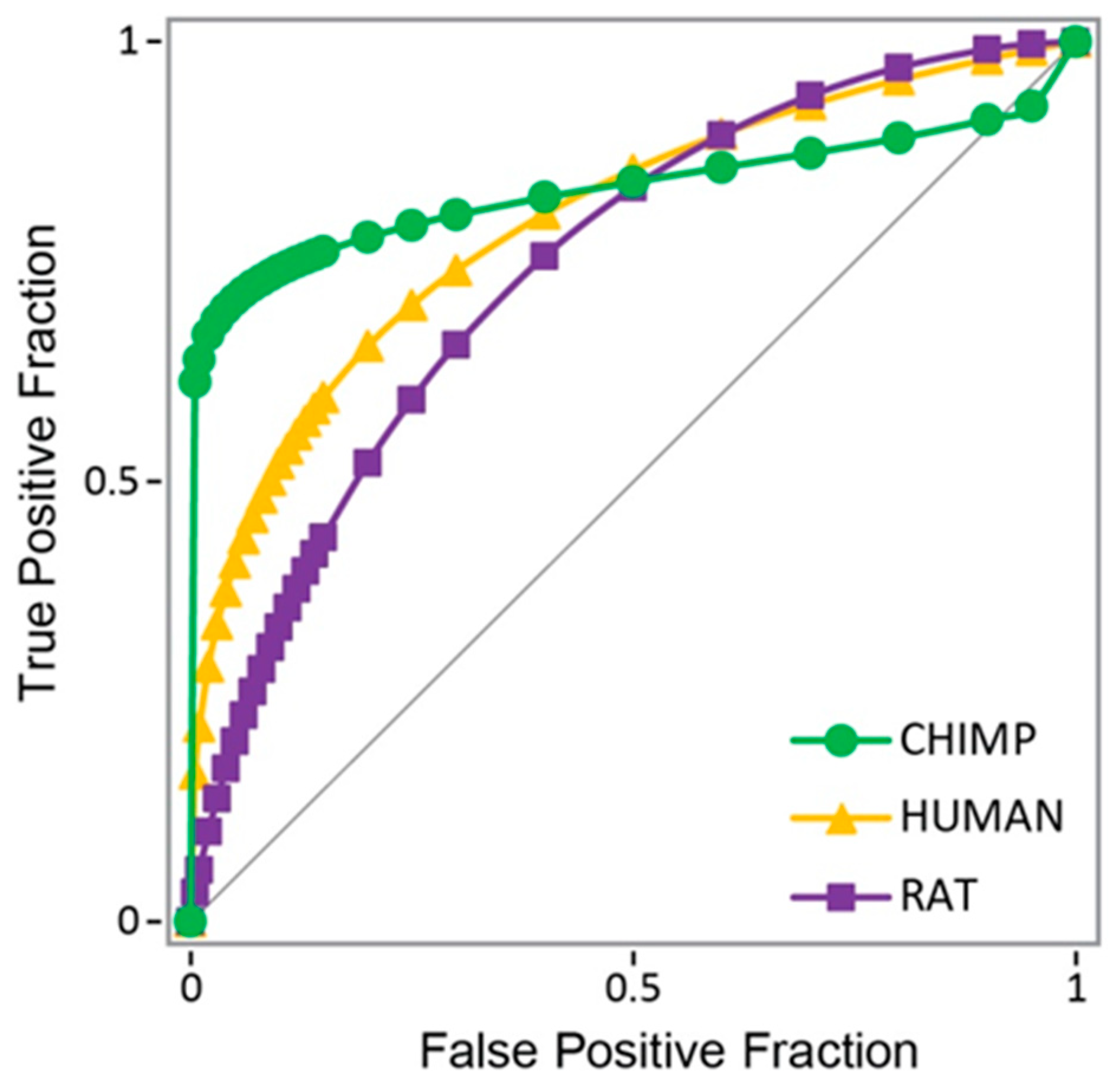
3.1. Androgen Receptor from Chimp as Model Protein
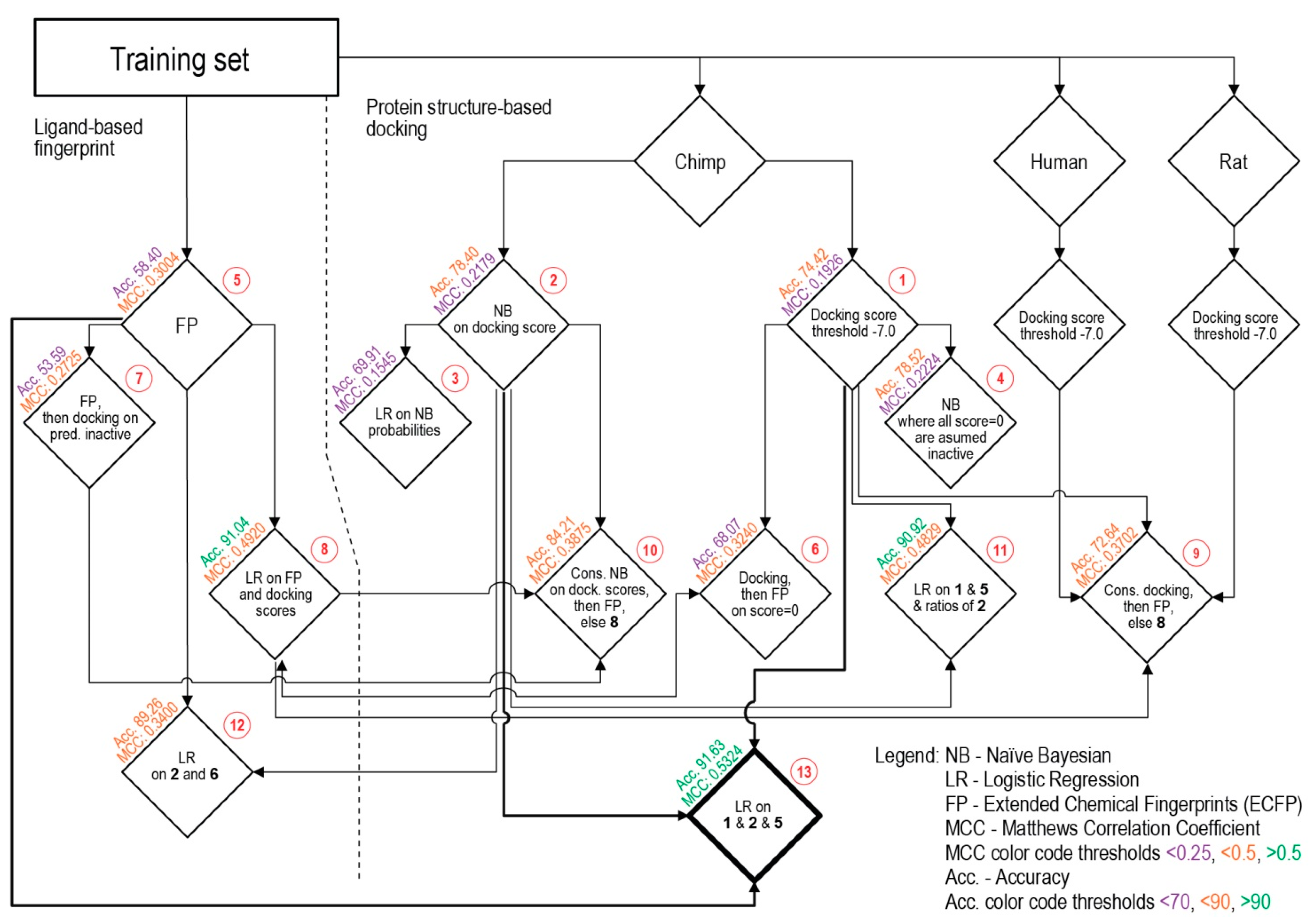
3.2. Consensus Methods for Best Performance
3.3. Validation of the Best Model
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Gray, L.E., Jr.; Ostby, J.F.; Furr, J.; Wolf, C.J.; Lambright, C.; Parks, L.; Veeramachaneni, D.N.; Wilson, V.; Price, M.; Hotchkiss, A.; et al. Effects of environmental antiandrogens on reproductive development in experimental animals. Hum. Reprod Update 2001, 7, 248–264. [Google Scholar] [CrossRef] [PubMed]
- Rider, C.V.; Furr, J.R.; Wilson, V.S.; Gray, L.E., Jr. Cumulative effects of in utero administration of mixtures of reproductive toxicants that disrupt common target tissues via diverse mechanisms of toxicity. Int. J. Androl. 2010, 33, 443–462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- LaLone, C.A.; Villeneuve, D.L.; Cavallin, J.E.; Kahl, M.D.; Durhan, E.J.; Makynen, E.A.; Jensen, K.M.; Stevens, K.E.; Severso, M.N.; Blanksma, C.A.; et al. Cross-species sensitivity to a novel androgen receptor agonist of potential environmental concern, spironolactone. Environ. Toxicol. Chem. 2013, 32, 2528–2541. [Google Scholar] [CrossRef] [PubMed]
- Tue Nguyen, H.M.; Heidenberg, D.J.; Sikka, S.C. Androgen receptor modulators: The impact of environment and lifestyle choices on reproduction. In Bioenvironmental Issues Affecting Men’s Reproductive and Sexual Health; Suresh, C.S., Hellstrom, W.J.G., Eds.; Academic Press: Cambridge, MA, USA, 2018; pp. 277–292. [Google Scholar] [CrossRef]
- Sifakis, D.; Androutsopoulos, V.P.; Tsatsakis, A.M.; Spandidos, D.A. Human exposure to endocrine disrupting chemicals: Effects on the male and female reproductive systems. Environ. Toxicol. Pharmacol. 2017, 51, 56–70. [Google Scholar] [CrossRef] [PubMed]
- Cheung, A.; Zajac, J.; Grossmann, M. Muscle and Bone Effects of Androgen Deprivation Therapy: Current and Emerging Therapies. Endocr Relat Cancer 2014, 21, R371–R394. [Google Scholar] [CrossRef] [PubMed]
- Manolagas, S.C.; O’Brien, C.A.; Almeida, M. The role of estrogen and androgen receptors in bone health and disease. Nat. Rev. Endocrinol. 2019, 9, 699–712. [Google Scholar] [CrossRef] [PubMed]
- Mendelsohn, M.E.; Karas, R.H. Molecular and cellular basis of cardiovascular gender differences. Science 2005, 308, 1583–1587. [Google Scholar] [CrossRef] [Green Version]
- Liu, P.Y.; Death, A.K.; Handelsman, D.J. Androgens and cardiovascular disease. Endocr. Rev. 2003, 24, 313–340. [Google Scholar] [CrossRef]
- Wu, F.C.W.; von Eckardstein, A. Androgens and coronary artery disease. Endocr. Rev. 2003, 24, 183–217. [Google Scholar] [CrossRef]
- Pennell, L.M.; Galligan, C.L.; Fish, E.N. Sex affects immunity. J. Autoimmun. 2012, 38, J282–J291. [Google Scholar] [CrossRef]
- Sellau, J.; Groneberg, M.; Lotter, H. Androgen-dependent immune modulation in parasitic infection. Semin. Immunopathol. 2019, 41, 213–224. [Google Scholar] [CrossRef]
- de Oliveira Barros, E.G.; Da Costa, N.M.; Palmero, C.Y.; Pinto, L.F.R.; Nasciutti, L.E.; Palumbo, A. Malignant invasion of the central nervous system: The hidden face of a poorly understood outcome of prostate cancer. World J. Urol. 2018, 36, 2009–2019. [Google Scholar] [CrossRef] [PubMed]
- Hussain, R.; Ghoumari, A.M.; Bielecki, B.; Steibel, J.; Boehm, N.; Liere, O.; Macklin, W.B.; Kumar, N.; Habert, R.; Mhaouty-Kodja, S.; et al. The neural androgen receptor: A therapeutic target for myelin repair in chronic demyelination. Brain 2013, 136, 132–146. [Google Scholar] [CrossRef] [PubMed]
- Swift-Gallant, A.; Monks, D.A. Androgenic mechanisms of sexual differentiation of the nervous system and behavior. Front. Neuroendocr. 2017, 46, 32–45. [Google Scholar] [CrossRef] [PubMed]
- Chute, J.P.; Ross, J.R.; McDonnell, D.P. Minireview: Nuclear receptors, hematopoiesis, and stem cells. Mol. Endocrinol. 2010, 24, 1–10. [Google Scholar] [CrossRef]
- Huang, C.-K.; Luo, J.; Lee, S.O.; Chang, C. Concise review: Androgen receptor differential roles in stem/progenitor cells including prostate, embryonic, stromal, and hematopoietic lineages. Stem Cells 2014, 32, 2299–2308. [Google Scholar] [CrossRef]
- Tan, M.H.E.; Li, J.; Xu, H.E.; Melcher, K.; Yong, E.-L. Androgen receptor: Structure, role in prostate cancer and drug discovery. Acta Pharm. Sin. 2014, 36, 3–23. [Google Scholar] [CrossRef] [Green Version]
- Vera-Badillo, F.E.; Templeton, A.J.; De Gouveia, P.; Diaz-Padilla, I.; Bedard, P.L.; Al-Mubarak, M.; Seruga, B.; Tannock, I.F.; Ocana, A.; Amir, E. Androgen receptor expression and outcomes in early breast cancer: A systematic review and meta-analysis. J. Natl. Cancer Inst. 2014, 106, djt319. [Google Scholar] [CrossRef]
- Lee, A.; Djamgoz, M.B.A. Triple negative breast cancer: Emerging therapeutic modalities and novel combination therapies. Cancer Treat. Rev. 2016, 26, 110–122. [Google Scholar] [CrossRef]
- Dobruch, J.; Daneshmand, S.; Fisch, M.; Lotan, Y.; Noon, A.P.; Resnick, M.J.; Shariat, S.F.; Zlotta, A.R.; Boorjian, S.A. Gender and bladder cancer: A collaborative review of etiology, biology, and outcomes. Eur. Urol. 2016, 69, 300–310. [Google Scholar] [CrossRef]
- Ma, W.-L.; Lai, H.-C.; Yeh, S.; Cai, X.; Chang, C. Androgen receptor roles in hepatocellular carcinoma, fatty liver, cirrhosis and hepatitis. Endocr Relat Cancer 2014, 21, R165–R182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, W.; Li, L.; He, D.; Xie, H.; Chen, J.; Yeh, C.-R.; Chang, L.S.-S.; Yeh, S.; Chang, C. Infiltrating neutrophils promote renal cell carcinoma (RCC) proliferation via modulating androgen receptor (AR) → c-Myc signals. Cancer Lett. 2015, 368, 71–78. [Google Scholar] [CrossRef] [PubMed]
- Verma, M.K.; Miki, Y.; Sasano, H. Sex steroid receptors in human lung diseases. J. Steroid Biochem Mol. Biol. 2011, 127, 216–222. [Google Scholar] [CrossRef]
- Chang, C.; Lee, S.O.; Yeh, S.; Chang, T.M. Androgen receptor (AR) differential roles in hormone-related tumors including prostate, bladder, kidney, lung, breast and liver. Oncogene 2013, 33, 3225–3234. [Google Scholar] [CrossRef] [Green Version]
- Schug, T.T.; Janesick, A.; Blumberg, B.; Heindel, J.J. Endocrine disrupting chemicals and disease susceptibility. J. Steroid Biochem. Mol. Biol. 2011, 127, 204–215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luccio-Camelo, D.C.; Prins, G.S. Disruption of androgen receptor signaling in males by environmental chemicals. J. Steroid Biochem. Mol. Biol. 2011, 127, 74–82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fisher, J.S. Environmental anti-androgens and male reproductive health: Focus on phthalates and testicular dysgenesis syndrome. Reproduction 2004, 127, 305–315. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.-J.; Park, Y.I.; Dong, M.-S. Effects of 2,4-D and DCP on the DHT-induced androgenic action in human prostate cancer cells. Toxicol. Sci. 2005, 88, 52–59. [Google Scholar] [CrossRef] [Green Version]
- Davey, R.A.; Grossmann, M. Androgen receptor structure, function and biology: From bench to bedside. Clin. Biochem. Rev. 2006, 37, 3–15. [Google Scholar]
- Nyrönen, T.; Söderholm, A.A. Structural basis for computational screening of non-steroidal androgen receptor ligands. Exp. Op. Drug Discov. 2010, 5, 5–20. [Google Scholar] [CrossRef]
- Unwalla, R.; Mousseau, J.J.; Fadeyi, O.O.; Choi, C.; Parris, K.; Hu, B.; Kenney, T.; Chippari, S.; McNally, C.; Vishwanathan, K.; et al. Structure-based approach to identify 5-[4-hydroxyphenyl]pyrrole-2-carbonitrile derivatives as potent and tissue selective androgen receptor modulators. J. Med. Chem. 2017, 60, 6451–6457. [Google Scholar] [CrossRef]
- Glisic, S.; Sencanski, M.; Perovic, V.; Stevanovic, S.; García-Sosa, A.T. Arginase flavonoid anti-leishmanial in silico inhibitors flagged against anti-targets. Molecules 2016, 21, 589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- García-Sosa, A.T.; Maran, U. Improving the use of ranking in virtual screening against HIV-1 integrase with triangular numbers and including ligand profiling with anti-targets. J. Chem. Inf. Model. 2014, 54, 3172–3185. [Google Scholar] [CrossRef]
- Vinggaard, A.M.; Niemelä, J.; Wedebye, E.B.; Jensen, G.E. Screening of 397 chemicals and development of a quantitative structure−activity relationship model for androgen receptor antagonism. Chem. Res. Toxicol. 2008, 21, 813–823. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Gramatica, P. Classification and virtual screening of androgen receptor antagonists. J. Chem. Inf. Model. 2010, 50, 861–874. [Google Scholar] [CrossRef]
- Todorov, M.; Mombelli, E.; Aït-Aïssa, S.; Mekenyan, O. Androgen receptor binding affinity: A QSAR evaluation. SAR QSAR Environ. Res. 2011, 22, 265–291. [Google Scholar] [CrossRef]
- Jensen, G.E.; Nikolov, N.G.; Wedebye, E.B.; Ringsted, T.; Niemelä, J.R. QSAR models for anti-androgenic effect—A preliminary study. SAR QSAR Environ. Res. 2011, 22, 35–49. [Google Scholar] [CrossRef]
- Norinder, U.; Rybacka, A.; Andersson, P.L. Conformal prediction to define applicability domain—A case study on predicting ER and AR binding. SAR QSAR Environ. Res. 2016, 27, 303–316. [Google Scholar] [CrossRef] [PubMed]
- Mansouri, K.; Kleinstreuer, N.; Watt, E.; Harris, J.; Judson, R. CoMPARA: Collaborative modeling project for androgen receptor activity. In Proceedings of the SOT 56th Annual Meeting and ToxExpo, Baltimore, Maryland, 12–16 May 2017. [Google Scholar] [CrossRef]
- Kleinstreuer, N.C.; Ceger, P.; Watt, E.D.; Martin, M.; Houck, K.; Browne, P.; Thomas, R.S.; Casey, W.M.; Dix, D.J.; Allen, D.; et al. Development and Validation of a Computational Model for Androgen Receptor Activity. Chem. Res. Toxicol. 2017, 30, 946–964. [Google Scholar] [CrossRef]
- Mansouri, K.; Kleinstreuer, N.; Abdelaziz, A.M.; Alberga, D.; Alves, V.M.; Andersson, P.L.; Andrade, C.H.; Bai, F.; Balabin, I.; Ballabio, D.; et al. CoMPARA: Collaborative modeling project for androgen receptor activity. Environ. Health Perspect. 2020, 128, 027002. [Google Scholar] [CrossRef] [PubMed]
- Mansouri, K.; Abdelaziz, A.; Rybacka, A.; Roncaglioni, A.; Tropsha, A.; Varnek, A.; Zakharov, A.; Worth, A.; Richard, A.M.; Grulke, C.M.; et al. CERAPP: Collaborative estrogen receptor activity prediction project. Environ. Health Perspect. 2016, 124, 1023–1033. [Google Scholar] [CrossRef]
- Schrödinger, LLC. Glide Virtual Screening Workflow; Schrödinger Press: New York, NY, USA, 2017. [Google Scholar]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The protein data bank. Nucleic. Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [Green Version]
- Instant JChem Version 5.6.0; ChemAxon Ltd.: Budapest, Hungary, 2017; Available online: http://www.chemaxon.com (accessed on 29 June 2020).
- García-Sosa, A.T.; Maran, U. Drugs, nondrugs, and disease category specificity: Organ effects by ligand pharmacology. SAR QSAR Environ. Res. 2013, 24, 319–331. [Google Scholar] [CrossRef] [PubMed]
- García-Sosa, A.T.; Mancera, R.L.; Dean, P.M. WaterScore: A novel method for distinguishing between bound and displaceable water molecules in the crystal structure of the binding site of protein-ligand complexes. J. Mol. Model. 2003, 9, 172–182. [Google Scholar] [CrossRef]
- Swets, J.A. The relative operating characteristic in Psychology. Science 1973, 182, 990–1000. [Google Scholar] [CrossRef] [PubMed]
- Sokolova, M.; Lapalme, G. A systematic analysis of performance measures for classification tasks. Inf. Process. Manag. 2009, 45, 427–437. [Google Scholar] [CrossRef]
- Ruusmann, V.; Sild, S.; Maran, U. QSAR DataBank—An approach for the digital organization and archiving of QSAR model information. J. Cheminf. 2014, 6, 25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruusmann, V.; Sild, S.; Maran, U. QSAR DataBank repository: Open and linked qualitative and quantitative structure–activity relationship models. J. Cheminf. 2015, 7, 32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- QsarDB Repository. Available online: http://qsardb.org/ (accessed on 4 March 2021).
- Garcia-Sosa, A.T.; Maran, U. Data for: Combined Naïve Bayesian, Chemical Fingerprints, and Molecular Docking Classifiers to Codel and Predict Androgen Receptor Binding Activity Data for Environmentally- and Health-Sensitive Substances. QsarDB Repository 2021. QDB.235. [Google Scholar] [CrossRef]
- Watt, E.D.; Judson, R.S. Uncertainty quantification in ToxCast high throughput screening. PLoS ONE 2018, 13, e0196963. [Google Scholar] [CrossRef]
- Trisciuzzi, D.; Alberga, D.; Mansouri, K.; Judson, R.; Novellino, E.; Mangiatordi, G.F.; Nicolotti, O. Predictive structure-based toxicology approaches to assess the androgenic potential of chemicals. J. Chem. Inf. Model 2017, 57, 2874–2884. [Google Scholar] [CrossRef]
- Manganelli, S.; Roncaglioni, A.; Mansouri, K.; Judson, R.S.; Benfenati, E.; Manganaro, A.; Ruiz, P. Development, validation and integration of in silico models to identify androgen active chemicals. Chemosphere 2019, 220, 204–215. [Google Scholar] [CrossRef]
- Zhu, Q. On the performance of Matthews correlation coefficient (MCC) for imbalanced dataset. Pattern Recognit Lett. 2020, 136, 71–80. [Google Scholar] [CrossRef]
- Ferrari, T.; Gini, G.; Golbamaki Bakhtyari, N.; Benfenati, E. Mining structural alerts from SMILES: A new way to derive structure-activity relationships. In Proceedings of the 2011 IEEE Symposium Series on Computational Intelligence (CIDM), Paris, France, 11–15 April 2011; pp. 120–127. [Google Scholar] [CrossRef]
- Piir, G.; Sild, S.; Maran, U. Binary and multi-class classification for androgen receptor agonists, antagonists and binders. Chemosphere 2021, 262, 128313. [Google Scholar] [CrossRef] [PubMed]
- García-Sosa, A.T. Androgen Receptor Binding Category Prediction with Deep Neural Networks and Structure-, Ligand-, and Statistically-Based Features. Molecules 2021, 26, 1285. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
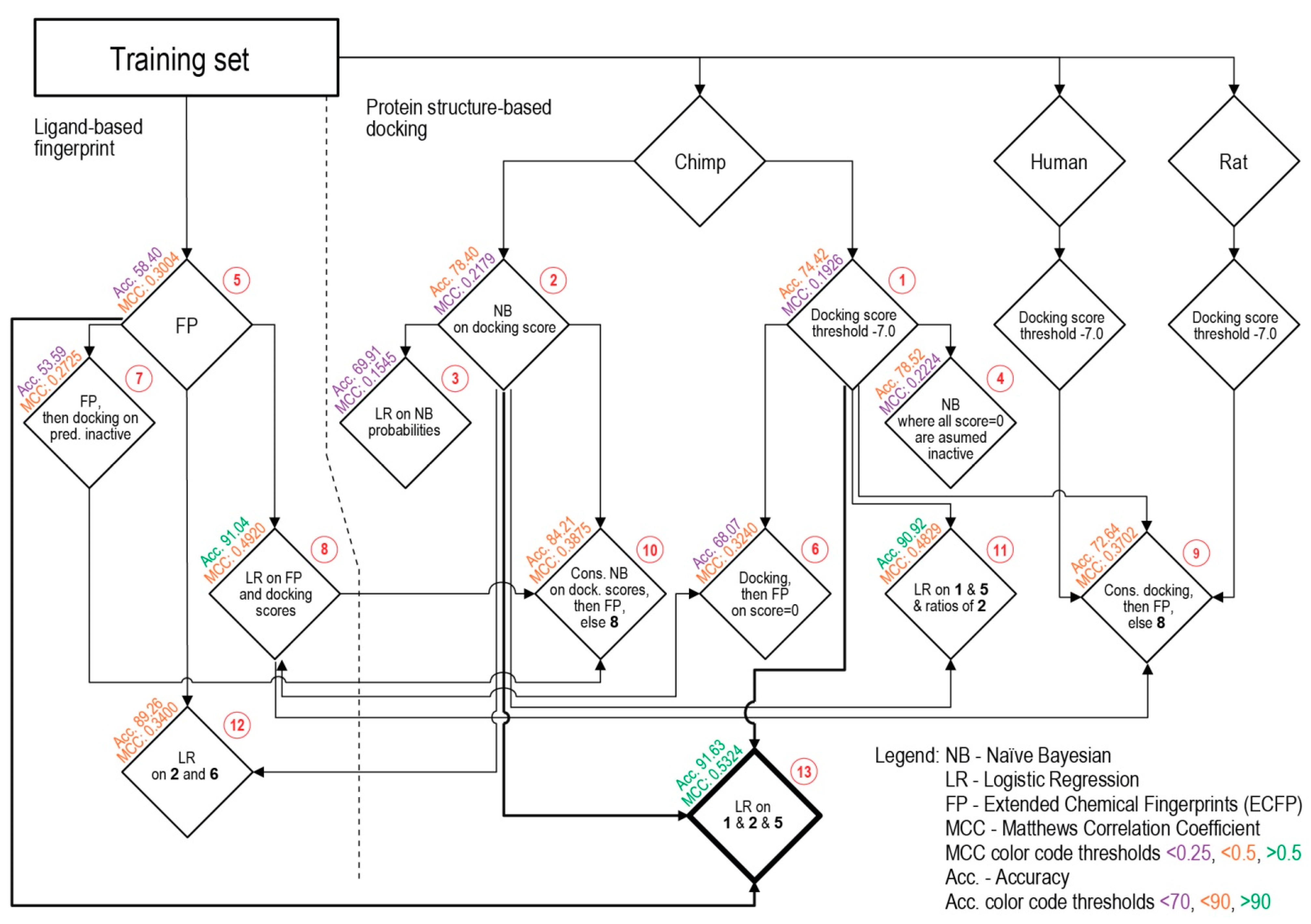{kind=link}
| Procedure | TP | TN | FP | FN | SP | SE | Acc. | MCC | |
|---|---|---|---|---|---|---|---|---|---|
| 1 | Docking score threshold | 97 | 1157 | 323 | 108 | 78.18 | 47.32 | 74.42 | 0.1926 |
| 2 | Bayesian on scores | 89 | 1232 | 248 | 116 | 83.24 | 43.41 | 78.40 | 0.2179 |
| 3 | Logistic regression on Bayesian | 100 | 1078 | 402 | 105 | 72.84 | 48.78 | 69.91 | 0.1545 |
| 4 | Modified Bayesian | 90 | 1233 | 247 | 115 | 83.31 | 43.90 | 78.52 | 0.2224 |
| 5 | Fingerprints (ECFP) | 189 | 795 | 685 | 16 | 53.72 | 92.20 | 58.40 | 0.3004 |
| 6 | Docking scores then ECFP | 169 | 978 | 502 | 36 | 66.08 | 82.44 | 68.07 | 0.3240 |
| 7 | ECFP then docking scores | 191 | 712 | 768 | 14 | 48.11 | 93.17 | 53.59 | 0.2725 |
| 8 | Logistic regr. on docking scores and ECFP | 71 | 1463 | 17 | 134 | 98.85 | 34.63 | 91.04 | 0.4920 |
| 9 | Consensus Docking and ECFP else 8 | 170 | 1054 | 426 | 35 | 71.22 | 82.93 | 72.64 | 0.3702 |
| 10 | Consensus Bayesian and ECFP else 8 | 117 | 1302 | 178 | 88 | 87.97 | 57.07 | 84.21 | 0.3875 |
| 11 | Logistic regr. on docking scores and ECFP and ratio of Bayesian | 69 | 1463 | 17 | 136 | 98.85 | 33.66 | 90.92 | 0.4829 |
| 12 | Logistic regression on and Bayesian avgs. and fingerprints | 42 | 1462 | 18 | 163 | 98.78 | 20.49 | 89.26 | 0.3400 |
| 13 | Logistic regression on docking scores and Bayesian avgs. and fingerprints | 75 | 1469 | 11 | 130 | 99.26 | 36.59 | 91.63 | 0.5324 |
| Procedure | NPV | PPV | +LR | −LR | BCR |
|---|---|---|---|---|---|
| 1 | 91.4145 | 19.8795 | 2.1133 | 0.7999 | 0.2774 |
| 2 | 91.3944 | 22.6107 | 2.1079 | 0.6793 | 0.4356 |
| 3 | 91.1243 | 19.9203 | 1.7959 | 0.7032 | 0.4618 |
| 4 | 91.4562 | 26.7062 | 2.6270 | 0.6735 | 0.3855 |
| 5 | 91.8699 | 87.2093 | 49.2239 | 0.6389 | 0.2535 |
| 6 | 98.0271 | 21.6247 | 1.9920 | 0.1453 | 0.4488 |
| 7 | 96.4497 | 25.1863 | 2.4305 | 0.2657 | 0.6211 |
| 8 | 98.0716 | 19.9166 | 1.7955 | 0.1420 | 0.3881 |
| 9 | 91.6093 | 80.6818 | 30.1521 | 0.6613 | 0.2388 |
| 10 | 96.7860 | 28.5235 | 2.8810 | 0.2397 | 0.6805 |
| 11 | 93.6691 | 39.6610 | 4.7454 | 0.4880 | 0.5011 |
| 12 | 91.4947 | 80.2326 | 29.3027 | 0.6711 | 0.2306 |
| 13 | 89.9692 | 70.0000 | 16.8455 | 0.8049 | 0.1294 |
| CAS | Name | Agonist | Antagonist | Predicted | Correct |
|---|---|---|---|---|---|
| 52806-53-8 | hydroxyflutamide | NA | Strong | 0 | X |
| 90357-06-5 | Bicalutamide | NA | Strong | 0 | X |
| 122-14-5 | Fenitrothion | NA | Strong | 0 | X |
| 63612-50-0 | Nilutamide | Negative | Moderate | 0 | X |
| 427-51-0 | cyproterone acetate | Weak | Moderate | 1 | Yes |
| 80-05-7 | bisphenol A | NA | Moderate/weak | 1 | Yes |
| 330-55-2 | Linuron | NA | Moderate/weak | 0 | X |
| 13311-84-7 | Flutamide | Negative | Moderate/weak | 0 | X |
| 67747-09-5 | Prochloraz | Negative | Moderate/weak | 0 | X |
| 789-02-6 | o,p′-DDT | Negative | Weak | 0 | Yes |
| 60168-88-9 | Fenarimol | Negative | Very weak | 0 | Yes |
| 58-18-4 | methyl testosterone | Strong | Negative | 1 | Yes |
| 58-22-0 | Testosterone | Strong | Negative | 1 | Yes |
| 63-05-8 | 4-androstenedione | Moderate | Negative | 1 | Yes |
| 1912-24-9 | Atrazine | Negative | Negative | 0 | Yes |
| 52918-63-5 | Deltamethrin | Negative | Negative | 0 | Yes |
| 10161-33-8 | 17b-trenbolone | Strong | NA | 1 | Yes |
| 797-63-7 | Levonorgestrel | Strong | NA | 1 | Yes |
| 68-22-4 | Norethindrone | Strong | NA | 1 | Yes |
| 521-18-6 | 5a-dihydrotestosterone | Strong | NA | 1 | Yes |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
García-Sosa, A.T.; Maran, U. Combined Naïve Bayesian, Chemical Fingerprints and Molecular Docking Classifiers to Model and Predict Androgen Receptor Binding Data for Environmentally- and Health-Sensitive Substances. Int. J. Mol. Sci. 2021, 22, 6695. https://doi.org/10.3390/ijms22136695
García-Sosa AT, Maran U. Combined Naïve Bayesian, Chemical Fingerprints and Molecular Docking Classifiers to Model and Predict Androgen Receptor Binding Data for Environmentally- and Health-Sensitive Substances. International Journal of Molecular Sciences. 2021; 22(13):6695. https://doi.org/10.3390/ijms22136695
Chicago/Turabian StyleGarcía-Sosa, Alfonso T., and Uko Maran. 2021. "Combined Naïve Bayesian, Chemical Fingerprints and Molecular Docking Classifiers to Model and Predict Androgen Receptor Binding Data for Environmentally- and Health-Sensitive Substances" International Journal of Molecular Sciences 22, no. 13: 6695. https://doi.org/10.3390/ijms22136695
APA StyleGarcía-Sosa, A. T., & Maran, U. (2021). Combined Naïve Bayesian, Chemical Fingerprints and Molecular Docking Classifiers to Model and Predict Androgen Receptor Binding Data for Environmentally- and Health-Sensitive Substances. International Journal of Molecular Sciences, 22(13), 6695. https://doi.org/10.3390/ijms22136695