
Brain Disposition of Antibody-Based Therapeutics: Dogma, Approaches and Perspectives




Abstract
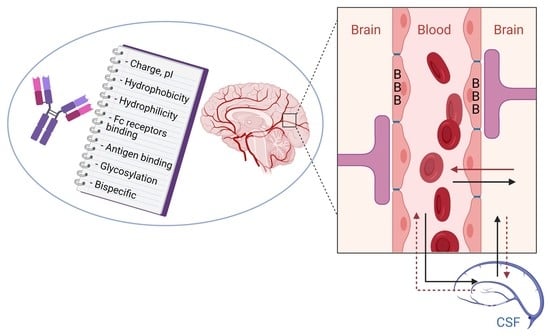
1. Introduction
2. Delivery of Antibodies into the Brain: Mechanism of Delivery
2.1. Physiology and Barriers of the CNS
2.2. BBB Structure
2.3. Pharmacokinetics and CNS Distribution of Antibodies
2.4. Mechanisms of Antibody Passage Across the BBB
3. Current In Vitro and In Vivo Methodologies for Measuring Brain Access of Antibodies: Advantages and Limitations
3.1. In Vitro Methods
3.2. In Vivo Methods
4. Approaches to Optimize BBB Internalization and Uptake of Antibodies
4.1. Modification of BBB Permeability
4.2. Physiological Approach to Transport Antibodies Across the BBB
4.3. Antibody Engineering Approaches to Increase Trans-BBB Transport
4.3.1. Role of Fc Receptors
4.3.2. Role of Antigen Binding
4.3.3. Role of Biophysical Properties
5. Conclusions and Outlook
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tucker, I.G. Drug delivery to the brain via the blood-brain barrier: A review of the literature and some recent patent disclosures. Ther. Deliv. 2011, 2, 311–327. [Google Scholar] [CrossRef]
- Alavijeh, M.S.; Chishty, M.; Qaiser, M.Z.; Palmer, A.M. Drug metabolism and pharmacokinetics, the blood-brain barrier, and central nervous system drug discovery. NeuroRx 2005, 2, 554–571. [Google Scholar] [CrossRef]
- Grabrucker, A.M.; Ruozi, B.; Belletti, D.; Pederzoli, F.; Forni, F.; Vandelli, M.A.; Tosi, G. Nanoparticle transport across the blood brain barrier. Tissue Barriers 2016, 4, e1153568. [Google Scholar] [CrossRef]
- Yu, Y.J.; Watts, R.J. Developing therapeutic antibodies for neurodegenerative disease. Neurotherapeutics 2013, 10, 459–472. [Google Scholar] [CrossRef]
- Dimitrov, D.S. Engineered CH2 domains (nanoantibodies). MAbs 2009, 1, 26–28. [Google Scholar] [CrossRef] [PubMed]
- Lajoie, J.M.; Shusta, E.V. Targeting receptor-mediated transport for delivery of biologics across the blood-brain barrier. Annu. Rev. Pharmacol. Toxicol. 2015, 55, 613–631. [Google Scholar] [CrossRef]
- Goulatis, L.I.; Shusta, E.V. Protein engineering approaches for regulating blood-brain barrier transcytosis. Curr. Opin. Struct. Biol. 2017, 45, 109–115. [Google Scholar] [CrossRef] [PubMed]
- Pardridge, W.M.; Boado, R.J. Reengineering biopharmaceuticals for targeted delivery across the blood-brain barrier. Methods Enzymol. 2012, 503, 269–292. [Google Scholar] [CrossRef]
- Gao, H. Progress and perspectives on targeting nanoparticles for brain drug delivery. Acta Pharm. Sin. B 2016, 6, 268–286. [Google Scholar] [CrossRef] [PubMed]
- Sweeney, M.D.; Sagare, A.P.; Zlokovic, B.V. Blood-brain barrier breakdown in Alzheimer disease and other neurodegenerative disorders. Nat. Rev. Neurol. 2018, 14, 133–150. [Google Scholar] [CrossRef] [PubMed]
- Salameh, T.S.; Banks, W.A. Delivery of therapeutic peptides and proteins to the CNS. Adv. Pharmacol. 2014, 71, 277–299. [Google Scholar] [CrossRef]
- Xiao, G.; Gan, L.S. Receptor-mediated endocytosis and brain delivery of therapeutic biologics. Int. J. Cell Biol. 2013, 2013, 703545. [Google Scholar] [CrossRef]
- Oller-Salvia, B.; Sanchez-Navarro, M.; Giralt, E.; Teixido, M. Blood-brain barrier shuttle peptides: An emerging paradigm for brain delivery. Chem. Soc. Rev. 2016, 45, 4690–4707. [Google Scholar] [CrossRef] [PubMed]
- Gabathuler, R. Approaches to transport therapeutic drugs across the blood-brain barrier to treat brain diseases. Neurobiol. Dis. 2010, 37, 48–57. [Google Scholar] [CrossRef]
- Pulgar, V.M. Transcytosis to Cross the Blood Brain Barrier, New Advancements and Challenges. Front. Neurosci. 2018, 12, 1019. [Google Scholar] [CrossRef] [PubMed]
- Strazielle, N.; Ghersi-Egea, J.F. Physiology of blood-brain interfaces in relation to brain disposition of small compounds and macromolecules. Mol. Pharm. 2013, 10, 1473–1491. [Google Scholar] [CrossRef] [PubMed]
- Vieira, D.B.; Gamarra, L.F. Getting into the brain: Liposome-based strategies for effective drug delivery across the blood-brain barrier. Int. J. Nanomed. 2016, 11, 5381–5414. [Google Scholar] [CrossRef]
- Papisov, M.I.; Belov, V.V.; Gannon, K.S. Physiology of the intrathecal bolus: The leptomeningeal route for macromolecule and particle delivery to CNS. Mol. Pharm. 2013, 10, 1522–1532. [Google Scholar] [CrossRef] [PubMed]
- Bagchi, S.; Chhibber, T.; Lahooti, B.; Verma, A.; Borse, V.; Jayant, R.D. In-Vitro blood-brain barrier models for drug screening and permeation studies: An overview. Drug Des. Devel. Ther. 2019, 13, 3591–3605. [Google Scholar] [CrossRef] [PubMed]
- Conner, K.P.; Devanaboyina, S.C.; Thomas, V.A.; Rock, D.A. The biodistribution of therapeutic proteins: Mechanism, implications for pharmacokinetics, and methods of evaluation. Pharmacol. Ther. 2020, 212, 107574. [Google Scholar] [CrossRef]
- Herda, L.M.; Polo, E.; Kelly, P.M.; Rocks, L.; Hudecz, D.; Dawson, K.A. Designing the future of nanomedicine: Current barriers to targeted brain therapeutics. Eur. J. Nanomed. 2014, 6, 127–139. [Google Scholar] [CrossRef]
- Cavaco, M.; Gaspar, D.; Arb Castanho, M.; Neves, V. Antibodies for the Treatment of Brain Metastases, a Dream or a Reality? Pharmaceutics 2020, 12, 62. [Google Scholar] [CrossRef]
- Modarres, H.P.; Janmaleki, M.; Novin, M.; Saliba, J.; El-Hajj, F.; RezayatiCharan, M.; Seyfoori, A.; Sadabadi, H.; Vandal, M.; Nguyen, M.D.; et al. In Vitro models and systems for evaluating the dynamics of drug delivery to the healthy and diseased brain. J. Control. Release 2018, 273, 108–130. [Google Scholar] [CrossRef] [PubMed]
- Hammarlund-Udenaes, M.; Paalzow, L.K.; de Lange, E.C. Drug equilibration across the blood-brain barrier—pharmacokinetic considerations based on the microdialysis method. Pharm. Res. 1997, 14, 128–134. [Google Scholar] [CrossRef]
- Taccola, C.; Barneoud, P.; Cartot-Cotton, S.; Valente, D.; Schussler, N.; Saubamea, B.; Chasseigneaux, S.; Cochois, V.; Mignon, V.; Curis, E.; et al. Modifications of physical and functional integrity of the blood-brain barrier in an inducible mouse model of neurodegeneration. Neuropharmacology 2021, 108588. [Google Scholar] [CrossRef] [PubMed]
- Darvesh, A.S.; Carroll, R.T.; Geldenhuys, W.J.; Gudelsky, G.A.; Klein, J.; Meshul, C.K.; Van der Schyf, C.J. In Vivo brain microdialysis: Advances in neuropsychopharmacology and drug discovery. Expert Opin. Drug Discov. 2011, 6, 109–127. [Google Scholar] [CrossRef]
- Chang, H.Y.; Morrow, K.; Bonacquisti, E.; Zhang, W.; Shah, D.K. Antibody pharmacokinetics in rat brain determined using microdialysis. MAbs 2018, 10, 843–853. [Google Scholar] [CrossRef]
- Chang, H.Y.; Wu, S.; Li, Y.; Zhang, W.; Burrell, M.; Webster, C.I.; Shah, D.K. Brain pharmacokinetics of anti-transferrin receptor antibody affinity variants in rats determined using microdialysis. MAbs 2021, 13, 1874121. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.Y.; Wu, S.; Meno-Tetang, G.; Shah, D.K. A translational platform PBPK model for antibody disposition in the brain. J. Pharmacokinet. Pharmacodyn. 2019, 46, 319–338. [Google Scholar] [CrossRef]
- Boswell, C.A.; Mundo, E.E.; Johnstone, B.; Ulufatu, S.; Schweiger, M.G.; Bumbaca, D.; Fielder, P.J.; Prabhu, S.; Khawli, L.A. Vascular physiology and protein disposition in a preclinical model of neurodegeneration. Mol. Pharm. 2013, 10, 1514–1521. [Google Scholar] [CrossRef] [PubMed]
- Tibbitts, J.; Canter, D.; Graff, R.; Smith, A.; Khawli, L.A. Key factors influencing ADME properties of therapeutic proteins: A need for ADME characterization in drug discovery and development. MAbs 2016, 8, 229–245. [Google Scholar] [CrossRef]
- Solon, E.G. Autoradiography techniques and quantification of drug distribution. Cell Tissue Res. 2015, 360, 87–107. [Google Scholar] [CrossRef] [PubMed]
- Pardridge, W.M. CSF, blood-bra in barrier, and brain drug delivery. Expert Opin. Drug Deliv. 2016, 13, 963–975. [Google Scholar] [CrossRef]
- Warnders, F.J.; Lub-de Hooge, M.N.; de Vries, E.G.E.; Kosterink, J.G.W. Influence of protein properties and protein modification on biodistribution and tumor uptake of anticancer antibodies, antibody derivatives, and non-Ig scaffolds. Med. Res. Rev. 2018, 38, 1837–1873. [Google Scholar] [CrossRef] [PubMed]
- Sehlin, D.; Fang, X.T.; Cato, L.; Antoni, G.; Lannfelt, L.; Syvanen, S. Antibody-based PET imaging of amyloid beta in mouse models of Alzheimer’s disease. Nat. Commun. 2016, 7, 10759. [Google Scholar] [CrossRef] [PubMed]
- Lesniak, W.G.; Chu, C.; Jablonska, A.; Behnam Azad, B.; Zwaenepoel, O.; Zawadzki, M.; Lisok, A.; Pomper, M.G.; Walczak, P.; Gettemans, J.; et al. PET imaging of distinct brain uptake of a nanobody and similarly-sized PAMAM dendrimers after intra-arterial administration. Eur. J. Nucl. Med. Mol. Imaging 2019, 46, 1940–1951. [Google Scholar] [CrossRef]
- Bien-Ly, N.; Yu, Y.J.; Bumbaca, D.; Elstrott, J.; Boswell, C.A.; Zhang, Y.; Luk, W.; Lu, Y.; Dennis, M.S.; Weimer, R.M.; et al. Transferrin receptor (TfR) trafficking determines brain uptake of TfR antibody affinity variants. J. Exp. Med. 2014, 211, 233–244. [Google Scholar] [CrossRef]
- Bien-Ly, N.; Boswell, C.A.; Jeet, S.; Beach, T.G.; Hoyte, K.; Luk, W.; Shihadeh, V.; Ulufatu, S.; Foreman, O.; Lu, Y.; et al. Lack of Widespread BBB Disruption in Alzheimer’s Disease Models: Focus on Therapeutic Antibodies. Neuron 2015, 88, 289–297. [Google Scholar] [CrossRef]
- Bayir, E.; Sendemir, A. In Vitro Human Blood-Brain Barrier Model for Drug Permeability Testing. Methods Mol. Biol. 2021. [Google Scholar] [CrossRef]
- Banks, W.A. From blood-brain barrier to blood-brain interface: New opportunities for CNS drug delivery. Nat. Rev. Drug Discov. 2016, 15, 275–292. [Google Scholar] [CrossRef]
- Murugan, K.; Choonara, Y.E.; Kumar, P.; Bijukumar, D.; du Toit, L.C.; Pillay, V. Parameters and characteristics governing cellular internalization and trans-barrier trafficking of nanostructures. Int. J. Nanomed. 2015, 10, 2191–2206. [Google Scholar] [CrossRef]
- Rankovic, Z. CNS drug design: Balancing physicochemical properties for optimal brain exposure. J. Med. Chem. 2015, 58, 2584–2608. [Google Scholar] [CrossRef]
- Boswell, C.A.; Tesar, D.B.; Mukhyala, K.; Theil, F.P.; Fielder, P.J.; Khawli, L.A. Effects of charge on antibody tissue distribution and pharmacokinetics. Bioconjug. Chem. 2010, 21, 2153–2163. [Google Scholar] [CrossRef]
- Patel, M.M.; Goyal, B.R.; Bhadada, S.V.; Bhatt, J.S.; Amin, A.F. Getting into the brain: Approaches to enhance brain drug delivery. CNS Drugs 2009, 23, 35–58. [Google Scholar] [CrossRef] [PubMed]
- Watts, R.J.; Dennis, M.S. Bispecific antibodies for delivery into the brain. Curr. Opin. Chem. Biol. 2013, 17, 393–399. [Google Scholar] [CrossRef] [PubMed]
- Coloma, M.J.; Lee, H.J.; Kurihara, A.; Landaw, E.M.; Boado, R.J.; Morrison, S.L.; Pardridge, W.M. Transport across the primate blood-brain barrier of a genetically engineered chimeric monoclonal antibody to the human insulin receptor. Pharm. Res. 2000, 17, 266–274. [Google Scholar] [CrossRef]
- Boado, R.J.; Hui, E.K.; Lu, J.Z.; Pardridge, W.M. Glycemic control and chronic dosing of rhesus monkeys with a fusion protein of iduronidase and a monoclonal antibody against the human insulin receptor. Drug Metab. Dispos. 2012, 40, 2021–2025. [Google Scholar] [CrossRef]
- Karim, R.; Palazzo, C.; Evrard, B.; Piel, G. Nanocarriers for the treatment of glioblastoma multiforme: Current state-of-the-art. J. Control. Release 2016, 227, 23–37. [Google Scholar] [CrossRef]
- Thom, G.; Hatcher, J.; Hearn, A.; Paterson, J.; Rodrigo, N.; Beljean, A.; Gurrell, I.; Webster, C. Isolation of blood-brain barrier-crossing antibodies from a phage display library by competitive elution and their ability to penetrate the central nervous system. MAbs 2018, 10, 304–314. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.T.; Li, W.; Meng, G.; Wang, P.; Liao, W. Strategies for transporting nanoparticles across the blood-brain barrier. Biomater. Sci. 2016, 4, 219–229. [Google Scholar] [CrossRef]
- Neuwelt, E.A.; Diehl, J.T.; Vu, L.H.; Hill, S.A.; Michael, A.J.; Frenkel, E.P. Monitoring of methotrexate delivery in patients with malignant brain tumors after osmotic blood-brain barrier disruption. Ann. Intern. Med. 1981, 94, 449–454. [Google Scholar] [CrossRef] [PubMed]
- Toman, P.; Lien, C.F.; Ahmad, Z.; Dietrich, S.; Smith, J.R.; An, Q.; Molnar, E.; Pilkington, G.J.; Gorecki, D.C.; Tsibouklis, J.; et al. Nanoparticles of alkylglyceryl-dextran-graft-poly(lactic acid) for drug delivery to the brain: Preparation and in vitro investigation. Acta Biomater. 2015, 23, 250–262. [Google Scholar] [CrossRef] [PubMed]
- Etame, A.B.; Diaz, R.J.; O’Reilly, M.A.; Smith, C.A.; Mainprize, T.G.; Hynynen, K.; Rutka, J.T. Enhanced delivery of gold nanoparticles with therapeutic potential into the brain using MRI-guided focused ultrasound. Nanomedicine 2012, 8, 1133–1142. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.L.; Hua, M.Y.; Yang, H.W.; Huang, C.Y.; Chu, P.C.; Wu, J.S.; Tseng, I.C.; Wang, J.J.; Yen, T.C.; Chen, P.Y.; et al. Magnetic resonance monitoring of focused ultrasound/magnetic nanoparticle targeting delivery of therapeutic agents to the brain. Proc. Natl. Acad. Sci. USA 2010, 107, 15205–15210. [Google Scholar] [CrossRef]
- Aryal, M.; Vykhodtseva, N.; Zhang, Y.Z.; McDannold, N. Multiple sessions of liposomal doxorubicin delivery via focused ultrasound mediated blood-brain barrier disruption: A safety study. J. Control. Release 2015, 204, 60–69. [Google Scholar] [CrossRef] [PubMed]
- Dennis, M.S.; Watts, R.J. Transferrin antibodies into the brain. Neuropsychopharmacology 2012, 37, 302–303. [Google Scholar] [CrossRef] [PubMed]
- Niewoehner, J.; Bohrmann, B.; Collin, L.; Urich, E.; Sade, H.; Maier, P.; Rueger, P.; Stracke, J.O.; Lau, W.; Tissot, A.C.; et al. Increased brain penetration and potency of a therapeutic antibody using a monovalent molecular shuttle. Neuron 2014, 81, 49–60. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.J.; Zhang, Y.; Kenrick, M.; Hoyte, K.; Luk, W.; Lu, Y.; Atwal, J.; Elliott, J.M.; Prabhu, S.; Watts, R.J.; et al. Boosting brain uptake of a therapeutic antibody by reducing its affinity for a transcytosis target. Sci. Transl. Med. 2011, 3, 84ra44. [Google Scholar] [CrossRef]
- Kariolis, M.S.; Wells, R.C.; Getz, J.A.; Kwan, W.; Mahon, C.S.; Tong, R.; Kim, D.J.; Srivastava, A.; Bedard, C.; Henne, K.R.; et al. Brain delivery of therapeutic proteins using an Fc fragment blood-brain barrier transport vehicle in mice and monkeys. Sci. Transl. Med. 2020, 12. [Google Scholar] [CrossRef]
- Atwal, J.K.; Chen, Y.; Chiu, C.; Mortensen, D.L.; Meilandt, W.J.; Liu, Y.; Heise, C.E.; Hoyte, K.; Luk, W.; Lu, Y.; et al. A therapeutic antibody targeting BACE1 inhibits amyloid-beta production in vivo. Sci. Transl. Med. 2011, 3, 84ra43. [Google Scholar] [CrossRef] [PubMed]
- Haqqani, A.S.; Caram-Salas, N.; Ding, W.; Brunette, E.; Delaney, C.E.; Baumann, E.; Boileau, E.; Stanimirovic, D. Multiplexed evaluation of serum and CSF pharmacokinetics of brain-targeting single-domain antibodies using a NanoLC-SRM-ILIS method. Mol. Pharm. 2013, 10, 1542–1556. [Google Scholar] [CrossRef]
- Tanha, J.; Muruganandam, A.; Stanimirovic, D. Phage display technology for identifying specific antigens on brain endothelial cells. Methods Mol. Med. 2003, 89, 435–449. [Google Scholar] [CrossRef]
- Demeule, M.; Currie, J.C.; Bertrand, Y.; Che, C.; Nguyen, T.; Regina, A.; Gabathuler, R.; Castaigne, J.P.; Beliveau, R. Involvement of the low-density lipoprotein receptor-related protein in the transcytosis of the brain delivery vector angiopep-2. J. Neurochem. 2008, 106, 1534–1544. [Google Scholar] [CrossRef]
- Huile, G.; Shuaiqi, P.; Zhi, Y.; Shijie, C.; Chen, C.; Xinguo, J.; Shun, S.; Zhiqing, P.; Yu, H. A cascade targeting strategy for brain neuroglial cells employing nanoparticles modified with angiopep-2 peptide and EGFP-EGF1 protein. Biomaterials 2011, 32, 8669–8675. [Google Scholar] [CrossRef]
- Muruganandam, A.; Tanha, J.; Narang, S.; Stanimirovic, D. Selection of phage-displayed llama single-domain antibodies that transmigrate across human blood-brain barrier endothelium. FASEB J. 2002, 16, 240–242. [Google Scholar] [CrossRef] [PubMed]
- Boado, R.J.; Zhang, Y.; Zhang, Y.; Wang, Y.; Pardridge, W.M. GDNF fusion protein for targeted-drug delivery across the human blood-brain barrier. Biotechnol. Bioeng. 2008, 100, 387–396. [Google Scholar] [CrossRef] [PubMed]
- Herz, J.; Strickland, D.K. LRP: A multifunctional scavenger and signaling receptor. J. Clin. Investig. 2001, 108, 779–784. [Google Scholar] [CrossRef] [PubMed]
- Paterson, J.; Webster, C.I. Exploiting transferrin receptor for delivering drugs across the blood-brain barrier. Drug Discov. Today Technol. 2016, 20, 49–52. [Google Scholar] [CrossRef] [PubMed]
- Zvonova, E.A.; Tyurin, A.A.; Soloviev, A.A.; Goldenkova-Pavlova, I.V. Strategies for Modulation of Pharmacokinetics of Recombinant Therapeutic Proteins. Biol. Bull. Rev. 2018, 8, 124–141. [Google Scholar] [CrossRef]
- Kuramochi, T.; Igawa, T.; Tsunoda, H.; Hattori, K. Humanization and simultaneous optimization of monoclonal antibody. Methods Mol. Biol. 2014, 1060, 123–137. [Google Scholar] [CrossRef]
- Carter, P.J. Potent antibody therapeutics by design. Nat. Rev. Immunol. 2006, 6, 343–357. [Google Scholar] [CrossRef]
- Khawli, L.A.; Biela, B.; Hu, P.; Epstein, A.L. Comparison of recombinant derivatives of chimeric TNT-3 antibody for the radioimaging of solid tumors. Hybrid Hybridomics 2003, 22, 1–9. [Google Scholar] [CrossRef]
- Sehlin, D.; Syvanen, S.; Faculty, M. Engineered antibodies: New possibilities for brain PET? Eur. J. Nucl. Med. Mol. Imaging 2019, 46, 2848–2858. [Google Scholar] [CrossRef]
- Yeung, Y.A.; Leabman, M.K.; Marvin, J.S.; Qiu, J.; Adams, C.W.; Lien, S.; Starovasnik, M.A.; Lowman, H.B. Engineering human IgG1 affinity to human neonatal Fc receptor: Impact of affinity improvement on pharmacokinetics in primates. J. Immunol. 2009, 182, 7663–7671. [Google Scholar] [CrossRef]
- Roopenian, D.C.; Christianson, G.J.; Sproule, T.J.; Brown, A.C.; Akilesh, S.; Jung, N.; Petkova, S.; Avanessian, L.; Choi, E.Y.; Shaffer, D.J.; et al. The MHC class I-like IgG receptor controls perinatal IgG transport, IgG homeostasis, and fate of IgG-Fc-coupled drugs. J. Immunol. 2003, 170, 3528–3533. [Google Scholar] [CrossRef]
- Deane, R.; Sagare, A.; Hamm, K.; Parisi, M.; LaRue, B.; Guo, H.; Wu, Z.; Holtzman, D.M.; Zlokovic, B.V. IgG-assisted age-dependent clearance of Alzheimer’s amyloid beta peptide by the blood-brain barrier neonatal Fc receptor. J. Neurosci. 2005, 25, 11495–11503. [Google Scholar] [CrossRef]
- Pardridge, W.M. Blood-Brain Barrier and Delivery of Protein and Gene Therapeutics to Brain. Front Aging Neurosci. 2019, 11, 373. [Google Scholar] [CrossRef]
- Abuqayyas, L.; Balthasar, J.P. Investigation of the role of FcgammaR and FcRn in mAb distribution to the brain. Mol. Pharm. 2013, 10, 1505–1513. [Google Scholar] [CrossRef] [PubMed]
- Garg, A.; Balthasar, J.P. Investigation of the influence of FcRn on the distribution of IgG to the brain. AAPS J. 2009, 11, 553–557. [Google Scholar] [CrossRef] [PubMed]
- Chen, N.; Wang, W.; Fauty, S.; Fang, Y.; Hamuro, L.; Hussain, A.; Prueksaritanont, T. The effect of the neonatal Fc receptor on human IgG biodistribution in mice. MAbs 2014, 6, 502–508. [Google Scholar] [CrossRef] [PubMed]
- Eigenmann, M.J.; Fronton, L.; Grimm, H.P.; Otteneder, M.B.; Krippendorff, B.F. Quantification of IgG monoclonal antibody clearance in tissues. MAbs 2017, 9, 1007–1015. [Google Scholar] [CrossRef] [PubMed]
- Cooper, P.R.; Ciambrone, G.J.; Kliwinski, C.M.; Maze, E.; Johnson, L.; Li, Q.; Feng, Y.; Hornby, P.J. Efflux of monoclonal antibodies from rat brain by neonatal Fc receptor, FcRn. Brain Res. 2013, 1534, 13–21. [Google Scholar] [CrossRef]
- Schlachetzki, F.; Zhu, C.; Pardridge, W.M. Expression of the neonatal Fc receptor (FcRn) at the blood-brain barrier. J. Neurochem. 2002, 81, 203–206. [Google Scholar] [CrossRef] [PubMed]
- Ruano-Salguero, J.S.; Lee, K.H. Antibody transcytosis across brain endothelial-like cells occurs nonspecifically and independent of FcRn. Sci. Rep. 2020, 10, 3685. [Google Scholar] [CrossRef] [PubMed]
- Friden, P.M.; Walus, L.R.; Musso, G.F.; Taylor, M.A.; Malfroy, B.; Starzyk, R.M. Anti-transferrin receptor antibody and antibody-drug conjugates cross the blood-brain barrier. Proc. Natl. Acad. Sci. USA 1991, 88, 4771–4775. [Google Scholar] [CrossRef] [PubMed]
- Sade, H.; Baumgartner, C.; Hugenmatter, A.; Moessner, E.; Freskgard, P.O.; Niewoehner, J. A human blood-brain barrier transcytosis assay reveals antibody transcytosis influenced by pH-dependent receptor binding. PLoS ONE 2014, 9, e96340. [Google Scholar] [CrossRef]
- Levin, V.A. Relationship of octanol/water partition coefficient and molecular weight to rat brain capillary permeability. J. Med. Chem. 1980, 23, 682–684. [Google Scholar] [CrossRef]
- Pan, W.; Kastin, A.J. Changing the chemokine gradient: CINC1 crosses the blood-brain barrier. J. Neuroimmunol. 2001, 115, 64–70. [Google Scholar] [CrossRef]
- Heyl, D.L.; Sefler, A.M.; He, J.X.; Sawyer, T.K.; Wustrow, D.J.; Akunne, H.C.; Davis, M.D.; Pugsley, T.A.; Heffner, T.G.; Corbin, A.E.; et al. Structure-activity and conformational studies of a series of modified C-terminal hexapeptide neurotensin analogues. Int. J. Pept. Protein Res. 1994, 44, 233–238. [Google Scholar] [CrossRef]
- Gentry, C.L.; Egleton, R.D.; Gillespie, T.; Abbruscato, T.J.; Bechowski, H.B.; Hruby, V.J.; Davis, T.P. The effect of halogenation on blood-brain barrier permeability of a novel peptide drug. Peptides 1999, 20, 1229–1238. [Google Scholar] [CrossRef]
- Sola, R.J.; Griebenow, K. Glycosylation of therapeutic proteins: An effective strategy to optimize efficacy. BioDrugs 2010, 24, 9–21. [Google Scholar] [CrossRef]
- Poduslo, J.F.; Curran, G.L. Glycation increases the permeability of proteins across the blood-nerve and blood-brain barriers. Brain Res. Mol. Brain Res. 1994, 23, 157–162. [Google Scholar] [CrossRef]
- Bickel, U.; Lee, V.M.Y.; Pardridge, W.M. Pharmacokinetic differences between111In- and125I-Labeled cationized monoclonal antibody against β-Amyloid in mouse and dog. Drug Delivery 2008, 2, 128–135. [Google Scholar] [CrossRef]
- Triguero, D.; Buciak, J.B.; Yang, J.; Pardridge, W.M. Blood-brain barrier transport of cationized immunoglobulin G: Enhanced delivery compared to native protein. Proc. Natl. Acad. Sci. USA 1989, 86, 4761–4765. [Google Scholar] [CrossRef]
- Kang, Y.S.; Pardridge, W.M. Brain delivery of biotin bound to a conjugate of neutral avidin and cationized human albumin. Pharm. Res. 1994, 11, 1257–1264. [Google Scholar] [CrossRef]
- Pardridge, W.M.; Triguero, D.; Buciak, J.L. Beta-endorphin chimeric peptides: Transport through the blood-brain barrier in vivo and cleavage of disulfide linkage by brain. Endocrinology 1990, 126, 977–984. [Google Scholar] [CrossRef] [PubMed]
- Nau, R.; Sorgel, F.; Eiffert, H. Penetration of drugs through the blood-cerebrospinal fluid/blood-brain barrier for treatment of central nervous system infections. Clin Microbiol. Rev. 2010, 23, 858–883. [Google Scholar] [CrossRef]
- Vendel, E.; Rottschafer, V.; de Lange, E.C.M. The 3D Brain Unit Network Model to Study Spatial Brain Drug Exposure under Healthy and Pathological Conditions. Pharm. Res. 2020, 37, 137. [Google Scholar] [CrossRef] [PubMed]
- Tanha, J.; Dubuc, G.; Hirama, T.; Narang, S.A.; MacKenzie, C.R. Selection by phage display of llama conventional V(H) fragments with heavy chain antibody V(H)H properties. J. Immunol. Methods 2002, 263, 97–109. [Google Scholar] [CrossRef]
- Abulrob, A.; Sprong, H.; Van Bergen en Henegouwen, P.; Stanimirovic, D. The blood-brain barrier transmigrating single domain antibody: Mechanisms of transport and antigenic epitopes in human brain endothelial cells. J. Neurochem. 2005, 95, 1201–1214. [Google Scholar] [CrossRef] [PubMed]
- Chacko, A.M.; Li, C.; Pryma, D.A.; Brem, S.; Coukos, G.; Muzykantov, V. Targeted delivery of antibody-based therapeutic and imaging agents to CNS tumors: Crossing the blood-brain barrier divide. Expert Opin. Drug Deliv. 2013, 10, 907–926. [Google Scholar] [CrossRef]
- Bumbaca Yadav, D.; Sharma, V.K.; Boswell, C.A.; Hotzel, I.; Tesar, D.; Shang, Y.; Ying, Y.; Fischer, S.K.; Grogan, J.L.; Chiang, E.Y.; et al. Evaluating the Use of Antibody Variable Region (Fv) Charge as a Risk Assessment Tool for Predicting Typical Cynomolgus Monkey Pharmacokinetics. J. Biol. Chem. 2015, 290, 29732–29741. [Google Scholar] [CrossRef]
- Naseri Kouzehgarani, G.; Feldsien, T.; Engelhard, H.H.; Mirakhur, K.K.; Phipps, C.; Nimmrich, V.; Clausznitzer, D.; Lefebvre, D.R. Harnessing cerebrospinal fluid circulation for drug delivery to brain tissues. Adv. Drug Deliv. Rev. 2021, 173, 20–59. [Google Scholar] [CrossRef]
- Boado, R.J.; Hui, E.K.; Lu, J.Z.; Sumbria, R.K.; Pardridge, W.M. Blood-brain barrier molecular trojan horse enables imaging of brain uptake of radioiodinated recombinant protein in the rhesus monkey. Bioconjug. Chem. 2013, 24, 1741–1749. [Google Scholar] [CrossRef]
- Hultqvist, G.; Syvanen, S.; Fang, X.T.; Lannfelt, L.; Sehlin, D. Bivalent Brain Shuttle Increases Antibody Uptake by Monovalent Binding to the Transferrin Receptor. Theranostics 2017, 7, 308–318. [Google Scholar] [CrossRef] [PubMed]
- He, Q.; Liu, J.; Liang, J.; Liu, X.; Li, W.; Liu, Z.; Ding, Z.; Tuo, D. Towards Improvements for Penetrating the Blood-Brain Barrier-Recent Progress from a Material and Pharmaceutical Perspective. Cells 2018, 7, 24. [Google Scholar] [CrossRef] [PubMed]
- Vallianatou, T.; Giaginis, C.; Tsantili-Kakoulidou, A. The impact of physicochemical and molecular properties in drug design: Navigation in the “drug-like” chemical space. Adv. Exp. Med. Biol. 2015, 822, 187–194. [Google Scholar] [CrossRef]
- Gadkar, K.; Yadav, D.B.; Zuchero, J.Y.; Couch, J.A.; Kanodia, J.; Kenrick, M.K.; Atwal, J.K.; Dennis, M.S.; Prabhu, S.; Watts, R.J.; et al. Mathematical PKPD and safety model of bispecific TfR/BACE1 antibodies for the optimization of antibody uptake in brain. Eur. J. Pharm. Biopharm. 2016, 101, 53–61. [Google Scholar] [CrossRef] [PubMed]
- Kanodia, J.S.; Gadkar, K.; Bumbaca, D.; Zhang, Y.; Tong, R.K.; Luk, W.; Hoyte, K.; Lu, Y.; Wildsmith, K.R.; Couch, J.A.; et al. Prospective Design of Anti-Transferrin Receptor Bispecific Antibodies for Optimal Delivery into the Human Brain. CPT Pharmacomet. Syst. Pharmacol. 2016, 5, 283–291. [Google Scholar] [CrossRef]
- Pearlstein, R.A.; McKay, D.J.J.; Hornak, V.; Dickson, C.; Golosov, A.; Harrison, T.; Velez-Vega, C.; Duca, J. Building New Bridges between In Vitro and In Vivo in Early Drug Discovery: Where Molecular Modeling Meets Systems Biology. Curr. Top Med. Chem. 2017, 17, 2642–2662. [Google Scholar] [CrossRef]
- Morales, J.F.; Montoto, S.S.; Fagiolino, P.; Ruiz, M.E. Current State and Future Perspectives in QSAR Models to Predict Blood- Brain Barrier Penetration in Central Nervous System Drug R&D. Mini. Rev. Med. Chem. 2017, 17, 247–257. [Google Scholar] [CrossRef]
- Wang, Y.; Xing, J.; Xu, Y.; Zhou, N.; Peng, J.; Xiong, Z.; Liu, X.; Luo, X.; Luo, C.; Chen, K.; et al. In silico ADME/T modelling for rational drug design. Q Rev. Biophys. 2015, 48, 488–515. [Google Scholar] [CrossRef]
- Blair, L.J.; Frauen, H.D.; Zhang, B.; Nordhues, B.A.; Bijan, S.; Lin, Y.C.; Zamudio, F.; Hernandez, L.D.; Sabbagh, J.J.; Selenica, M.L.; et al. Tau depletion prevents progressive blood-brain barrier damage in a mouse model of tauopathy. Acta Neuropathol. Commun. 2015, 3, 8. [Google Scholar] [CrossRef] [PubMed]
- Lalatsa, A.; Leite, D.M. Single-Domain Antibodies for Brain Targeting. Biopharm. Int. 2014, 27, 20–26. [Google Scholar]
- Zuchero, Y.J.; Chen, X.; Bien-Ly, N.; Bumbaca, D.; Tong, R.K.; Gao, X.; Zhang, S.; Hoyte, K.; Luk, W.; Huntley, M.A.; et al. Discovery of Novel Blood-Brain Barrier Targets to Enhance Brain Uptake of Therapeutic Antibodies. Neuron 2016, 89, 70–82. [Google Scholar] [CrossRef]
- Lu, Y.; Khawli, L.A.; Purushothama, S.; Theil, F.P.; Partridge, M.A. Recent Advances in Assessing Immunogenicity of Therapeutic Proteins: Impact on Biotherapeutic Development. J. Immunol. Res. 2016, 2016, 8141269. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.; Manoli, H.; Jaw, S.; Frutoz, K.; Epstein, A.L.; Khawli, L.A.; Theil, F.P. Unraveling the Effect of Immunogenicity on the PK/PD, Efficacy, and Safety of Therapeutic Proteins. J. Immunol. Res. 2016, 2016, 2342187. [Google Scholar] [CrossRef]
- Khawli, L.A.; Prabhu, S. Drug delivery across the blood-brain barrier. Mol. Pharm. 2013, 10, 1471–1472. [Google Scholar] [CrossRef]
- Deo, A.K.; Theil, F.P.; Nicolas, J.M. Confounding parameters in preclinical assessment of blood-brain barrier permeation: An overview with emphasis on species differences and effect of disease states. Mol. Pharm. 2013, 10, 1581–1595. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.T.; Zhao, Y.Z.; Wong, H.L.; Cai, J.; Peng, L.; Tian, X.Q. Current approaches to enhance CNS delivery of drugs across the brain barriers. Int. J. Nanomed. 2014, 9, 2241–2257. [Google Scholar] [CrossRef]
- Terstappen, G.C.; Meyer, A.H.; Bell, R.D.; Zhang, W. Strategies for delivering therapeutics across the blood-brain barrier. Nat. Rev. Drug Discov. 2021, 20, 362–383. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Key Classes and Functions of Biomolecules |
|---|
|
|
|
|
|
|
|
|
|
|
|
|
| Receptors |
|---|
|
|
|
|
|
|
|
|
|
| Biomolecules |
|---|
|
|
|
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kouhi, A.; Pachipulusu, V.; Kapenstein, T.; Hu, P.; Epstein, A.L.; Khawli, L.A. Brain Disposition of Antibody-Based Therapeutics: Dogma, Approaches and Perspectives. Int. J. Mol. Sci. 2021, 22, 6442. https://doi.org/10.3390/ijms22126442
Kouhi A, Pachipulusu V, Kapenstein T, Hu P, Epstein AL, Khawli LA. Brain Disposition of Antibody-Based Therapeutics: Dogma, Approaches and Perspectives. International Journal of Molecular Sciences. 2021; 22(12):6442. https://doi.org/10.3390/ijms22126442
Chicago/Turabian StyleKouhi, Aida, Vyshnavi Pachipulusu, Talya Kapenstein, Peisheng Hu, Alan L. Epstein, and Leslie A. Khawli. 2021. "Brain Disposition of Antibody-Based Therapeutics: Dogma, Approaches and Perspectives" International Journal of Molecular Sciences 22, no. 12: 6442. https://doi.org/10.3390/ijms22126442
APA StyleKouhi, A., Pachipulusu, V., Kapenstein, T., Hu, P., Epstein, A. L., & Khawli, L. A. (2021). Brain Disposition of Antibody-Based Therapeutics: Dogma, Approaches and Perspectives. International Journal of Molecular Sciences, 22(12), 6442. https://doi.org/10.3390/ijms22126442