
Anakinra Activates Superoxide Dismutase 2 to Mitigate Inflammasome Activity

 , , ,
, , ,  , , , and
, , , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Anakinra Prevents Mitochondrial Oxidative Stress via PGC1alpha and SOD2
2.2. Anakinra Prevents SOD2 Degradation via USP36-COPS3
2.3. Anakinra and SOD2 Ameliorates the Oxidative Inflammatory Pathology In Vivo
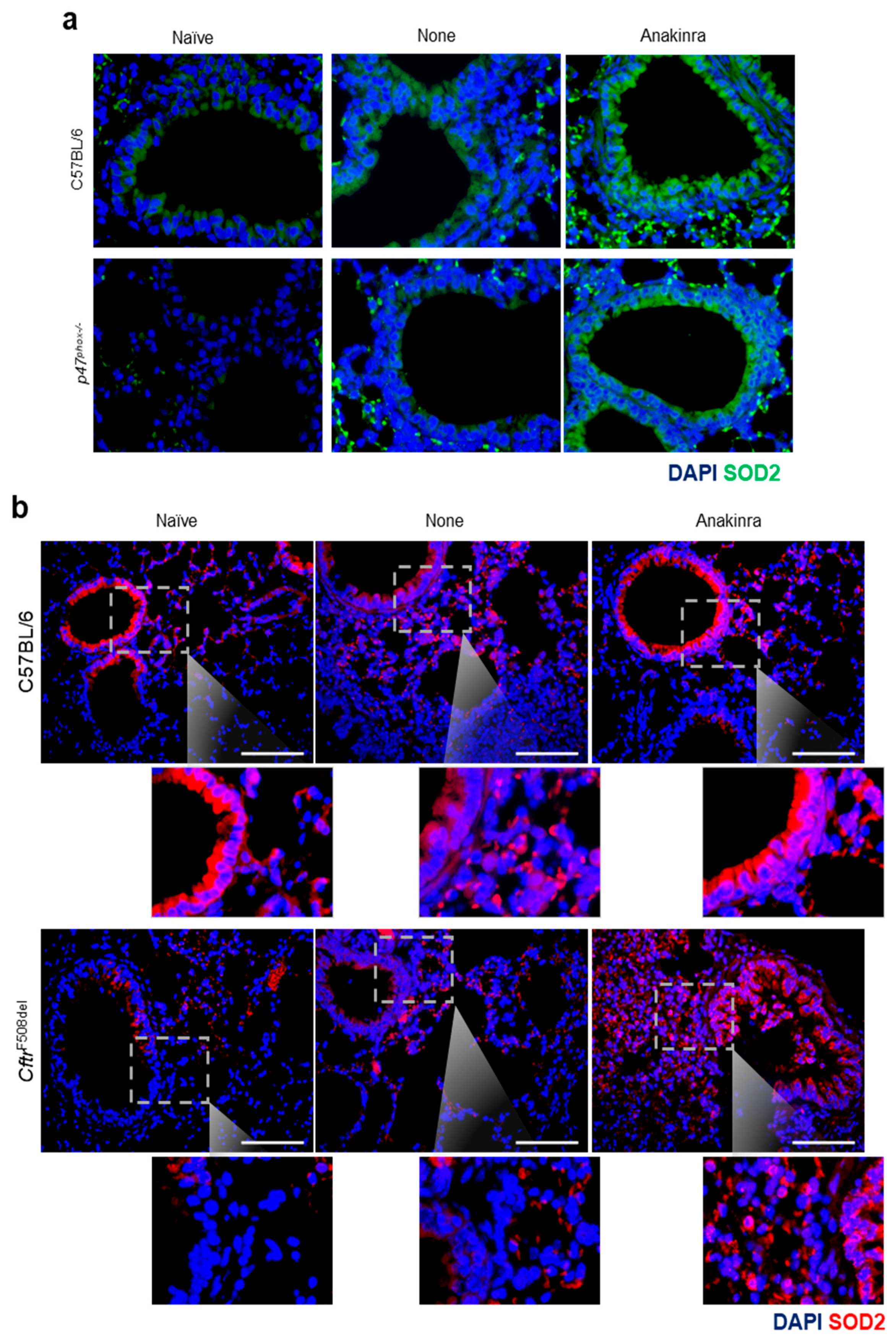
2.4. Anakinra Activates SOD2 in Murine CGD and CF
3. Discussion
4. Materials and Methods
4.1. Cells
4.2. Transcription Factors Activation Profiling Analysis
4.3. Scanning Electron Microscopy
4.4. siRNA Design and Delivery
4.5. Western Blot Analysis and Immunoprecipitation
4.6. Catalase Activity
4.7. ELISA and Real-Time PCR
4.8. Mice, Infections and Treatments
4.9. Immunofluorescence
4.10. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| CGD | chronic granulomatous disease |
| COP9 | Constitutive photomorphogenesis 9 |
| COPS3 | COP9 Signalosome Subunit 3 |
| CF | Cystic fibrosis |
| CHX | Cycloheximide |
| IL-1 | Interleukin-1 |
| IL-1Ra | IL-1 receptor antagonist |
| IL1R1 | IL-1 receptor 1 |
| IP | Immunoprecipitation |
| mtROS | mitochondrial reactive oxidative species |
| NLRP3 | NLR family pyrin domain containing 3 |
| PGC-1α | PPAR Gamma Coactivator-1alpha |
| PPAR | peroxisome proliferation-activated receptor |
| ROS | reactive oxidative species |
| SIRT3 | Sirtuin 3 |
| SOD2 | superoxide dismutase |
| TXN-2 | Thioredoxin-2 |
| USP36 | Ubiquitin Specific Peptidase 36 |
| WCL | Whole Cell Lysate |
References
- Evavold, C.L.; Kagan, J.C. Inflammasomes: Threat-Assessment Organelles of the Innate Immune System. Immunity 2019, 51, 609–624. [Google Scholar] [CrossRef] [PubMed]
- Liston, A.; Masters, S.L. Homeostasis-altering molecular processes as mechanisms of inflammasome activation. Nat. Rev. Immunol. 2017, 17, 208–214. [Google Scholar] [CrossRef] [PubMed]
- Mangan, M.S.J.; Olhava, E.J.; Roush, W.R.; Seidel, H.M.; Glick, G.D.; Latz, E. Targeting the NLRP3 inflammasome in inflammatory diseases. Nat. Rev. Drug Discov. 2018, 17, 588–606. [Google Scholar] [CrossRef] [PubMed]
- Swanson, K.V.; Deng, M.; Ting, J.P. The NLRP3 inflammasome: Molecular activation and regulation to therapeutics. Nat. Rev. Immunol. 2019, 19, 477–489. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Dinarello, C.A.; Molgora, M.; Garlanda, C. Interleukin-1 and Related Cytokines in the Regulation of Inflammation and Immunity. Immunity 2019, 50, 778–795. [Google Scholar] [CrossRef] [PubMed]
- Dinarello, C.A.; Simon, A.; van der Meer, J.W. Treating inflammation by blocking interleukin-1 in a broad spectrum of diseases. Nat. Rev. Drug Discov. 2012, 11, 633–652. [Google Scholar] [CrossRef]
- Cavalli, G.; Dinarello, C.A. Anakinra Therapy for Non-cancer Inflammatory Diseases. Front. Pharmacol. 2018, 9, 1157. [Google Scholar] [CrossRef]
- de Luca, A.; Smeekens, S.P.; Casagrande, A.; Iannitti, R.; Conway, K.L.; Gresnigt, M.S.; Begun, J.; Plantinga, T.S.; Joosten, L.A.; van der Meer, J.W.; et al. IL-1 receptor blockade restores autophagy and reduces inflammation in chronic granulomatous disease in mice and in humans. Proc. Natl. Acad. Sci. USA 2014, 111, 3526–3531. [Google Scholar] [CrossRef]
- Iannitti, R.G.; Napolioni, V.; Oikonomou, V.; De Luca, A.; Galosi, C.; Pariano, M.; Massi-Benedetti, C.; Borghi, M.; Puccetti, M.; Lucidi, V.; et al. IL-1 receptor antagonist ameliorates inflammasome-dependent inflammation in murine and human cystic fibrosis. Nat. Commun. 2016, 7, 10791. [Google Scholar] [CrossRef]
- Cao, Z.; Wang, Y.; Long, Z.; He, G. Interaction between autophagy and the NLRP3 inflammasome. Acta Biochim. Biophys. Sin. (Shanghai) 2019, 51, 1087–1095. [Google Scholar] [CrossRef] [PubMed]
- Dubois, V.; Eeckhoute, J.; Lefebvre, P.; Staels, B. Distinct but complementary contributions of PPAR isotypes to energy homeostasis. J. Clin. Investig. 2017, 127, 1202–1214. [Google Scholar] [CrossRef] [PubMed]
- Rius-Perez, S.; Torres-Cuevas, I.; Millan, I.; Ortega, A.L.; Perez, S. PGC-1alpha, Inflammation, and Oxidative Stress: An Integrative View in Metabolism. Oxid. Med. Cell Longev. 2020, 2020, 1452696. [Google Scholar] [CrossRef]
- Kim, M.S.; Ramakrishna, S.; Lim, K.H.; Kim, J.H.; Baek, K.H. Protein stability of mitochondrial superoxide dismutase SOD2 is regulated by USP36. J. Cell Biochem. 2011, 112, 498–508. [Google Scholar] [CrossRef]
- Banda, N.K.; Guthridge, C.; Sheppard, D.; Cairns, K.S.; Muggli, M.; Bech-Otschir, D.; Dubiel, W.; Arend, W.P. Intracellular IL-1 receptor antagonist type 1 inhibits IL-1-induced cytokine production in keratinocytes through binding to the third component of the COP9 signalosome. J. Immunol. 2005, 174, 3608–3616. [Google Scholar] [CrossRef]
- Li, N.; Ragheb, K.; Lawler, G.; Sturgis, J.; Rajwa, B.; Melendez, J.A.; Robinson, J.P. Mitochondrial complex I inhibitor rotenone induces apoptosis through enhancing mitochondrial reactive oxygen species production. J. Biol. Chem. 2003, 278, 8516–8525. [Google Scholar] [CrossRef] [PubMed]
- Sundqvist, M.; Christenson, K.; Bjornsdottir, H.; Osla, V.; Karlsson, A.; Dahlgren, C.; Speert, D.P.; Fasth, A.; Brown, K.L.; Bylund, J. Elevated Mitochondrial Reactive Oxygen Species and Cellular Redox Imbalance in Human NADPH-Oxidase-Deficient Phagocytes. Front. Immunol. 2017, 8, 1828. [Google Scholar] [CrossRef] [PubMed]
- Hector, A.; Griese, M.; Hartl, D. Oxidative stress in cystic fibrosis lung disease: An early event, but worth targeting? Eur. Respir. J. 2014, 44, 17–19. [Google Scholar] [CrossRef]
- Favia, M.; de Bari, L.; Bobba, A.; Atlante, A. An Intriguing Involvement of Mitochondria in Cystic Fibrosis. J. Clin. Med. 2019, 8, 1890. [Google Scholar] [CrossRef]
- Seo, Y.S.; Kim, H.S.; Lee, A.Y.; Chun, J.M.; Kim, S.B.; Moon, B.C.; Kwon, B.I. Codonopsis lanceolata attenuates allergic lung inflammation by inhibiting Th2 cell activation and augmenting mitochondrial ROS dismutase (SOD2) expression. Sci. Rep. 2019, 9, 2312. [Google Scholar] [CrossRef]
- Siedlinski, M.; van Diemen, C.C.; Postma, D.S.; Vonk, J.M.; Boezen, H.M. Superoxide dismutases, lung function and bronchial responsiveness in a general population. Eur. Respir. J. 2009, 33, 986–992. [Google Scholar] [CrossRef]
- He, C.; Hart, P.C.; Germain, D.; Bonini, M.G. SOD2 and the Mitochondrial UPR: Partners Regulating Cellular Phenotypic Transitions. Trends Biochem. Sci. 2016, 41, 568–577. [Google Scholar] [CrossRef]
- Song, J.Q.; Jiang, L.Y.; Fu, C.P.; Wu, X.; Liu, Z.L.; Xie, L.; Wu, X.D.; Hao, S.Y.; Li, S.Q. Heterozygous SOD2 deletion deteriorated chronic intermittent hypoxia-induced lung inflammation and vascular remodeling through mtROS-NLRP3 signaling pathway. Acta Pharmacol. Sin. 2020, 41, 1197–1207. [Google Scholar] [CrossRef]
- Traba, J.; Geiger, S.S.; Kwarteng-Siaw, M.; Han, K.; Ra, O.H.; Siegel, R.M.; Gius, D.; Sack, M.N. Prolonged fasting suppresses mitochondrial NLRP3 inflammasome assembly and activation via SIRT3-mediated activation of superoxide dismutase 2. J. Biol. Chem. 2017, 292, 12153–12164. [Google Scholar] [CrossRef] [PubMed]
- Hart, P.C.; Mao, M.; de Abreu, A.L.; Ansenberger-Fricano, K.; Ekoue, D.N.; Ganini, D.; Kajdacsy-Balla, A.; Diamond, A.M.; Minshall, R.D.; Consolaro, M.E.; et al. MnSOD upregulation sustains the Warburg effect via mitochondrial ROS and AMPK-dependent signalling in cancer. Nat. Commun. 2015, 6, 6053. [Google Scholar] [CrossRef]
- Tao, R.; Coleman, M.C.; Pennington, J.D.; Ozden, O.; Park, S.H.; Jiang, H.; Kim, H.S.; Flynn, C.R.; Hill, S.; Hayes McDonald, W.; et al. Sirt3-mediated deacetylation of evolutionarily conserved lysine 122 regulates MnSOD activity in response to stress. Mol. Cell 2010, 40, 893–904. [Google Scholar] [CrossRef] [PubMed]
- Shen, X.; Zheng, S.; Metreveli, N.S.; Epstein, P.N. Protection of cardiac mitochondria by overexpression of MnSOD reduces diabetic cardiomyopathy. Diabetes 2006, 55, 798–805. [Google Scholar] [CrossRef]
- Green, D.R.; Galluzzi, L.; Kroemer, G. Mitochondria and the autophagy-inflammation-cell death axis in organismal aging. Science 2011, 333, 1109–1112. [Google Scholar] [CrossRef]
- Liang, H.; Ward, W.F. PGC-1alpha: A key regulator of energy metabolism. Adv. Physiol. Educ. 2006, 30, 145–151. [Google Scholar] [CrossRef]
- Kinnula, V.L.; Crapo, J.D. Superoxide dismutases in the lung and human lung diseases. Am. J. Respir. Crit. Care Med. 2003, 167, 1600–1619. [Google Scholar] [CrossRef]
- Marzetti, E.; Csiszar, A.; Dutta, D.; Balagopal, G.; Calvani, R.; Leeuwenburgh, C. Role of mitochondrial dysfunction and altered autophagy in cardiovascular aging and disease: From mechanisms to therapeutics. Am. J. Physiol. Heart Circ. Physiol. 2013, 305, H459–H476. [Google Scholar] [CrossRef] [PubMed]
- Mathieu, E.; Bernard, A.S.; Delsuc, N.; Quevrain, E.; Gazzah, G.; Lai, B.; Chain, F.; Langella, P.; Bachelet, M.; Masliah, J.; et al. A Cell-Penetrant Manganese Superoxide Dismutase (MnSOD) Mimic Is Able To Complement MnSOD and Exerts an Antiinflammatory Effect on Cellular and Animal Models of Inflammatory Bowel Diseases. Inorg. Chem. 2017, 56, 2545–2555. [Google Scholar] [CrossRef] [PubMed]
- Batinic-Haberle, I.; Tome, M.E. Thiol regulation by Mn porphyrins, commonly known as SOD mimics. Redox Biol. 2019, 25, 101139. [Google Scholar] [CrossRef] [PubMed]
- Elfawy, H.A.; Das, B. Crosstalk between mitochondrial dysfunction, oxidative stress, and age related neurodegenerative disease: Etiologies and therapeutic strategies. Life Sci. 2019, 218, 165–184. [Google Scholar] [CrossRef]
- De Luca, A.; Iannitti, R.G.; Bozza, S.; Beau, R.; Casagrande, A.; D’Angelo, C.; Moretti, S.; Cunha, C.; Giovannini, G.; Massi-Benedetti, C.; et al. CD4(+) T cell vaccination overcomes defective cross-presentation of fungal antigens in a mouse model of chronic granulomatous disease. J. Clin. Investig. 2012, 122, 1816–1831. [Google Scholar] [CrossRef]
- van Doorninck, J.H.; French, P.J.; Verbeek, E.; Peters, R.H.; Morreau, H.; Bijman, J.; Scholte, B.J. A mouse model for the cystic fibrosis delta F508 mutation. EMBO J. 1995, 14, 4403–4411. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pariano, M.; Pieroni, S.; De Luca, A.; Iannitti, R.G.; Borghi, M.; Puccetti, M.; Giovagnoli, S.; Renga, G.; D’Onofrio, F.; Bellet, M.M.; et al. Anakinra Activates Superoxide Dismutase 2 to Mitigate Inflammasome Activity. Int. J. Mol. Sci. 2021, 22, 6531. https://doi.org/10.3390/ijms22126531
Pariano M, Pieroni S, De Luca A, Iannitti RG, Borghi M, Puccetti M, Giovagnoli S, Renga G, D’Onofrio F, Bellet MM, et al. Anakinra Activates Superoxide Dismutase 2 to Mitigate Inflammasome Activity. International Journal of Molecular Sciences. 2021; 22(12):6531. https://doi.org/10.3390/ijms22126531
Chicago/Turabian StylePariano, Marilena, Stefania Pieroni, Antonella De Luca, Rossana G. Iannitti, Monica Borghi, Matteo Puccetti, Stefano Giovagnoli, Giorgia Renga, Fiorella D’Onofrio, Marina M. Bellet, and et al. 2021. "Anakinra Activates Superoxide Dismutase 2 to Mitigate Inflammasome Activity" International Journal of Molecular Sciences 22, no. 12: 6531. https://doi.org/10.3390/ijms22126531
APA StylePariano, M., Pieroni, S., De Luca, A., Iannitti, R. G., Borghi, M., Puccetti, M., Giovagnoli, S., Renga, G., D’Onofrio, F., Bellet, M. M., Stincardini, C., Della-Fazia, M. A., Servillo, G., van de Veerdonk, F. L., Costantini, C., & Romani, L. (2021). Anakinra Activates Superoxide Dismutase 2 to Mitigate Inflammasome Activity. International Journal of Molecular Sciences, 22(12), 6531. https://doi.org/10.3390/ijms22126531