
Regulation of Caspase-8 Activity at the Crossroads of Pro-Inflammation and Anti-Inflammation


Abstract
1. Introduction
2. Anti-Inflammatory Functions of Caspase-8
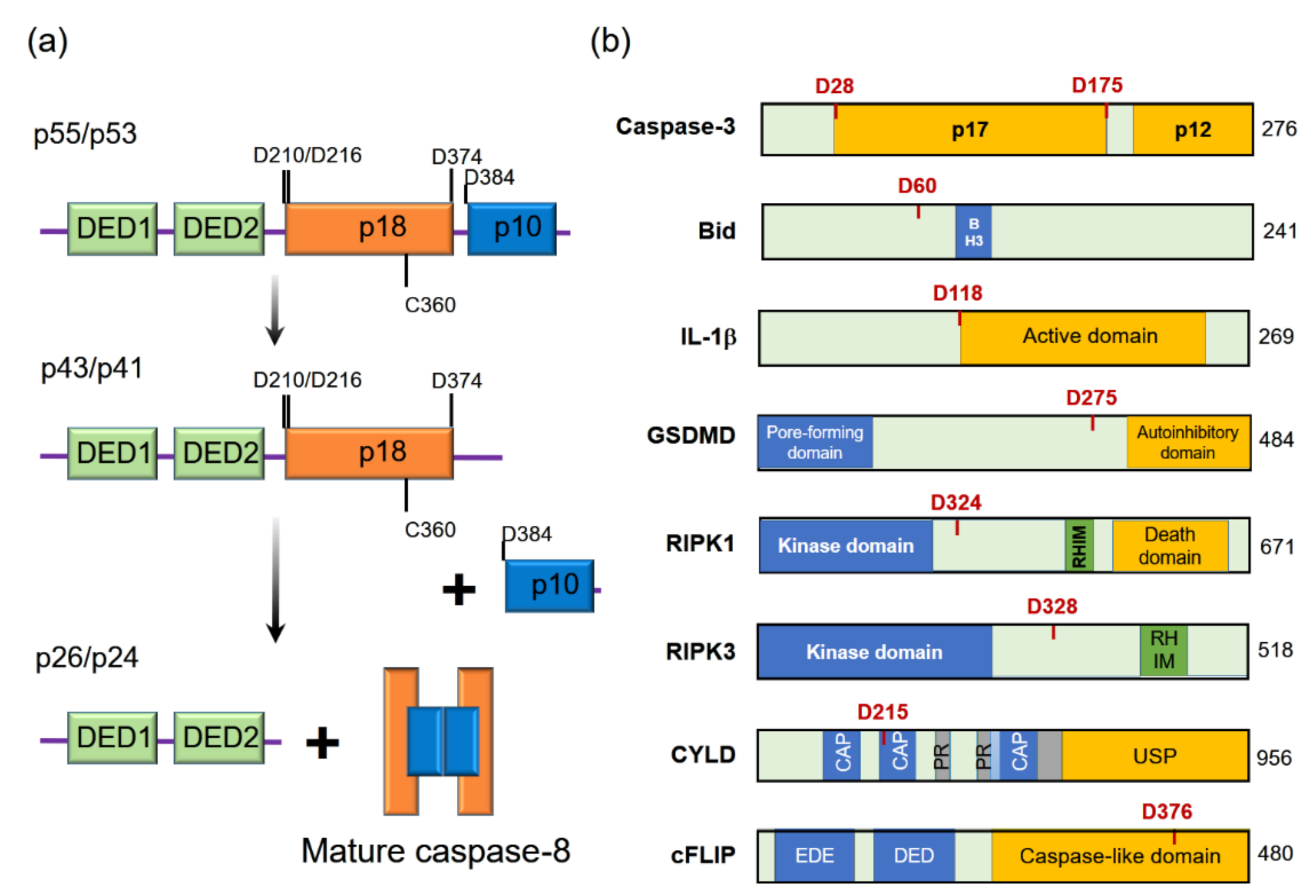
2.1. Induction of Apoptosis
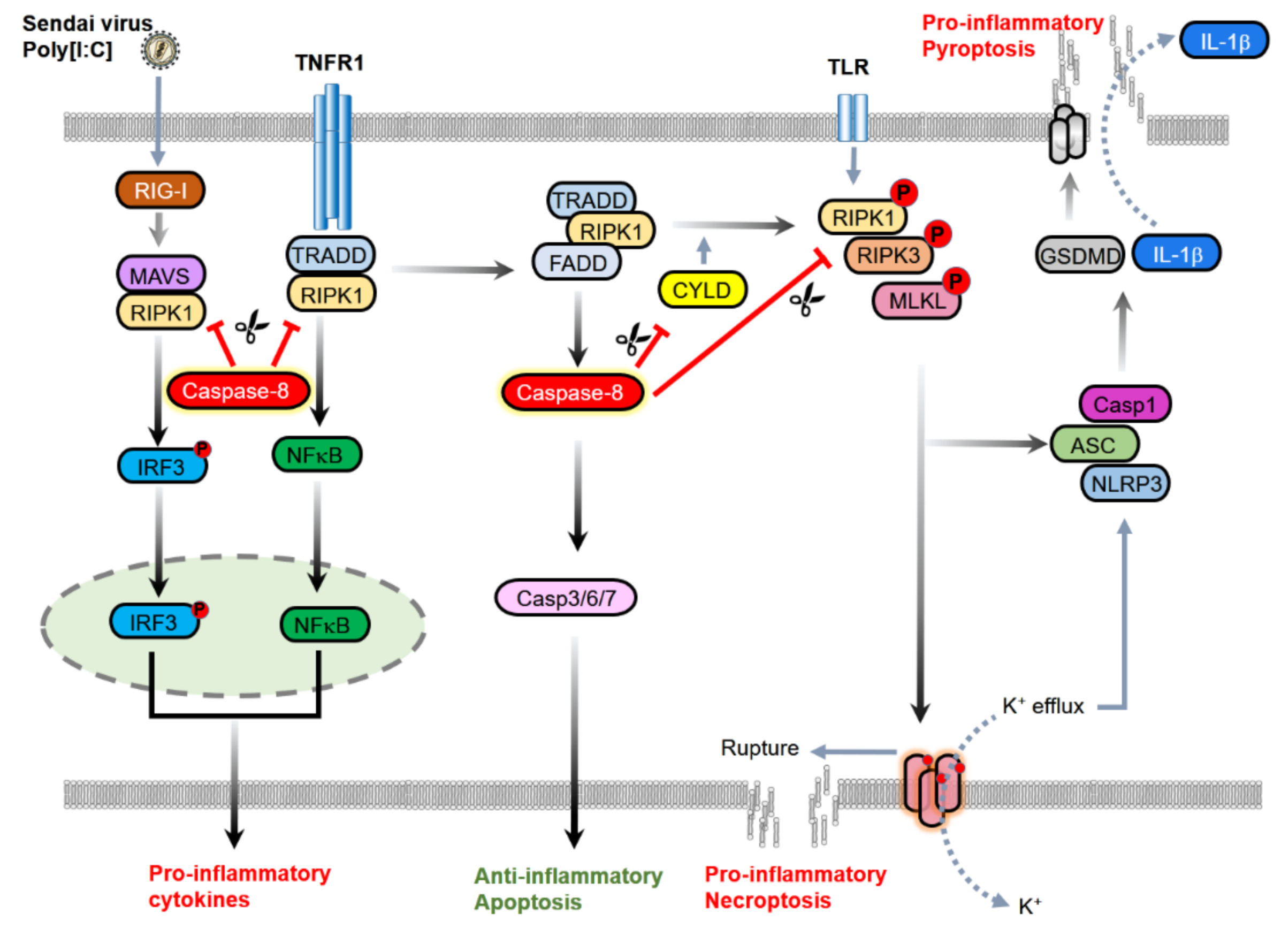
2.2. The Inhibition of Necroptosis
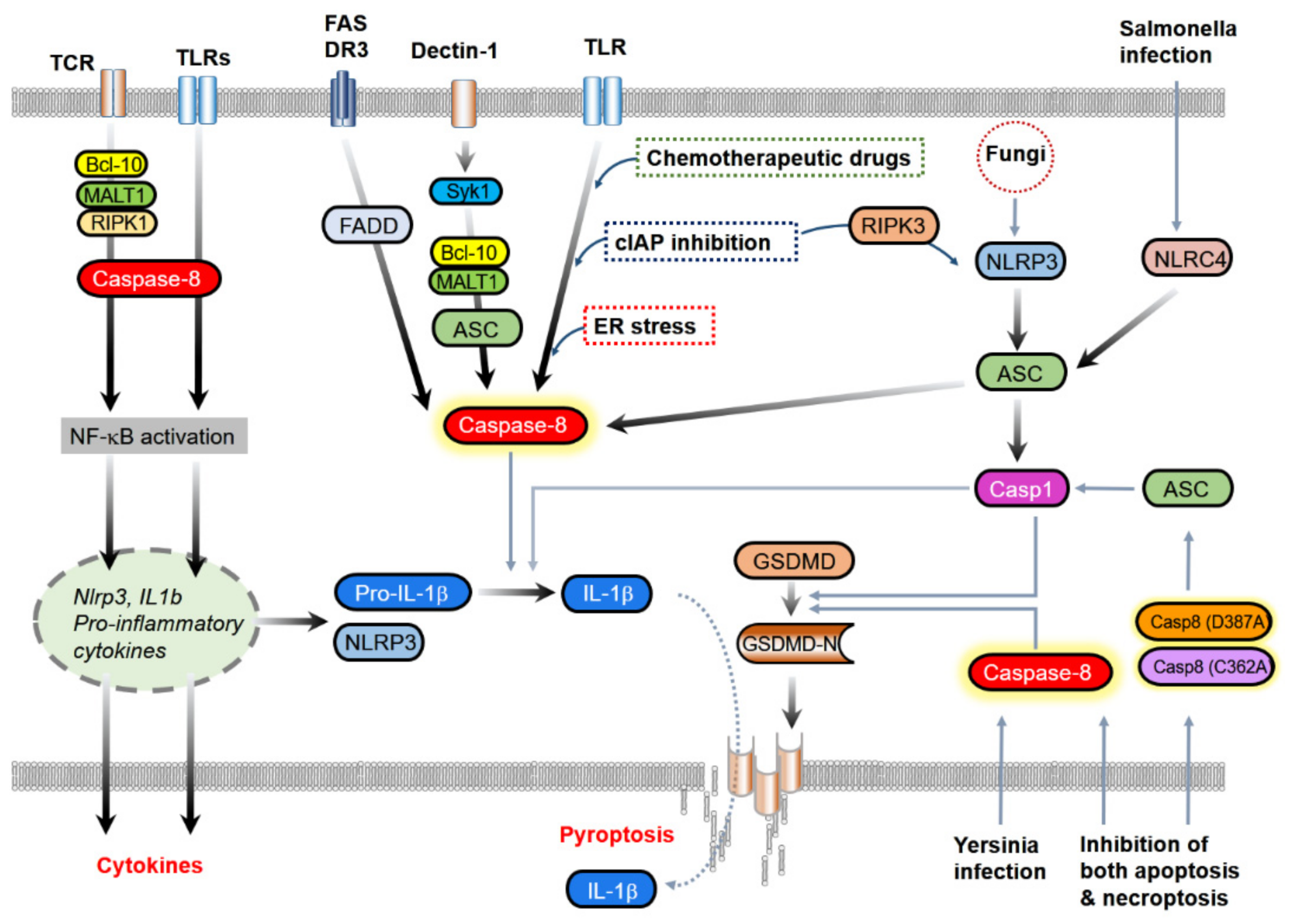
2.3. Inhibition of Inflammasomes
2.4. Inhibition of Inflammatory Signaling Complex
3. Proinflammatory Functions of Caspase-8
3.1. Contribution to NF-κB Activation and Cytokine Induction
3.2. IL-1β Processing
3.2.1. The Association with Classical Inflammasome Components
3.2.2. Direct Processing of IL-1β
3.3. Induction of Pyroptosis
4. Other Inflammation-Related Processes in which Caspase-8 Might Be Involved
4.1. Autophagy
4.2. Ferroptosis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Galluzzi, L.; Lopez-Soto, A.; Kumar, S.; Kroemer, G. Caspases Connect Cell-Death Signaling to Organismal Homeostasis. Immunity 2016, 44, 221–231. [Google Scholar] [CrossRef] [PubMed]
- Elmore, S. Apoptosis: A Review of Programmed Cell Death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef]
- Pop, C.; Fitzgerald, P.; Green, D.R.; Salvesen, G.S. Role of Proteolysis in Caspase-8 Activation and Stabilization. Biochemistry 2007, 46, 4398–4407. [Google Scholar] [CrossRef]
- Salvesen, G.S.; Dixit, V.M. Caspase Activation: The Induced-Proximity Model. Proc. Natl. Acad. Sci. USA 1999, 96, 10964–10967. [Google Scholar] [CrossRef] [PubMed]
- Chang, D.W.; Xing, Z.; Capacio, V.L.; Peter, M.E.; Yang, X. Interdimer Processing Mechanism of Procaspase-8 Activation. EMBO J. 2003, 22, 4132–4142. [Google Scholar] [CrossRef]
- Li, H.; Zhu, H.; Xu, C.J.; Yuan, J. Cleavage of BID by Caspase 8 Mediates the Mitochondrial Damage in the Fas Pathway of Apoptosis. Cell 1998, 94, 491–501. [Google Scholar] [CrossRef]
- Luo, X.; Budihardjo, I.; Zou, H.; Slaughter, C.; Wang, X. Bid, a Bcl2 Interacting Protein, Mediates Cytochrome C Release from Mitochondria in Response to Activation of Cell Surface Death Receptors. Cell 1998, 94, 481–490. [Google Scholar] [CrossRef]
- Hung, C.L.; Chang, H.H.; Lee, S.W.; Chiang, Y.W. Stepwise Activation of the Pro-Apoptotic Protein Bid at Mitochondrial Membranes. Cell Death Differ. 2021. [Google Scholar] [CrossRef]
- Maelfait, J.; Vercammen, E.; Janssens, S.; Schotte, P.; Haegman, M.; Magez, S.; Beyaert, R. Stimulation of Toll-Like Receptor 3 and 4 Induces Interleukin-1beta Maturation by Caspase-8. J. Exp. Med. 2008, 205, 1967–1973. [Google Scholar] [CrossRef]
- Lieberman, J.; Wu, H.; Kagan, J.C. Gasdermin D Activity in Inflammation and Host Defense. Sci. Immunol. 2019, 4. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Devin, A.; Rodriguez, Y.; Liu, Z.G. Cleavage of the Death Domain Kinase RIP by Caspase-8 Prompts TNF-Induced Apoptosis. Genes Dev. 1999, 13, 2514–2526. [Google Scholar] [CrossRef] [PubMed]
- Feng, S.; Yang, Y.; Mei, Y.; Ma, L.; Zhu, D.E.; Hoti, N.; Castanares, M.; Wu, M. Cleavage of RIP3 Inactivates its Caspase-Independent Apoptosis Pathway by Removal of Kinase Domain. Cell. Signal. 2007, 19, 2056–2067. [Google Scholar] [CrossRef] [PubMed]
- O’Donnell, M.A.; Perez-Jimenez, E.; Oberst, A.; Ng, A.; Massoumi, R.; Xavier, R.; Green, D.R.; Ting, A.T. Caspase 8 Inhibits Programmed Necrosis by Processing CYLD. Nat. Cell Biol. 2011, 13, 1437–1442. [Google Scholar] [CrossRef] [PubMed]
- Verstraeten, S.V.; Zago, M.P.; MacKenzie, G.G.; Keen, C.L.; Oteiza, P.I. Influence of Zinc Deficiency on Cell-Membrane Fluidity in Jurkat, 3T3 and IMR-32 Cells. Biochem. J. 2004, 378, 579–587. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, M.; Imamura, R.; Motani, K.; Nishiuchi, T.; Matsumoto, N.; Kinoshita, T.; Suda, T. Mechanism and Repertoire of ASC-Mediated Gene Expression. J. Immunol. 2009, 182, 7655–7662. [Google Scholar] [CrossRef]
- Silke, J. The Regulation of TNF Signalling: What a Tangled Web we Weave. Curr. Opin. Immunol. 2011, 23, 620–626. [Google Scholar] [CrossRef]
- Ashkenazi, A.; Dixit, V.M. Death Receptors: Signaling and Modulation. Science 1998, 281, 1305–1308. [Google Scholar] [CrossRef]
- Peter, M.E.; Krammer, P.H. Mechanisms of CD95 (APO-1/Fas)-Mediated Apoptosis. Curr. Opin. Immunol. 1998, 10, 545–551. [Google Scholar] [CrossRef]
- Suliman, A.; Lam, A.; Datta, R.; Srivastava, R.K. Intracellular Mechanisms of TRAIL: Apoptosis through Mitochondrial-Dependent and -Independent Pathways. Oncogene 2001, 20, 2122–2133. [Google Scholar] [CrossRef]
- Rubio-Moscardo, F.; Blesa, D.; Mestre, C.; Siebert, R.; Balasas, T.; Benito, A.; Rosenwald, A.; Climent, J.; Martinez, J.I.; Schilhabel, M.; et al. Characterization of 8p21.3 Chromosomal Deletions in B-Cell Lymphoma: TRAIL-R1 and TRAIL-R2 as Candidate Dosage-Dependent Tumor Suppressor Genes. Blood 2005, 106, 3214–3222. [Google Scholar] [CrossRef]
- Fu, T.M.; Li, Y.; Lu, A.; Li, Z.; Vajjhala, P.R.; Cruz, A.C.; Srivastava, D.B.; DiMaio, F.; Penczek, P.A.; Siegel, R.M.; et al. Cryo-EM Structure of Caspase-8 Tandem DED Filament Reveals Assembly and Regulation Mechanisms of the Death-Inducing Signaling Complex. Mol. Cell 2016, 64, 236–250. [Google Scholar] [CrossRef] [PubMed]
- Schleich, K.; Warnken, U.; Fricker, N.; Ozturk, S.; Richter, P.; Kammerer, K.; Schnolzer, M.; Krammer, P.H.; Lavrik, I.N. Stoichiometry of the CD95 Death-Inducing Signaling Complex: Experimental and Modeling Evidence for a Death Effector Domain Chain Model. Mol. Cell 2012, 47, 306–319. [Google Scholar] [CrossRef]
- Fox, J.L.; Hughes, M.A.; Meng, X.; Sarnowska, N.A.; Powley, I.R.; Jukes-Jones, R.; Dinsdale, D.; Ragan, T.J.; Fairall, L.; Schwabe, J.W.R.; et al. Cryo-EM Structural Analysis of FADD:Caspase-8 Complexes Defines the Catalytic Dimer Architecture for Co-Ordinated Control of Cell Fate. Nat. Commun. 2021, 12, 819. [Google Scholar] [CrossRef] [PubMed]
- Shen, C.; Pei, J.; Guo, X.; Zhou, L.; Li, Q.; Quan, J. Structural Basis for Dimerization of the Death Effector Domain of the F122A Mutant of Caspase-8. Sci. Rep. 2018, 8, 16723. [Google Scholar]
- Chang, D.W.; Xing, Z.; Pan, Y.; Algeciras-Schimnich, A.; Barnhart, B.C.; Yaish-Ohad, S.; Peter, M.E.; Yang, X. C-FLIP(L) is a Dual Function Regulator for Caspase-8 Activation and CD95-Mediated Apoptosis. EMBO J. 2002, 21, 3704–3714. [Google Scholar] [CrossRef] [PubMed]
- Micheau, O.; Thome, M.; Schneider, P.; Holler, N.; Tschopp, J.; Nicholson, D.W.; Briand, C.; Grutter, M.G. The Long Form of FLIP is an Activator of Caspase-8 at the Fas Death-Inducing Signaling Complex. J. Biol. Chem. 2002, 277, 45162–45171. [Google Scholar] [CrossRef]
- Kang, T.B.; Oh, G.S.; Scandella, E.; Bolinger, B.; Ludewig, B.; Kovalenko, A.; Wallach, D. Mutation of a Self-Processing Site in Caspase-8 Compromises its Apoptotic but Not its Nonapoptotic Functions in Bacterial Artificial Chromosome-Transgenic Mice. J. Immunol. 2008, 181, 2522–2532. [Google Scholar] [CrossRef]
- Oberst, A.; Dillon, C.P.; Weinlich, R.; McCormick, L.L.; Fitzgerald, P.; Pop, C.; Hakem, R.; Salvesen, G.S.; Green, D.R. Catalytic Activity of the Caspase-8-FLIP(L) Complex Inhibits RIPK3-Dependent Necrosis. Nature 2011, 471, 363–367. [Google Scholar] [CrossRef] [PubMed]
- Hughes, M.A.; Powley, I.R.; Jukes-Jones, R.; Horn, S.; Feoktistova, M.; Fairall, L.; Schwabe, J.W.; Leverkus, M.; Cain, K.; MacFarlane, M. Co-Operative and Hierarchical Binding of C-FLIP and Caspase-8: A Unified Model Defines how C-FLIP Isoforms Differentially Control Cell Fate. Mol. Cell 2016, 61, 834–849. [Google Scholar] [CrossRef]
- Syntichaki, P.; Tavernarakis, N. Death by Necrosis. Uncontrollable Catastrophe, or is there Order Behind the Chaos? EMBO Rep. 2002, 3, 604–609. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.W.; Shao, J.; Lin, J.; Zhang, N.; Lu, B.J.; Lin, S.C.; Dong, M.Q.; Han, J. RIP3, an Energy Metabolism Regulator that Switches TNF-Induced Cell Death from Apoptosis to Necrosis. Science 2009, 325, 332–336. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.S.; Challa, S.; Moquin, D.; Genga, R.; Ray, T.D.; Guildford, M.; Chan, F.K. Phosphorylation-Driven Assembly of the RIP1-RIP3 Complex Regulates Programmed Necrosis and Virus-Induced Inflammation. Cell 2009, 137, 1112–1123. [Google Scholar] [CrossRef]
- Kaiser, W.J.; Upton, J.W.; Long, A.B.; Livingston-Rosanoff, D.; Daley-Bauer, L.P.; Hakem, R.; Caspary, T.; Mocarski, E.S. RIP3 Mediates the Embryonic Lethality of Caspase-8-Deficient Mice. Nature 2011, 471, 368–372. [Google Scholar] [CrossRef]
- Dillon, C.P.; Oberst, A.; Weinlich, R.; Janke, L.J.; Kang, T.B.; Ben-Moshe, T.; Mak, T.W.; Wallach, D.; Green, D.R. Survival Function of the FADD-CASPASE-8-cFLIP(L) Complex. Cell. Rep. 2012, 1, 401–407. [Google Scholar] [CrossRef]
- Scaffidi, P.; Misteli, T.; Bianchi, M.E. Release of Chromatin Protein HMGB1 by Necrotic Cells Triggers Inflammation. Nature 2002, 418, 191–195. [Google Scholar] [CrossRef] [PubMed]
- Vanden Berghe, T.; Hassannia, B.; Vandenabeele, P. An Outline of Necrosome Triggers. Cell Mol. Life Sci. 2016, 73, 2137–2152. [Google Scholar] [CrossRef] [PubMed]
- Kang, T.B.; Ben-Moshe, T.; Varfolomeev, E.E.; Pewzner-Jung, Y.; Yogev, N.; Jurewicz, A.; Waisman, A.; Brenner, O.; Haffner, R.; Gustafsson, E.; et al. Caspase-8 Serves both Apoptotic and Nonapoptotic Roles. J. Immunol. 2004, 173, 2976–2984. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Wang, H.; Wang, Z.; He, S.; Chen, S.; Liao, D.; Wang, L.; Yan, J.; Liu, W.; Lei, X.; et al. Mixed Lineage Kinase Domain-Like Protein Mediates Necrosis Signaling Downstream of RIP3 Kinase. Cell 2012, 148, 213–227. [Google Scholar] [CrossRef]
- McQuade, T.; Cho, Y.; Chan, F.K. Positive and Negative Phosphorylation Regulates RIP1- and RIP3-Induced Programmed Necrosis. Biochem. J. 2013, 456, 409–415. [Google Scholar] [CrossRef]
- Zhao, J.; Jitkaew, S.; Cai, Z.; Choksi, S.; Li, Q.; Luo, J.; Liu, Z.G. Mixed Lineage Kinase Domain-Like is a Key Receptor Interacting Protein 3 Downstream Component of TNF-Induced Necrosis. Proc. Natl. Acad. Sci. USA 2012, 109, 5322–5327. [Google Scholar] [CrossRef]
- Newton, K.; Wickliffe, K.E.; Dugger, D.L.; Maltzman, A.; Roose-Girma, M.; Dohse, M.; Komuves, L.; Webster, J.D.; Dixit, V.M. Cleavage of RIPK1 by Caspase-8 is Crucial for Limiting Apoptosis and Necroptosis. Nature 2019, 574, 428–431. [Google Scholar] [CrossRef] [PubMed]
- Kovalenko, A.; Kim, J.C.; Kang, T.B.; Rajput, A.; Bogdanov, K.; Dittrich-Breiholz, O.; Kracht, M.; Brenner, O.; Wallach, D. Caspase-8 Deficiency in Epidermal Keratinocytes Triggers an Inflammatory Skin Disease. J. Exp. Med. 2009, 206, 2161–2177. [Google Scholar] [CrossRef]
- Lee, P.; Lee, D.J.; Chan, C.; Chen, S.W.; Ch’en, I.; Jamora, C. Dynamic Expression of Epidermal Caspase 8 Simulates a Wound Healing Response. Nature 2009, 458, 519–523. [Google Scholar] [CrossRef]
- Gunther, C.; Martini, E.; Wittkopf, N.; Amann, K.; Weigmann, B.; Neumann, H.; Waldner, M.J.; Hedrick, S.M.; Tenzer, S.; Neurath, M.F.; et al. Caspase-8 Regulates TNF-Alpha-Induced Epithelial Necroptosis and Terminal Ileitis. Nature 2011, 477, 335–339. [Google Scholar] [CrossRef]
- Alvarez-Diaz, S.; Dillon, C.P.; Lalaoui, N.; Tanzer, M.C.; Rodriguez, D.A.; Lin, A.; Lebois, M.; Hakem, R.; Josefsson, E.C.; O’Reilly, L.A.; et al. The Pseudokinase MLKL and the Kinase RIPK3 have Distinct Roles in Autoimmune Disease Caused by Loss of Death-Receptor-Induced Apoptosis. Immunity 2016, 45, 513–526. [Google Scholar] [CrossRef]
- Lehle, A.S.; Farin, H.F.; Marquardt, B.; Michels, B.E.; Magg, T.; Li, Y.; Liu, Y.; Ghalandary, M.; Lammens, K.; Hollizeck, S.; et al. Intestinal Inflammation and Dysregulated Immunity in Patients with Inherited Caspase-8 Deficiency. Gastroenterology 2019, 156, 275–278. [Google Scholar] [CrossRef] [PubMed]
- Martinon, F.; Burns, K.; Tschopp, J. The Inflammasome: A Molecular Platform Triggering Activation of Inflammatory Caspases and Processing of proIL-Beta. Mol. Cell 2002, 10, 417–426. [Google Scholar] [CrossRef]
- Schroder, K.; Tschopp, J. The Inflammasomes. Cell 2010, 140, 821–832. [Google Scholar] [CrossRef] [PubMed]
- Place, D.E.; Kanneganti, T.D. Recent Advances in Inflammasome Biology. Curr. Opin. Immunol. 2018, 50, 32–38. [Google Scholar] [CrossRef]
- Shi, J.; Zhao, Y.; Wang, K.; Shi, X.; Wang, Y.; Huang, H.; Zhuang, Y.; Cai, T.; Wang, F.; Shao, F. Cleavage of GSDMD by Inflammatory Caspases Determines Pyroptotic Cell Death. Nature 2015, 526, 660–665. [Google Scholar] [CrossRef]
- Kayagaki, N.; Stowe, I.B.; Lee, B.L.; O’Rourke, K.; Anderson, K.; Warming, S.; Cuellar, T.; Haley, B.; Roose-Girma, M.; Phung, Q.T.; et al. Caspase-11 Cleaves Gasdermin D for Non-Canonical Inflammasome Signalling. Nature 2015, 526, 666–671. [Google Scholar] [CrossRef]
- Fink, S.L.; Cookson, B.T. Pillars Article: Caspase-1-Dependent Pore Formation during Pyroptosis Leads to Osmotic Lysis of Infected Host Macrophages. Cell Microbiol. 2006, 8, 1812–1825, J. Immunol.2019, 202, 1913–1926. [Google Scholar]
- Wang, Z.; Zhang, S.; Xiao, Y.; Zhang, W.; Wu, S.; Qin, T.; Yue, Y.; Qian, W.; Li, L. NLRP3 Inflammasome and Inflammatory Diseases. Oxid. Med. Cell. Longev 2020, 2020, 4063562. [Google Scholar] [CrossRef] [PubMed]
- Kang, T.B.; Yang, S.H.; Toth, B.; Kovalenko, A.; Wallach, D. Caspase-8 Blocks Kinase RIPK3-Mediated Activation of the NLRP3 Inflammasome. Immunity 2013, 38, 27–40. [Google Scholar] [CrossRef]
- Cuda, C.M.; Misharin, A.V.; Gierut, A.K.; Saber, R.; Haines, G.K., 3rd; Hutcheson, J.; Hedrick, S.M.; Mohan, C.; Budinger, G.S.; Stehlik, C.; et al. Caspase-8 Acts as a Molecular Rheostat to Limit RIPK1- and MyD88-Mediated Dendritic Cell Activation. J. Immunol. 2014, 192, 5548–5560. [Google Scholar] [CrossRef]
- Kang, S.; Fernandes-Alnemri, T.; Rogers, C.; Mayes, L.; Wang, Y.; Dillon, C.; Roback, L.; Kaiser, W.; Oberst, A.; Sagara, J.; et al. Caspase-8 Scaffolding Function and MLKL Regulate NLRP3 Inflammasome Activation Downstream of TLR3. Nat. Commun. 2015, 6, 7515. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez, K.D.; Davis, M.A.; Daniels, B.P.; Olsen, T.M.; Ralli-Jain, P.; Tait, S.W.; Gale, M., Jr.; Oberst, A. MLKL Activation Triggers NLRP3-Mediated Processing and Release of IL-1beta Independently of Gasdermin-D. J. Immunol. 2017, 198, 2156–2164. [Google Scholar] [CrossRef] [PubMed]
- Fritsch, M.; Gunther, S.D.; Schwarzer, R.; Albert, M.C.; Schorn, F.; Werthenbach, J.P.; Schiffmann, L.M.; Stair, N.; Stocks, H.; Seeger, J.M.; et al. Caspase-8 is the Molecular Switch for Apoptosis, Necroptosis and Pyroptosis. Nature 2019, 575, 683–687. [Google Scholar] [CrossRef]
- Rebe, C.; Cathelin, S.; Launay, S.; Filomenko, R.; Prevotat, L.; L’Ollivier, C.; Gyan, E.; Micheau, O.; Grant, S.; Dubart-Kupperschmitt, A.; et al. Caspase-8 Prevents Sustained Activation of NF-κB in Monocytes Undergoing Macrophagic Differentiation. Blood 2007, 109, 1442–1450. [Google Scholar] [CrossRef] [PubMed]
- Rajput, A.; Kovalenko, A.; Bogdanov, K.; Yang, S.H.; Kang, T.B.; Kim, J.C.; Du, J.; Wallach, D. RIG-I RNA Helicase Activation of IRF3 Transcription Factor is Negatively Regulated by Caspase-8-Mediated Cleavage of the RIP1 Protein. Immunity 2011, 34, 340–351. [Google Scholar] [CrossRef]
- Kang, T.B.; Jeong, J.S.; Yang, S.H.; Kovalenko, A.; Wallach, D. Caspase-8 Deficiency in Mouse Embryos Triggers Chronic RIPK1-Dependent Activation of Inflammatory Genes, Independently of RIPK3. Cell Death Differ. 2018, 25, 1107–1117. [Google Scholar] [CrossRef]
- Lalaoui, N.; Boyden, S.E.; Oda, H.; Wood, G.M.; Stone, D.L.; Chau, D.; Liu, L.; Stoffels, M.; Kratina, T.; Lawlor, K.E.; et al. Mutations that Prevent Caspase Cleavage of RIPK1 Cause Autoinflammatory Disease. Nature 2020, 577, 103–108. [Google Scholar] [CrossRef]
- Tao, P.; Sun, J.; Wu, Z.; Wang, S.; Wang, J.; Li, W.; Pan, H.; Bai, R.; Zhang, J.; Wang, Y.; et al. A Dominant Autoinflammatory Disease Caused by Non-Cleavable Variants of RIPK1. Nature 2020, 577, 109–114. [Google Scholar] [CrossRef]
- Chaudhary, P.M.; Eby, M.T.; Jasmin, A.; Kumar, A.; Liu, L.; Hood, L. Activation of the NF-κB Pathway by Caspase 8 and its Homologs. Oncogene 2000, 19, 4451–4460. [Google Scholar] [CrossRef]
- Hu, W.H.; Johnson, H.; Shu, H.B. Activation of NF-κB by FADD, Casper, and Caspase-8. J. Biol. Chem. 2000, 275, 10838–10844. [Google Scholar] [CrossRef] [PubMed]
- Su, H.; Bidere, N.; Zheng, L.; Cubre, A.; Sakai, K.; Dale, J.; Salmena, L.; Hakem, R.; Straus, S.; Lenardo, M. Requirement for Caspase-8 in NF-κB Activation by Antigen Receptor. Science 2005, 307, 1465–1468. [Google Scholar] [CrossRef]
- Salmena, L.; Lemmers, B.; Hakem, A.; Matysiak-Zablocki, E.; Murakami, K.; Au, P.Y.; Berry, D.M.; Tamblyn, L.; Shehabeldin, A.; Migon, E.; et al. Essential Role for Caspase 8 in T-Cell Homeostasis and T-Cell-Mediated Immunity. Genes Dev. 2003, 17, 883–895. [Google Scholar] [CrossRef]
- Chun, H.J.; Zheng, L.; Ahmad, M.; Wang, J.; Speirs, C.K.; Siegel, R.M.; Dale, J.K.; Puck, J.; Davis, J.; Hall, C.G.; et al. Pleiotropic Defects in Lymphocyte Activation Caused by Caspase-8 Mutations Lead to Human Immunodeficiency. Nature 2002, 419, 395–399. [Google Scholar] [CrossRef]
- Lemmers, B.; Salmena, L.; Bidere, N.; Su, H.; Matysiak-Zablocki, E.; Murakami, K.; Ohashi, P.S.; Jurisicova, A.; Lenardo, M.; Hakem, R.; et al. Essential Role for Caspase-8 in Toll-Like Receptors and NFkappaB Signaling. J. Biol. Chem. 2007, 282, 7416–7423. [Google Scholar] [CrossRef] [PubMed]
- Henry, C.M.; Martin, S.J. Caspase-8 Acts in a Non-Enzymatic Role as a Scaffold for Assembly of a Pro-Inflammatory “FADDosome” Complex upon TRAIL Stimulation. Mol. Cell 2017, 65, 715–729.e5. [Google Scholar] [CrossRef] [PubMed]
- Hartwig, T.; Montinaro, A.; von Karstedt, S.; Sevko, A.; Surinova, S.; Chakravarthy, A.; Taraborrelli, L.; Draber, P.; Lafont, E.; Arce Vargas, F.; et al. The TRAIL-Induced Cancer Secretome Promotes a Tumor-Supportive Immune Microenvironment via CCR2. Mol. Cell 2017, 65, 730–742.e5. [Google Scholar] [CrossRef] [PubMed]
- Varfolomeev, E.; Maecker, H.; Sharp, D.; Lawrence, D.; Renz, M.; Vucic, D.; Ashkenazi, A. Molecular Determinants of Kinase Pathway Activation by Apo2 Ligand/Tumor Necrosis Factor-Related Apoptosis-Inducing Ligand. J. Biol. Chem. 2005, 280, 40599–40608. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, I.; Matsuo, K.; Matsushita, Y.; Haruna, Y.; Niwa, M.; Kataoka, T. The C-Terminal Domain of the Long Form of Cellular FLICE-Inhibitory Protein (C-FLIPL) Inhibits the Interaction of the Caspase 8 Prodomain with the Receptor-Interacting Protein 1 (RIP1) Death Domain and Regulates Caspase 8-Dependent Nuclear Factor κB (NF-κB) Activation. J. Biol. Chem. 2014, 289, 3876–3887. [Google Scholar]
- Philip, N.H.; DeLaney, A.; Peterson, L.W.; Santos-Marrero, M.; Grier, J.T.; Sun, Y.; Wynosky-Dolfi, M.A.; Zwack, E.E.; Hu, B.; Olsen, T.M.; et al. Activity of Uncleaved Caspase-8 Controls Anti-Bacterial Immune Defense and TLR-Induced Cytokine Production Independent of Cell Death. PLoS Pathog. 2016, 12, e1005910. [Google Scholar] [CrossRef]
- Hasegawa, M.; Imamura, R.; Kinoshita, T.; Matsumoto, N.; Masumoto, J.; Inohara, N.; Suda, T. ASC-Mediated NF-κB Activation Leading to Interleukin-8 Production Requires Caspase-8 and is Inhibited by CLARP. J. Biol. Chem. 2005, 280, 15122–15130. [Google Scholar] [CrossRef]
- Gurung, P.; Anand, P.K.; Malireddi, R.K.; Vande Walle, L.; Van Opdenbosch, N.; Dillon, C.P.; Weinlich, R.; Green, D.R.; Lamkanfi, M.; Kanneganti, T.D. FADD and Caspase-8 Mediate Priming and Activation of the Canonical and Noncanonical Nlrp3 Inflammasomes. J. Immunol. 2014, 192, 1835–1846. [Google Scholar] [CrossRef] [PubMed]
- Man, S.M.; Tourlomousis, P.; Hopkins, L.; Monie, T.P.; Fitzgerald, K.A.; Bryant, C.E. Salmonella Infection Induces Recruitment of Caspase-8 to the Inflammasome to Modulate IL-1beta Production. J. Immunol. 2013, 191, 5239–5246. [Google Scholar] [CrossRef]
- Tian, Y.; Li, H.; Liu, X.; Xie, L.; Huang, Z.; Li, W.; Li, Z.; Pan, Y.; Chen, X.; Su, W. Pharmacological Inhibition of Caspase-8 Suppresses Inflammation-Induced Angiogenesis in the Cornea. Biomolecules 2020, 10, 210. [Google Scholar] [CrossRef]
- Pereira, L.M.N.; Assis, P.A.; de Araujo, N.M.; Durso, D.F.; Junqueira, C.; Ataide, M.A.; Pereira, D.B.; Lien, E.; Fitzgerald, K.A.; Zamboni, D.S.; et al. Caspase-8 Mediates Inflammation and Disease in Rodent Malaria. Nat. Commun. 2020, 11, 1–13. [Google Scholar] [CrossRef]
- Golks, A.; Brenner, D.; Krammer, P.H.; Lavrik, I.N. The C-FLIP-NH2 Terminus (p22-FLIP) Induces NF-κB Activation. J. Exp. Med. 2006, 203, 1295–1305. [Google Scholar] [CrossRef] [PubMed]
- DeLaney, A.A.; Berry, C.T.; Christian, D.A.; Hart, A.; Bjanes, E.; Wynosky-Dolfi, M.A.; Li, X.; Tummers, B.; Udalova, I.A.; Chen, Y.H.; et al. Caspase-8 Promotes C-Rel-Dependent Inflammatory Cytokine Expression and Resistance Against Toxoplasma Gondii. Proc. Natl. Acad. Sci. USA 2019, 116, 11926–11935. [Google Scholar] [CrossRef] [PubMed]
- Koenig, A.; Buskiewicz, I.A.; Fortner, K.A.; Russell, J.Q.; Asaoka, T.; He, Y.W.; Hakem, R.; Eriksson, J.E.; Budd, R.C. The C-FLIPL Cleavage Product p43FLIP Promotes Activation of Extracellular Signal-Regulated Kinase (ERK), Nuclear Factor κB (NF-κB), and Caspase-8 and T Cell Survival. J. Biol. Chem. 2014, 289, 1183–1191. [Google Scholar] [CrossRef]
- Kataoka, T.; Tschopp, J. N-Terminal Fragment of C-FLIP(L) Processed by Caspase 8 Specifically Interacts with TRAF2 and Induces Activation of the NF-κB Signaling Pathway. Mol. Cell. Biol. 2004, 24, 2627–2636. [Google Scholar] [CrossRef]
- Gitlin, A.D.; Heger, K.; Schubert, A.F.; Reja, R.; Yan, D.; Pham, V.C.; Suto, E.; Zhang, J.; Kwon, Y.C.; Freund, E.C.; et al. Integration of Innate Immune Signalling by Caspase-8 Cleavage of N4BP1. Nature 2020, 587, 275–280. [Google Scholar] [CrossRef]
- Allam, R.; Lawlor, K.E.; Yu, E.C.; Mildenhall, A.L.; Moujalled, D.M.; Lewis, R.S.; Ke, F.; Mason, K.D.; White, M.J.; Stacey, K.J.; et al. Mitochondrial Apoptosis is Dispensable for NLRP3 Inflammasome Activation but Non-Apoptotic Caspase-8 is Required for Inflammasome Priming. EMBO Rep. 2014, 15, 982–990. [Google Scholar] [CrossRef] [PubMed]
- Dinarello, C.A. Biologic Basis for Interleukin-1 in Disease. Blood 1996, 87, 2095–2147. [Google Scholar] [CrossRef] [PubMed]
- Sagulenko, V.; Thygesen, S.J.; Sester, D.P.; Idris, A.; Cridland, J.A.; Vajjhala, P.R.; Roberts, T.L.; Schroder, K.; Vince, J.E.; Hill, J.M.; et al. AIM2 and NLRP3 Inflammasomes Activate both Apoptotic and Pyroptotic Death Pathways via ASC. Cell Death Differ. 2013, 20, 1149–1160. [Google Scholar] [CrossRef] [PubMed]
- Antonopoulos, C.; Russo, H.M.; El Sanadi, C.; Martin, B.N.; Li, X.; Kaiser, W.J.; Mocarski, E.S.; Dubyak, G.R. Caspase-8 as an Effector and Regulator of NLRP3 Inflammasome Signaling. J. Biol. Chem. 2015, 290, 20167–20184. [Google Scholar] [CrossRef]
- Chen, M.; Xing, Y.; Lu, A.; Fang, W.; Sun, B.; Chen, C.; Liao, W.; Meng, G. Internalized Cryptococcus Neoformans Activates the Canonical Caspase-1 and the Noncanonical Caspase-8 Inflammasomes. J. Immunol. 2015, 195, 4962–4972. [Google Scholar] [CrossRef] [PubMed]
- Vajjhala, P.R.; Lu, A.; Brown, D.L.; Pang, S.W.; Sagulenko, V.; Sester, D.P.; Cridland, S.O.; Hill, J.M.; Schroder, K.; Stow, J.L.; et al. The Inflammasome Adaptor ASC Induces Procaspase-8 Death Effector Domain Filaments. J. Biol. Chem. 2015, 290, 29217–29230. [Google Scholar] [CrossRef]
- Lawlor, K.E.; Khan, N.; Mildenhall, A.; Gerlic, M.; Croker, B.A.; D’Cruz, A.A.; Hall, C.; Kaur Spall, S.; Anderton, H.; Masters, S.L.; et al. RIPK3 Promotes Cell Death and NLRP3 Inflammasome Activation in the Absence of MLKL. Nat. Commun. 2015, 6, 6282. [Google Scholar] [CrossRef]
- Antonopoulos, C.; El Sanadi, C.; Kaiser, W.J.; Mocarski, E.S.; Dubyak, G.R. Proapoptotic Chemotherapeutic Drugs Induce Noncanonical Processing and Release of IL-1beta Via Caspase-8 in Dendritic Cells. J. Immunol. 2013, 191, 4789–4803. [Google Scholar] [CrossRef] [PubMed]
- Tummers, B.; Mari, L.; Guy, C.S.; Heckmann, B.L.; Rodriguez, D.A.; Ruhl, S.; Moretti, J.; Crawford, J.C.; Fitzgerald, P.; Kanneganti, T.D.; et al. Caspase-8-Dependent Inflammatory Responses are Controlled by its Adaptor, FADD, and Necroptosis. Immunity 2020, 52, 994–1006.e8. [Google Scholar] [CrossRef] [PubMed]
- Gringhuis, S.I.; Kaptein, T.M.; Wevers, B.A.; Theelen, B.; van der Vlist, M.; Boekhout, T.; Geijtenbeek, T.B. Dectin-1 is an Extracellular Pathogen Sensor for the Induction and Processing of IL-1beta Via a Noncanonical Caspase-8 Inflammasome. Nat. Immunol. 2012, 13, 246–254. [Google Scholar] [CrossRef] [PubMed]
- Ganesan, S.; Rathinam, V.A.K.; Bossaller, L.; Army, K.; Kaiser, W.J.; Mocarski, E.S.; Dillon, C.P.; Green, D.R.; Mayadas, T.N.; Levitz, S.M.; et al. Caspase-8 Modulates Dectin-1 and Complement Receptor 3-Driven IL-1beta Production in Response to Beta-Glucans and the Fungal Pathogen, Candida Albicans. J. Immunol. 2014, 193, 2519–2530. [Google Scholar] [CrossRef]
- Hedl, M.; Abraham, C. A TNFSF15 Disease-Risk Polymorphism Increases Pattern-Recognition Receptor-Induced Signaling through Caspase-8-Induced IL-1. Proc. Natl. Acad. Sci. USA 2014, 111, 13451–13456. [Google Scholar] [CrossRef] [PubMed]
- Bossaller, L.; Chiang, P.I.; Schmidt-Lauber, C.; Ganesan, S.; Kaiser, W.J.; Rathinam, V.A.; Mocarski, E.S.; Subramanian, D.; Green, D.R.; Silverman, N.; et al. Cutting Edge: FAS (CD95) Mediates Noncanonical IL-1beta and IL-18 Maturation Via Caspase-8 in an RIP3-Independent Manner. J. Immunol. 2012, 189, 5508–5512. [Google Scholar] [CrossRef]
- Zhang, C.J.; Jiang, M.; Zhou, H.; Liu, W.; Wang, C.; Kang, Z.; Han, B.; Zhang, Q.; Chen, X.; Xiao, J.; et al. TLR-Stimulated IRAKM Activates Caspase-8 Inflammasome in Microglia and Promotes Neuroinflammation. J. Clin. Investig. 2018, 128, 5399–5412. [Google Scholar] [CrossRef] [PubMed]
- Moriwaki, K.; Bertin, J.; Gough, P.J.; Chan, F.K. A RIPK3-Caspase 8 Complex Mediates Atypical Pro-IL-1beta Processing. J. Immunol. 2015, 194, 1938–1944. [Google Scholar] [CrossRef]
- Stammler, D.; Eigenbrod, T.; Menz, S.; Frick, J.S.; Sweet, M.J.; Shakespear, M.R.; Jantsch, J.; Siegert, I.; Wolfle, S.; Langer, J.D.; et al. Inhibition of Histone Deacetylases Permits Lipopolysaccharide-Mediated Secretion of Bioactive IL-1beta via a Caspase-1-Independent Mechanism. J. Immunol. 2015, 195, 5421–5431. [Google Scholar] [CrossRef]
- Shenderov, K.; Riteau, N.; Yip, R.; Mayer-Barber, K.D.; Oland, S.; Hieny, S.; Fitzgerald, P.; Oberst, A.; Dillon, C.P.; Green, D.R.; et al. Cutting Edge: Endoplasmic Reticulum Stress Licenses Macrophages to Produce Mature IL-1beta in Response to TLR4 Stimulation through a Caspase-8- and TRIF-Dependent Pathway. J. Immunol. 2014, 192, 2029–2033. [Google Scholar] [CrossRef]
- Vince, J.E.; Wong, W.W.; Gentle, I.; Lawlor, K.E.; Allam, R.; O’Reilly, L.; Mason, K.; Gross, O.; Ma, S.; Guarda, G.; et al. Inhibitor of Apoptosis Proteins Limit RIP3 Kinase-Dependent Interleukin-1 Activation. Immunity 2012, 36, 215–227. [Google Scholar] [CrossRef]
- Sarhan, J.; Liu, B.C.; Muendlein, H.I.; Li, P.; Nilson, R.; Tang, A.Y.; Rongvaux, A.; Bunnell, S.C.; Shao, F.; Green, D.R.; et al. Caspase-8 Induces Cleavage of Gasdermin D to Elicit Pyroptosis during Yersinia Infection. Proc. Natl. Acad. Sci. USA 2018, 115, E10888–E10897. [Google Scholar] [CrossRef]
- Orning, P.; Weng, D.; Starheim, K.; Ratner, D.; Best, Z.; Lee, B.; Brooks, A.; Xia, S.; Wu, H.; Kelliher, M.A.; et al. Pathogen Blockade of TAK1 Triggers Caspase-8-Dependent Cleavage of Gasdermin D and Cell Death. Science 2018, 362, 1064–1069. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, S.; Keitany, G.; Li, Y.; Wang, Y.; Ball, H.L.; Goldsmith, E.J.; Orth, K. Yersinia YopJ Acetylates and Inhibits Kinase Activation by Blocking Phosphorylation. Science 2006, 312, 1211–1214. [Google Scholar] [CrossRef] [PubMed]
- Orth, K.; Palmer, L.E.; Bao, Z.Q.; Stewart, S.; Rudolph, A.E.; Bliska, J.B.; Dixon, J.E. Inhibition of the Mitogen-Activated Protein Kinase Kinase Superfamily by a Yersinia Effector. Science 1999, 285, 1920–1923. [Google Scholar] [CrossRef]
- Philip, N.H.; Dillon, C.P.; Snyder, A.G.; Fitzgerald, P.; Wynosky-Dolfi, M.A.; Zwack, E.E.; Hu, B.; Fitzgerald, L.; Mauldin, E.A.; Copenhaver, A.M.; et al. Caspase-8 Mediates Caspase-1 Processing and Innate Immune Defense in Response to Bacterial Blockade of NF-κB and MAPK Signaling. Proc. Natl. Acad. Sci. USA 2014, 111, 7385–7390. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.W.; Demarco, B.; Heilig, R.; Shkarina, K.; Boettcher, A.; Farady, C.J.; Pelczar, P.; Broz, P. Extrinsic and Intrinsic Apoptosis Activate Pannexin-1 to Drive NLRP3 Inflammasome Assembly. EMBO J. 2019, 38. [Google Scholar] [CrossRef] [PubMed]
- Schwarzer, R.; Jiao, H.; Wachsmuth, L.; Tresch, A.; Pasparakis, M. FADD and Caspase-8 Regulate Gut Homeostasis and Inflammation by Controlling MLKL- and GSDMD-Mediated Death of Intestinal Epithelial Cells. Immunity 2020, 52, 978–993.e6. [Google Scholar] [CrossRef]
- Dikic, I.; Elazar, Z. Mechanism and Medical Implications of Mammalian Autophagy. Nat. Rev. Mol. Cell Biol. 2018, 19, 349–364. [Google Scholar] [CrossRef]
- Pan, H.; Chen, L.; Xu, Y.; Han, W.; Lou, F.; Fei, W.; Liu, S.; Jing, Z.; Sui, X. Autophagy-Associated Immune Responses and Cancer Immunotherapy. Oncotarget 2016, 7, 21235–21246. [Google Scholar] [CrossRef]
- Zhong, Z.; Sanchez-Lopez, E.; Karin, M. Autophagy, Inflammation, and Immunity: A Troika Governing Cancer and its Treatment. Cell 2016, 166, 288–298. [Google Scholar] [CrossRef]
- Martinez-Lopez, N.; Athonvarangkul, D.; Singh, R. Autophagy and Aging. Adv. Exp. Med. Biol. 2015, 847, 73–87. [Google Scholar]
- Perrotta, C.; Cattaneo, M.G.; Molteni, R.; De Palma, C. Autophagy in the Regulation of Tissue Differentiation and Homeostasis. Front. Cell. Dev. Biol. 2020, 8, 602901. [Google Scholar] [CrossRef] [PubMed]
- Tsapras, P.; Nezis, I.P. Caspase Involvement in Autophagy. Cell Death Differ. 2017, 24, 1369–1379. [Google Scholar] [CrossRef]
- Qian, M.; Fang, X.; Wang, X. Autophagy and Inflammation. Clin. Transl. Med. 2017, 6, 24. [Google Scholar] [CrossRef] [PubMed]
- Pu, Q.; Gan, C.; Li, R.; Li, Y.; Tan, S.; Li, X.; Wei, Y.; Lan, L.; Deng, X.; Liang, H.; et al. Atg7 Deficiency Intensifies Inflammasome Activation and Pyroptosis in Pseudomonas Sepsis. J. Immunol. 2017, 198, 3205–3213. [Google Scholar] [CrossRef]
- Saitoh, T.; Fujita, N.; Jang, M.H.; Uematsu, S.; Yang, B.G.; Satoh, T.; Omori, H.; Noda, T.; Yamamoto, N.; Komatsu, M.; et al. Loss of the Autophagy Protein Atg16L1 Enhances Endotoxin-Induced IL-1beta Production. Nature 2008, 456, 264–268. [Google Scholar] [CrossRef] [PubMed]
- Crisan, T.O.; Plantinga, T.S.; van de Veerdonk, F.L.; Farcas, M.F.; Stoffels, M.; Kullberg, B.J.; van der Meer, J.W.; Joosten, L.A.; Netea, M.G. Inflammasome-Independent Modulation of Cytokine Response by Autophagy in Human Cells. PLoS ONE 2011, 6, e18666. [Google Scholar] [CrossRef] [PubMed]
- Harris, J.; Hartman, M.; Roche, C.; Zeng, S.G.; O’Shea, A.; Sharp, F.A.; Lambe, E.M.; Creagh, E.M.; Golenbock, D.T.; Tschopp, J.; et al. Autophagy Controls IL-1beta Secretion by Targeting Pro-IL-1beta for Degradation. J. Biol. Chem. 2011, 286, 9587–9597. [Google Scholar] [CrossRef]
- Bell, B.D.; Leverrier, S.; Weist, B.M.; Newton, R.H.; Arechiga, A.F.; Luhrs, K.A.; Morrissette, N.S.; Walsh, C.M. FADD and Caspase-8 Control the Outcome of Autophagic Signaling in Proliferating T Cells. Proc. Natl. Acad. Sci. USA 2008, 105, 16677–16682. [Google Scholar] [CrossRef]
- You, M.; Savaraj, N.; Kuo, M.T.; Wangpaichitr, M.; Varona-Santos, J.; Wu, C.; Nguyen, D.M.; Feun, L. TRAIL Induces Autophagic Protein Cleavage through Caspase Activation in Melanoma Cell Lines Under Arginine Deprivation. Mol. Cell. Biochem. 2013, 374, 181–190. [Google Scholar] [CrossRef] [PubMed]
- Wirawan, E.; Vande Walle, L.; Kersse, K.; Cornelis, S.; Claerhout, S.; Vanoverberghe, I.; Roelandt, R.; De Rycke, R.; Verspurten, J.; Declercq, W.; et al. Caspase-Mediated Cleavage of Beclin-1 Inactivates Beclin-1-Induced Autophagy and Enhances Apoptosis by Promoting the Release of Proapoptotic Factors from Mitochondria. Cell. Death Dis. 2010, 1, e18. [Google Scholar] [CrossRef]
- Oral, O.; Oz-Arslan, D.; Itah, Z.; Naghavi, A.; Deveci, R.; Karacali, S.; Gozuacik, D. Cleavage of Atg3 Protein by Caspase-8 Regulates Autophagy during Receptor-Activated Cell Death. Apoptosis 2012, 17, 810–820. [Google Scholar] [CrossRef] [PubMed]
- Laussmann, M.A.; Passante, E.; Dussmann, H.; Rauen, J.A.; Wurstle, M.L.; Delgado, M.E.; Devocelle, M.; Prehn, J.H.; Rehm, M. Proteasome Inhibition can Induce an Autophagy-Dependent Apical Activation of Caspase-8. Cell Death Differ. 2011, 18, 1584–1597. [Google Scholar] [CrossRef] [PubMed]
- Tang, D.; Kepp, O.; Kroemer, G. Ferroptosis Becomes Immunogenic: Implications for Anticancer Treatments. Oncoimmunology 2020, 10, 1862949. [Google Scholar]
- Li, J.; Cao, F.; Yin, H.L.; Huang, Z.J.; Lin, Z.T.; Mao, N.; Sun, B.; Wang, G. Ferroptosis: Past, Present and Future. Cell. Death Dis. 2020, 11, 88. [Google Scholar] [CrossRef] [PubMed]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An Iron-Dependent Form of Nonapoptotic Cell Death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef]
- Zhou, Y.; Shen, Y.; Chen, C.; Sui, X.; Yang, J.; Wang, L.; Zhou, J. The Crosstalk between Autophagy and Ferroptosis: What can we Learn to Target Drug Resistance in Cancer? Cancer Biol. Med. 2019, 16, 630–646. [Google Scholar]
- Hong, S.H.; Lee, D.H.; Lee, Y.S.; Jo, M.J.; Jeong, Y.A.; Kwon, W.T.; Choudry, H.A.; Bartlett, D.L.; Lee, Y.J. Molecular Crosstalk between Ferroptosis and Apoptosis: Emerging Role of ER Stress-Induced p53-Independent PUMA Expression. Oncotarget 2017, 8, 115164–115178. [Google Scholar] [CrossRef]
- Muller, T.; Dewitz, C.; Schmitz, J.; Schroder, A.S.; Brasen, J.H.; Stockwell, B.R.; Murphy, J.M.; Kunzendorf, U.; Krautwald, S. Necroptosis and Ferroptosis are Alternative Cell Death Pathways that Operate in Acute Kidney Failure. Cell Mol. Life Sci. 2017, 74, 3631–3645. [Google Scholar] [CrossRef] [PubMed]
- Gunther, C.; Buchen, B.; He, G.W.; Hornef, M.; Torow, N.; Neumann, H.; Wittkopf, N.; Martini, E.; Basic, M.; Bleich, A.; et al. Caspase-8 Controls the Gut Response to Microbial Challenges by Tnf-Alpha-Dependent and Independent Pathways. Gut 2015, 64, 601–610. [Google Scholar] [CrossRef] [PubMed]
- Weng, D.; Marty-Roix, R.; Ganesan, S.; Proulx, M.K.; Vladimer, G.I.; Kaiser, W.J.; Mocarski, E.S.; Pouliot, K.; Chan, F.K.; Kelliher, M.A.; et al. Caspase-8 and RIP Kinases Regulate Bacteria-Induced Innate Immune Responses and Cell Death. Proc. Natl. Acad. Sci. USA 2014, 111, 7391–7396. [Google Scholar] [CrossRef] [PubMed]
- Zewinger, S.; Reiser, J.; Jankowski, V.; Alansary, D.; Hahm, E.; Triem, S.; Klug, M.; Schunk, S.J.; Schmit, D.; Kramann, R.; et al. Apolipoprotein C3 Induces Inflammation and Organ Damage by Alternative Inflammasome Activation. Nat. Immunol. 2020, 21, 30–41. [Google Scholar] [CrossRef]
- Donado, C.A.; Cao, A.B.; Simmons, D.P.; Croker, B.A.; Brennan, P.J.; Brenner, M.B. A Two-Cell Model for IL-1beta Release Mediated by Death-Receptor Signaling. Cell. Rep. 2020, 31, 107466. [Google Scholar] [CrossRef]
- Newton, K.; Wickliffe, K.E.; Maltzman, A.; Dugger, D.L.; Reja, R.; Zhang, Y.; Roose-Girma, M.; Modrusan, Z.; Sagolla, M.S.; Webster, J.D.; et al. Activity of Caspase-8 Determines Plasticity between Cell Death Pathways. Nature 2019, 575, 679–682. [Google Scholar] [CrossRef]
- Demarco, B.; Grayczyk, J.P.; Bjanes, E.; Le Roy, D.; Tonnus, W.; Assenmacher, C.A.; Radaelli, E.; Fettrelet, T.; Mack, V.; Linkermann, A.; et al. Caspase-8-Dependent Gasdermin D Cleavage Promotes Antimicrobial Defense but Confers Susceptibility to TNF-Induced Lethality. Sci. Adv. 2020, 6. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Roles | Comments | References |
|---|---|---|
| Anti-inflammation | ||
| Inhibition of necroptosis | Caspase-8 limits necroptosis by cleavage of RIPK1, RIPK3 and CYLD. | [10,11,12] |
| Caspase-8 maintains the gut barrier by preventing necroptosis of infected cells | [93,109,132] | |
| Caspase-8 deficiency or lack of its activity promotes TNF-induced necroptosis. | [38,39,40] | |
| Inhibition of inflammasome | The deficiency of caspase-8 causes LPS-induced inflammasome activation mediated by RIPK1-RIPK3-MLKL | [54,55] |
| Inhibition of inflammatory signaling | The expression of non-cleavable RIPK1 by caspase-8 in human results in auto-inflammatory disease | [62,63] |
| The deficiency of caspase-8 can facilitate the activation of proinflammatory genes in a necroptosis independent manner | [61] | |
| Caspase-8 suppresses the cytosolic RNA sensor RIG-I-induced proinflammatory gene expression through limiting RIPK1 function. | [60] | |
| Caspase-8 can suppress an inflammatory pathway downstream of cytosolic innate DNA receptor in cultured keratinocytes | [42] | |
| Proinflammation | ||
| Inflammasome activation | Internalized bacteria induce caspase-8-mediated NLRP3 inflammasome activation | [89] |
| Caspase-8 is required for pathogens (Yersinia, Salmonella), YopJ, cytoplasmic dsRNA, or apolipoprotein C3-induced NLRP3 inflammasome activation | [77,107,133,134] | |
| LPS triggering in cIAP KO cells induces caspase-8 promoted NLRP3 activation | [91] | |
| IL-1β processing | TLR stimulation under stress conditions (cIAP inhibition, a chemotherapeutic drug, ER stress) leads to caspase-8-dependent IL-1β processing | [92,101,102] |
| Fas or Dectin-1 activation induces caspase-8-mediated IL-1β processing | [95,135] | |
| In the EAE mouse model, caspase-8 processes IL-1β by forming IRAKM-casp8-ASC complex in microglia | [98] | |
| Pyroptosis induction | Catalytic dead-caspase-8 causes pyroptosis -dependent perinatal mouse death. | [58,136] |
| Yersinia infection or TAK1 blocking trigger caspase-8-mediated GSDMD processing and pyroptosis induction | [103,104,137] | |
| NF-κB activation, cytokine production | Caspase-8 is required for NF-κB activation and cytokine production by antigen receptor, TLR triggering or TRAIL stimulation. | [55,66,70,78,81] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Han, J.-H.; Park, J.; Kang, T.-B.; Lee, K.-H. Regulation of Caspase-8 Activity at the Crossroads of Pro-Inflammation and Anti-Inflammation. Int. J. Mol. Sci. 2021, 22, 3318. https://doi.org/10.3390/ijms22073318
Han J-H, Park J, Kang T-B, Lee K-H. Regulation of Caspase-8 Activity at the Crossroads of Pro-Inflammation and Anti-Inflammation. International Journal of Molecular Sciences. 2021; 22(7):3318. https://doi.org/10.3390/ijms22073318
Chicago/Turabian StyleHan, Jun-Hyuk, Jooho Park, Tae-Bong Kang, and Kwang-Ho Lee. 2021. "Regulation of Caspase-8 Activity at the Crossroads of Pro-Inflammation and Anti-Inflammation" International Journal of Molecular Sciences 22, no. 7: 3318. https://doi.org/10.3390/ijms22073318
APA StyleHan, J.-H., Park, J., Kang, T.-B., & Lee, K.-H. (2021). Regulation of Caspase-8 Activity at the Crossroads of Pro-Inflammation and Anti-Inflammation. International Journal of Molecular Sciences, 22(7), 3318. https://doi.org/10.3390/ijms22073318