
Human α-Galactosidase A Mutants: Priceless Tools to Develop Novel Therapies for Fabry Disease

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Fabry Disease
2. The Human Gla Enzyme
2.1. Structure of GLA Enzyme
2.2. GLA Synthesis and Trafficking
2.3. Catalytic Mechanism of GLA
3. Current Treatments For FD
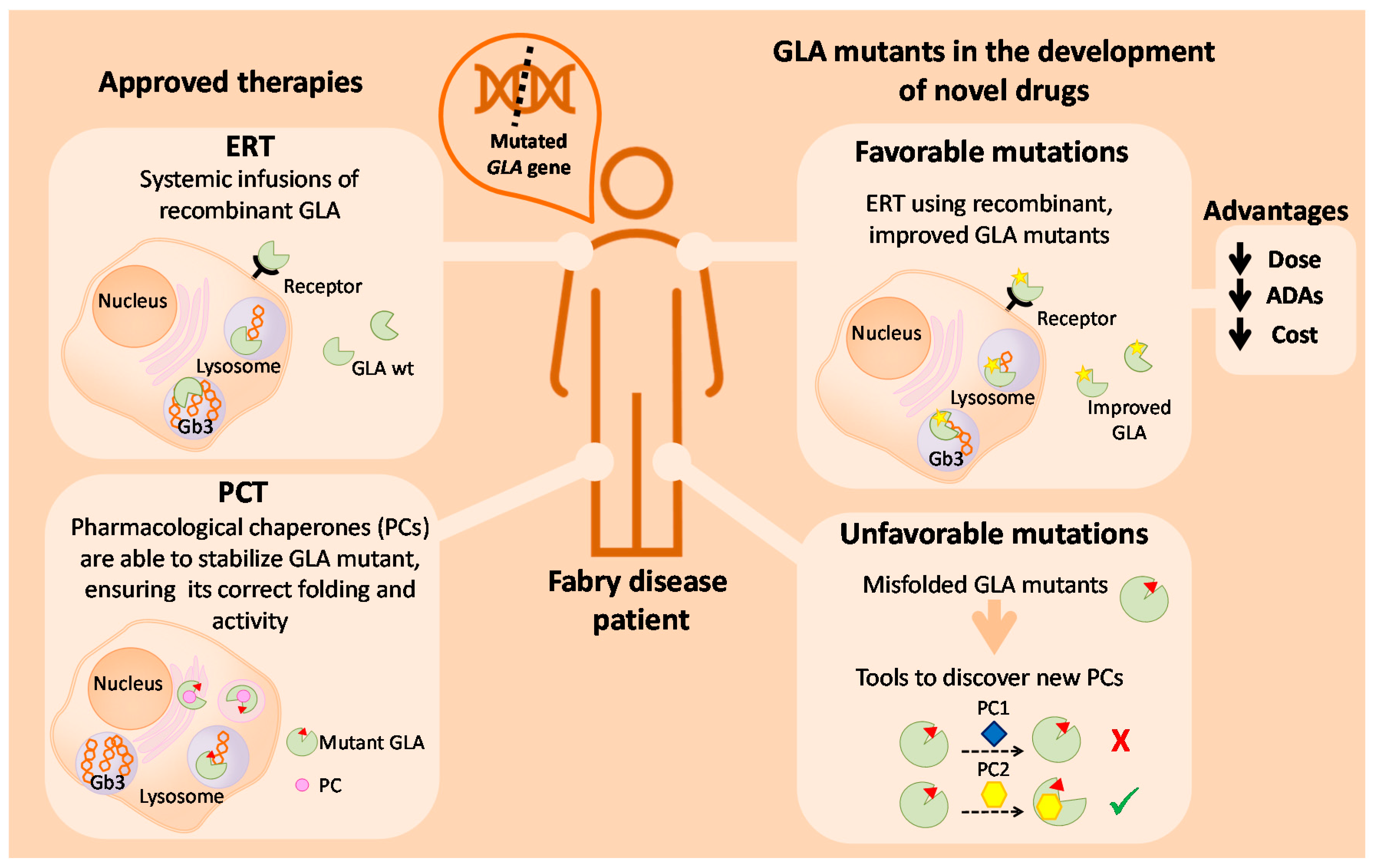
3.1. Enzyme Replacement Therapy
3.2. Pharmacological Chaperones
4. GLA Mutations
4.1. Most Frequent GLA Mutant Variants
4.2. Most Frequently Mutated Residues
4.3. Nonsense Mutations
4.4. Location-Dependent Biological Effect of Mutations
4.5. GLA Mutants with Low Activity and/or Stability
Mutants as Tools to Develop Pharmacological Chaperones
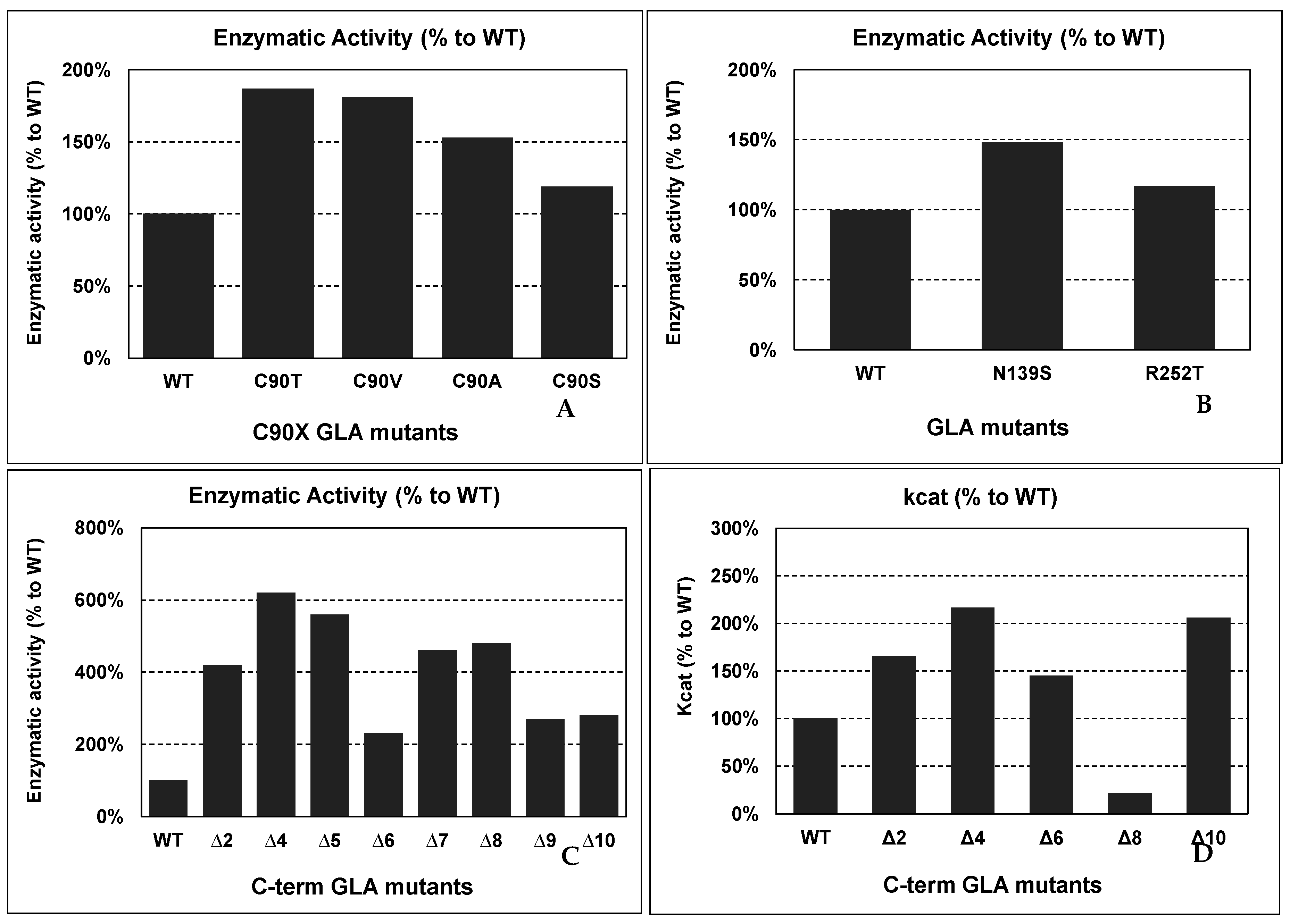
4.6. GLA Mutants with Improved Activity and/or Stability
Putative Applications of Improved Mutants in Innovative FD Therapies
4.7. Other Modifications in Recombinant GLAs in Innovative FD Therapies
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Acknowledgments
Conflicts of Interest
References
- Germain, D.P. Fabry disease. Orphanet J. Rare Dis. 2010, 5, 30. [Google Scholar] [CrossRef] [PubMed]
- Bernardes, T.P.; Foresto, R.D.; Kirsztajn, G.M. Fabry disease: Genetics, pathology, and treatment. Rev. Assoc. Med. Bras. 2020, 66, s10–s16. [Google Scholar] [CrossRef] [PubMed]
- Stenson, P.D.; Ball, E.V.; Mort, M.; Phillips, A.D.; Shiel, J.A.; Thomas, N.S.T.; Abeysinghe, S.; Krawczak, M.; Cooper, D.N. Human Gene Mutation Database (HGMD®): 2003 update: HGMD 2003 UPDATE. Hum. Mutat. 2003, 21, 577–581. [Google Scholar] [CrossRef]
- Simonetta, I.; Tuttolomondo, A.; Di Chiara, T.; Miceli, S.; Vogiatzis, D.; Corpora, F.; Pinto, A. Genetics and Gene Therapy of Anderson-Fabry Disease. Curr. Gene Ther. 2018, 18, 96–106. [Google Scholar] [CrossRef]
- Sakuraba, H.; Oshima, A.; Fukuhara, Y.; Shimmoto, M.; Nagao, Y.; Bishop, D.F.; Desnick, R.J.; Suzuki, Y. Identification of point mutations in the alpha-galactosidase A gene in classical and atypical hemizygotes with Fabry disease. Am. J. Hum. Genet. 1990, 47, 784–789. [Google Scholar] [PubMed]
- Andrade, J.; Waters, P.J.; Singh, R.S.; Levin, A.; Toh, B.-C.; Vallance, H.D.; Sirrs, S. Screening for Fabry disease in patients with chronic kidney disease: Limitations of plasma alpha-galactosidase assay as a screening test. Clin. J. Am. Soc. Nephrol. 2008, 3, 139–145. [Google Scholar] [CrossRef] [PubMed]
- Beck, M. The Mainz Severity Score Index (MSSI): Development and validation of a system for scoring the signs and symptoms of Fabry disease. Acta Paediatr. 2006, 95, 43–46. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.J.; Kanack, A.J.; Dahms, N.M. Progress in the understanding and treatment of Fabry disease. Biochim. Biophys. Acta Gen. Subj. 2020, 1864, 129437. [Google Scholar] [CrossRef]
- Havndrup, O.; Christiansen, M.; Stoevring, B.; Jensen, M.; Hoffman-Bang, J.; Andersen, P.S.; Hasholt, L.; Nørremølle, A.; Feldt-Rasmussen, U.; Køber, L.; et al. Fabry disease mimicking hypertrophic cardiomyopathy: Genetic screening needed for establishing the diagnosis in women. Eur. J. Heart Fail. 2010, 12, 535–540. [Google Scholar] [CrossRef]
- Aerts, J.M.; Groener, J.E.; Kuiper, S.; Donker-Koopman, W.E.; Strijland, A.; Ottenhoff, R.; van Roomen, C.; Mirzaian, M.; Wijburg, F.A.; Linthorst, G.E.; et al. Elevated globotriaosylsphingosine is a hallmark of Fabry disease. Proc. Natl. Acad. Sci. USA 2008, 105, 2812–2817. [Google Scholar] [CrossRef]
- Bichet, D.G.; Aerts, J.M.; Auray-Blais, C.; Maruyama, H.; Mehta, A.B.; Skuban, N.; Krusinska, E.; Schiffmann, R. Assessment of plasma lyso-Gb(3) for clinical monitoring of treatment response in migalastat-treated patients with Fabry disease. Genet. Med. 2021, 23, 192–201. [Google Scholar] [CrossRef]
- Arends, M.; Biegstraaten, M.; Hughes, D.A.; Mehta, A.; Elliott, P.M.; Oder, D.; Watkinson, O.T.; Vaz, F.M.; van Kuilenburg, A.B.P.; Wanner, C.; et al. Retrospective study of long-term outcomes of enzyme replacement therapy in Fabry disease: Analysis of prognostic factors. PLoS ONE 2017, 12, e0182379. [Google Scholar] [CrossRef]
- Cammarata, G.; Scalia, S.; Colomba, P.; Zizzo, C.; Pisani, A.; Riccio, E.; Montalbano, M.; Alessandro, R.; Giordano, A.; Duro, G. A pilot study of circulating microRNAs as potential biomarkers of Fabry disease. Oncotarget 2018, 9, 27333–27345. [Google Scholar] [CrossRef] [PubMed]
- Boutin, M.; Auray-Blais, C. Metabolomic Discovery of Novel Urinary Galabiosylceramide Analogs as Fabry Disease Biomarkers. J. Am. Soc. Mass Spectrom. 2015, 26, 499–510. [Google Scholar] [CrossRef] [PubMed]
- Matafora, V.; Cuccurullo, M.; Beneduci, A.; Petrazzuolo, O.; Simeone, A.; Anastasio, P.; Mignani, R.; Feriozzi, S.; Pisani, A.; Comotti, C.; et al. Early markers of Fabry disease revealed by proteomics. Mol. Biosyst. 2015, 11, 1543–1551. [Google Scholar] [CrossRef] [PubMed]
- Bishop, D.F.; Kornreich, R.; Desnick, R.J. Structural organization of the human alpha-galactosidase A gene: Further evidence for the absence of a 3’ untranslated region. Proc. Natl. Acad. Sci. USA 1988, 85, 3903–3907. [Google Scholar] [CrossRef]
- Garman, S.C.; Garboczi, D.N. The molecular defect leading to Fabry disease: Structure of human alpha-galactosidase. J. Mol. Biol. 2004, 337, 319–335. [Google Scholar] [CrossRef]
- Ioannou, Y.A.; Zeidner, K.M.; Grace, M.E.; Desnick, R.J. Human alpha-galactosidase A: Glycosylation site 3 is essential for enzyme solubility. Biochem. J. 1998, 332, 789–797. [Google Scholar] [CrossRef]
- Matsuura, F.; Ohta, M.; Ioannou, Y.A.; Desnick, R.J. Human -galactosidase A: Characterization of the N-linked oligosaccharides on the intracellular and secreted glycoforms overexpressed by Chinese hamster ovary cells. Glycobiology 1998, 8, 329–339. [Google Scholar] [CrossRef]
- Poole, L.B. The basics of thiols and cysteines in redox biology and chemistry. Free Radic. Biol. Med. 2015, 80, 148–157. [Google Scholar] [CrossRef]
- Nakaniwa, T.; Fukada, H.; Inoue, T.; Gouda, M.; Nakai, R.; Kirii, Y.; Adachi, M.; Tamada, T.; Segawa, S.; Kuroki, R.; et al. Seven Cysteine-Deficient Mutants Depict the Interplay between Thermal and Chemical Stabilities of Individual Cysteine Residues in Mitogen-Activated Protein Kinase c-Jun N-Terminal Kinase 1. Biochemistry 2012, 51, 8410–8421. [Google Scholar] [CrossRef]
- Saftig, P. Physiology of the lysosome. In Fabry Disease: Perspectives from 5 Years of FOS; Mehta, A., Beck, M., Sunder-Plassmann, G., Eds.; Oxford PharmaGenesis: Oxford, UK, 2006. [Google Scholar]
- Koshland, D.E. Stereochemistry and the Mechanism of Enzymatic Reactions. Biol. Rev. 1953, 28, 416–436. [Google Scholar] [CrossRef]
- Guce, A.I.; Clark, N.E.; Salgado, E.N.; Ivanen, D.R.; Kulminskaya, A.A.; Brumer, H.; Garman, S.C. Catalytic Mechanism of Human α-Galactosidase. J. Biol. Chem. 2010, 285, 3625–3632. [Google Scholar] [CrossRef] [PubMed]
- Ohashi, T. Current status and future prospect of enzyme replacement therapy for Fabry disease. Rinsho Shinkeigaku 2019, 59, 335–338. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Stappers, F.; Scharnetzki, D.; Schmitz, B.; Manikowski, D.; Brand, S.; Grobe, K.; Lenders, M.; Brand, E. Neutralising anti-drug antibodies in Fabry disease can inhibit endothelial enzyme uptake and activity. J. Inherit. Metab. Dis. 2020, 43, 334–347. [Google Scholar] [CrossRef]
- Liguori, L.; Monticelli, M.; Allocca, M.; Hay Mele, B.; Lukas, J.; Cubellis, M.V.; Andreotti, G. Pharmacological Chaperones: A Therapeutic Approach for Diseases Caused by Destabilizing Missense Mutations. Int. J. Mol. Sci. 2020, 21, 489. [Google Scholar] [CrossRef]
- Ishii, S.; Chang, H.-H.; Kawasaki, K.; Yasuda, K.; Wu, H.-L.; Garman, S.C.; Fan, J.-Q. Mutant α-galactosidase A enzymes identified in Fabry disease patients with residual enzyme activity: Biochemical characterization and restoration of normal intracellular processing by 1-deoxygalactonojirimycin. Biochem. J. 2007, 406, 285–295. [Google Scholar] [CrossRef]
- Lenders, M.; Stappers, F.; Brand, E. In Vitro and In Vivo Amenability to Migalastat in Fabry Disease. Mol. Ther. Methods Clin. Dev. 2020, 19, 24–34. [Google Scholar] [CrossRef]
- Okumiya, T.; Ishii, S.; Takenaka, T.; Kase, R.; Kamei, S.; Sakuraba, H.; Suzuki, Y. Galactose Stabilizes Various Missense Mutants of α-Galactosidase in Fabry Disease. Biochem. Biophys. Res. Commun. 1995, 214, 1219–1224. [Google Scholar] [CrossRef]
- Fan, J.-Q.; Ishii, S.; Asano, N.; Suzuki, Y. Accelerated transport and maturation of lysosomal α–galactosidase A in Fabry lymphoblasts by an enzyme inhibitor. Nat. Med. 1999, 5, 112–115. [Google Scholar] [CrossRef]
- Asano, N.; Ishii, S.; Ikeda, K.; Kizu, H.; Yasuda, K.; Kato, A.; Martin, O.R.; Fan, J.-Q. In vitro inhibition and intracellular enhancement of lysosomal α-galactosidase A activity in Fabry lymphoblasts by 1-deoxygalactonojirimycin and its derivatives: Enhancement of α-Gal A in Fabry lymphoblasts. Eur. J. Biochem. 2000, 267, 4179–4186. [Google Scholar] [CrossRef]
- Guce, A.I.; Clark, N.E.; Rogich, J.J.; Garman, S.C. The Molecular Basis of Pharmacological Chaperoning in Human α-Galactosidase. Chem. Biol. 2011, 18, 1521–1526. [Google Scholar] [CrossRef] [PubMed]
- van der Tol, L.; Smid, B.E.; Poorthuis, B.J.H.M.; Biegstraaten, M.; Deprez, R.H.L.; Linthorst, G.E.; Hollak, C.E.M. A systematic review on screening for Fabry disease: Prevalence of individuals with genetic variants of unknown significance. J. Med. Genet. 2014, 51, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Mehta, A.; Hughes, D.A. Fabry Disease. In GeneReviews®; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Stephens, K., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Germain, D.P.; Poenaru, L. Fabry Disease: Identification of Novel Alpha-Galactosidase A Mutations and Molecular Carrier Detection by Use of Fluorescent Chemical Cleavage of Mismatches. Biochem. Biophys. Res. Commun. 1999, 257, 708–713. [Google Scholar] [CrossRef]
- Qiu, H.; Honey, D.M.; Kingsbury, J.S.; Park, A.; Boudanova, E.; Wei, R.R.; Pan, C.Q.; Edmunds, T. Impact of cysteine variants on the structure, activity, and stability of recombinant human α-galactosidase A: Cysteine Variants of Human α-Galactosidase A. Protein Sci. 2015, 24, 1401–1411. [Google Scholar] [CrossRef]
- Lee, B.H.; Heo, S.H.; Kim, G.-H.; Park, J.-Y.; Kim, W.-S.; Kang, D.-H.; Choe, K.H.; Kim, W.-H.; Yang, S.H.; Yoo, H.-W. Mutations of the GLA gene in Korean patients with Fabry disease and frequency of the E66Q allele as a functional variant in Korean newborns. J. Hum. Genet. 2010, 55, 512–517. [Google Scholar] [CrossRef] [PubMed]
- Serebrinsky, G.; Calvo, M.; Fernandez, S.; Saito, S.; Ohno, K.; Wallace, E.; Warnock, D.; Sakuraba, H.; Politei, J. Late onset variants in Fabry disease: Results in high risk population screenings in Argentina. Mol. Genet. Metab. Rep. 2015, 4, 19–24. [Google Scholar] [CrossRef] [PubMed]
- Meng, Y.; Zhang, W.-M.; Shi, H.-P.; Wei, M.; Huang, S.-Z. Clinical manifestations and mutation study in 16 Chinese patients with Fabry disease. Zhonghua Yi Xue Za Zhi 2010, 90, 551–554. [Google Scholar]
- Garman, S.C.; Garboczi, D.N. Structural basis of Fabry disease. Mol. Genet. Metab. 2002, 77, 3–11. [Google Scholar] [CrossRef]
- Guffon, N.; Froissart, R.; Chevalier-Porst, F.; Maire, I. Mutation analysis in 11 French patients with Fabry disease. Hum. Mutat. 1998, 11, S288–S290. [Google Scholar] [CrossRef]
- Ebrahim, H.Y.; Baker, R.J.; Mehta, A.B.; Hughes, D.A. Functional analysis of variant lysosomal acid glycosidases of Anderson-Fabry and Pompe disease in a human embryonic kidney epithelial cell line (HEK 293 T). J. Inherit. Metab. Dis. 2012, 35, 325–334. [Google Scholar] [CrossRef]
- Nakao, S.; Takenaka, T.; Maeda, M.; Kodama, C.; Tanaka, A.; Tahara, M.; Yoshida, A.; Kuriyama, M.; Hayashibe, H.; Sakuraba, H.; et al. An Atypical Variant of Fabry’s Disease in Men with Left Ventricular Hypertrophy. N. Engl. J. Med. 1995, 333, 288–293. [Google Scholar] [CrossRef]
- Germain, D.P.; Brand, E.; Burlina, A.; Cecchi, F.; Garman, S.C.; Kempf, J.; Laney, D.A.; Linhart, A.; Maródi, L.; Nicholls, K.; et al. Phenotypic characteristics of the p.Asn215Ser (p.N215S) GLA mutation in male and female patients with Fabry disease: A multicenter Fabry Registry study. Mol. Genet. Genom. Med. 2018, 6, 492–503. [Google Scholar] [CrossRef] [PubMed]
- Meghdari, M.; Gao, N.; Abdullahi, A.; Stokes, E.; Calhoun, D.H. Carboxyl-Terminal Truncations Alter the Activity of the Human α-Galactosidase A. PLoS ONE 2015, 10, e0118341. [Google Scholar] [CrossRef] [PubMed]
- Miyamura, N.; Araki, E.; Matsuda, K.; Yoshimura, R.; Furukawa, N.; Tsuruzoe, K.; Shirotani, T.; Kishikawa, H.; Yamaguchi, K.; Shichiri, M. A carboxy-terminal truncation of human alpha-galactosidase A in a heterozygous female with Fabry disease and modification of the enzymatic activity by the carboxy-terminal domain. Increased, reduced, or absent enzyme activity depending on number of amino. J. Clin. Investig. 1996, 98, 1809–1817. [Google Scholar] [CrossRef] [PubMed]
- Garman, S.C. Structure-function relationships in α-galactosidase A: Structure-function relationships in α-galactosidase A. Acta Paediatr. 2007, 96, 6–16. [Google Scholar] [CrossRef]
- Wu, X.; Katz, E.; Valle, M.C.D.; Mascioli, K.; Flanagan, J.J.; Castelli, J.P.; Schiffmann, R.; Boudes, P.; Lockhart, D.J.; Valenzano, K.J.; et al. A pharmacogenetic approach to identify mutant forms of α-galactosidase a that respond to a pharmacological chaperone for Fabry disease. Hum. Mutat. 2011, 32, 965–977. [Google Scholar] [CrossRef] [PubMed]
- Citro, V.; Cammisa, M.; Liguori, L.; Cimmaruta, C.; Lukas, J.; Cubellis, M.; Andreotti, G. The Large Phenotypic Spectrum of Fabry Disease Requires Graduated Diagnosis and Personalized Therapy: A Meta-Analysis Can Help to Differentiate Missense Mutations. Int. J. Mol. Sci. 2016, 17, 2010. [Google Scholar] [CrossRef]
- Yu, Y.; Mena-Barragán, T.; Higaki, K.; Johnson, J.L.; Drury, J.E.; Lieberman, R.L.; Nakasone, N.; Ninomiya, H.; Tsukimura, T.; Sakuraba, H.; et al. Molecular Basis of 1-Deoxygalactonojirimycin Arylthiourea Binding to Human α-Galactosidase A: Pharmacological Chaperoning Efficacy on Fabry Disease Mutants. ACS Chem. Biol. 2014, 9, 1460–1469. [Google Scholar] [CrossRef]
- Mena-Barragán, T.; Narita, A.; Matias, D.; Tiscornia, G.; Nanba, E.; Ohno, K.; Suzuki, Y.; Higaki, K.; Garcia Fernández, J.M.; Ortiz Mellet, C. pH-Responsive Pharmacological Chaperones for Rescuing Mutant Glycosidases. Angew. Chem. Int. Ed. 2015, 54, 11696–11700. [Google Scholar] [CrossRef]
- Citro, V.; Peña-García, J.; den-Haan, H.; Pérez-Sánchez, H.; Del Prete, R.; Liguori, L.; Cimmaruta, C.; Lukas, J.; Cubellis, M.V.; Andreotti, G. Identification of an Allosteric Binding Site on Human Lysosomal Alpha-Galactosidase Opens the Way to New Pharmacological Chaperones for Fabry Disease. PLoS ONE 2016, 11, e0165463. [Google Scholar] [CrossRef] [PubMed]
- Hay Mele, B.; Citro, V.; Andreotti, G.; Cubellis, M.V. Drug repositioning can accelerate discovery of pharmacological chaperones. Orphanet J. Rare Dis. 2015, 10, 55. [Google Scholar] [CrossRef]
- Rigat, B.; Mahuran, D. Diltiazem, a L-type Ca2+ channel blocker, also acts as a pharmacological chaperone in Gaucher patient cells. Mol. Genet. Metab. 2009, 96, 225–232. [Google Scholar] [CrossRef] [PubMed]
- Lukas, J.; Pockrandt, A.-M.; Seemann, S.; Sharif, M.; Runge, F.; Pohlers, S.; Zheng, C.; Gläser, A.; Beller, M.; Rolfs, A.; et al. Enzyme Enhancers for the Treatment of Fabry and Pompe Disease. Mol. Ther. 2015, 23, 456–464. [Google Scholar] [CrossRef] [PubMed]
- Maegawa, G.H.B.; Tropak, M.B.; Buttner, J.D.; Rigat, B.A.; Fuller, M.; Pandit, D.; Tang, L.; Kornhaber, G.J.; Hamuro, Y.; Clarke, J.T.R.; et al. Identification and Characterization of Ambroxol as an Enzyme Enhancement Agent for Gaucher Disease. J. Biol. Chem. 2009, 284, 23502–23516. [Google Scholar] [CrossRef] [PubMed]
- Calamini, B.; Silva, M.C.; Madoux, F.; Hutt, D.M.; Khanna, S.; Chalfant, M.A.; Saldanha, S.A.; Hodder, P.; Tait, B.D.; Garza, D.; et al. Small-molecule proteostasis regulators for protein conformational diseases. Nat. Chem. Biol. 2012, 8, 185–196. [Google Scholar] [CrossRef]
- Muntau, A.C.; Leandro, J.; Staudigl, M.; Mayer, F.; Gersting, S.W. Innovative strategies to treat protein misfolding in inborn errors of metabolism: Pharmacological chaperones and proteostasis regulators. J. Inherit. Metab. Dis. 2014, 37, 505–523. [Google Scholar] [CrossRef]
- Seemann, S.; Ernst, M.; Cimmaruta, C.; Struckmann, S.; Cozma, C.; Koczan, D.; Knospe, A.-M.; Haake, L.R.; Citro, V.; Bräuer, A.U.; et al. Proteostasis regulators modulate proteasomal activity and gene expression to attenuate multiple phenotypes in Fabry disease. Biochem. J. 2020, 477, 359–380. [Google Scholar] [CrossRef]
- Mu, T.-W.; Ong, D.S.T.; Wang, Y.-J.; Balch, W.E.; Yates, J.R.; Segatori, L.; Kelly, J.W. Chemical and Biological Approaches Synergize to Ameliorate Protein-Folding Diseases. Cell 2008, 134, 769–781. [Google Scholar] [CrossRef]
- Lukas, J.; Knospe, A.-M.; Seemann, S.; Citro, V.; Cubellis, M.V.; Rolfs, A. In Vitro Enzyme Measurement to Test Pharmacological Chaperone Responsiveness in Fabry and Pompe Disease. J. Vis. Exp. 2017, 130, 56550. [Google Scholar] [CrossRef]
- Cammisa, M.; Correra, A.; Andreotti, G.; Cubellis, M. Fabry_CEP: A tool to identify Fabry mutations responsive to pharmacological chaperones. Orphanet J. Rare Dis. 2013, 8, 111. [Google Scholar] [CrossRef] [PubMed]
- Deegan, P.B. Fabry disease, enzyme replacement therapy and the significance of antibody responses. J. Inherit. Metab. Dis. 2012, 35, 227–243. [Google Scholar] [CrossRef] [PubMed]
- Vedder, A.C.; Breunig, F.; Donker-Koopman, W.E.; Mills, K.; Young, E.; Winchester, B.; Ten Berge, I.J.M.; Groener, J.E.M.; Aerts, J.M.F.G.; Wanner, C.; et al. Treatment of Fabry disease with different dosing regimens of agalsidase: Effects on antibody formation and GL-3. Mol. Genet. Metab. 2008, 94, 319–325. [Google Scholar] [CrossRef] [PubMed]
- Howey, D.C.; Bowsher, R.R.; Brunelle, R.L.; Woodworth, J.R. [Lys(B28), Pro(B29)]-Human Insulin: A Rapidly Absorbed Analogue of Human Insulin. Diabetes 1994, 43, 396–402. [Google Scholar] [CrossRef]
- Lee, K.; Jin, X.; Zhang, K.; Copertino, L.; Andrews, L.; Baker-Malcolm, J.; Geagan, L.; Qiu, H.; Seiger, K.; Barngrover, D.; et al. A biochemical and pharmacological comparison of enzyme replacement therapies for the glycolipid storage disorder Fabry disease. Glycobiology 2003, 13, 305–313. [Google Scholar] [CrossRef]
- Sohn, Y.; Lee, J.M.; Park, H.-R.; Jung, S.-C.; Park, T.H.; Oh, D.-B. Enhanced sialylation and in vivo efficacy of recombinant human α-galactosidase through in vitro glycosylation. BMB Rep. 2013, 46, 157–162. [Google Scholar] [CrossRef]
- Ruderfer, I.; Shulman, A.; Kizhner, T.; Azulay, Y.; Nataf, Y.; Tekoah, Y.; Shaaltiel, Y. Development and Analytical Characterization of Pegunigalsidase Alfa, a Chemically Cross-Linked Plant Recombinant Human α-Galactosidase-A for Treatment of Fabry Disease. Bioconjug. Chem. 2018, 29, 1630–1639. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Modrego, A.; Amaranto, M.; Godino, A.; Mendoza, R.; Barra, J.L.; Corchero, J.L. Human α-Galactosidase A Mutants: Priceless Tools to Develop Novel Therapies for Fabry Disease. Int. J. Mol. Sci. 2021, 22, 6518. https://doi.org/10.3390/ijms22126518
Modrego A, Amaranto M, Godino A, Mendoza R, Barra JL, Corchero JL. Human α-Galactosidase A Mutants: Priceless Tools to Develop Novel Therapies for Fabry Disease. International Journal of Molecular Sciences. 2021; 22(12):6518. https://doi.org/10.3390/ijms22126518
Chicago/Turabian StyleModrego, Andrea, Marilla Amaranto, Agustina Godino, Rosa Mendoza, José Luis Barra, and José Luis Corchero. 2021. "Human α-Galactosidase A Mutants: Priceless Tools to Develop Novel Therapies for Fabry Disease" International Journal of Molecular Sciences 22, no. 12: 6518. https://doi.org/10.3390/ijms22126518
APA StyleModrego, A., Amaranto, M., Godino, A., Mendoza, R., Barra, J. L., & Corchero, J. L. (2021). Human α-Galactosidase A Mutants: Priceless Tools to Develop Novel Therapies for Fabry Disease. International Journal of Molecular Sciences, 22(12), 6518. https://doi.org/10.3390/ijms22126518