
Molecular Pathophysiology of Autosomal Recessive Polycystic Kidney Disease


Abstract
1. Introduction
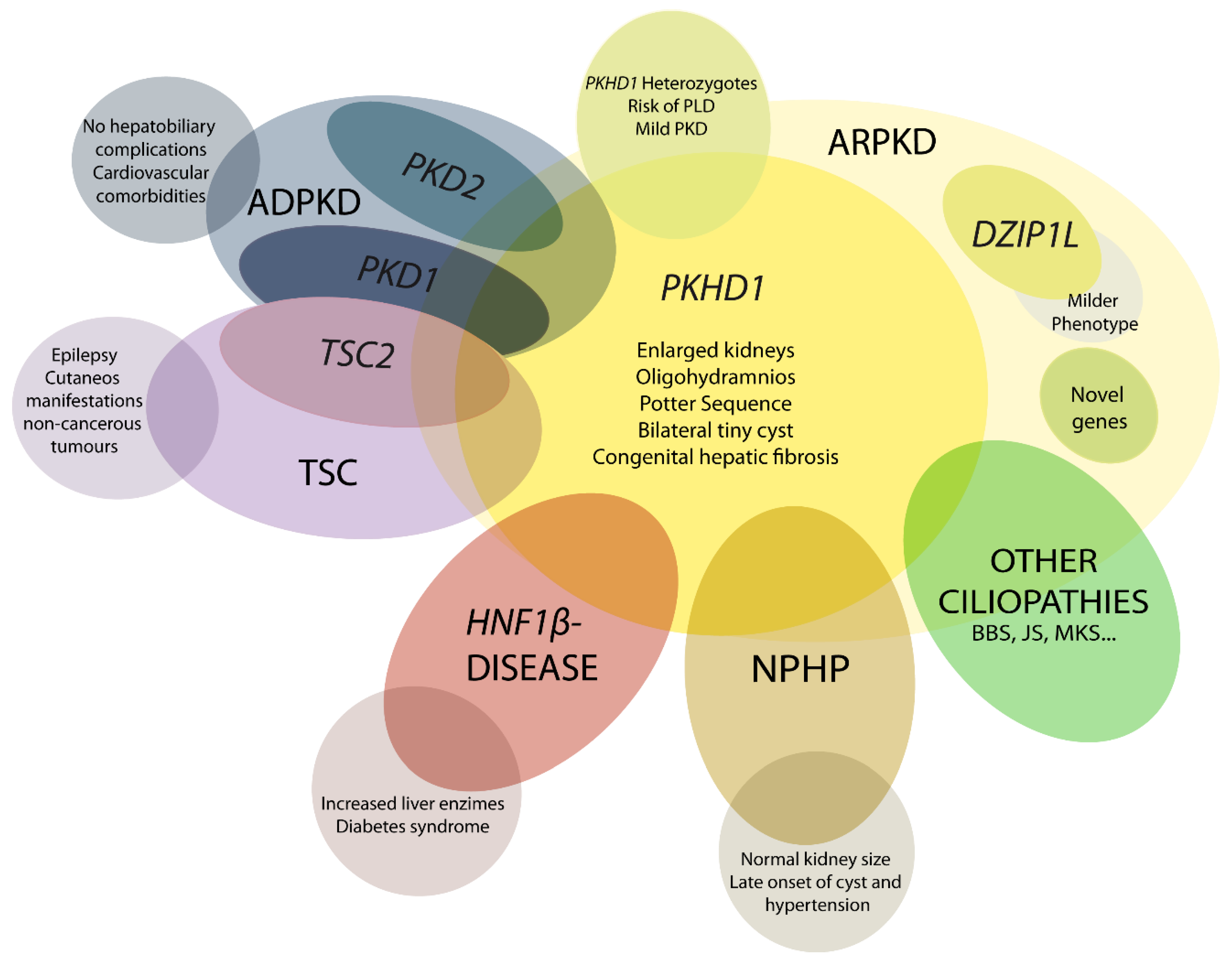
2. Autosomal Recessive Polycystic Kidney Disease Clinical Presentation
3. Diagnosis
4. Differential Diagnosis
4.1. DZIP1L-Related Polycystic Kidney Disease
4.2. Early and Severe Autosomal Dominant Polycystic Kidney Disease (ADPKD)
4.3. Early and Severe PKD due to TSC2-PKD1
4.4. HNF1β-Related Disease
4.5. Nephronophthisis (NPHP)
4.6. Mutations in Other Ciliary Genes
5. Genetics of ARPKD
Genotype-Phenotype Correlation
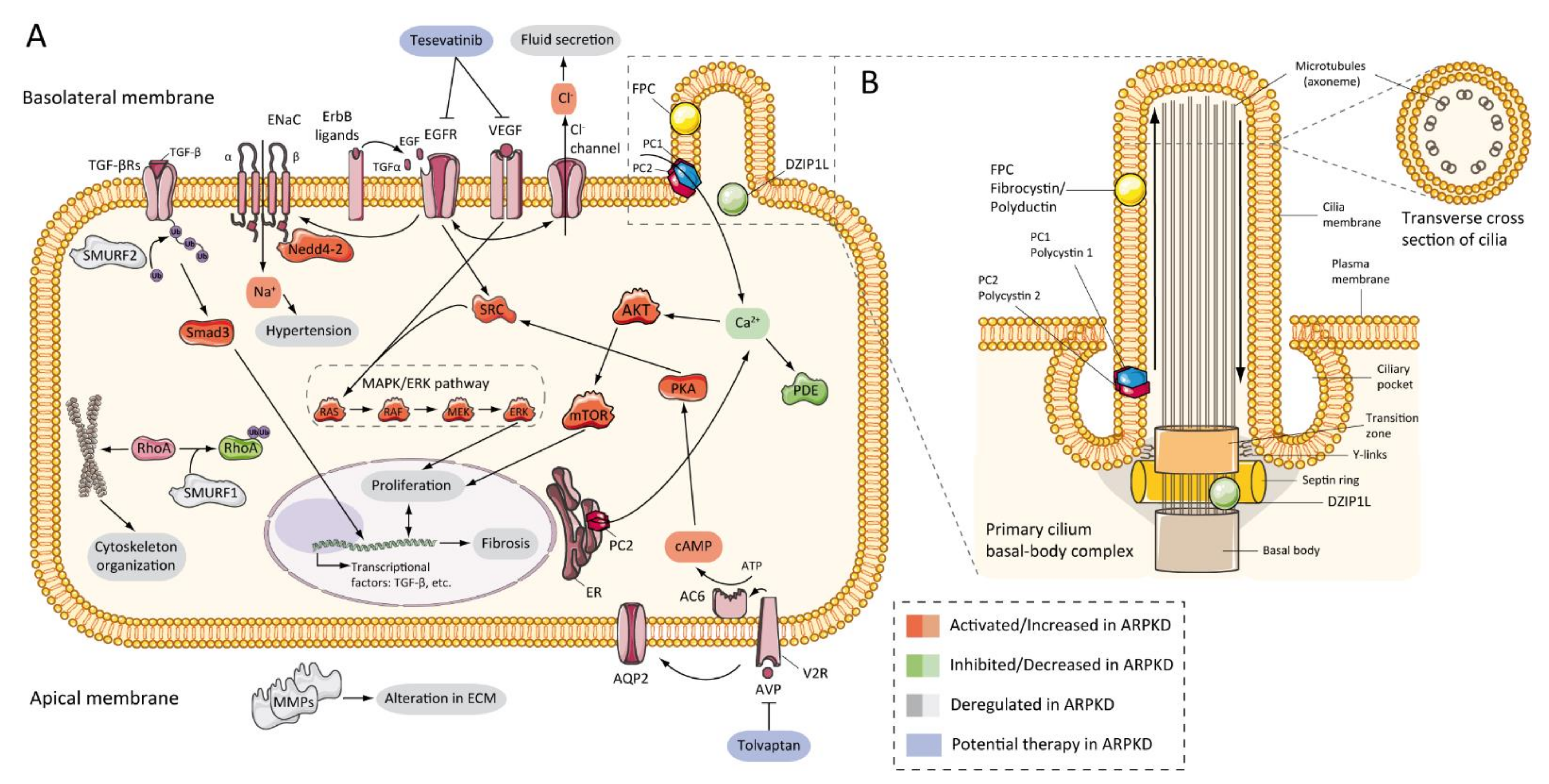
6. ARPKD Proteins: Structure and Function
7. Pathogenesis of ARPKD/Molecular Basis/Disease Mechanism
7.1. ARPKD Rodent Models: Lessons from Animal Models

{kind=link}
{kind=link}
{kind=link}
| Model Name | Specie | Gene | Allele Type (Mutation Type) | Liver Phenotype | Kidney Phenotype | Other Phenotypes | Ref. |
|---|---|---|---|---|---|---|---|
| Animal models that mimics ARPKD | |||||||
| cpk (Cys1cpk) * | Mouse | Cys1 | Spontaneous | ∙Liver cysts ∙Dilated bile duct ∙Hepatic fibrosis | ∙Kidney cysts ∙Enlarged kidney | ∙Pancreas cysts | [77,93,94,95,96] |
| pcy (DBA/2-pcy/pcy) (Nphp3pcy) * | Mouse | Nphp3 | Spontaneous | None | ∙Kidney cysts ∙Enlarged kidney ∙Renal fibrosis | ∙Intracranial aneurysm | [79,80,97] |
| bpk (Bicc1jcpk-bpk) * | Mouse | Bicc1 | Spontaneous | ∙Enlarged bile duct | ∙Kidney cysts ∙Enlarged kidney | ∙Premature death ∙Postnatal lethality | [81] |
| jck (Nek8jck) * | Mouse | Nek8 | Spontaneous | ∙None | ∙Kidney cysts ∙Enlarged kidney | ∙Premature death | [82] |
| orpk (Ift88Tg737Rpw) * | Mouse | Ift88 | Transgenic (insertion) | ∙Abnormal bile duct morphology ∙Liver fibrosis | ∙Kidney cysts ∙Enlarged kidney | ∙Pancreas cysts ∙Polydactyly | [87,88] |
| jcpk | Mouse | Bicc1 | Chemical induction | ∙Dilated bile duct | ∙Kidney cysts ∙Dilated renal tubules | ∙Dilated pancreatic ducts | [85] |
| ARPKD models | |||||||
| PCK | Rat | Pkhd1 | Spontaneous (splicing mutation) | ∙Liver cysts ∙Dilated bile duct ∙Hepatic fibrosis | ∙Kidney cysts ∙Enlarged kidney ∙Renal fibrosis | ∙Pancreas cysts | [89,98] |
| Pkhd1ex40 | Mouse | Pkhd1 | Targeted (KO by insertion) | ∙Liver cysts ∙Hepatic fibrosis | ∙None | ∙Portal hypertension | [90] |
| Pkhd1del2/del2 (Pkhd1tm1Cjwa) * | Mouse | Pkhd1 | Targeted (KO by deletion) | ∙Liver cysts ∙Dilated bile duct ∙Hepatic fibrosis | ∙Kidney cysts ∙Dilated renal tubules | ∙Pancreas cysts ∙Pancreatic duct abnormalities | [99] |
| Pkhd1del3−4 (Pkhd1tm1.1Ggg) * | Mouse | Pkhd1 | Targeted (KO by deletion) | ∙Liver cysts ∙Hepatic fibrosis | ∙Kidney cysts ∙Renal fibrosis | ∙Pancreas cysts ∙Choledochal cyst ∙Ascending cholangitis | [60] |
| Pkhd1del4/del4 (Pkhd1tm1Som)* | Mouse | Pkhd1 | Targeted (KO by deletion) | ∙Liver cysts ∙Biliary cysts ∙Hepatic fibrosis | ∙None | ∙Pancreas cysts ∙Pancreas fibrosis ∙Enlarged spleen | [100] |
| Pkhd1e15GFP∆16 (Pkhd1tm1Gwu) * | Mouse | Pkhd1 | Targeted (KO by deletion) | ∙Liver cysts ∙Hepatic fibrosis | ∙Kidney cysts ∙Dilated renal tubules ∙Renal fibrosis | ∙Pancreas cysts ∙Dilated pancreatic duct ∙Gastrointestinal ulcer | [101] |
| Pkhd1lacZ (Pkhd1tm1Sswi) * | Mouse | Pkhd1 | Targeted (KO by deletion) | ∙Dilated bile duct ∙Hepatic fibrosis | ∙Kidney cysts ∙Dilated renal tubules ∙Renal fibrosis | ∙Pancreas cysts | [102] |
| Pkhd1LSL(-) (Pkhd1tm2Cjwa) * | Mouse | Pkhd1 | Targeted (KO by insertion) | ∙Liver cysts ∙Hepatic fibrosis | ∙Kidney cysts ∙Dilated renal tubules | ∙Unknown | [103] |
| Pkhd1Δ67 | Mouse | Pkhd1 | Targeted (KO by deletion) | ∙None | ∙None | ∙None | [67] |
| Dzip1lwpy/wpy (Dzip1lwarpy) * | Mouse | Dzip1l | Targeted (KO by single point mutation) | ∙Abnormal bile duct morphology | ∙Kidney cysts ∙Dilated renal tubules | ∙Polydactyly ∙Abnormal eye morphology ∙Cleft upper lip ∙Cleft palate | [37] |
| Pkhd1C642* (Pkhd1em1Mrug) * | Mouse | Pkhd1 | Targeted (KO by deletion) | ∙Liver cysts ∙Biliary cysts ∙Hepatic fibrosis | ∙Dilated renal tubules ∙Proximal tubule ectasia | ∙Unknown | [67] |
7.2. Abnormalities of EGFR-Axis Expression and Fluid Secretion
7.3. cAMP and Proliferation
7.4. Other Pathways Involved in ARPKD Physiopathology
7.5. Role of Cilia
8. Clinical Trials
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Guay-Woodford, L.M.; Muecher, G.; Hopkins, S.D.; Avner, E.D.; Germino, G.G.; Guillot, A.P.; Herrin, J.; Holleman, R.; Irons, D.A.; Primack, W.; et al. The severe perinatal form of autosomal recessive polycystic kidney disease maps to chromosome 6p21.1-p12: Implications for genetic counseling. Am. J. Hum. Genet. 1995, 56, 1101–1107. [Google Scholar] [PubMed]
- Kaariainen, H. Polycystic kidney disease in children: A genetic and epidemiological study of 82 Finnish patients. J. Med. Genet. 1987, 24, 474–481. [Google Scholar] [CrossRef] [PubMed]
- Alzarka, B.; Morizono, H.; Bollman, J.W.; Kim, D.; Guay-Woodford, L.M. Design and implementation of the hepatorenal fibrocystic disease core center clinical database: A centralized resource for characterizing autosomal recessive polycystic kidney disease and other hepatorenal fibrocystic diseases. Front. Pediatr. 2017, 5. [Google Scholar] [CrossRef]
- Bergmann, C. Genetics of Autosomal Recessive Polycystic Kidney Disease and Its Differential Diagnoses. Front. Pediatr. 2018, 5, 1–13. [Google Scholar] [CrossRef]
- Cordido, A.; Besada-Cerecedo, L.; García-González, M.A. The Genetic and Cellular Basis of Autosomal Dominant Polycystic Kidney Disease—A Primer for Clinicians. Front. Pediatr. 2017, 5, 279. [Google Scholar] [CrossRef]
- Bergmann, C.; Guay-Woodford, L.M.; Harris, P.C.; Horie, S.; Peters, D.J.M.; Torres, V.E. Polycystic kidney disease. Nat. Rev. Dis. Prim. 2018, 4, 50. [Google Scholar] [CrossRef] [PubMed]
- Bergmann, C.; Senderek, J.; Schneider, F.; Dornia, C.; Küpper, F.; Eggermann, T.; Rudnik-Schöneborn, S.; Kirfel, J.; Moser, M.; Büttner, R.; et al. PKHD1 Mutations in Families Requesting Prenatal Diagnosis for Autosomal Recessive Polycystic Kidney Disease (ARPKD). Hum. Mutat. 2004, 23, 487–495. [Google Scholar] [CrossRef]
- Guay-Woodford, L.M.; Desmond, R.A. Autosomal recessive polycystic kidney disease: The clinical experience in North America. Pediatrics 2003, 111, 1072–1080. [Google Scholar] [CrossRef]
- Adeva, M.; El-Youssef, M.; Rossetti, S.; Kamath, P.S.; Kubly, V.; Consugar, M.B.; Milliner, D.M.; King, B.F.; Torres, V.E.; Harris, P.C. Clinical and molecular characterization defines a broadened spectrum of autosomal recessive polycystic kidney disease (ARPKD). Medicine 2006, 85, 1–21. [Google Scholar] [CrossRef]
- Gunay-Aygun, M.; Avner, E.D.; Bacallao, R.L.; Choyke, P.L.; Flynn, J.T.; Germino, G.G.; Guay-Woodford, L.; Harris, P.; Heller, T.; Ingelfinger, J.; et al. Autosomal recessive polycystic kidney disease and congenital hepatic fibrosis: Summary statement of a First National Institutes of Health/Office of Rare Diseases conference. J. Pediatr. 2006, 149, 159–164. [Google Scholar] [CrossRef]
- Liebau, M.C. Early clinical management of autosomal recessive polycystic kidney disease. Pediatr. Nephrol. 2021. [Google Scholar] [CrossRef] [PubMed]
- Bergmann, C.; Senderek, J.; Windelen, E.; Küpper, F.; Middeldorf, I.; Schneider, F.; Dornia, C.; Rudnik-Schöneborn, S.; Konrad, M.; Schmitt, C.P.; et al. Clinical consequences of PKHD1 mutations in 164 patients with autosomal-recessive polycystic kidney disease (ARPKD). Kidney Int. 2005, 67, 829–848. [Google Scholar] [CrossRef]
- Erger, F.; Brüchle, N.O.; Gembruch, U.; Zerres, K. Prenatal ultrasound, genotype, and outcome in a large cohort of prenatally affected patients with autosomal-recessive polycystic kidney disease and other hereditary cystic kidney diseases. Arch. Gynecol. Obstet. 2017, 295, 897–906. [Google Scholar] [CrossRef]
- Burgmaier, K.; Kilian, S.; Bammens, B.; Benzing, T.; Billing, H.; Büscher, A.; Galiano, M.; Grundmann, F.; Klaus, G.; Mekahli, D.; et al. Clinical courses and complications of young adults with Autosomal Recessive Polycystic Kidney Disease (ARPKD). Sci. Rep. 2019, 9. [Google Scholar] [CrossRef] [PubMed]
- Belin, S.; Delco, C.; Parvex, P.; Hanquinet, S.; Fokstuen, S.; De Tejada, B.M.; Eperon, I. Management of delivery of a fetus with autosomal recessive polycystic kidney disease: A case report of abdominal dystocia and review of the literature. J. Med. Case Rep. 2019, 13. [Google Scholar] [CrossRef]
- Fonck, C.; Chauveau, D.; Gagnadoux, M.F.; Pirson, Y.; Grünfeld, J.P. Autosomal recessive polycystic kidney disease. Nephrol. Dial. Transplant. 2001, 16, 1648–1652. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Rubio San Simón, A.; Carbayo Jiménez, T.; Vara Martín, J.; Alonso Díaz, C.; Espino Hernández, M. Autosomal recessive polycystic kidney disease in the 21st century: Long-term follow up and outcomes. An. Pediatr. 2018, 91, 120–122. [Google Scholar] [CrossRef] [PubMed]
- Burgmaier, K.; Brandt, J.; Shroff, R.; Witters, P.; Weber, L.T.; Dötsch, J.; Schaefer, F.; Mekahli, D.; Liebau, M.C. Gastrostomy tube insertion in pediatric patients with autosomal recessive polycystic kidney disease (ARPKD): Current practice. Front. Pediatr. 2018, 6. [Google Scholar] [CrossRef]
- Cole, B.R.; Conley, S.B.; Stapleton, F.B. Polycystic kidney disease in the first year of life. J. Pediatr. 1987, 111, 693–699. [Google Scholar] [CrossRef]
- Avni, F.E.; Guissard, G.; Hall, M.; Janssen, F.; DeMaertelaer, V.; Rypens, F. Hereditary polycystic kidney diseases in children: Changing sonographic patterns through childhood. Pediatr. Radiol. 2002, 32, 169–174. [Google Scholar] [CrossRef]
- Bergmann, C. Early and Severe Polycystic Kidney Disease and Related Ciliopathies: An Emerging Field of Interest. Nephron 2019, 141, 50–60. [Google Scholar] [CrossRef]
- Rivero, P.C.; Ecuador, U.R.D.C.L.; Andrade, R.E.C.; Baquero, S.M.; Espirel, V.M.; Ecuador, U.T.D.N. Polycystic kidney disease. Annu. Rev. Med. 2009, 60, 321–337. [Google Scholar] [CrossRef]
- Gunay-Aygun, M.; Font-Montgomery, E.; Lukose, L.; Tuchman Gerstein, M.; Piwnica-Worms, K.; Choyke, P.; Daryanani, K.T.; Turkbey, B.; Fischer, R.; Bernardini, I.; et al. Characteristics of congenital hepatic fibrosis in a large cohort of patients with autosomal recessive polycystic kidney disease. Gastroenterology 2013, 144. [Google Scholar] [CrossRef]
- Turkbey, B.; Ocak, I.; Daryanani, K.; Font-Montgomery, E.; Lukose, L.; Bryant, J.; Tuchman, M.; Mohan, P.; Heller, T.; Gahl, W.A.; et al. Autosomal recessive polycystic kidney disease and congenital hepatic fibrosis. Pediatr. Radiol. 2009, 39, 100–111. [Google Scholar] [CrossRef]
- Hartung, E.A.; Wen, J.; Poznick, L.; Furth, S.L.; Darge, K. Ultrasound Elastography to Quantify Liver Disease Severity in Autosomal Recessive Polycystic Kidney Disease. J. Pediatr. 2019, 209, 107–115.e5. [Google Scholar] [CrossRef] [PubMed]
- Guay-Woodford, L.M.; Bissler, J.J.; Braun, M.C.; Bockenhauer, D.; Cadnapaphornchai, M.A.; Dell, K.M.; Kerecuk, L.; Liebau, M.C.; Alonso-Peclet, M.H.; Shneider, B.; et al. Consensus expert recommendations for the diagnosis and management of autosomal recessive polycystic kidney disease: Report of an international conference. J. Pediatr. 2014, 165, 611–617. [Google Scholar] [CrossRef]
- Gately, R.; Lock, G.; Patel, C.; Clouston, J.; Hawley, C.; Mallett, A. Multiple Cerebral Aneurysms in an Adult With Autosomal Recessive Polycystic Kidney Disease. Kidney Int. Rep. 2021, 6, 219–223. [Google Scholar] [CrossRef]
- Perez, J.L.; McDowell, M.M.; Zussman, B.; Jadhav, A.P.; Miyashita, Y.; McKiernan, P.; Greene, S. Ruptured intracranial aneurysm in a patient with autosomal recessive polycystic kidney disease. J. Neurosurg. Pediatr. 2019, 23, 75–79. [Google Scholar] [CrossRef]
- Blyth, H.; Ockenden, B.G. Polycystic disease of kidney and liver presenting in childhood. J. Med. Genet. 1971, 8, 257–284. [Google Scholar] [CrossRef] [PubMed]
- Besse, W.; Dong, K.; Choi, J.; Punia, S.; Fedeles, S.V.; Choi, M.; Gallagher, A.-R.R.; Huang, E.B.; Gulati, A.; Knight, J.; et al. Isolated polycystic liver disease genes define effectors of polycystin-1 function. J. Clin. Investig. 2017. [Google Scholar] [CrossRef] [PubMed]
- Gunay-aygun, M.; Turkbey, B.I.; Bryant, J.; Daryanani, K.T.; Tuchman, M.; Piwnica-worms, K.; Choyke, P.; Heller, T.; Gahl, W.A. Hepatorenal findings in obligate heterozygotes for autosomal recessive polycystic kidney disease. Mol. Genet. Metab. 2011, 104, 677–681. [Google Scholar] [CrossRef]
- Guay-Woodford, L.M. Autosomal recessive polycystic kidney disease: The prototype of the hepato-renal fibrocystic diseases. J. Pediatr. Genet. 2014, 3, 89–101. [Google Scholar] [CrossRef]
- Raina, R.; Chakraborty, R.; Sethi, S.K.; Kumar, D.; Gibson, K.; Bergmann, C. Diagnosis and Management of Renal Cystic Disease of the Newborn: Core Curriculum 2021. Am. J. Kidney Dis. 2021. [Google Scholar] [CrossRef]
- Onuchic, L.F.; Furu, L.; Nagasawa, Y.; Hou, X.; Eggermann, T.; Ren, Z.; Bergmann, C.; Senderek, J.; Esquivel, E.; Zeltner, R.; et al. PKHD1, the polycystic kidney and hepatic disease 1 gene, encodes a novel large protein containing multiple immunoglobulin-like plexin-transcription-factor domains and parallel beta-helix 1 repeats. Am. J. Hum. Genet. 2002, 70, 1305–1317. [Google Scholar] [CrossRef]
- Obeidova, L.; Seeman, T.; Fencl, F.; Blahova, K.; Hojny, J.; Elisakova, V.; Reiterova, J.; Stekrova, J. Results of targeted next-generation sequencing in children with cystic kidney diseases often change the clinical diagnosis. PLoS ONE 2020, 15. [Google Scholar] [CrossRef]
- Bergmann, C. ARPKD and early manifestations of ADPKD: The original polycystic kidney disease and phenocopies. Pediatr. Nephrol. 2015, 30, 15–30. [Google Scholar] [CrossRef]
- Lu, H.; Galeano, M.C.R.; Ott, E.; Kaeslin, G.; Kausalya, P.J.; Kramer, C.; Ortiz-Brüchle, N.; Hilger, N.; Metzis, V.; Hiersche, M.; et al. Mutations in DZIP1L, which encodes a ciliary-transition-zone protein, cause autosomal recessive polycystic kidney disease. Nat. Genet. 2017, 49, 1025–1034. [Google Scholar] [CrossRef]
- Fedeles, S.V.; Tian, X.; Gallagher, A.R.; Mitobe, M.; Nishio, S.; Lee, S.H.; Cai, Y.; Geng, L.; Crews, C.M.; Somlo, S. A genetic interaction network of five genes for human polycystic kidney and liver diseases defines polycystin-1 as the central determinant of cyst formation. Nat. Genet. 2011, 43, 639–647. [Google Scholar] [CrossRef] [PubMed]
- Torres, V.E.; Harris, P.C.; Pirson, Y. Autosomal dominant polycystic kidney disease. Lancet 2007, 369, 1287–1301. [Google Scholar] [CrossRef]
- Schepis, C. The tuberous sclerosis complex. Dermatol. Cryosurg. Cryother. 2016, 615–617. [Google Scholar] [CrossRef]
- Shepherd, C.W.; Gomez, M.R.; Lie, J.; Crowson, C.S. Causes of Death in Patients With Tuberous Sclerosis. Mayo Clin. Proc. 1991, 66, 792–796. [Google Scholar] [CrossRef]
- Decramer, S.; Parant, O.; Beaufils, S.; Clauin, S.; Guillou, C.; Kessler, S.; Aziza, J.; Bandin, F.; Schanstra, J.P.; Bellanné-Chantelot, C. Anomalies of the TCF2 Gene Are the Main Cause of Fetal Bilateral Hyperechogenic Kidneys. J. Am. Soc. Nephrol. 2007, 18, 923–933. [Google Scholar] [CrossRef]
- Verhave, J.C.; Bech, A.P.; Wetzels, J.F.M.; Nijenhuis, T. Hepatocyte nuclear factor 1β-associated kidney disease: More than renal cysts and diabetes. J. Am. Soc. Nephrol. 2016, 27, 345–353. [Google Scholar] [CrossRef]
- Hoff, S.; Halbritter, J.; Epting, D.; Frank, V.; Nguyen, T.M.T.; Van Reeuwijk, J.; Boehlke, C.; Schell, C.; Yasunaga, T.; Helmstädter, M.; et al. ANKS6 is a central component of a nephronophthisis module linking NEK8 to INVS and NPHP3. Nat. Genet. 2013, 45, 951–956. [Google Scholar] [CrossRef]
- Bergmann, C.; Fliegauf, M.; Brüchle, N.O.; Frank, V.; Olbrich, H.; Kirschner, J.; Schermer, B.; Schmedding, I.; Kispert, A.; Kränzlin, B.; et al. Loss of Nephrocystin-3 Function Can Cause Embryonic Lethality, Meckel-Gruber-like Syndrome, Situs Inversus, and Renal-Hepatic-Pancreatic Dysplasia. Am. J. Hum. Genet. 2008, 82, 959–970. [Google Scholar] [CrossRef] [PubMed]
- Huber, T.B.; Walz, G.; Kuehn, E.W. MTOR and rapamycin in the kidney: Signaling and therapeutic implications beyond immunosuppression. Kidney Int. 2011, 79, 502–511. [Google Scholar] [CrossRef]
- Zerres, K.; Mücher, G.; Becker, J.; Steinkamm, C.; Rudnik-Schöneborn, S.; Heikkilä, P.; Rapola, J.; Salonen, R.; Germino, G.G.; Onuchic, L.; et al. Prenatal diagnosis of autosomal recessive polycystic kidney disease (ARPKD): Molecular genetics, clinical experience, and fetal morphology. Am. J. Med. Genet. 1998, 76, 137–144. [Google Scholar] [CrossRef]
- Ward, C.J.; Hogan, M.C.; Rossetti, S.; Walker, D.; Sneddon, T.; Wang, X.; Kubly, V.; Cunningham, J.M.; Bacallao, R.; Ishibashi, M.; et al. The gene mutated in autosomal recessive polycystic kidney disease encodes a large, receptor-like protein. Nat. Genet. 2002, 30, 259–269. [Google Scholar] [CrossRef]
- Zerres, K.; Rudnik-Schöneborn, S.; Deget, F.; Holtkamp, U.; Brodehl, J.; Geisert, J.; Schärer, K. Autosomal recessive polycystic kidney disease in 115 children: Clinical presentation, course and influence of gender. Arbeitsgemeinschaft für Pädiatrische, Nephrologie. Acta Paediatr. 1996, 85, 437–445. [Google Scholar] [CrossRef]
- Deget, F.; Rudnik-Schöneborn, S.; Zerres, K. Course of autosomal recessive polycystic kidney disease (ARPKD) in siblings: A clinical comparison of 20 sibships. Clin. Genet. 1995, 47, 248–253. [Google Scholar] [CrossRef]
- Consugar, M.B.; Anderson, S.A.; Rossetti, S.; Pankratz, V.S.; Ward, C.J.; Torra, R.; Coto, E.; El-youssef, M.; Kantarci, S.; Utsch, B.; et al. Haplotype Analysis Improves Molecular Diagnostics of Autosomal Recessive Polycystic Kidney Disease. Am. J. Kidney Dis. 2005, 45, 77–87. [Google Scholar] [CrossRef]
- Perugorria, M.J.; Banales, J.M. Genetics: Novel causative genes for polycystic liver disease. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 391–392. [Google Scholar] [CrossRef]
- Hartung, E.A.; Guay-Woodford, L.M. Polycystic kidney disease: DZIP1L defines a new functional zip code for autosomal recessive PKD. Nat. Rev. Nephrol. 2017, 13, 519–520. [Google Scholar] [CrossRef]
- Bergmann, C.; Senderek, J.; Sedlacek, B.; Pegiazoglou, I.; Puglia, P.; Eggermann, T.; Rudnik-Schöneborn, S.; Furu, L.; Onuchic, L.F.; De Baca, M.; et al. Spectrum of mutations in the gene for autosomal recessive polycystic kidney disease (ARPKD/PKHD1). J. Am. Soc. Nephrol. 2003, 14, 76–89. [Google Scholar] [CrossRef] [PubMed]
- Benz, E.G.; Hartung, E.A. Predictors of progression in autosomal dominant and autosomal recessive polycystic kidney disease. Pediatr. Nephrol. 2021. [Google Scholar] [CrossRef]
- Ebner, K.; Dafinger, C.; Ortiz-Bruechle, N.; Koerber, F.; Schermer, B.; Benzing, T.; Dötsch, J.; Zerres, K.; Weber, L.T.; Beck, B.B.; et al. Challenges in establishing genotype–phenotype correlations in ARPKD: Case report on a toddler with two severe PKHD1 mutations. Pediatr. Nephrol. 2017, 32, 1269–1273. [Google Scholar] [CrossRef] [PubMed]
- Furu, L.; Onuchic, L.F.; Gharavi, A.; Hou, X.; Esquivel, E.L.; Nagasawa, Y.; Bergmann, C.; Senderek, J.; Avner, E.; Zerres, K.; et al. Milder Presentation of Recessive Polycystic Kidney Disease Requires Presence of Amino Acid Substitution Mutations. J. Am. Soc. Nephrol. 2003. [Google Scholar] [CrossRef]
- Burgmaier, K.; Kunzmann, K.; Ariceta, G.; Bergmann, C.; Buescher, A.K.; Burgmaier, M.; Dursun, I.; Duzova, A.; Eid, L.; Erger, F.; et al. Risk Factors for Early Dialysis Dependency in Autosomal Recessive Polycystic Kidney Disease. J. Pediatr. 2018, 199, 22–28.e6. [Google Scholar] [CrossRef]
- Stevanovic, R.; Glumac, S.; Trifunovic, J.; Medjo, B.; Nastasovic, T.; Markovic-Lipkovski, J. Autosomal recessive polycystic kidney disease: Case report. Clin. Genet. 1995, 47, 248–253. [Google Scholar] [CrossRef]
- Garcia-Gonzalez, M.A.; Menezes, L.F.; Piontek, K.B.; Kaimori, J.; Huso, D.L.; Watnick, T.; Onuchic, L.F.; Guay-Woodford, L.M.; Germino, G.G. Genetic interaction studies link autosomal dominant and recessive polycystic kidney disease in a common pathway. Hum. Mol. Genet. 2007, 16, 1940–1950. [Google Scholar] [CrossRef]
- Bergmann, C.; von Bothmer, J.; Ortiz Brü chle, N.; Venghaus, A.; Frank, V.; Fehrenbach, H.; Hampel, T.; Pape, L.; Buske, A.; Jonsson, J.; et al. Mutations in Multiple PKD Genes May Explain Early and Severe Polycystic Kidney Disease. J. Am. Soc. Nephrol. 2011, 22, 2047–2056. [Google Scholar] [CrossRef] [PubMed]
- Nagasawa, Y.; Matthiesen, S.; Onuchic, L.F.; Hou, X.; Bergmann, C.; Esquivel, E.; Senderek, J.; Ren, Z.; Zeltner, R.; Furu, L.; et al. Identification and Characterization of Pkhd1, the Mouse Orthologue of the Human ARPKD Gene. J. Am. Soc. Nephrol. 2002, 13, 2246–2258. [Google Scholar] [CrossRef]
- Igarashi, P.; Somlo, S. Genetics and Pathogenesis of Polycystic Kidney Disease. J. Am. Soc. Nephrol. 2002, 13, 2384–2398. [Google Scholar] [CrossRef] [PubMed]
- Hogan, M.C.; Griffin, M.D.; Rossetti, S.; Torres, V.E.; Ward, C.J.; Harris, P.C. PKHDL1, a homolog of the autosomal recessive polycystic kidney disease gene, encodes a receptor with inducible T lymphocyte expression. Hum. Mol. Genet. 2003, 12, 685–698. [Google Scholar] [CrossRef]
- Menezes, L.F.C.; Cai, Y.; Nagasawa, Y.; Silva, A.M.G.; Watkins, M.L.; Da Silva, A.M.; Somlo, S.; Guay-Woodford, L.M.; Germino, G.G.; Onuchic, L.F. Polyductin, the PKHD1 gene product, comprises isoforms expressed in plasma membrane, primary cilium, and cytoplasm. Kidney Int. 2004, 66, 1345–1355. [Google Scholar] [CrossRef]
- Kaimori, J.-Y.; Nagasawa, Y.; Menezes, L.F.; Garcia-Gonzalez, M.A.; Deng, J.; Imai, E.; Onuchic, L.F.; Guay-Woodford, L.M.; Germino, G.G. Polyductin undergoes notch-like processing and regulated release from primary cilia. Hum. Mol. Genet. 2007, 16, 942–956. [Google Scholar] [CrossRef]
- Outeda, P.; Menezes, L.; Hartung, E.A.; Bridges, S.; Zhou, F.; Zhu, X.; Xu, H.; Huang, Q.; Yao, Q.; Qian, F.; et al. A novel model of autosomal recessive polycystic kidney questions the role of the fibrocystin C-terminus in disease mechanism. Kidney Int. 2017, 92, 1130–1144. [Google Scholar] [CrossRef]
- Ward, C.J.; Yuan, D.; Masyuk, T.V.; Wang, X.; Punyashthiti, R.; Whelan, S.; Bacallao, R.; Torra, R.; LaRusso, N.F.; Torres, V.E.; et al. Cellular and subcellular localization of the ARPKD protein; fibrocystin is expressed on primary cilia. Hum. Mol. Genet. 2003, 12, 2703–2710. [Google Scholar] [CrossRef]
- Wang, S.; Luo, Y.; Wilson, P.D.; Witman, G.B.; Zhou, J. The Autosomal Recessive Polycystic Kidney Disease Protein Is Localized to Primary Cilia, with Concentration in the Basal Body Area. J. Am. Soc. Nephrol. 2004, 15, 592–602. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.-Z.; Mai, W.; Li, C.; Cho, S.; Hao, C.; Moeckel, G.; Zhao, R.; Kim, I.; Wang, J.; Xiong, H.; et al. PKHD1 protein encoded by the gene for autosomal recessive polycystic kidney disease associates with basal bodies and primary cilia in renal epithelial cells. Proc. Natl. Acad. Sci. USA 2004, 101, 2311–2316. [Google Scholar] [CrossRef]
- Masyuk, T.V.; Huang, B.Q.; Ward, C.J.; Masyuk, A.I.; Yuan, D.; Splinter, P.L.; Punyashthiti, R.; Ritman, E.L.; Torres, V.E.; Harris, P.C.; et al. Defects in Cholangiocyte Fibrocystin Expression and Ciliary Structure in the PCK Rat. Gastroenterology 2003, 125, 1303–1310. [Google Scholar] [CrossRef] [PubMed]
- Sweeney, W.E.; Avner, E.D. Molecular and cellular pathophysiology of autosomal recessive polycystic kidney disease (ARPKD). Cell Tissue Res. 2006, 326, 671–685. [Google Scholar] [CrossRef] [PubMed]
- Wilson, P.D. Polycystic Kidney Disease. N. Engl. J. Med. 2004, 350, 151–164. [Google Scholar] [CrossRef] [PubMed]
- Follit, J.A.; Li, X.; Vucica, Y.; Pazour, G.J. The cytoplasmic tail of fibrocystin contains a ciliary targeting sequence. J. Cell Biol. 2010, 188, 21–28. [Google Scholar] [CrossRef]
- Glazer, A.M.; Wilkinson, A.W.; Backer, C.B.; Lapan, S.W.; Gutzman, J.H.; Cheeseman, I.M.; Reddien, P.W. The Zn Finger protein Iguana impacts Hedgehog signaling by promoting ciliogenesis. Dev. Biol. 2010, 337, 148–156. [Google Scholar] [CrossRef]
- Guay-Woodford, L.M. Murine models of polycystic kidney disease: Molecular and therapeutic insights. Am. J. Physiol. Ren. Physiol. 2003, 285, 1034–1049. [Google Scholar] [CrossRef]
- Fry, J.L.; Koch, W.E.; Jennette, J.C.; McFarland, E.; Fried, F.A.; Mandell, J. A genetically determined murine model of infantile polycystic kidney disease. J. Urol. 1985, 134, 828–833. [Google Scholar] [CrossRef]
- Schieren, G.; Pey, R.; Bach, J.; Hafner, M.; Gretz, N. Murine models of polycystic kidney disease. Nephrol. Dial. Transplant. 1996, 11, 38–45. [Google Scholar] [CrossRef]
- Takahashi, H.; Calvet, J.P.; Dittemore-Hoover, D.; Yoshida, K.; Grantham, J.J.; Gattone, V.H. A hereditary model of slowly progressive polycystic kidney disease in the mouse. J. Am. Soc. Nephrol. 1991, 1, 980–989. [Google Scholar] [CrossRef]
- Woo, D.D.L.; Nguyen, D.K.P.; Khatibi, N.; Olsen, P. Genetic identification of two major modifier loci of polycystic kidney disease progression in pcy mice. J. Clin. Investig. 1997, 100, 1934–1940. [Google Scholar] [CrossRef][Green Version]
- Nauta, J.; Ozawa, Y.; Sweeney, W.E.; Rutledge, J.C.; Avner, E.D. Renal and biliary abnormalities in a new murine model of autosomal recessive polycystic kidney disease. Pediatr. Nephrol. 1993, 7, 163–172. [Google Scholar] [CrossRef] [PubMed]
- Atala, A.; Freeman, M.R.; Mandell, J.; Beier, D.R. Juvenile cystic kidneys (jck): A new mouse mutation which causes polycystic kidneys. Kidney Int. 1993, 43, 1081–1085. [Google Scholar] [CrossRef][Green Version]
- Cogswell, C.; Price, S.J.; Hou, X.; Guay-Woodford, L.M.; Flaherty, L.; Bryda, E.C. Positional cloning of jcpk/bpk locus of the mouse. Mamm. Genome 2003, 14, 242–249. [Google Scholar] [CrossRef]
- Liu, S.; Lu, W.; Obara, T.; Kuida, S.; Lehoczky, J.; Dewar, K.; Drummond, I.A.; Beier, D.R. A defect in a novel Nek-family kinase causes cystic kidney disease in the mouse and in zebrafish. Development 2002, 129, 5839–5846. [Google Scholar] [CrossRef]
- Flaherty, L.; Bryda, E.C.; Collins, D.; Rudofsky, U.; Montgomery, J.C. New mouse model for polycystic kidney disease with both recessive and dominant gene effects. Kidney Int. 1995, 47, 552–558. [Google Scholar] [CrossRef] [PubMed]
- Guay-Woodford, L.M.; Bryda, E.C.; Christine, B.; Lindsey, J.R.; Collier, W.R.; Avner, E.D.; D’eustachio, P.; Flaherty, L. Evidence that two phenotypically distinct mouse PKD mutations, bpk and jcpk, are allelic. Kidney Int. 1996, 50, 1158–1165. [Google Scholar] [CrossRef][Green Version]
- Moyer, J.H.; Lee-Tischler, M.J.; Kwon, H.Y.; Schrick, J.J.; Avner, E.D.; Sweeney, W.E.; Godfrey, V.L.; Cacheiro, N.L.A.; Wilkinson, J.E.; Woychik, R.P. Candidate gene associated with a mutation causing recessive polycystic kidney disease in mice. Science 1994, 264, 1329–1333. [Google Scholar] [CrossRef] [PubMed]
- Sweeney, W.E.; Avner, E.D. Functional activity of epidermal growth factor receptors in autosomal recessive polycystic kidney disease. Am. J. Physiol. Ren. Physiol. 1998, 275, F387–F394. [Google Scholar] [CrossRef]
- Lager, D.J.; Qian, Q.; Bengal, R.J.; Ishibashi, M.; Torres, V.E. The pck rat: A new model that resembles human autosomal dominant polycystic kidney and liver disease. Kidney Int. 2001, 59, 126–136. [Google Scholar] [CrossRef]
- Moser, M.; Matthiesen, S.; Kirfel, J.; Schorle, H.; Bergmann, G.; Senderek, J.; Rudnik-Schöneborn, S.; Zerres, K.; Buettner, R. A mouse model for cystic biliary dysgenesis in autosomal recessive polycystic kidney disease (ARPKD). Hepatology 2005, 41, 1113–1121. [Google Scholar] [CrossRef]
- Al-Bhalal, L.; Akhtar, M. Molecular basis of autosomal recessive polycystic kidney disease (ARPKD). Adv. Anat. Pathol. 2008, 15, 54–58. [Google Scholar] [CrossRef] [PubMed]
- Sweeney, W.E.; Avner, E.D. Pathophysiology of childhood polycystic kidney diseases: New insights into disease-specific therapy. Pediatr. Res. 2014, 75, 148–157. [Google Scholar] [CrossRef]
- Guay-Woodford, L.M.; D’eustachio, P.; Bruns, G.A.P. Identification of the syntenic human linkage group for the mouse congenital polycystic kidney (cpk) locus (Abstract). J. Am. Soc. Nephrol. 1993, 4, 814. [Google Scholar]
- Orellana, S.A.; Sweeney, W.E.; Neff, C.D.; Avner, E.D. Epidermal growth factor receptor expression in abnormal in murine polycystic kidney. Kidney Int. 1995, 47, 490–499. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Gattone, V.H.; MacNaughton, K.A.; Kraybill, A.L. Murine autosomal recessive polycystic kidney disease with multiorgan involvement induced by the cpk gene. Anat. Rec. 1996, 245, 488–499. [Google Scholar] [CrossRef]
- Ricker, J.; Gattone, V.H.; Calvet, J.P.; Rankin, C.A. Development of Autosomal Recessive Polycystic Kidney Disease in BALB/c-cpk/cpk Mice | American Society of Nephrology. J. Am. Soc. Nephrol. 2000, 10, 1837–1847. [Google Scholar] [CrossRef]
- Olbrich, H.; Fliegauf, M.; Hoefele, J.; Kispert, A.; Otto, E.; Volz, A.; Wolf, M.T.; Sasmaz, G.; Trauer, U.; Reinhardt, R.; et al. Mutations in a novel gene, NPHP3, cause adolescent nephronophthisis, tapeto-retinal degeneration and hepatic fibrosis. Nat. Genet. 2003, 34, 455–459. [Google Scholar] [CrossRef]
- Muff, M.A.; Masyuk, T.V.; Stroope, A.J.; Huang, B.Q.; Splinter, P.L.; Lee, S.O.; LaRusso, N.F. Development and characterization of a cholangiocyte cell line from the PCK rat, an animal model of Autosomal Recessive Polycystic Kidney Disease. Lab. Investig. 2006, 86, 940–950. [Google Scholar] [CrossRef]
- Woollard, J.R.; Punyashtiti, R.; Richardson, S.; Masyuk, T.V.; Whelan, S.; Huang, B.Q.; Lager, D.J.; Vandeursen, J.; Torres, V.E.; Gattone, V.H.; et al. A mouse model of autosomal recessive polycystic kidney disease with biliary duct and proximal tubule dilatation. Kidney Int. 2007, 72, 328–336. [Google Scholar] [CrossRef]
- Gallagher, A.R.; Esquivel, E.L.; Briere, T.S.; Tian, X.; Mitobe, M.; Menezes, L.F.; Markowitz, G.S.; Jain, D.; Onuchic, L.F.; Somlo, S. Biliary and pancreatic dysgenesis in mice harboring a mutation in Pkhd1. Am. J. Pathol. 2008, 172, 417–429. [Google Scholar] [CrossRef]
- Kim, I.; Fu, Y.; Hui, K.; Moeckel, G.; Mai, W.; Li, C.; Liang, D.; Zhao, P.; Ma, J.; Chen, X.-Z.; et al. Fibrocystin/Polyductin Modulates Renal Tubular Formation by Regulating Polycystin-2 Expression and Function. J. Am. Soc. Nephrol. 2008, 19, 455–468. [Google Scholar] [CrossRef]
- Williams, S.S.; Cobo-Stark, P.; James, L.R.; Somlo, S.; Igarashi, P. Kidney cysts, pancreatic cysts, and biliary disease in a mouse model of autosomal recessive polycystic kidney disease. Pediatr. Nephrol. 2008, 23, 733–741. [Google Scholar] [CrossRef] [PubMed]
- Bakeberg, J.L.; Tammachote, R.; Woollard, J.R.; Hogan, M.C.; Tuan, H.F.; Li, M.; Van Deursen, J.M.; Wu, Y.; Huang, B.Q.; Torres, V.E.; et al. Epitope-tagged Pkhd1 tracks the processing, secretion, and localization of fibrocystin. J. Am. Soc. Nephrol. 2011, 22, 2266–2277. [Google Scholar] [CrossRef]
- Bult, C.J.; Kadin, J.A.; Richardson, J.E.; Blake, J.A.; Eppig, J.T. The mouse genome database: Enhancements and updates. Nucleic Acids Res. 2009, 38. [Google Scholar] [CrossRef]
- Neufeld, T.K.; Douglass, D.; Grant, M.; Ye, M.; Silva, F.; Nadasdy, T.; Grantham, J.J. In vitro formation and expansion of cysts derived from human renal cortex epithelial cells. Kidney Int. 1992, 41, 1222–1236. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ye, M.; Grant, M.; Sharma, M.; Elzinga, L.; Swan, S.; Torres, V.E.; Grantham, J.J. Cyst fluid from human autosomal dominant polycystic kidneys promotes cyst formation and expansion by renal epithelial cells in vitro. J. Am. Soc. Nephrol. 1992, 3, 984–994. [Google Scholar] [CrossRef]
- Lakshmanan, J.; Fisher, D.A. An inborn error in epidermal growth factor prohormone metabolism in a mouse model of autosomal recessive polycystic kidney disease. Biochem. Biophys. Res. Commun. 1993, 196, 892–901. [Google Scholar] [CrossRef]
- Pugh, J.L.; Sweeney, W.E.; Avner, E.D. Tyrosine kinase activity of the EGF receptor in murine metanephric organ culture. Kidney Int. 1995, 47, 774–781. [Google Scholar] [CrossRef] [PubMed]
- Dell, K.M.R.; Nemo, R.; Sweeney, W.E.; Avner, E.D. EGF-related growth factors in the pathogenesis of murine ARPKD. Kidney Int. 2004, 65, 2018–2029. [Google Scholar] [CrossRef] [PubMed]
- Richards, W.G.; Sweeney, W.E.; Yoder, B.K.; Wilkinson, J.E.; Woychik, R.P.; Avner, E.D. Epidermal growth factor receptor activity mediates renal cyst formation in polycystic kidney disease. J. Clin. Investig. 1998, 101, 935–939. [Google Scholar] [CrossRef]
- Arbeiter, A.; Büscher, R.; Bonzel, K.E.; Wingen, A.M.; Vester, U.; Wohlschläger, J.; Zerres, K.; Nürnberger, J.; Bergmann, C.; Hoyer, P.F. Nephrectomy in an autosomal recessive polycystic kidney disease (ARPKD) patient with rapid kidney enlargement and increased expression of EGFR. Nephrol. Dial. Transplant. 2008, 23, 3026–3029. [Google Scholar] [CrossRef] [PubMed]
- Rohatgi, R.; Zavilowitz, B.; Vergara, M.; Woda, C.; Kim, P.; Satlin, L.M. Cyst fluid composition in human autosomal recessive polycystic kidney disease. Pediatr. Nephrol. 2005, 20, 552–553. [Google Scholar] [CrossRef] [PubMed]
- Gattone, V.H.; Calvet, J.P. Murine Infantile Polycystic Kidney Disease: A Role for Reduced Renal Epidermal Growth Factor. Am. J. Kidney Dis. 1991, 17, 606–607. [Google Scholar] [CrossRef]
- Nakanishi, K.; Gattone, V.H.; Sweeney, W.E.; Avner, E.D. Renal dysfunction but not cystic change is ameliorated by neonatal epidermal growth factor in bpk mice. Pediatr. Nephrol. 2001, 16, 45–50. [Google Scholar] [CrossRef]
- Lowden, D.A.; Lindemann, G.W.; Merlino, G.; Barash, B.D.; Calvet, J.P.; Gattone, V.H. Renal cysts in transgenic mice expressing transforming gorwth factor-alpha. J. Lab. Clin. Med. 1994, 124, 386–394. [Google Scholar] [PubMed]
- Zheleznova, N.N.; Wilson, P.D.; Staruschenko, A. Epidermal growth factor-mediated proliferation and sodium transport in normal and PKD epithelial cells. Biochim. Biophys. Acta Mol. Basis Dis. 2011, 1812, 1301–1313. [Google Scholar] [CrossRef] [PubMed]
- Nauta, J.; Sweeney, W.E.; Rutledge, J.C.; Avner, E.D. Biliary epithelial cells from mice with congenital polycystic kidney disease are hyperresponsive to epidermal growth factor. Pediatr. Res. 1995, 37, 755–763. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Luetteke, N.C.; Phillips, H.K.; Qiu, T.H.; Copeland, N.G.; Shelton Earp, H.; Jenkins, N.A.; Lee, D.C. The mouse waved-2 phenotype results from a point mutation in the EGF receptor tyrosine kinase. Genes Dev. 1994, 8, 399–413. [Google Scholar] [CrossRef]
- Sweeney, W.E.; Chen, Y.; Nakanishi, K.; Frost, P.; Avner, E.D. Treatment of polycystic kidney disease with a novel tyrosine kinase inhibitor. Kidney Int. 2000, 57, 33–40. [Google Scholar] [CrossRef]
- Sweeney, W.E.; Hamahira, K.; Sweeney, J.; Garcia-Gatrell, M.; Frost, P.; Avner, E.D. Combination treatment of PKD utilizing dual inhibition of EGF-receptor activity and ligand bioavailability. Kidney Int. 2003, 64, 1310–1319. [Google Scholar] [CrossRef][Green Version]
- Nemo, R.; Murcia, N.; Dell, K.M. Transforming Growth Factor Alpha (TGF-) and Other Targets of Tumor Necrosis Factor-Alpha Converting Enzyme (TACE) in Murine Polycystic Kidney Disease. Pediatr. Res. 2005, 57, 732–737. [Google Scholar] [CrossRef]
- Torres, V.E.; Sweeney, W.E.; Wang, X.; Qian, Q.; Harris, P.C.; Frost, P.; Avner, E.D. Epidermal growth factor receptor tyrosine kinase inhibition is not protective in PCK rats. Kidney Int. 2004, 66, 1766–1773. [Google Scholar] [CrossRef] [PubMed]
- Veizis, E.I.; Carlin, C.R.; Cotton, C.U. Decreased amiloride-sensitive Na+ absorption in collecting duct principal cells isolated from BPK ARPKD mice. Am. J. Physiol. Ren. Physiol. 2004, 286. [Google Scholar] [CrossRef]
- Veizis, I.E.; Cotton, C.U. Abnormal EGF-dependent regulation of sodium absorption in ARPKD collecting duct cells. Am. J. Physiol. Ren. Physiol. 2005, 288. [Google Scholar] [CrossRef] [PubMed]
- Rohatgi, R.; Greenberg, A.; Burrow, C.R.; Wilson, P.D.; Satlin, L.M. Na transport in autosomal recessive polycystic kidney disease (ARPKD) cyst lining epithelial cells. J. Am. Soc. Nephrol. 2003, 14, 827–836. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Olteanu, D.; Yoder, B.K.; Liu, W.; Croyle, M.J.; Welty, E.A.; Rosborough, K.; Wyss, J.M.; Bell, P.D.; Guay-Woodford, L.M.; Bevensee, M.O.; et al. Heightened epithelial Na+ channel-mediated Na+ absorption in a murine polycystic kidney disease model epithelium lacking apical monocilia. Am. J. Physiol. Cell Physiol. 2006, 290. [Google Scholar] [CrossRef]
- Kaimori, J.; Lin, C.-C.; Outeda, P.; Garcia-Gonzalez, M.A.; Menezes, L.F.; Hartung, E.A.; Li, A.; Wu, G.; Fujita, H.; Sato, Y.; et al. NEDD4-family E3 ligase dysfunction due to PKHD1/Pkhd1 defects suggests a mechanistic model for ARPKD pathobiology. Sci. Rep. 2017, 7, 7733. [Google Scholar] [CrossRef] [PubMed]
- Davidow, C.J.; Maser, R.L.; Rome, L.A.; Calvet, J.P.; Grantham, J.J. The cystic fibrosis transmembrane conductance regulator mediates transepithelial fluid secretion by human autosomal dominant polycystic kidney disease epithelium in vitro. Kidney Int. 1996, 50, 208–218. [Google Scholar] [CrossRef]
- Jouret, F.; Devuyst, O. Targeting chloride transport in autosomal dominant polycystic kidney disease. Cell. Signal. 2020, 73, 109703. [Google Scholar] [CrossRef]
- Nakanishi, K.; Sweeney, W.E.; Dell, K.M.; Cotton, C.U.; Avner, E.D. Role of CFTR in Autosomal Recessive Polycystic Kidney Disease. J. Am. Soc. Nephrol. 2001, 12, 719–725. [Google Scholar] [CrossRef]
- Yamaguchi, T.; Nagao, S.; Kasahara, M.; Takahashi, H.; Grantham, J.J. Renal accumulation and excretion of cyclic adenosine monophosphate in a murine model of slowly progressive polycystic kidney disease. Am. J. Kidney Dis. 1997, 30, 703–709. [Google Scholar] [CrossRef]
- Smith, L.A.; Bukanov, N.O.; Husson, H.; Russo, R.J.; Barry, T.C.; Taylor, A.L.; Beier, D.R.; Ibraghimov-Beskrovnaya, O. Development of polycystic kidney disease in juvenile cystic kidney mice: Insights into pathogenesis, ciliary abnormalities, and common features with human disease. J. Am. Soc. Nephrol. 2006, 17, 2821–2831. [Google Scholar] [CrossRef]
- Belibi, F.A.; Reif, G.; Wallace, D.P.; Yamaguchi, T.; Olsen, L.; Li, H.; Helmkamp, G.M.; Grantham, J.J. Cyclic AMP promotes growth and secretion in human polycystic kidney epithelial cells. Kidney Int. 2004, 66, 964–973. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, T.; Pelling, J.C.; Ramaswamy, N.T.; Eppler, J.W.; Wallace, D.P.; Nagao, S.; Rome, L.A.; Sullivan, L.P.; Grantham, J.J. cAMP stimulates the in vitro proliferation of renal cyst epithelial cells by activating the extracellular signal-regulated kinase pathway. Kidney Int. 2000, 57, 1460–1471. [Google Scholar] [CrossRef]
- Yamaguchi, T.; Nagao, S.; Wallace, D.P.; Belibi, F.A.; Cowley, B.D.; Pelling, J.C.; Grantham, J.J. Cyclic AMP activates B-Raf and ERK in cyst epithelial cells from autosomal-dominant polycystic kidneys. Kidney Int. 2003, 63, 1983–1994. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, T.; Wallace, D.P.; Magenheimer, B.S.; Hempson, S.J.; Grantham, J.J.; Calvet, J.P. Calcium restriction allows cAMP activation of the B-Raf/ERK pathway, switching cells to a cAMP-dependent growth-stimulated phenotype. J. Biol. Chem. 2004, 279, 40419–40430. [Google Scholar] [CrossRef]
- Wang, X. Effectiveness of Vasopressin V2 Receptor Antagonists OPC-31260 and OPC-41061 on Polycystic Kidney Disease Development in the PCK Rat. J. Am. Soc. Nephrol. 2005, 16, 846–851. [Google Scholar] [CrossRef]
- Wang, X.; Wu, Y.; Ward, C.J.; Harris, P.C.; Torres, V.E. Vasopressin directly regulates cyst growth in polycystic kidney disease. J. Am. Soc. Nephrol. 2008, 19, 102–108. [Google Scholar] [CrossRef] [PubMed]
- Zheng, R.; Wang, L.; Fan, J.; Zhou, Q. Inhibition of PKHD1 may cause S-phase entry via mTOR signaling pathway. Cell Biol. Int. 2009, 33, 926–933. [Google Scholar] [CrossRef]
- Fischer, D.C.; Jacoby, U.; Pape, L.; Ward, C.J.; Kuwertz-Broeking, E.; Renken, C.; Nizze, H.; Querfeld, U.; Rudolph, B.; Mueller-Wiefel, D.E.; et al. Activation of the AKTmTOR pathway in autosomal recessive polycystic kidney disease (ARPKD). Nephrol. Dial. Transplant. 2009, 24, 1819–1827. [Google Scholar] [CrossRef]
- Ren, X.S.; Sato, Y.; Harada, K.; Sasaki, M.; Furubo, S.; Song, J.Y.; Nakanuma, Y. Activation of the PI3K/mTOR Pathway Is Involved in Cystic Proliferation of Cholangiocytes of the PCK Rat. PLoS ONE 2014, 9, e87660. [Google Scholar] [CrossRef] [PubMed]
- Hovater, M.B.; Olteanu, D.; Hanson, E.L.; Cheng, N.-L.; Siroky, B.; Fintha, A.; Komlosi, P.; Liu, W.; Satlin, L.M.; Darwin Bell, P.; et al. Loss of apical monocilia on collecting duct principal cells impairs ATP secretion across the apical cell surface and ATP-dependent and flow-induced calcium signals. Purinergic Signal. 2007. [Google Scholar] [CrossRef]
- Yamaguchi, T.; Hempson, S.J.; Reif, G.A.; Hedge, A.M.; Wallace, D.P. Calcium restores a normal proliferation phenotype in human polycystic kidney disease epithelial cells. J. Am. Soc. Nephrol. 2006, 17, 178–187. [Google Scholar] [CrossRef]
- Sweeney, W.E.; von Vigier, R.O.; Frost, P.; Avner, E.D. Src inhibition ameliorates polycystic kidney disease. J. Am. Soc. Nephrol. 2008, 19, 1331–1341. [Google Scholar] [CrossRef]
- Goel, M.; Zuo, C.-D.; Schilling, W.P. Role of cAMP/PKA signaling cascade in vasopressin-induced trafficking of TRPC3 channels in principal cells of the collecting duct. Am. J. Physiol. Ren. Physiol. 2010, 298. [Google Scholar] [CrossRef] [PubMed]
- Chebib, F.T.; Sussman, C.R.; Wang, X.; Harris, P.C.; Torres, V.E. Vasopressin and disruption of calcium signalling in polycystic kidney disease. Nat. Rev. Nephrol. 2015, 11, 451–464. [Google Scholar] [CrossRef] [PubMed]
- Gattone, V.H.; Wang, X.; Harris, P.C.; Torres, V.E. Inhibition of renal cystic disease development and progression by a vasopressin V2 receptor antagonist. Nat. Med. 2003, 9, 1323–1326. [Google Scholar] [CrossRef] [PubMed]
- Torres, V.E.; Wang, X.; Qian, Q.; Somlo, S.; Harris, P.C.; Gattone, V.H. Effective treatment of an orthologous model of autosomal dominant polycystic kidney disease. Nat. Med. 2004, 10, 363–364. [Google Scholar] [CrossRef]
- Torres, V.E.; Chapman, A.B.; Devuyst, O.; Gansevoort, R.T.; Grantham, J.J.; Higashihara, E.; Perrone, R.D.; Krasa, H.B.; Ouyang, J.; Czerwiec, F.S. Tolvaptan in patients with autosomal dominant polycystic kidney disease. N. Engl. J. Med. 2012, 367, 2407–2418. [Google Scholar] [CrossRef]
- Gansevoort, R.T.; Arici, M.; Benzing, T.; Birn, H.; Capasso, G.; Covic, A.; Devuyst, O.; Drechsler, C.; Eckardt, K.U.; Emma, F.; et al. Recommendations for the use of tolvaptan in autosomal dominant polycystic kidney disease: A position statement on behalf of the ERA-EDTA Working Groups on Inherited Kidney Disorders and European Renal Best Practice. Nephrol. Dial. Transplant. 2016, 31, 337–348. [Google Scholar] [CrossRef]
- Sweeney, W.E.; Avner, E.D. Emerging Therapies for Childhood Polycystic Kidney Disease. Front. Pediatr. 2017, 5, 1–7. [Google Scholar] [CrossRef]
- Liu, B.; Li, C.; Liu, Z.; Dai, Z.; Tao, Y. Increasing extracellular matrix collagen level and MMP activity induces cyst development in polycystic kidney disease. BMC Nephrol. 2012, 13, 1–8. [Google Scholar] [CrossRef]
- Urribarri, A.D.; Munoz-Garrido, P.; Perugorria, M.J.; Erice, O.; Merino-Azpitarte, M.; Arbelaiz, A.; Lozano, E.; Hijona, E.; Jiménez-Agüero, R.; Fernandez-Barrena, M.G.; et al. Inhibition of metalloprotease hyperactivity in cystic cholangiocytes halts the development of polycystic liver diseases. Gut 2014, 63, 1658–1667. [Google Scholar] [CrossRef]
- Dai, B.; Liu, Y.; Mei, C.; Fu, L.; Xiong, X.; Zhang, Y.; Shen, X.; Hua, Z. Rosiglitazone attenuates development of polycystic kidney disease and prolongs survival in Han:SPRD rats. Clin. Sci. 2010, 119, 323–333. [Google Scholar] [CrossRef] [PubMed]
- Yoshihara, D.; Kugita, M.; Yamaguchi, T.; Aukema, H.M.; Kurahashi, H.; Morita, M.; Hiki, Y.; Calvet, J.P.; Wallace, D.P.; Toyohara, T.; et al. Global gene expression profiling in PPAR-γ agonist-treated kidneys in an orthologous rat model of human autosomal recessive polycystic kidney disease. PPAR Res. 2012. [Google Scholar] [CrossRef] [PubMed]
- Riwanto, M.; Kapoor, S.; Rodriguez, D.; Edenhofer, I.; Segerer, S.; Wüthrich, R.P. Inhibition of aerobic glycolysis attenuates disease progression in polycystic kidney disease. PLoS ONE 2016, 11. [Google Scholar] [CrossRef] [PubMed]
- Fischer, E.; Legue, E.; Doyen, A.; Nato, F.; Nicolas, J.F.; Torres, V.; Yaniv, M.; Pontoglio, M. Defective planar cell polarity in polycystic kidney disease. Nat. Genet. 2006, 38, 21–23. [Google Scholar] [CrossRef] [PubMed]
- Nishio, S.; Tian, X.; Gallagher, A.R.; Yu, Z.; Patel, V.; Igarashi, P.; Somlo, S. Loss of Oriented Cell Division Does not Initiate Cyst Formation. J. Am. Soc. Nephrol. 2010, 21, 295–302. [Google Scholar] [CrossRef] [PubMed]
- Hildebrandt, F.; Benzing, T.; Katsanis, N. Ciliopathies. N. Engl. J. Med. 2011, 364, 1533–1543. [Google Scholar] [CrossRef] [PubMed]
- Busch, T.; Köttgen, M.; Hofherr, A. TRPP2 ion channels: Critical regulators of organ morphogenesis in health and disease. Cell Calcium 2017, 66, 25–32. [Google Scholar] [CrossRef]
- Hanaoka, K.; Qian, F.; Boletta, A.; Bhunia, A.K.; Piontek, K.; Tsiokas, L.; Sukhatme, V.P.; Guggino, W.B.; Germino, G.G. Co-assembly of polycystin-1 and -2 produces unique cation-permeable currents. Nature 2000, 408, 990–994. [Google Scholar] [CrossRef]
- Nauli, S.M.; Alenghat, F.J.; Luo, Y.; Williams, E.; Vassilev, P.; Li, X.; Elia, A.E.H.; Lu, W.; Brown, E.M.; Quinn, S.J.; et al. Polycystins 1 and 2 mediate mechanosensation in the primary cilium of kidney cells. Nat. Genet. 2003, 33, 129–137. [Google Scholar] [CrossRef]
- Wang, Z.; Ng, C.; Liu, X.; Wang, Y.; Li, B.; Kashyap, P.; Chaudhry, H.A.; Castro, A.; Kalontar, E.M.; Ilyayev, L.; et al. The ion channel function of polycystin-1 in the polycystin-1/polycystin-2 complex. EMBO Rep. 2019, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Dai, X.-Q.Q.; Li, Q.; Chen, C.X.; Mai, W.; Hussain, Z.; Long, W.; Montalbetti, N.; Li, G.; Glynne, R.; et al. Kinesin-2 mediates physical and functional interactions between polycystin-2 and fibrocystin. Hum. Mol. Genet. 2006, 15, 3280–3292. [Google Scholar] [CrossRef]
- Wang, S.; Zhang, J.; Nauli, S.M.; Li, X.; Starremans, P.G.; Luo, Y.; Roberts, K.A.; Zhou, J. Fibrocystin/Polyductin, Found in the Same Protein Complex with Polycystin-2, Regulates Calcium Responses in Kidney Epithelia Fibrocystin/Polyductin, Found in the Same Protein Complex with Polycystin-2, Regulates Calcium Responses in Kidney Epithel. Mol. Cell. Biol. 2007, 27, 3241–3252. [Google Scholar] [CrossRef]
- Allison, S.J. Polycystic kidney disease: FPC in ARPKD. Nat. Rev. Nephrol. 2017, 13, 597. [Google Scholar] [CrossRef] [PubMed]
- Olson, R.J.; Hopp, K.; Wells, H.; Smith, J.M.; Furtado, J.; Constans, M.M.; Escobar, D.L.; Geurts, A.M.; Torres, V.E.; Harris, P.C. Synergistic Genetic Interactions between Pkhd1 and Pkd1 Result in an ARPKD-Like Phenotype in Murine Models. J. Am. Soc. Nephrol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Kaimori, J.; Germino, G.G. ARPKD and ADPKD: First cousins or more distant relatives? J. Am. Soc. Nephrol. 2008, 19, 416–418. [Google Scholar] [CrossRef] [PubMed]
- Ma, M.; Tian, X.; Igarashi, P.; Pazour, G.J.; Somlo, S. Loss of cilia suppresses cyst growth in genetic models of autosomal dominant polycystic kidney disease. Nat. Genet. 2013, 45, 1004–1012. [Google Scholar] [CrossRef]
- Gallagher, A.R.; Somlo, S. Loss of cilia does not slow liver disease progression in mouse models of autosomal recessive polycystic kidney disease. Kidney360 2020, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Palander, O.; El-Zeiry, M.; Trimble, W.S. Uncovering the Roles of Septins in Cilia. Front. Cell Dev. Biol. 2017, 5, 36. [Google Scholar] [CrossRef]
- Torres, V.E.; Chapman, A.B.; Devuyst, O.; Gansevoort, R.T.; Perrone, R.D.; Koch, G.; Ouyang, J.; McQuade, R.D.; Blais, J.D.; Czerwiec, F.S.; et al. Tolvaptan in Later-Stage Autosomal Dominant Polycystic Kidney Disease. N. Engl. J. Med. 2017, 377, 1930–1942. [Google Scholar] [CrossRef]
- Edwards, M.E.; Chebib, F.T.; Irazabal, M.V.; Ofstie, T.G.; Bungum, L.A.; Metzger, A.J.; Senum, S.R.; Hogan, M.C.; El-Zoghby, Z.M.; Kline, T.L.; et al. Long-term administration of tolvaptan in autosomal dominant polycystic kidney disease. Clin. J. Am. Soc. Nephrol. 2018, 13, 1153–1161. [Google Scholar] [CrossRef] [PubMed]
- Talbot, J.J.; Song, X.; Wang, X.; Rinschen, M.M.; Doerr, N.; LaRiviere, W.B.; Schermer, B.; Pei, Y.P.; Torres, V.E.; Weimbs, T. The cleaved cytoplasmic tail of polycystin-1 regulates Src-dependent STAT3 activation. J. Am. Soc. Nephrol. 2014, 25, 1737–1748. [Google Scholar] [CrossRef] [PubMed]
- Sweeney, W.E.; Frost, P.; Avner, E.D. Tesevatinib ameliorates progression of polycystic kidney disease in rodent models of autosomal recessive polycystic kidney disease. World J. Nephrol. 2017, 6, 188–200. [Google Scholar] [CrossRef] [PubMed]



| Identifier | Intervention | Study Design and Characteristics | Study Description | Sponsor |
|---|---|---|---|---|
| NCT04782258 | Tolvaptan | ∙Study type: interventional ∙Primary purpose: treatment ∙Period: April 2021–June 2025 (estimated) ∙Patients: 20 (estimated, not yet recruitment) ∙Allocation: non-randomized ∙Intervention model: parallel assignment ∙Masking: none (open label) | ∙The primary objective of this phase 3 trial is to evaluate the safety of Tolvaptan (OPC-41061) in infants and children, 8 days to less than 18 years old of age, with ARPKD. ∙Participants in this study will be assigned to Tolvaptan for 18 months and closely monitored over the course of the study. | Otsuka Pharmaceutical Development & Commercialization, Inc. Princeton, New Jersey, USA. |
| NCT04786574 | Tolvaptan | ∙Study type: interventional ∙Primary purpose: treatment ∙Period: April 2021–July 2025 (estimated) ∙Patients: 20 (estimated, not yet recruitment) ∙Allocation: N/A ∙Intervention model: single group assignment ∙Masking: none (open label) | ∙In this Phase 3 trial, the primary objective is to evaluate safety, tolerability, and efficacy of Tolvaptan (OPC-41061) in pediatric subjects, 28 days to less than 12 weeks of age, with ARPKD. ∙Participants in this trial will be assigned to Tolvaptan for 24 months and closely monitored over the course of the study. | Otsuka Pharmaceutical Development & Commercialization, Inc. Princeton, New Jersey, USA. |
| NCT03096080 | Tesevatinib | ∙Study type: interventional ∙Primary purpose: treatment ∙Period: March 2017–October 2019 (completed) ∙Patients: 10 ∙Allocation: Non-randomized ∙Intervention model: sequential assignment ∙Masking: none (open label) | ∙This trial in Phase 1 evaluates safety and tolerability of a single ascending dose of a Tesevatinib (KD019, XL647) liquid formulation administered to pediatric subjects (child with age 5–12 years) with ARPKD. ∙To determine safety of the Tesevatinib liquid formulation in pediatric subjects with ARPKD, all participants receive active study drug on Day 1 of the study enrollment. | Kadmon Corporation, LLC Philadelphia, Pennsylvania, USA. Milwaukee, Wisconsin, USA. |
| NCT01401998 | Observational | ∙Study type: observational ∙Period: July 2011–December 2022 (recruitment) ∙Patients: 200 (estimated) ∙Observational model: cohort ∙Time perspective: retrospective assignment | ∙This study captures clinical and genetic information of ARPKD patients to expand the knowledge of disease. ∙The primary goal of this trial is create a clinical and mutational databases including clinical information and identifying genetic mutations from all patients enrolled in the study. ∙Mutational database will be useful to facilitate genetic research as genotype-phenotype correlations, new disease gene studies, or modifier gene studies. ∙Create a tissue resource with human tissue from both affected and controls individuals. | Lisa M. Guay-Woodford (Collaborator: National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK)). Washington, District of Columbia, USA. |
| NCT00068224 | Observational | ∙Study type: observational ∙Period: September 2003–February 2021 (completed) ∙Patients: 374 ∙Observational model: cohort ∙Time perspective: prospective | ∙This study evaluated patients with ciliopathies, including ARPKD. ∙ The goal of the study is to better understand the medical complications of these disorders and identify characteristics that can help in the design of new treatments. | National Human Genome Research Institute (NHGRI). Bethesda, Maryland, USA. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cordido, A.; Vizoso-Gonzalez, M.; Garcia-Gonzalez, M.A. Molecular Pathophysiology of Autosomal Recessive Polycystic Kidney Disease. Int. J. Mol. Sci. 2021, 22, 6523. https://doi.org/10.3390/ijms22126523
Cordido A, Vizoso-Gonzalez M, Garcia-Gonzalez MA. Molecular Pathophysiology of Autosomal Recessive Polycystic Kidney Disease. International Journal of Molecular Sciences. 2021; 22(12):6523. https://doi.org/10.3390/ijms22126523
Chicago/Turabian StyleCordido, Adrian, Marta Vizoso-Gonzalez, and Miguel A. Garcia-Gonzalez. 2021. "Molecular Pathophysiology of Autosomal Recessive Polycystic Kidney Disease" International Journal of Molecular Sciences 22, no. 12: 6523. https://doi.org/10.3390/ijms22126523
APA StyleCordido, A., Vizoso-Gonzalez, M., & Garcia-Gonzalez, M. A. (2021). Molecular Pathophysiology of Autosomal Recessive Polycystic Kidney Disease. International Journal of Molecular Sciences, 22(12), 6523. https://doi.org/10.3390/ijms22126523