
Emerin Represses STAT3 Signaling through Nuclear Membrane-Based Spatial Control


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Emerin Represses STAT3 Transcriptional Activity
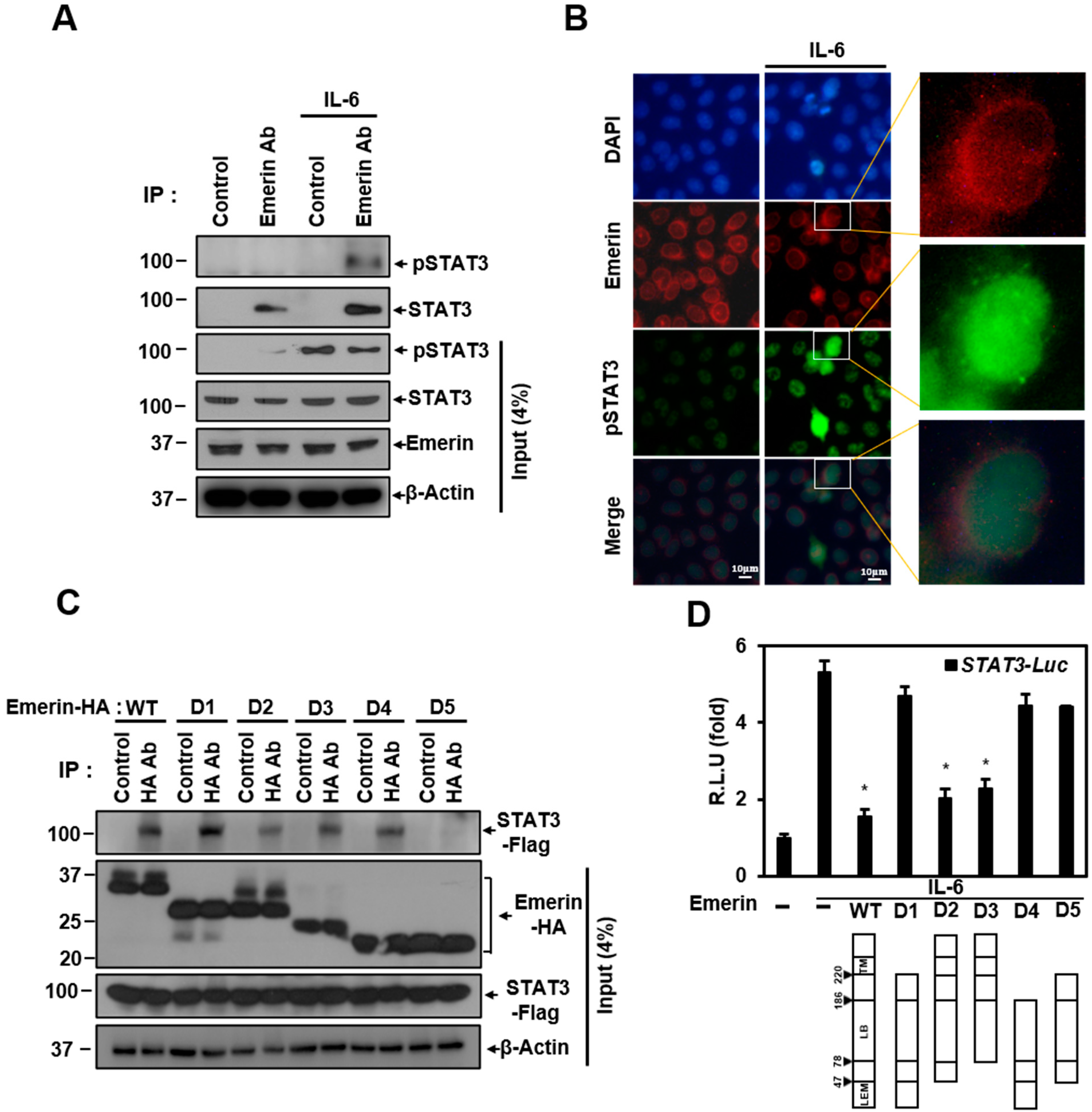
2.2. Emerin Interacts with STAT3 Proteins
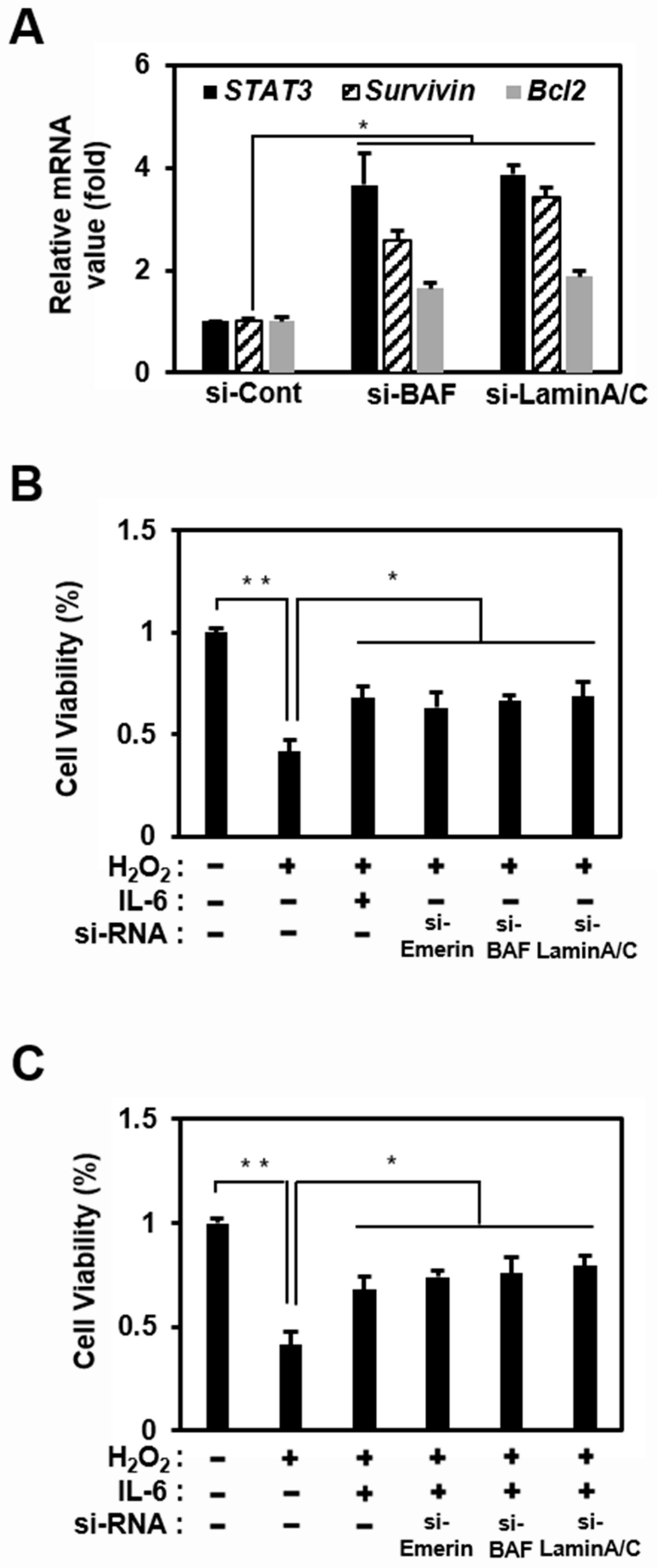
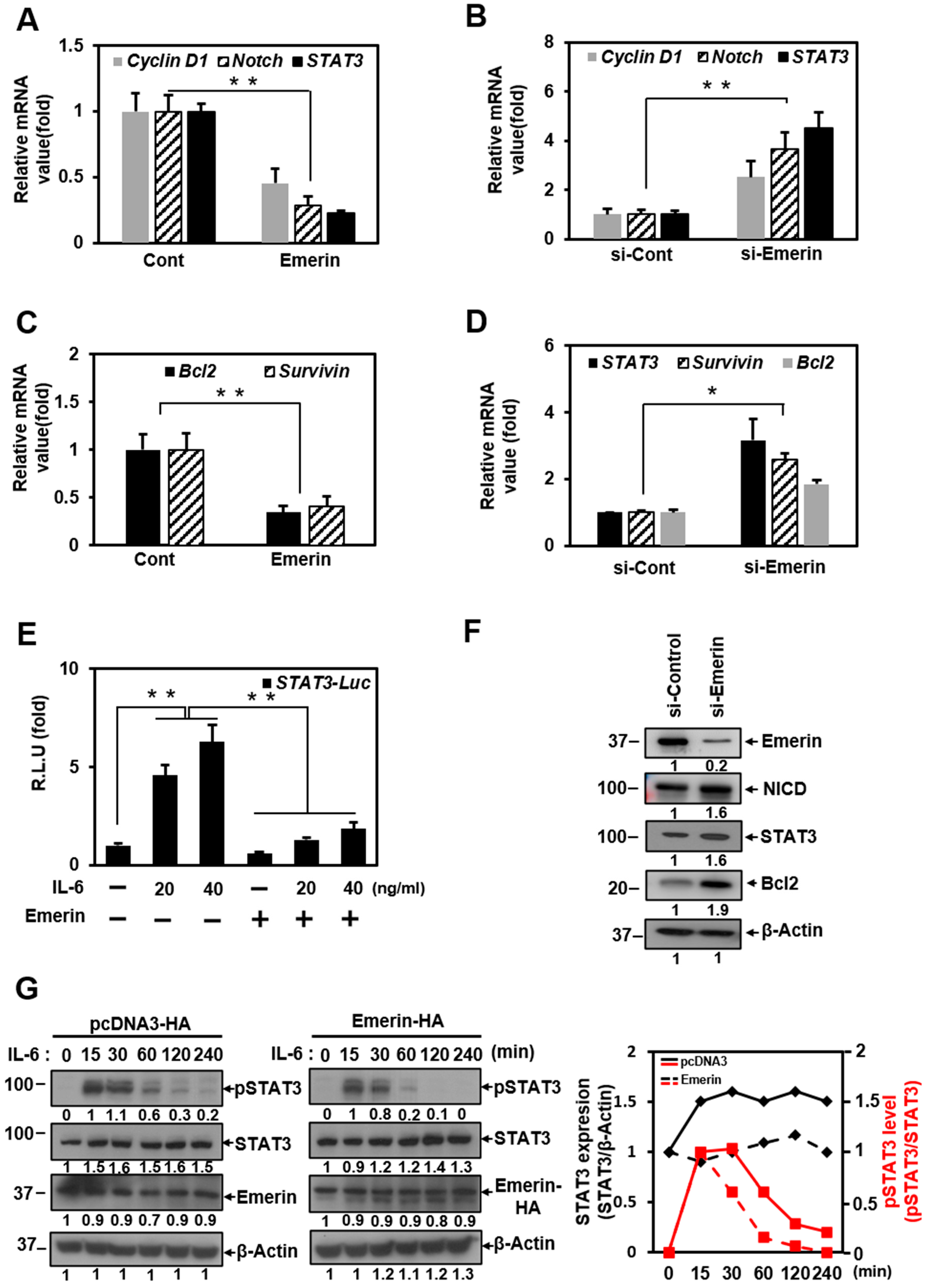
2.3. Emerin-Network Affects Cell Survival Through Modulating STAT3 Signal
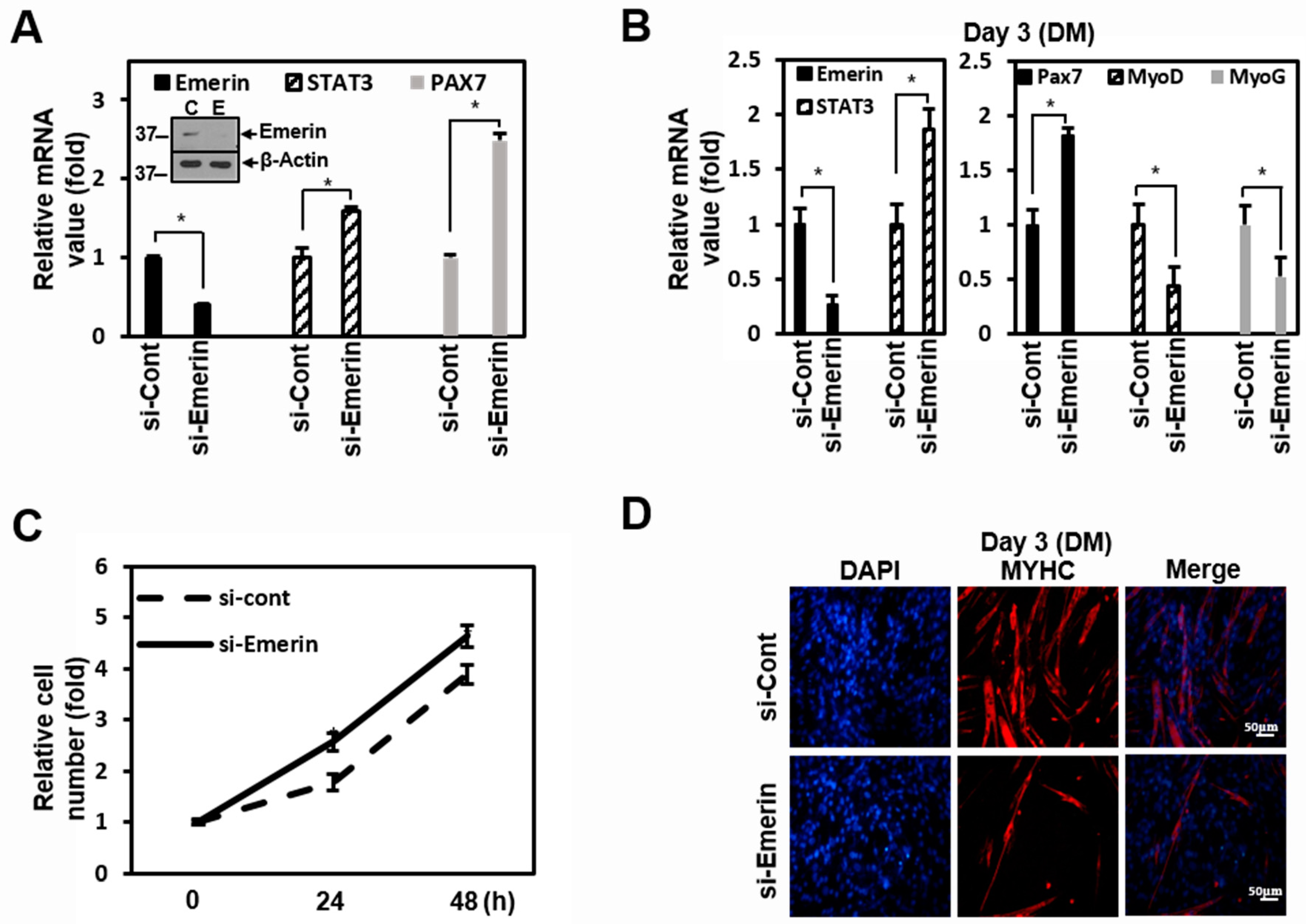
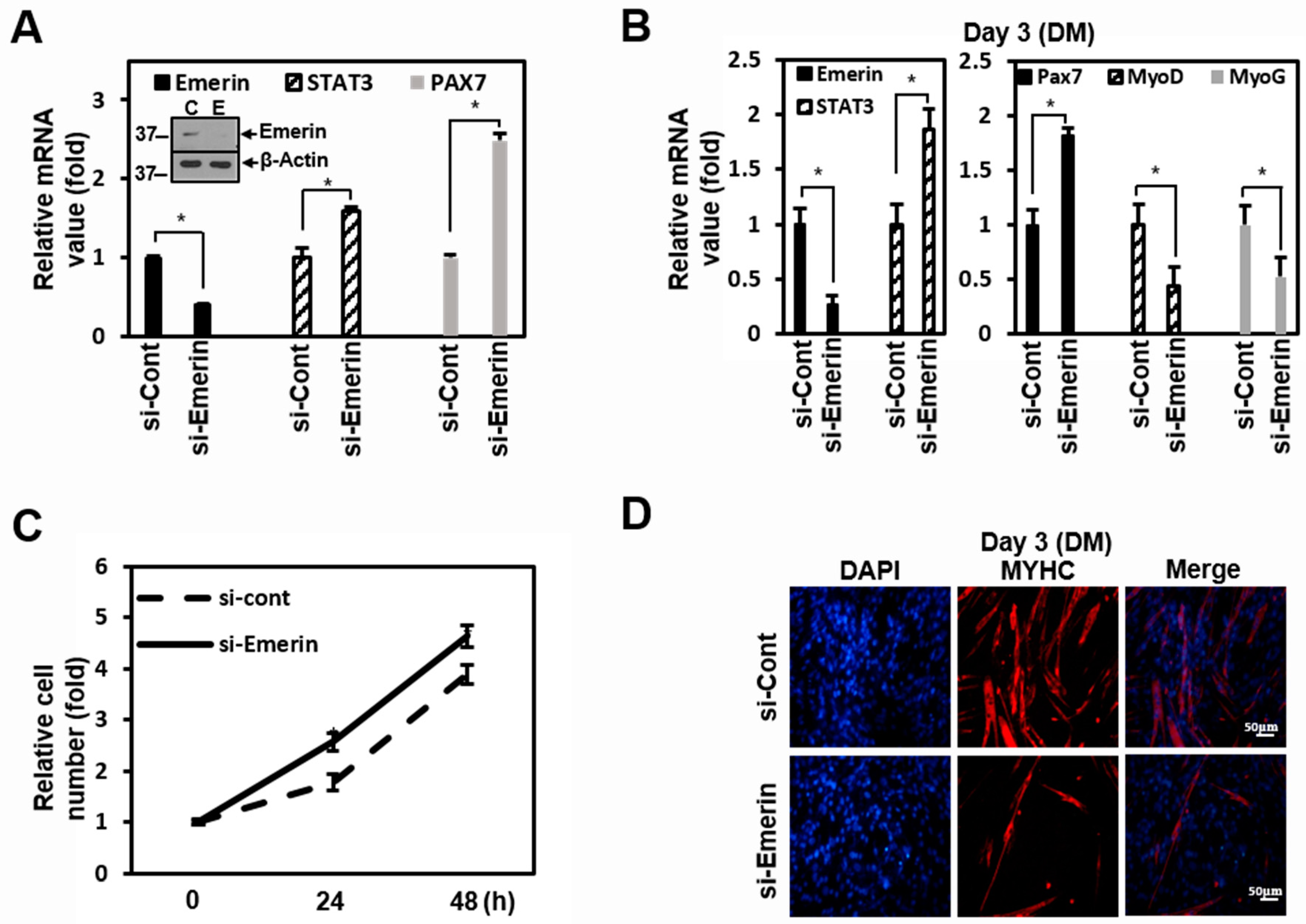
2.4. Emerin Regulates Muscle Cell Proliferation Through STAT3 Signaling
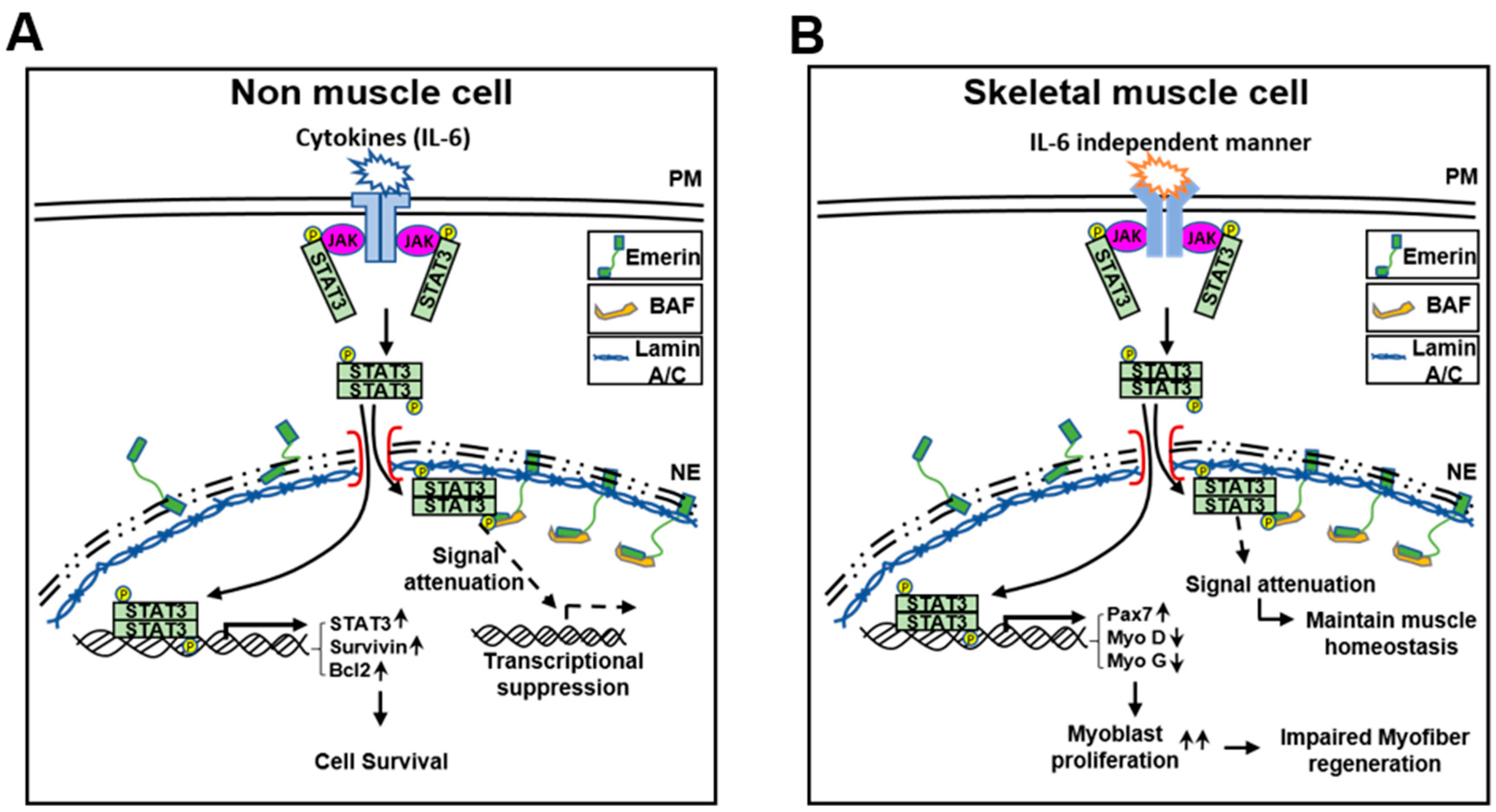
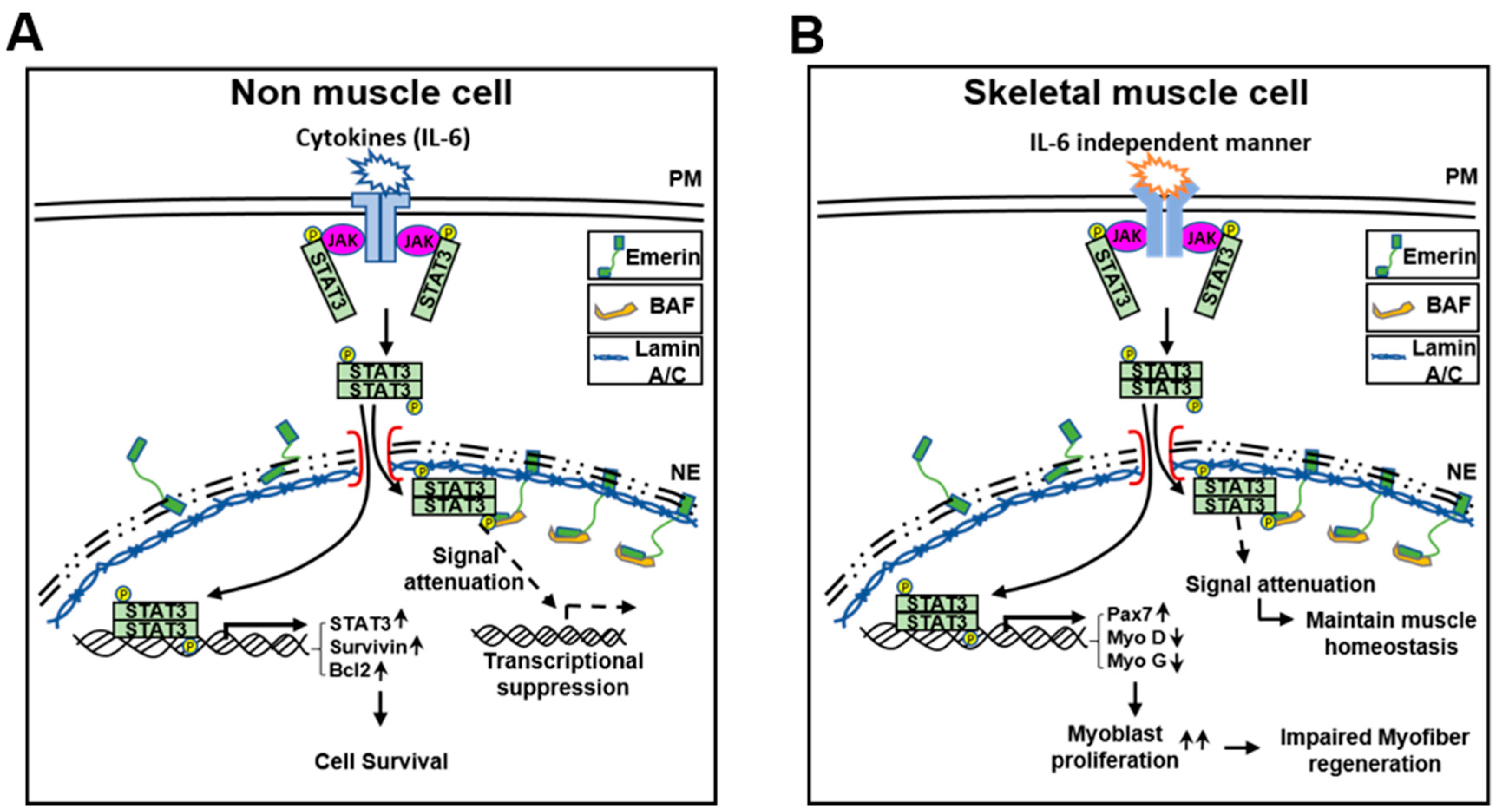
3. Discussion
4. Materials and Methods
4.1. Antibodies and Chemicals
4.2. Cell Culture and Transfection
4.3. Plasmid Constructs
4.4. Transcription Factor Profiling Assay
4.5. Luciferase Reporter Assay
4.6. Immunoblotting
4.7. Immunoprecipitation
4.8. Immunocytochemistry (ICC)
4.9. Quantitative RT-PCR (qRT-PCR)
4.10. Small Interfering RNA
4.11. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| STAT3 | Signal transducer and activators of transcription3 |
| JAK | Janus-kinase |
| LEM | LAP2-emerin-MAN1 |
| TM | Transmembrane |
| PM | Plasma membrane |
| NE | Nuclear envelope |
| EDMD | Emery–Dreifuss muscular dystrophy |
References
- Gerace, L.; Huber, M.D. Nuclear lamina at the crossroads of the cytoplasm and nucleus. J. Struct. Biol 2012, 177, 24–31. [Google Scholar] [CrossRef] [Green Version]
- Davidson, P.M.; Lammerding, J. Broken nuclei—Lamins, nuclear mechanics, and disease. Trends Cell Biol. 2014, 24, 247–256. [Google Scholar] [CrossRef] [Green Version]
- Schirmer, E.C.; Gerace, L. The nuclear membrane proteome: Extending the envelope. Trends Biochem. Sci. 2005, 30, 551–558. [Google Scholar] [CrossRef] [Green Version]
- Korfali, N.; Wilkie, G.S.; Swanson, S.K.; Srsen, V.; de Las Heras, J.; Batrakou, D.G.; Malik, P.; Zuleger, N.; Kerr, A.R.; Florens, L.; et al. The nuclear envelope proteome differs notably between tissues. Nucleus 2012, 3, 552–564. [Google Scholar] [CrossRef] [Green Version]
- Barton, L.J.; Soshnev, A.A.; Geyer, P.K. Networking in the nucleus: A spotlight on LEM-domain proteins. Curr. Opin. Cell Biol. 2015, 34, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Shimi, T.; Butin-Israeli, V.; Adam, S.A.; Goldman, R.D. Nuclear lamins in cell regulation and disease. Cold Spring Harb. Symp. Quant. Biol. 2010, 75, 525–531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, K.K.; Haraguchi, T.; Lee, R.S.; Koujin, T.; Hiraoka, Y.; Wilson, K.L. Distinct functional domains in emerin bind lamin A and DNA-bridging protein BAF. J. Cell Sci. 2001, 114, 4567–4573. [Google Scholar] [CrossRef] [PubMed]
- Mislow, J.M.; Holaska, J.M.; Kim, M.S.; Lee, K.K.; Segura-Totten, M.; Wilson, K.L.; McNally, E.M. Nesprin-1alpha self-associates and binds directly to emerin and lamin A in vitro. FEBS Lett. 2002, 525, 135–140. [Google Scholar] [CrossRef] [Green Version]
- Holaska, J.M.; Wilson, K.L.; Mansharamani, M. The nuclear envelope, lamins and nuclear assembly. Curr. Opin. Cell Biol. 2002, 14, 357–364. [Google Scholar] [CrossRef]
- Dahl, K.N.; Ribeiro, A.J.; Lammerding, J. Nuclear shape, mechanics, and mechanotransduction. Circ. Res. 2008, 102, 1307–1318. [Google Scholar] [CrossRef] [Green Version]
- Markiewicz, E.; Tilgner, K.; Barker, N.; van de Wetering, M.; Clevers, H.; Dorobek, M.; Hausmanowa-Petrusewicz, I.; Ramaekers, F.C.; Broers, J.L.; Blankesteijn, W.M.; et al. The inner nuclear membrane protein emerin regulates beta-catenin activity by restricting its accumulation in the nucleus. EMBO J. 2006, 25, 3275–3285. [Google Scholar] [CrossRef]
- Holaska, J.M.; Lee, K.K.; Kowalski, A.K.; Wilson, K.L. Transcriptional repressor germ cell-less (GCL) and barrier to autointegration factor (BAF) compete for binding to emerin in vitro. J. Biol. Chem. 2003, 278, 6969–6975. [Google Scholar] [CrossRef] [Green Version]
- Dedeic, Z.; Cetera, M.; Cohen, T.V.; Holaska, J.M. Emerin inhibits Lmo7 binding to the Pax3 and MyoD promoters and expression of myoblast proliferation genes. J. Cell Sci. 2011, 124, 1691–1702. [Google Scholar] [CrossRef] [Green Version]
- Nagase, T.; Seki, N.; Ishikawa, K.; Tanaka, A.; Nomura, N. Prediction of the coding sequences of unidentified human genes. V. The coding sequences of 40 new genes (KIAA0161-KIAA0200) deduced by analysis of cDNA clones from human cell line KG-1. DNA Res. 1996, 3, 17–24. [Google Scholar] [CrossRef]
- Lee, B.; Lee, T.H.; Shim, J. Emerin suppresses Notch signaling by restricting the Notch intracellular domain to the nuclear membrane. Biochim. Biophys. Acta Mol. Cell Res. 2017, 1864, 303–313. [Google Scholar] [CrossRef]
- Haraguchi, T.; Holaska, J.M.; Yamane, M.; Koujin, T.; Hashiguchi, N.; Mori, C.; Wilson, K.L.; Hiraoka, Y. Emerin binding to Btf, a death-promoting transcriptional repressor, is disrupted by a missense mutation that causes Emery-Dreifuss muscular dystrophy. Eur. J. Biochem. 2004, 271, 1035–1045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, X.; Zheng, C.; Chen, X.; Tang, Y.; Zhang, H.; Yan, C.; Ma, H.; Li, X. Targeted next-generation sequencing identified a known EMD mutation in a Chinese patient with Emery-Dreifuss muscular dystrophy. Hum. Genome Var. 2019, 6, 42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koch, A.J.; Holaska, J.M. Emerin in health and disease. Semin Cell Dev. Biol. 2014, 29, 95–106. [Google Scholar] [CrossRef] [Green Version]
- Raffaele Di Barletta, M.; Ricci, E.; Galluzzi, G.; Tonali, P.; Mora, M.; Morandi, L.; Romorini, A.; Voit, T.; Orstavik, K.H.; Merlini, L.; et al. Different mutations in the LMNA gene cause autosomal dominant and autosomal recessive Emery-Dreifuss muscular dystrophy. Am. J. Hum. Genet. 2000, 66, 1407–1412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jimenez-Escrig, A.; Gobernado, I.; Garcia-Villanueva, M.; Sanchez-Herranz, A. Autosomal recessive Emery-Dreifuss muscular dystrophy caused by a novel mutation (R225Q) in the lamin A/C gene identified by exome sequencing. Muscle Nerve 2012, 45, 605–610. [Google Scholar] [CrossRef]
- Helbling-Leclerc, A.; Bonne, G.; Schwartz, K. Emery-Dreifuss muscular dystrophy. Eur J. Hum. Genet. 2002, 10, 157–161. [Google Scholar] [CrossRef] [Green Version]
- Bione, S.; Maestrini, E.; Rivella, S.; Mancini, M.; Regis, S.; Romeo, G.; Toniolo, D. Identification of a novel X-linked gene responsible for Emery-Dreifuss muscular dystrophy. Nat. Genet. 1994, 8, 323–327. [Google Scholar] [CrossRef] [PubMed]
- Emery, A.E. Emery-Dreifuss muscular dystrophy—A 40 year retrospective. Neuromuscul. Disord. 2000, 10, 228–232. [Google Scholar] [CrossRef]
- Aggarwal, B.B.; Vijayalekshmi, R.V.; Sung, B. Targeting inflammatory pathways for prevention and therapy of cancer: Short-term friend, long-term foe. Clin. Cancer Res. 2009, 15, 425–430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aggarwal, B.B.; Sethi, G.; Ahn, K.S.; Sandur, S.K.; Pandey, M.K.; Kunnumakkara, A.B.; Sung, B.; Ichikawa, H. Targeting signal-transducer-and-activator-of-transcription-3 for prevention and therapy of cancer: Modern target but ancient solution. Ann. N. Y. Acad. Sci. 2006, 1091, 151–169. [Google Scholar] [CrossRef] [Green Version]
- Darnell, J.E., Jr.; Kerr, I.M.; Stark, G.R. Jak-STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. Science 1994, 264, 1415–1421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heinrich, P.C.; Behrmann, I.; Muller-Newen, G.; Schaper, F.; Graeve, L. Interleukin-6-type cytokine signalling through the gp130/Jak/STAT pathway. Biochem. J. 1998, 334 Pt 2, 297–314. [Google Scholar] [CrossRef] [Green Version]
- Quaglino, A.; Schere-Levy, C.; Romorini, L.; Meiss, R.P.; Kordon, E.C. Mouse mammary tumors display Stat3 activation dependent on leukemia inhibitory factor signaling. Breast Cancer Res. 2007, 9, R69. [Google Scholar] [CrossRef] [Green Version]
- Stout, B.A.; Bates, M.E.; Liu, L.Y.; Farrington, N.N.; Bertics, P.J. IL-5 and granulocyte-macrophage colony-stimulating factor activate STAT3 and STAT5 and promote Pim-1 and cyclin D3 protein expression in human eosinophils. J. Immunol. 2004, 173, 6409–6417. [Google Scholar] [CrossRef] [Green Version]
- Kordula, T.; Bugno, M.; Goldstein, J.; Travis, J. Activation of signal transducer and activator of transcription-3 (Stat3) expression by interferon-gamma and interleukin-6 in hepatoma cells. Biochem. Biophys. Res. Commun. 1995, 216, 999–1005. [Google Scholar] [CrossRef]
- Miscia, S.; Marchisio, M.; Grilli, A.; Di Valerio, V.; Centurione, L.; Sabatino, G.; Garaci, F.; Zauli, G.; Bonvini, E.; Di Baldassarre, A. Tumor necrosis factor alpha (TNF-alpha) activates Jak1/Stat3-Stat5B signaling through TNFR-1 in human B cells. Cell Growth Differ. 2002, 13, 13–18. [Google Scholar]
- Park, O.K.; Schaefer, T.S.; Nathans, D. In vitro activation of Stat3 by epidermal growth factor receptor kinase. Proc. Natl. Acad. Sci. USA 1996, 93, 13704–13708. [Google Scholar] [CrossRef] [Green Version]
- Vignais, M.L.; Sadowski, H.B.; Watling, D.; Rogers, N.C.; Gilman, M. Platelet-derived growth factor induces phosphorylation of multiple JAK family kinases and STAT proteins. Mol. Cell Biol. 1996, 16, 1759–1769. [Google Scholar] [CrossRef] [Green Version]
- Gurzov, E.N.; Stanley, W.J.; Pappas, E.G.; Thomas, H.E.; Gough, D.J. The JAK/STAT pathway in obesity and diabetes. FEBS J. 2016, 283, 3002–3015. [Google Scholar] [CrossRef] [Green Version]
- Tkach, M.; Rosemblit, C.; Rivas, M.A.; Proietti, C.J.; Diaz Flaque, M.C.; Mercogliano, M.F.; Beguelin, W.; Maronna, E.; Guzman, P.; Gercovich, F.G.; et al. p42/p44 MAPK-mediated Stat3Ser727 phosphorylation is required for progestin-induced full activation of Stat3 and breast cancer growth. Endocr. Relat. Cancer 2013, 20, 197–212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silva, C.M. Role of STATs as downstream signal transducers in Src family kinase-mediated tumorigenesis. Oncogene 2004, 23, 8017–8023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, C.P.; Cao, X. Structure, function, and regulation of STAT proteins. Mol. Biosyst. 2006, 2, 536–550. [Google Scholar] [CrossRef]
- Yu, H.; Jove, R. The STATs of cancer--new molecular targets come of age. Nat. Rev. Cancer 2004, 4, 97–105. [Google Scholar] [CrossRef] [PubMed]
- Pensa, S.; Regis, G.; Boselli, D.; Novelli, F.; Poli, V. STAT1 and STAT3 in Tumorigenesis: Two Sides of the Same Coin? In Madame Curie Bioscience Database; Landes Bioscience: Austin, TX, USA, 2013. [Google Scholar]
- Takeda, K.; Kaisho, T.; Yoshida, N.; Takeda, J.; Kishimoto, T.; Akira, S. Stat3 activation is responsible for IL-6-dependent T cell proliferation through preventing apoptosis: Generation and characterization of T cell-specific Stat3-deficient mice. J. Immunol. 1998, 161, 4652–4660. [Google Scholar] [CrossRef] [Green Version]
- Xiong, H.; Zhang, Z.G.; Tian, X.Q.; Sun, D.F.; Liang, Q.C.; Zhang, Y.J.; Lu, R.; Chen, Y.X.; Fang, J.Y. Inhibition of JAK1, 2/STAT3 signaling induces apoptosis, cell cycle arrest, and reduces tumor cell invasion in colorectal cancer cells. Neoplasia 2008, 10, 287–297. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Shaw, P.E. Autocrine-mediated activation of STAT3 correlates with cell proliferation in breast carcinoma lines. J. Biol Chem. 2002, 277, 17397–17405. [Google Scholar] [CrossRef] [Green Version]
- Prendergast, G.C. Mechanisms of apoptosis by c-Myc. Oncogene 1999, 18, 2967–2987. [Google Scholar] [CrossRef] [Green Version]
- Kanda, N.; Seno, H.; Konda, Y.; Marusawa, H.; Kanai, M.; Nakajima, T.; Kawashima, T.; Nanakin, A.; Sawabu, T.; Uenoyama, Y.; et al. STAT3 is constitutively activated and supports cell survival in association with survivin expression in gastric cancer cells. Oncogene 2004, 23, 4921–4929. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Debnath, B.; Xu, S.; Neamati, N. Small molecule inhibitors of signal transducer and activator of transcription 3 (Stat3) protein. J. Med. Chem. 2012, 55, 6645–6668. [Google Scholar] [CrossRef] [PubMed]
- Dechow, T.N.; Pedranzini, L.; Leitch, A.; Leslie, K.; Gerald, W.L.; Linkov, I.; Bromberg, J.F. Requirement of matrix metalloproteinase-9 for the transformation of human mammary epithelial cells by Stat3-C. Proc. Natl. Acad. Sci. USA 2004, 101, 10602–10607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsareva, S.A.; Moriggl, R.; Corvinus, F.M.; Wiederanders, B.; Schutz, A.; Kovacic, B.; Friedrich, K. Signal transducer and activator of transcription 3 activation promotes invasive growth of colon carcinomas through matrix metalloproteinase induction. Neoplasia 2007, 9, 279–291. [Google Scholar] [CrossRef] [Green Version]
- Steinman, R.A.; Wentzel, A.; Lu, Y.; Stehle, C.; Grandis, J.R. Activation of Stat3 by cell confluence reveals negative regulation of Stat3 by cdk2. Oncogene 2003, 22, 3608–3615. [Google Scholar] [CrossRef] [Green Version]
- Vultur, A.; Cao, J.; Arulanandam, R.; Turkson, J.; Jove, R.; Greer, P.; Craig, A.; Elliott, B.; Raptis, L. Cell-to-cell adhesion modulates Stat3 activity in normal and breast carcinoma cells. Oncogene 2004, 23, 2600–2616. [Google Scholar] [CrossRef] [Green Version]
- Schaefer, L.K.; Ren, Z.; Fuller, G.N.; Schaefer, T.S. Constitutive activation of Stat3alpha in brain tumors: Localization to tumor endothelial cells and activation by the endothelial tyrosine kinase receptor (VEGFR-2). Oncogene 2002, 21, 2058–2065. [Google Scholar] [CrossRef] [Green Version]
- Niu, G.; Wright, K.L.; Huang, M.; Song, L.; Haura, E.; Turkson, J.; Zhang, S.; Wang, T.; Sinibaldi, D.; Coppola, D.; et al. Constitutive Stat3 activity up-regulates VEGF expression and tumor angiogenesis. Oncogene 2002, 21, 2000–2008. [Google Scholar] [CrossRef] [Green Version]
- Miller, A.; McLeod, L.; Alhayyani, S.; Szczepny, A.; Watkins, D.N.; Chen, W.; Enriori, P.; Ferlin, W.; Ruwanpura, S.; Jenkins, B.J. Blockade of the IL-6 trans-signalling/STAT3 axis suppresses cachexia in Kras-induced lung adenocarcinoma. Oncogene 2017, 36, 3059–3066. [Google Scholar] [CrossRef] [PubMed]
- Zimmers, T.A.; Fishel, M.L.; Bonetto, A. STAT3 in the systemic inflammation of cancer cachexia. Semin. Cell Dev. Biol. 2016, 54, 28–41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nunes, A.M.; Wuebbles, R.D.; Sarathy, A.; Fontelonga, T.M.; Deries, M.; Burkin, D.J.; Thorsteinsdottir, S. Impaired fetal muscle development and JAK-STAT activation mark disease onset and progression in a mouse model for merosin-deficient congenital muscular dystrophy. Hum. Mol. Genet. 2017, 26, 2018–2033. [Google Scholar] [CrossRef] [PubMed]
- Wada, E.; Tanihata, J.; Iwamura, A.; Takeda, S.; Hayashi, Y.K.; Matsuda, R. Treatment with the anti-IL-6 receptor antibody attenuates muscular dystrophy via promoting skeletal muscle regeneration in dystrophin-/utrophin-deficient mice. Skelet Muscle 2017, 7, 23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, F.; Li, T.; Azuelos, I.; Giordano, C.; Liang, H.; Hussain, S.N.; Matecki, S.; Petrof, B.J. Ventilator-induced diaphragmatic dysfunction in MDX mice. Muscle Nerve 2018, 57, 442–448. [Google Scholar] [CrossRef] [PubMed]
- Guadagnin, E.; Mazala, D.; Chen, Y.W. STAT3 in Skeletal Muscle Function and Disorders. Int. J. Mol. Sci. 2018, 19, 2265. [Google Scholar] [CrossRef] [Green Version]
- Muchir, A.; Pavlidis, P.; Bonne, G.; Hayashi, Y.K.; Worman, H.J. Activation of MAPK in hearts of EMD null mice: Similarities between mouse models of X-linked and autosomal dominant Emery Dreifuss muscular dystrophy. Hum. Mol. Genet. 2007, 16, 1884–1895. [Google Scholar] [CrossRef] [Green Version]
- Haraguchi, T.; Koujin, T.; Segura-Totten, M.; Lee, K.K.; Matsuoka, Y.; Yoneda, Y.; Wilson, K.L.; Hiraoka, Y. BAF is required for emerin assembly into the reforming nuclear envelope. J. Cell Sci. 2001, 114, 4575–4585. [Google Scholar] [CrossRef] [PubMed]
- Segura-Totten, M.; Wilson, K.L. BAF: Roles in chromatin, nuclear structure and retrovirus integration. Trends Cell Biol. 2004, 14, 261–266. [Google Scholar] [CrossRef]
- Puente, X.S.; Quesada, V.; Osorio, F.G.; Cabanillas, R.; Cadinanos, J.; Fraile, J.M.; Ordonez, G.R.; Puente, D.A.; Gutierrez-Fernandez, A.; Fanjul-Fernandez, M.; et al. Exome sequencing and functional analysis identifies BANF1 mutation as the cause of a hereditary progeroid syndrome. Am. J. Hum. Genet. 2011, 88, 650–656. [Google Scholar] [CrossRef] [Green Version]
- Cheung, C.H.A.; Chang, Y.C.; Lin, T.Y.; Cheng, S.M.; Leung, E. Anti-apoptotic proteins in the autophagic world: An update on functions of XIAP, Survivin, and BRUCE. J. Biomed. Sci. 2020, 27, 31. [Google Scholar] [CrossRef]
- Czabotar, P.E.; Lessene, G.; Strasser, A.; Adams, J.M. Control of apoptosis by the BCL-2 protein family: Implications for physiology and therapy. Nat. Rev. Mol. Cell Biol. 2014, 15, 49–63. [Google Scholar] [CrossRef]
- Tajbakhsh, S. Skeletal muscle stem cells in developmental versus regenerative myogenesis. J. Intern. Med. 2009, 266, 372–389. [Google Scholar] [CrossRef] [PubMed]
- Relaix, F.; Montarras, D.; Zaffran, S.; Gayraud-Morel, B.; Rocancourt, D.; Tajbakhsh, S.; Mansouri, A.; Cumano, A.; Buckingham, M. Pax3 and Pax7 have distinct and overlapping functions in adult muscle progenitor cells. J. Cell Biol. 2006, 172, 91–102. [Google Scholar] [CrossRef]
- Tajbakhsh, S.; Rocancourt, D.; Cossu, G.; Buckingham, M. Redefining the genetic hierarchies controlling skeletal myogenesis: Pax-3 and Myf-5 act upstream of MyoD. Cell 1997, 89, 127–138. [Google Scholar] [CrossRef] [Green Version]
- Brack, A.S.; Conboy, I.M.; Conboy, M.J.; Shen, J.; Rando, T.A. A temporal switch from notch to Wnt signaling in muscle stem cells is necessary for normal adult myogenesis. Cell Stem Cell 2008, 2, 50–59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tierney, M.T.; Aydogdu, T.; Sala, D.; Malecova, B.; Gatto, S.; Puri, P.L.; Latella, L.; Sacco, A. STAT3 signaling controls satellite cell expansion and skeletal muscle repair. Nat. Med. 2014, 20, 1182–1186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Price, F.D.; von Maltzahn, J.; Bentzinger, C.F.; Dumont, N.A.; Yin, H.; Chang, N.C.; Wilson, D.H.; Frenette, J.; Rudnicki, M.A. Inhibition of JAK-STAT signaling stimulates adult satellite cell function. Nat. Med. 2014, 20, 1174–1181. [Google Scholar] [CrossRef] [Green Version]
- Bromberg, J.F.; Wrzeszczynska, M.H.; Devgan, G.; Zhao, Y.; Pestell, R.G.; Albanese, C.; Darnell, J.E., Jr. Stat3 as an oncogene. Cell 1999, 98, 295–303. [Google Scholar] [CrossRef] [Green Version]
- Wake, M.S.; Watson, C.J. STAT3 the oncogene—Still eluding therapy? FEBS J. 2015, 282, 2600–2611. [Google Scholar] [CrossRef] [Green Version]
- Sullivan, T.; Escalante-Alcalde, D.; Bhatt, H.; Anver, M.; Bhat, N.; Nagashima, K.; Stewart, C.L.; Burke, B. Loss of A-type lamin expression compromises nuclear envelope integrity leading to muscular dystrophy. J. Cell Biol. 1999, 147, 913–920. [Google Scholar] [CrossRef] [PubMed] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, B.; Lee, S.; Lee, Y.; Park, Y.; Shim, J. Emerin Represses STAT3 Signaling through Nuclear Membrane-Based Spatial Control. Int. J. Mol. Sci. 2021, 22, 6669. https://doi.org/10.3390/ijms22136669
Lee B, Lee S, Lee Y, Park Y, Shim J. Emerin Represses STAT3 Signaling through Nuclear Membrane-Based Spatial Control. International Journal of Molecular Sciences. 2021; 22(13):6669. https://doi.org/10.3390/ijms22136669
Chicago/Turabian StyleLee, Byongsun, Seungjae Lee, Younggwang Lee, Yongjin Park, and Jaekyung Shim. 2021. "Emerin Represses STAT3 Signaling through Nuclear Membrane-Based Spatial Control" International Journal of Molecular Sciences 22, no. 13: 6669. https://doi.org/10.3390/ijms22136669
APA StyleLee, B., Lee, S., Lee, Y., Park, Y., & Shim, J. (2021). Emerin Represses STAT3 Signaling through Nuclear Membrane-Based Spatial Control. International Journal of Molecular Sciences, 22(13), 6669. https://doi.org/10.3390/ijms22136669