
Studying SARS-CoV-2 with Fluorescence Microscopy


Abstract
1. Introduction
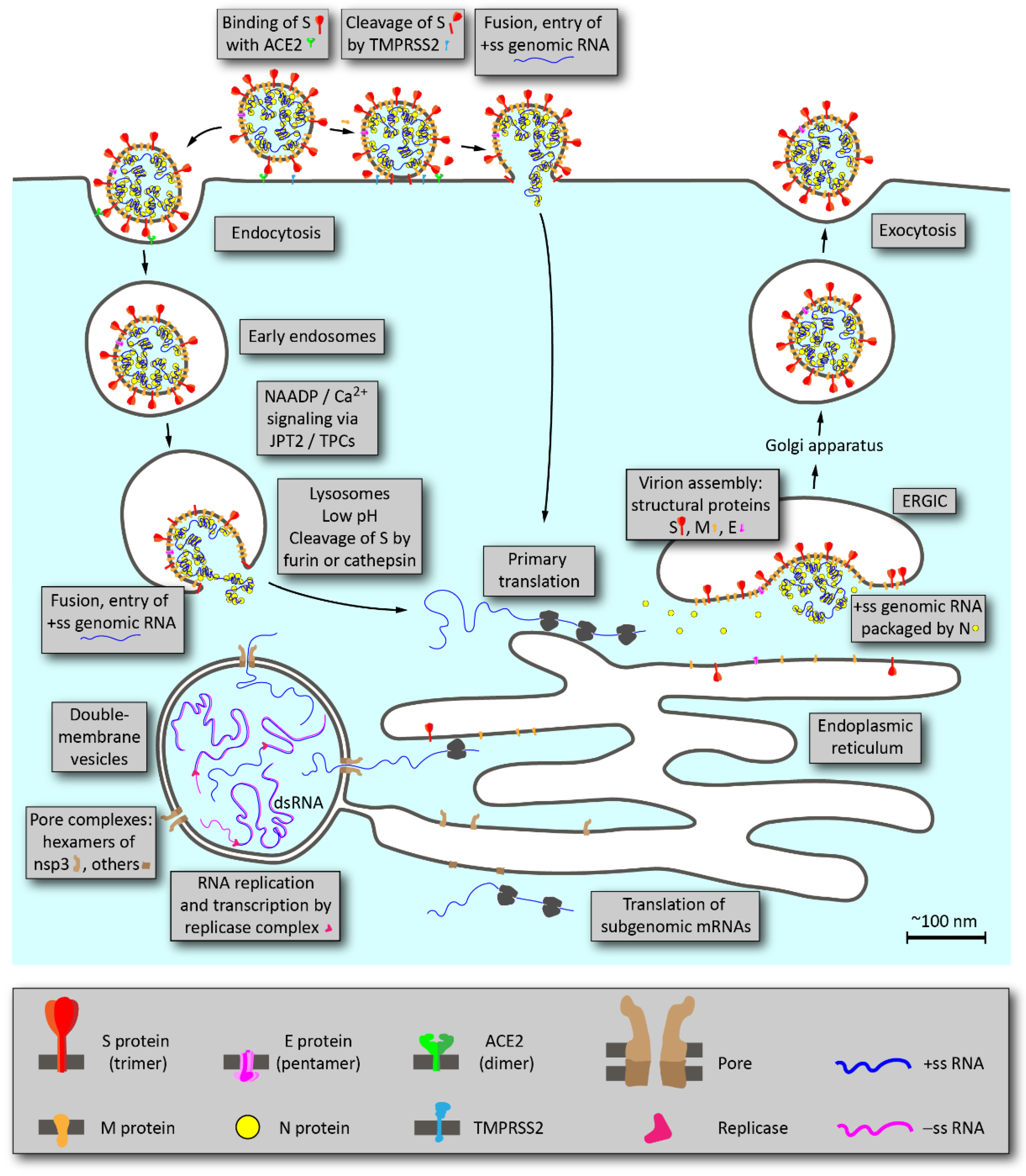
2. Overview of SARS-CoV-2 Life Cycle
3. Fluorescence Microscopy-Based Studies of Viral Life Cycle
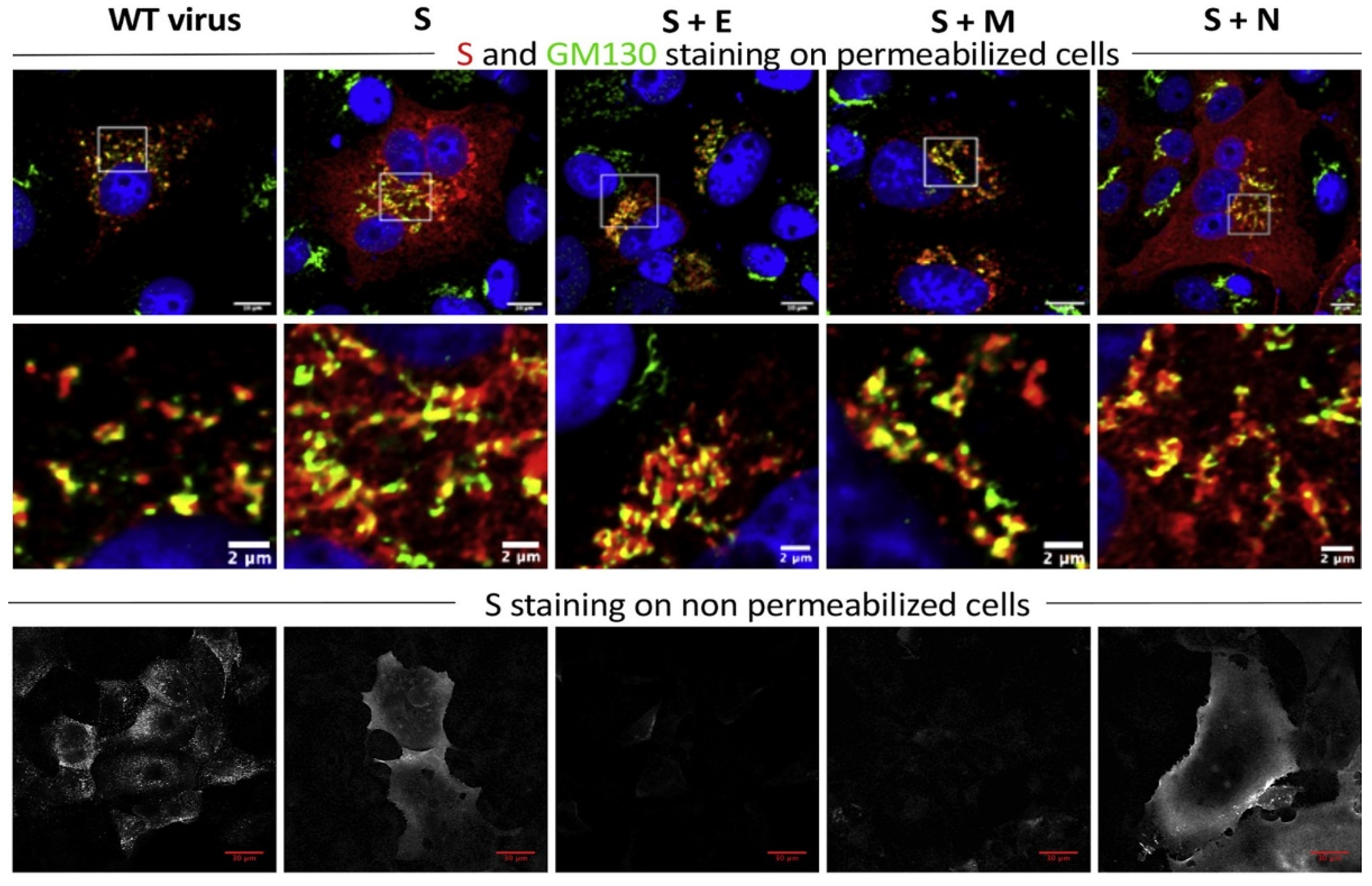
4. Influence of Viral Proteins on the Host Cell
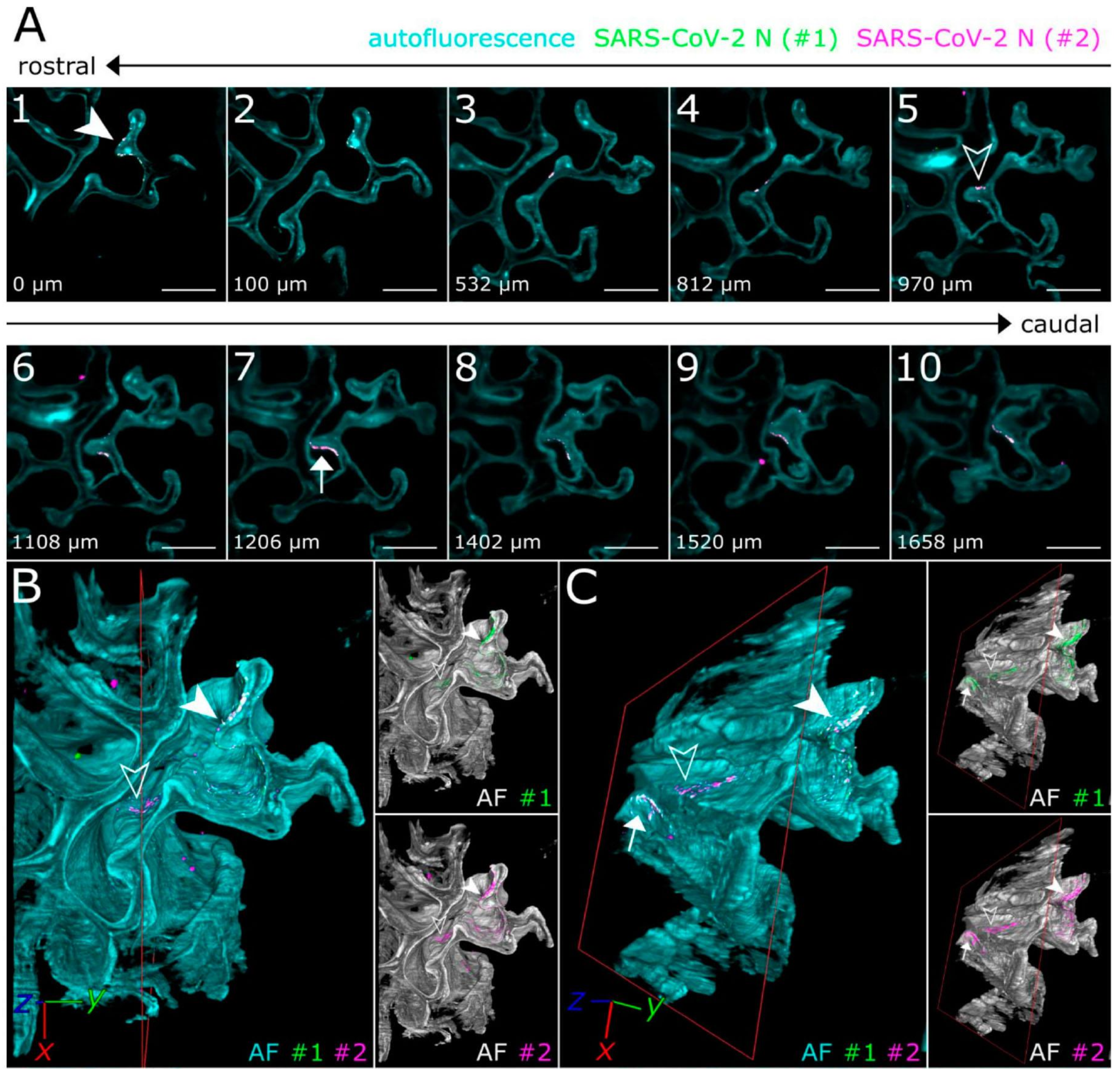
5. RNA Tracking
6. SARS-CoV-2 Drug Screening and Inhibitor Testing
7. Recombinant SARS-CoV-2 Expressing Reporter Genes
8. Perspectives
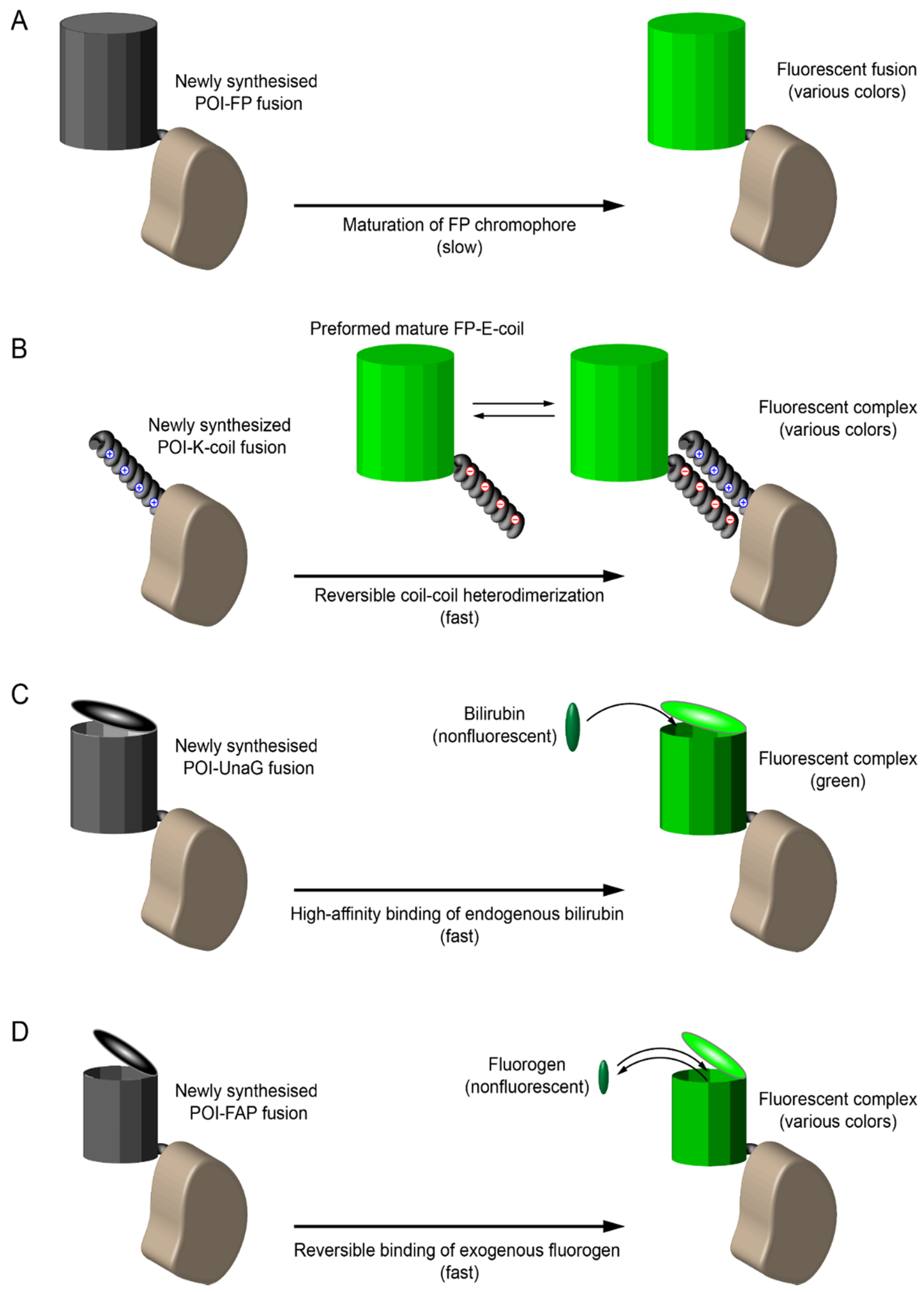
8.1. Early Labeling and Tracking of Viral Proteins
8.2. Aptamer-Based RNA Labeling
8.3. Monitoring Cell Physiology with Fluorescent Sensors
8.4. Correlative Fluorescence and Electron Microscopy
8.5. Super-Resolution Fluorescence Microscopy
8.6. Expansion Microscopy
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Beilstein, F.; Cohen, G.H.; Eisenberg, R.J.; Nicolas, V.; Esclatine, A.; Pasdeloup, D. Dynamic organization of herpesvirus glycoproteins on the viral envelope revealed by super-resolution microscopy. PLoS Pathog. 2019, 15, e1008209. [Google Scholar] [CrossRef] [PubMed]
- Dáder, B.; Burckbuchler, M.; Macia, J.-L.; Alcon, C.; Curie, C.; Gargani, D.; Zhou, J.S.; Ng, J.C.K.; Brault, V.; Drucker, M. Split green fluorescent protein as a tool to study infection with a plant pathogen, cauliflower mosaic virus. PLoS ONE 2019, 14, e0213087. [Google Scholar] [CrossRef]
- Kumar, A.; Kim, J.H.; Ranjan, P.; Metcalfe, M.G.; Cao, W.; Mishina, M.; Gangappa, S.; Guo, Z.; Boyden, E.S.; Zaki, S.; et al. Influenza Virus Exploits Tunneling Nanotubes for Cell-to-Cell Spread. Sci. Rep. 2017, 7, 40360. [Google Scholar] [CrossRef]
- Mazumder, N.; Lyn, R.K.; Singaravelu, R.; Ridsdale, A.; Moffatt, D.J.; Hu, C.-W.; Tsai, H.-R.; McLauchlan, J.; Stolow, A.; Kao, F.-J.; et al. Fluorescence lifetime imaging of alterations to cellular metabolism by domain 2 of the Hepatitis C virus core protein. PLoS ONE 2013, 8, e66738. [Google Scholar] [CrossRef][Green Version]
- Zgheib, S.; Lysova, I.; Réal, E.; Dukhno, O.; Vauchelles, R.; Pires, M.; Anton, H.; Mély, Y. Quantitative monitoring of the cytoplasmic release of NCp7 proteins from individual HIV-1 viral cores during the early steps of infection. Sci. Rep. 2019, 9, 945. [Google Scholar] [CrossRef]
- V’kovski, P.; Kratzel, A.; Steiner, S.; Stalder, H.; Thiel, V. Coronavirus biology and replication: Implications for SARS-CoV-2. Nat. Rev. Microbiol. 2021, 19, 155–170. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, M.; Kleine-Weber, H.; Pöhlmann, S. A Multibasic cleavage site in the spike protein of SARS-CoV-2 is essential for infection of human lung cells. Mol. Cell 2020, 78, 779–784.e5. [Google Scholar] [CrossRef] [PubMed]
- Ziebuhr, J.; Snijder, E.J.; Gorbalenya, A.E. Virus-encoded proteinases and proteolytic processing in the nidovirales. J. Gen. Virol. 2000, 81, 853–879. [Google Scholar] [CrossRef] [PubMed]
- Jin, Z.; Du, X.; Xu, Y.; Deng, Y.; Liu, M.; Zhao, Y.; Zhang, B.; Li, X.; Zhang, L.; Peng, C.; et al. Structure of Mpro from SARS-CoV-2 and discovery of its inhibitors. Nature 2020, 582, 289–293. [Google Scholar] [CrossRef]
- Gordon, D.E.; Jang, G.M.; Bouhaddou, M.; Xu, J.; Obernier, K.; White, K.M.; O’Meara, M.J.; Rezelj, V.V.; Guo, J.Z.; Swaney, D.L.; et al. A SARS-CoV-2 protein interaction map reveals targets for drug repurposing. Nature 2020, 583, 459–468. [Google Scholar] [CrossRef]
- Krijnse-Locker, J.; Ericsson, M.; Rottier, P.J.; Griffiths, G. Characterization of the budding compartment of mouse hepatitis virus: Evidence that transport from the RER to the Golgi complex requires only one vesicular transport step. J. Cell Biol. 1994, 124, 55–70. [Google Scholar] [CrossRef]
- Fehr, A.R.; Perlman, S. Coronaviruses: An overview of their replication and pathogenesis. Methods Mol. Biol. 2015, 1282, 1–23. [Google Scholar]
- Alvarez, E.; DeDiego, M.L.; Nieto-Torres, J.L.; Jiménez-Guardeño, J.M.; Marcos-Villar, L.; Enjuanes, L. The envelope protein of severe acute respiratory syndrome Coronavirus interacts with the non-structural Protein 3 and is ubiquitinated. Virology 2010, 402, 281–291. [Google Scholar] [CrossRef]
- Woo, J.; Lee, E.Y.; Lee, M.; Kim, T.; Cho, Y.-E. An in vivo cell-based assay for investigating the specific interaction between the SARS-CoV N-protein and its Viral RNA packaging sequence. Biochem. Biophys. Res. Commun. 2019, 520, 499–506. [Google Scholar] [CrossRef]
- Cortese, M.; Laketa, V. Advanced microscopy technologies enable rapid response to SARS-CoV-2 pandemic. Cell. Microbiol. 2021, e13319. [Google Scholar] [CrossRef]
- Wang, H.; Yang, P.; Liu, K.; Guo, F.; Zhang, Y.; Zhang, G.; Jiang, C. SARS Coronavirus entry into host cells through a novel Clathrin- and caveolae-independent endocytic pathway. Cell Res. 2008, 18, 290–301. [Google Scholar] [CrossRef]
- Yeung, M.L.; Teng, J.L.L.; Jia, L.; Zhang, C.; Huang, C.; Cai, J.-P.; Zhou, R.; Chan, K.-H.; Zhao, H.; Zhu, L.; et al. Soluble ACE2-mediated cell entry of SARS-CoV-2 via interaction with proteins related to the renin-angiotensin system. Cell 2021, 184, 2212–2228.e12. [Google Scholar] [CrossRef]
- Yang, J.; Petitjean, S.J.L.; Koehler, M.; Zhang, Q.; Dumitru, A.C.; Chen, W.; Derclaye, S.; Vincent, S.P.; Soumillion, P.; Alsteens, D. Molecular interaction and inhibition of SARS-CoV-2 binding to the ACE2 receptor. Nat. Commun. 2020, 11, 4541. [Google Scholar] [CrossRef]
- Schoof, M.; Faust, B.; Saunders, R.A.; Sangwan, S.; Rezelj, V.; Hoppe, N.; Boone, M.; Billesbølle, C.B.; Puchades, C.; Azumaya, C.M.; et al. An ultrapotent synthetic nanobody neutralizes SARS-CoV-2 by stabilizing inactive spike. Science 2020, 370, 1473–1479. [Google Scholar] [CrossRef]
- Ou, X.; Liu, Y.; Lei, X.; Li, P.; Mi, D.; Ren, L.; Guo, L.; Guo, R.; Chen, T.; Hu, J.; et al. Characterization of Spike glycoprotein of SARS-CoV-2 on virus entry and its immune cross-reactivity with SARS-CoV. Nat. Commun. 2020, 11, 1620. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.-L.; Chou, Y.-Y.; Rothlauf, P.W.; Liu, Z.; Soh, T.K.; Cureton, D.; Case, J.B.; Chen, R.E.; Diamond, M.S.; Whelan, S.P.J.; et al. Inhibition of PIKfyve kinase prevents infection by Zaire Ebolavirus and SARS-CoV-2. Proc. Natl. Acad. Sci. USA 2020, 117, 20803–20813. [Google Scholar] [CrossRef]
- Zhang, Q.; Chen, C.Z.; Swaroop, M.; Xu, M.; Wang, L.; Lee, J.; Wang, A.Q.; Pradhan, M.; Hagen, N.; Chen, L.; et al. Heparan sulfate assists SARS-CoV-2 in cell entry and can be targeted by approved drugs in vitro. Cell Discov. 2020, 6, 80. [Google Scholar] [CrossRef] [PubMed]
- Cantuti-Castelvetri, L.; Ojha, R.; Pedro, L.D.; Djannatian, M.; Franz, J.; Kuivanen, S.; van der Meer, F.; Kallio, K.; Kaya, T.; Anastasina, M.; et al. Neuropilin-1 facilitates SARS-CoV-2 cell entry and infectivity. Science 2020, 370, 856–860. [Google Scholar] [CrossRef] [PubMed]
- Lv, J.; Wang, Z.; Qu, Y.; Zhu, H.; Zhu, Q.; Tong, W.; Bao, L.; Lv, Q.; Cong, J.; Li, D.; et al. Distinct uptake, amplification, and release of SARS-CoV-2 by M1 and M2 alveolar macrophages. Cell Discov. 2021, 7, 24. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Li, C.; Liu, X.; Chiu, M.C.; Zhao, X.; Wang, D.; Wei, Y.; Lee, A.; Zhang, A.J.; Chu, H.; et al. Infection of bat and human intestinal organoids by SARS-CoV-2. Nat. Med. 2020, 26, 1077–1083. [Google Scholar] [CrossRef]
- Zaeck, L.M.; Scheibner, D.; Sehl, J.; Müller, M.; Hoffmann, D.; Beer, M.; Abdelwhab, E.M.; Mettenleiter, T.C.; Breithaupt, A.; Finke, S. Light sheet microscopy-assisted 3D analysis of SARS-CoV-2 infection in the respiratory tract of the ferret model. Viruses 2021, 13, 529. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Liu, D.X.; Tam, J.P. Lipid rafts are involved in SARS-CoV entry into vero E6 cells. Biochem. Biophys. Res. Commun. 2008, 369, 344–349. [Google Scholar] [CrossRef]
- Li, X.; Zhu, W.; Fan, M.; Zhang, J.; Peng, Y.; Huang, F.; Wang, N.; He, L.; Zhang, L.; Holmdahl, R.; et al. Dependence of SARS-CoV-2 infection on cholesterol-rich lipid raft and endosomal acidification. Comput. Struct. Biotechnol. J. 2021, 19, 1933–1943. [Google Scholar] [CrossRef]
- Bayati, A.; Kumar, R.; Francis, V.; McPherson, P.S. SARS-CoV-2 infects cells following viral entry via Clathrin-mediated endocytosis. J. Biol. Chem. 2021, 296, 100306. [Google Scholar] [CrossRef]
- Gunaratne, G.S.; Brailoiu, E.; He, S.; Unterwald, E.M.; Patel, S.; Slama, J.T.; Walseth, T.F.; Marchant, J.S. Essential requirement for JPT2 in NAADP-evoked Ca2+ signaling. Sci. Signal. 2021, 14, eabd5605. [Google Scholar] [CrossRef]
- Gunaratne, G.S.; Yang, Y.; Li, F.; Walseth, T.F.; Marchant, J.S. NAADP-Dependent Ca2+ signaling regulates Middle East respiratory syndrome-Coronavirus pseudovirus translocation through the endolysosomal system. Cell Calcium 2018, 75, 30–41. [Google Scholar] [CrossRef]
- Yuan, X.; Li, J.; Shan, Y.; Yang, Z.; Zhao, Z.; Chen, B.; Yao, Z.; Dong, B.; Wang, S.; Chen, J.; et al. Subcellular localization and membrane association of SARS-CoV 3a protein. Virus Res. 2005, 109, 191–202. [Google Scholar] [CrossRef]
- Knoops, K.; Kikkert, M.; van den Worm, S.H.E.; Zevenhoven-Dobbe, J.C.; van der Meer, Y.; Koster, A.J.; Mommaas, A.M.; Snijder, E.J. SARS-Coronavirus replication is supported by a reticulovesicular network of modified endoplasmic reticulum. PLoS Biol. 2008, 6, e226. [Google Scholar] [CrossRef] [PubMed]
- Wolff, G.; Limpens, R.W.A.L.; Zevenhoven-Dobbe, J.C.; Laugks, U.; Zheng, S.; de Jong, A.W.M.; Koning, R.I.; Agard, D.A.; Grünewald, K.; Koster, A.J.; et al. A molecular pore spans the double membrane of the Coronavirus Replication Organelle. Science 2020, 369, 1395–1398. [Google Scholar] [CrossRef] [PubMed]
- Siu, Y.L.; Teoh, K.T.; Lo, J.; Chan, C.M.; Kien, F.; Escriou, N.; Tsao, S.W.; Nicholls, J.M.; Altmeyer, R.; Peiris, J.S.M.; et al. The M, E, and N structural proteins of the severe acute respiratory syndrome Coronavirus are required for efficient assembly, trafficking, and release of virus-like particles. J. Virol. 2008, 82, 11318–11330. [Google Scholar] [CrossRef]
- Boson, B.; Legros, V.; Zhou, B.; Siret, E.; Mathieu, C.; Cosset, F.-L.; Lavillette, D.; Denolly, S. The SARS-CoV-2 Envelope and membrane proteins modulate maturation and retention of the Spike protein, allowing assembly of virus-like particles. J. Biol. Chem. 2020, 296, 100111. [Google Scholar] [CrossRef]
- Nieva, J.L.; Madan, V.; Carrasco, L. Viroporins: Structure and biological functions. Nat. Rev. Microbiol. 2012, 10, 563–574. [Google Scholar] [CrossRef]
- Castaño-Rodriguez, C.; Honrubia, J.M.; Gutiérrez-Álvarez, J.; DeDiego, M.L.; Nieto-Torres, J.L.; Jimenez-Guardeño, J.M.; Regla-Nava, J.A.; Fernandez-Delgado, R.; Verdia-Báguena, C.; Queralt-Martín, M.; et al. Role of severe acute respiratory syndrome Coronavirus Viroporins E, 3a, and 8a in replication and pathogenesis. MBio 2018, 9, e02325-17. [Google Scholar] [CrossRef] [PubMed]
- Yue, Y.; Nabar, N.R.; Shi, C.-S.; Kamenyeva, O.; Xiao, X.; Hwang, I.-Y.; Wang, M.; Kehrl, J.H. SARS-Coronavirus open reading Frame-3a drives multimodal necrotic cell death. Cell Death Dis. 2018, 9, 904. [Google Scholar] [CrossRef] [PubMed]
- Kern, D.M.; Sorum, B.; Hoel, C.M.; Sridharan, S.; Remis, J.P.; Toso, D.B.; Brohawn, S.G. Cryo-EM structure of the SARS-CoV-2 3a ion channel in lipid nanodiscs. bioRxiv 2020. [Google Scholar] [CrossRef]
- Cohen, J.R.; Lin, L.D.; Machamer, C.E. Identification of a Golgi complex-targeting signal in the cytoplasmic tail of the severe acute respiratory syndrome Coronavirus envelope protein. J. Virol. 2011, 85, 5794–5803. [Google Scholar] [CrossRef] [PubMed]
- Cabrera-Garcia, D.; Bekdash, R.; Abbott, G.W.; Yazawa, M.; Harrison, N.L. The envelope protein of SARS-CoV-2 increases intra-Golgi pH and forms a cation channel that is regulated by pH. J. Physiol. 2021. [Google Scholar] [CrossRef] [PubMed]
- Yuan, X.; Wu, J.; Shan, Y.; Yao, Z.; Dong, B.; Chen, B.; Zhao, Z.; Wang, S.; Chen, J.; Cong, Y. SARS Coronavirus 7a protein blocks cell cycle progression at G0/G1 phase via the cyclin D3/pRb pathway. Virology 2006, 346, 74–85. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, A.K.; Blanco, M.R.; Bruce, E.A.; Honson, D.D.; Chen, L.M.; Chow, A.; Bhat, P.; Ollikainen, N.; Quinodoz, S.A.; Loney, C.; et al. SARS-CoV-2 disrupts splicing, translation, and protein trafficking to suppress host defenses. Cell 2020, 183, 1325–1339.e21. [Google Scholar] [CrossRef] [PubMed]
- Ferrer, M.; Henriet, S.; Chamontin, C.; Lainé, S.; Mougel, M. From cells to virus particles: Quantitative methods to monitor RNA packaging. Viruses 2016, 8, 239. [Google Scholar] [CrossRef]
- Lu, S.; Ye, Q.; Singh, D.; Cao, Y.; Diedrich, J.K.; Yates, J.R., 3rd; Villa, E.; Cleveland, D.W.; Corbett, K.D. The SARS-CoV-2 nucleocapsid phosphoprotein forms mutually exclusive condensates with RNA and the membrane-associated M Protein. Nat. Commun. 2021, 12, 502. [Google Scholar] [CrossRef]
- van den Worm, S.H.E.; Knoops, K.; Zevenhoven-Dobbe, J.C.; Beugeling, C.; van der Meer, Y.; Mommaas, A.M.; Snijder, E.J. Development and RNA-synthesizing activity of Coronavirus replication structures in the absence of protein synthesis. J. Virol. 2011, 85, 5669–5673. [Google Scholar] [CrossRef]
- Von Stillfried, S.; Boor, P. Detection methods for SARS-CoV-2 in tissue. Pathologe 2021. [Google Scholar] [CrossRef]
- Rensen, E.; Pietropaoli, S.; Mueller, F.; Weber, C.; Souquere, S.; Isnard, P.; Rabant, M.; Gibier, J.-B.; Simon-Loriere, E.; Rameix-Welti, M.-A.; et al. Sensitive visualization of SARS-CoV-2 RNA with CoronaFISH. bioRxiv 2021. [Google Scholar] [CrossRef]
- Liu, J.; Babka, A.M.; Kearney, B.J.; Radoshitzky, S.R.; Kuhn, J.H.; Zeng, X. Molecular detection of SARS-CoV-2 in formalin-fixed, paraffin-embedded specimens. JCI Insight 2020, 5, e139042. [Google Scholar] [CrossRef]
- Kula-Pacurar, A.; Wadas, J.; Suder, A.; Szczepanski, A.; Milewska, A.; Ochman, M.; Stacel, T.; Pyrc, K. Visualization of SARS-CoV-2 using immuno RNA-fluorescence in situ hybridization. J. Vis. Exp. 2020. [Google Scholar] [CrossRef]
- Ogando, N.S.; Dalebout, T.J.; Zevenhoven-Dobbe, J.C.; Limpens, R.W.A.L.; van der Meer, Y.; Caly, L.; Druce, J.; de Vries, J.J.C.; Kikkert, M.; Bárcena, M.; et al. SARS-Coronavirus-2 replication in Vero E6 Cells: Replication kinetics, rapid adaptation and cytopathology. J. Gen. Virol. 2020, 101, 925–940. [Google Scholar] [CrossRef]
- Gorshkov, K.; Susumu, K.; Chen, J.; Xu, M.; Pradhan, M.; Zhu, W.; Hu, X.; Breger, J.C.; Wolak, M.; Oh, E. Quantum dot-conjugated SARS-CoV-2 Spike pseudo-virions enable tracking of angiotensin converting enzyme 2 binding and endocytosis. ACS Nano 2020, 14, 12234–12247. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Huang, W.-C.; Yang, C.-S.; Cheng, F.-J.; Chiu, Y.-F.; Chen, H.-F.; Huynh, T.K.; Huang, C.-F.; Chen, C.-H.; Wang, H.-C.; et al. Screening strategy of TMPRSS2 inhibitors by FRET-based enzymatic activity for TMPRSS2-based cancer and COVID-19 treatment. Am. J. Cancer Res. 2021, 11, 827–836. [Google Scholar]
- Froggatt, H.M.; Heaton, B.E.; Heaton, N.S. Development of a fluorescence-based, high-throughput SARS-CoV-2 3CLpro reporter assay. J. Virol. 2020, 94, e01265-20. [Google Scholar] [CrossRef]
- Xia, Z.; Sacco, M.D.; Ma, C.; Townsend, J.A.; Kitamura, N.; Hu, Y.; Ba, M.; Szeto, T.; Zhang, X.; Meng, X.; et al. Discovery of SARS-CoV-2 papain-like protease inhibitors through a combination of high-throughput screening and FlipGFP-based reporter assay. bioRxiv 2021. [Google Scholar] [CrossRef]
- Resnick, S.J.; Iketani, S.; Hong, S.J.; Zask, A.; Liu, H.; Kim, S.; Melore, S.; Lin, F.-Y.; Nair, M.S.; Huang, Y.; et al. Inhibitors of Coronavirus 3CL proteases protect cells from protease-mediated cytotoxicity. J. Virol. 2021, JVI-02374. [Google Scholar] [CrossRef]
- Shiaelis, N.; Tometzki, A.; Peto, L.; McMahon, A.; Hepp, C.; Bickerton, E.; Favard, C.; Muriaux, D.; Andersson, M.; Oakley, S.; et al. Virus detection and identification in minutes using single-particle imaging and deep learning. bioRxiv 2020. [Google Scholar] [CrossRef]
- Chiem, K.; Morales Vasquez, D.; Park, J.-G.; Platt, R.N.; Anderson, T.; Walter, M.R.; Kobie, J.J.; Ye, C.; Martinez-Sobrido, L. Generation and characterization of recombinant SARS-CoV-2 expressing reporter genes. J. Virol. 2021, 95, e02209-20. [Google Scholar] [CrossRef]
- Plescia, C.B.; David, E.A.; Patra, D.; Sengupta, R.; Amiar, S.; Su, Y.; Stahelin, R.V. SARS-CoV-2 Viral budding and entry can be modeled using BSL-2 level virus-like particles. J. Biol. Chem. 2020, 296, 100103. [Google Scholar] [CrossRef]
- Hou, Y.J.; Okuda, K.; Edwards, C.E.; Martinez, D.R.; Asakura, T.; Dinnon, K.H., 3rd; Kato, T.; Lee, R.E.; Yount, B.L.; Mascenik, T.M.; et al. SARS-CoV-2 reverse genetics reveals a variable infection gradient in the respiratory tract. Cell 2020, 182, 429–446.e14. [Google Scholar] [CrossRef] [PubMed]
- Xie, X.; Muruato, A.; Lokugamage, K.G.; Narayanan, K.; Zhang, X.; Zou, J.; Liu, J.; Schindewolf, C.; Bopp, N.E.; Aguilar, P.V.; et al. An infectious cDNA clone of SARS-CoV-2. Cell Host Microbe 2020, 27, 841–848.e3. [Google Scholar] [CrossRef]
- Sims, A.C.; Baric, R.S.; Yount, B.; Burkett, S.E.; Collins, P.L.; Pickles, R.J. Severe acute respiratory syndrome Coronavirus infection of human ciliated airway epithelia: Role of ciliated cells in viral spread in the conducting airways of the lungs. J. Virol. 2005, 79, 15511–15524. [Google Scholar] [CrossRef] [PubMed]
- Xie, X.; Muruato, A.E.; Zhang, X.; Lokugamage, K.G.; Fontes-Garfias, C.R.; Zou, J.; Liu, J.; Ren, P.; Balakrishnan, M.; Cihlar, T.; et al. A nanoluciferase SARS-CoV-2 for rapid neutralization testing and screening of anti-infective drugs for COVID-19. Nat. Commun. 2020, 11, 5214. [Google Scholar] [CrossRef] [PubMed]
- Thi Nhu Thao, T.; Labroussaa, F.; Ebert, N.; V’kovski, P.; Stalder, H.; Portmann, J.; Kelly, J.; Steiner, S.; Holwerda, M.; Kratzel, A.; et al. Rapid reconstruction of SARS-CoV-2 using a synthetic genomics platform. Nature 2020, 582, 561–565. [Google Scholar] [CrossRef] [PubMed]
- Perfilov, M.M.; Gurskaya, N.G.; Serebrovskaya, E.O.; Melnikov, P.A.; Kharitonov, S.L.; Lewis, T.R.; Arshavsky, V.Y.; Baklaushev, V.P.; Mishin, A.S.; Lukyanov, K.A. Highly photostable fluorescent labeling of proteins in live cells using exchangeable coiled coils heterodimerization. Cell. Mol. Life Sci. 2020, 77, 4429–4440. [Google Scholar] [CrossRef]
- Kumagai, A.; Ando, R.; Miyatake, H.; Greimel, P.; Kobayashi, T.; Hirabayashi, Y.; Shimogori, T.; Miyawaki, A. A Bilirubin-inducible fluorescent protein from eel muscle. Cell 2013, 153, 1602–1611. [Google Scholar] [CrossRef]
- Bozhanova, N.G.; Baranov, M.S.; Klementieva, N.V.; Sarkisyan, K.S.; Gavrikov, A.S.; Yampolsky, I.V.; Zagaynova, E.V.; Lukyanov, S.A.; Lukyanov, K.A.; Mishin, A.S. Protein labeling for live cell fluorescence microscopy with a highly photostable renewable signal. Chem. Sci. 2017, 8, 7138–7142. [Google Scholar] [CrossRef]
- Muslinkina, L.; Gavrikov, A.S.; Bozhanova, N.G.; Mishin, A.S.; Baranov, M.S.; Meiler, J.; Pletneva, N.V.; Pletnev, V.Z.; Pletnev, S. Structure-based rational design of two enhanced bacterial lipocalin Blc tags for protein-PAINT super-resolution microscopy. ACS Chem. Biol. 2020, 15, 2456–2465. [Google Scholar] [CrossRef]
- Dolgosheina, E.V.; Jeng, S.C.Y.; Panchapakesan, S.S.S.; Cojocaru, R.; Chen, P.S.K.; Wilson, P.D.; Hawkins, N.; Wiggins, P.A.; Unrau, P.J. RNA mango aptamer-fluorophore: A bright, high-affinity complex for RNA labeling and tracking. ACS Chem. Biol. 2014, 9, 2412–2420. [Google Scholar] [CrossRef]
- Li, X.; Kim, H.; Litke, J.L.; Wu, J.; Jaffrey, S.R. Fluorophore-promoted RNA folding and photostability enables imaging of single broccoli-tagged mRNAs in live mammalian cells. Angew. Chem. Int. Ed. Engl. 2020, 59, 4511–4518. [Google Scholar] [CrossRef] [PubMed]
- Cawte, A.D.; Unrau, P.J.; Rueda, D.S. Live cell imaging of single RNA molecules with fluorogenic mango II arrays. Nat. Commun. 2020, 11, 1283. [Google Scholar] [CrossRef]
- Wu, J.; Zaccara, S.; Khuperkar, D.; Kim, H.; Tanenbaum, M.E.; Jaffrey, S.R. Live imaging of mRNA using RNA-stabilized fluorogenic proteins. Nat. Methods 2019, 16, 862–865. [Google Scholar] [CrossRef] [PubMed]
- Nasu, Y.; Shen, Y.; Kramer, L.; Campbell, R.E. Structure- and mechanism-guided design of single fluorescent protein-based biosensors. Nat. Chem. Biol. 2021, 17, 509–518. [Google Scholar] [CrossRef]
- Bilan, D.S.; Belousov, V.V. HyPer family probes: State of the art. Antioxid. Redox Signal. 2016, 24, 731–751. [Google Scholar] [CrossRef] [PubMed]
- Pak, V.V.; Ezeriņa, D.; Lyublinskaya, O.G.; Pedre, B.; Tyurin-Kuzmin, P.A.; Mishina, N.M.; Thauvin, M.; Young, D.; Wahni, K.; Gache, S.A.M.; et al. Ultrasensitive genetically encoded indicator for hydrogen peroxide identifies roles for the oxidant in cell migration and mitochondrial function. Cell Metab. 2020, 31, 642–653.e6. [Google Scholar] [CrossRef]
- Siu, G.K.Y.; Zhou, F.; Yu, M.K.; Zhang, L.; Wang, T.; Liang, Y.; Chen, Y.; Chan, H.C.; Yu, S. Hepatitis C Virus NS5A Protein cooperates with phosphatidylinositol 4-kinase IIIα to induce mitochondrial fragmentation. Sci. Rep. 2016, 6, 23464. [Google Scholar] [CrossRef]
- Hammond, G.R.V.; Balla, T. Polyphosphoinositide binding domains: Key to inositol lipid biology. Biochim. Biophys. Acta 2015, 1851, 746–758. [Google Scholar] [CrossRef]
- Liu, Z.; Pouli, D.; Alonzo, C.A.; Varone, A.; Karaliota, S.; Quinn, K.P.; Münger, K.; Karalis, K.P.; Georgakoudi, I. Mapping metabolic changes by noninvasive, multiparametric, high-resolution imaging using endogenous contrast. Sci. Adv. 2018, 4, eaap9302. [Google Scholar] [CrossRef]
- Melia, C.E.; Peddie, C.J.; de Jong, A.W.M.; Snijder, E.J.; Collinson, L.M.; Koster, A.J.; van der Schaar, H.M.; van Kuppeveld, F.J.M.; Bárcena, M. Origins of enterovirus replication organelles established by whole-cell electron microscopy. MBio 2019, 10, e00951-19. [Google Scholar] [CrossRef]
- Westberg, M.; Bregnhøj, M.; Etzerodt, M.; Ogilby, P.R. No photon wasted: An efficient and selective singlet oxygen photosensitizing protein. J. Phys. Chem. B 2017, 121, 9366–9371. [Google Scholar] [CrossRef]
- Shu, X.; Lev-Ram, V.; Deerinck, T.J.; Qi, Y.; Ramko, E.B.; Davidson, M.W.; Jin, Y.; Ellisman, M.H.; Tsien, R.Y. A genetically encoded tag for correlated light and electron microscopy of intact cells, tissues, and organisms. PLoS Biol. 2011, 9, e1001041. [Google Scholar] [CrossRef] [PubMed]
- Mishin, A.S.; Lukyanov, K.A. Live-cell super-resolution fluorescence microscopy. Biochemistry 2019, 84, S19–S31. [Google Scholar] [CrossRef]
- Jacquemet, G.; Carisey, A.F.; Hamidi, H.; Henriques, R.; Leterrier, C. The cell biologist’s guide to super-resolution microscopy. J. Cell Sci. 2020, 133, jcs240713. [Google Scholar] [CrossRef]
- Balzarotti, F.; Eilers, Y.; Gwosch, K.C.; Gynnå, A.H.; Westphal, V.; Stefani, F.D.; Elf, J.; Hell, S.W. Nanometer resolution imaging and tracking of fluorescent molecules with minimal photon fluxes. Science 2017, 355, 606–612. [Google Scholar] [CrossRef]
- Gwosch, K.C.; Pape, J.K.; Balzarotti, F.; Hoess, P.; Ellenberg, J.; Ries, J.; Hell, S.W. MINFLUX nanoscopy delivers 3D multicolor nanometer resolution in cells. Nat. Methods 2020, 17, 217–224. [Google Scholar] [CrossRef] [PubMed]
- Arista-Romero, M.; Pujals, S.; Albertazzi, L. Towards a quantitative single particle characterization by super resolution microscopy: From virus structures to antivirals design. Front. Bioeng. Biotechnol. 2021, 9, 647874. [Google Scholar] [CrossRef] [PubMed]
- Stebbing, J.; Sánchez Nievas, G.; Falcone, M.; Youhanna, S.; Richardson, P.; Ottaviani, S.; Shen, J.X.; Sommerauer, C.; Tiseo, G.; Ghiadoni, L.; et al. JAK Inhibition reduces SARS-CoV-2 liver infectivity and modulates inflammatory responses to reduce morbidity and mortality. Sci. Adv. 2021, 7, eabe4724. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Tillberg, P.W.; Boyden, E.S. Optical imaging expansion microscopy. Science 2015, 347, 543–548. [Google Scholar] [CrossRef] [PubMed]
- Wassie, A.T.; Zhao, Y.; Boyden, E.S. Expansion microscopy: Principles and uses in biological research. Nat. Methods 2019, 16, 33–41. [Google Scholar] [CrossRef]
- Wang, Y.; Yu, Z.; Cahoon, C.K.; Parmely, T.; Thomas, N.; Unruh, J.R.; Slaughter, B.D.; Hawley, R.S. Combined expansion microscopy with structured illumination microscopy for analyzing protein complexes. Nat. Protoc. 2018, 13, 1869–1895. [Google Scholar] [CrossRef] [PubMed]
- Gao, M.; Maraspini, R.; Beutel, O.; Zehtabian, A.; Eickholt, B.; Honigmann, A.; Ewers, H. Expansion stimulated emission depletion microscopy (ExSTED). ACS Nano 2018, 12, 4178–4185. [Google Scholar] [CrossRef] [PubMed]
- Zwettler, F.U.; Reinhard, S.; Gambarotto, D.; Bell, T.D.M.; Hamel, V.; Guichard, P.; Sauer, M. Molecular resolution imaging by post-labeling expansion single-molecule localization microscopy (Ex-SMLM). Nat. Commun. 2020, 11, 3388. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.S.; Santiago, G.T.; Alvarez, M.M.; Schiff, S.J.; Boyden, E.S.; Khademhosseini, A. Expansion mini-microscopy: An enabling alternative in point-of-care diagnostics. Curr. Opin. Biomed. Eng. 2017, 1, 45–53. [Google Scholar] [CrossRef] [PubMed]
- Gao, R.; Yu, C.-C.J.; Gao, L.; Piatkevich, K.D.; Neve, R.L.; Munro, J.B.; Upadhyayula, S.; Boyden, E.S. A Highly homogeneous polymer composed of tetrahedron-like monomers for high-isotropy expansion microscopy. Nat. Nanotechnol. 2021, 16, 698–707. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Applications | Probes | Imaging Methods | References |
|---|---|---|---|
| Viral entry in cell lines | ACE2 fused with FP | Confocal and epifluorescence microscopy | [16,17,18,20,22] |
| VSV fused with FP | Spinning disk confocal microscopy, flow cytometry | [21] | |
| Cell filaments fused with FP | Confocal and TIRF microscopy | [22] | |
| Immunofluorescence | Confocal microscopy | [16,17,22] | |
| FD-curve-based AFM guided by fluorescence | Atomic force microscopy, fluorescence microscopy | [18] | |
| Viral entry in tissues | Immunofluorescence | Confocal microscopy | [23,24,25] |
| Immunofluorescence | Light-sheet microscopy, confocal microscopy | [26] | |
| Endocytosis of virus | Immunofluorescence | Confocal microscopy | [27,29,31] |
| ACE2 fused with FP (pseudoviruses) | Confocal microscopy | [28,29] | |
| TPC1 and TPS2 fused with FP | Spinning disk confocal microscopy | [31] | |
| Viral RNA release | 3a/nsp3 fused with FP | Confocal microscopy, cryo-EM | [32,34] |
| S, M, N fused with FP | Wide-field and confocal microscopy | [35,46] | |
| Staining with fluorescent dyes | Confocal microscopy | [32] | |
| Immunofluorescence | Confocal microscopy | [33,36] | |
| Influence of viral proteins on the host cell | Immunofluorescence | Confocal microscopy | [38,41,44] |
| 3a, E, 7a, nsp1, Rip3, Gal3, TFEB and LAMP1 fused with FP | 3a—confocal microscopy and cryo-EM; the rest—confocal microscopy | [39,40,42,43,44] | |
| RNA tracking | N protein with Cy5, ssRNA with 6-FAM | Confocal microscopy, photobleaching | [46] |
| FISH, immunofluorescence | Confocal microscopy | [46,47,48,49,50,51] | |
| SARS-CoV-2 drug screening and inhibitor testing | Immunofluorescence | Fluorescence microscopy | [52] |
| Quantum dots | Single-molecule imaging | [53] | |
| FRET-based reporter for TMPRSS2 detection | Microplate reader | [54] | |
| FlipGFP-based reporters for 3CLpro or PLpro detection | Fluorescence microscopy | [55,56] | |
| Fluorescence dye labeling | TIRF microscopy | [58] | |
| Recombinant SARS-CoV-2 expressing reporter genes | S, Rab5, PTS1, LAMP1 tagged with FP | Confocal microscopy | [60] |
| SARS-CoV-2 tagged with an FP | Confocal microscopy | [59,61,62,65] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Putlyaeva, L.V.; Lukyanov, K.A. Studying SARS-CoV-2 with Fluorescence Microscopy. Int. J. Mol. Sci. 2021, 22, 6558. https://doi.org/10.3390/ijms22126558
Putlyaeva LV, Lukyanov KA. Studying SARS-CoV-2 with Fluorescence Microscopy. International Journal of Molecular Sciences. 2021; 22(12):6558. https://doi.org/10.3390/ijms22126558
Chicago/Turabian StylePutlyaeva, Lidia V., and Konstantin A. Lukyanov. 2021. "Studying SARS-CoV-2 with Fluorescence Microscopy" International Journal of Molecular Sciences 22, no. 12: 6558. https://doi.org/10.3390/ijms22126558
APA StylePutlyaeva, L. V., & Lukyanov, K. A. (2021). Studying SARS-CoV-2 with Fluorescence Microscopy. International Journal of Molecular Sciences, 22(12), 6558. https://doi.org/10.3390/ijms22126558