
Regulatory SNPs: Altered Transcription Factor Binding Sites Implicated in Complex Traits and Diseases

Abstract
1. Introduction
2. Brief History of rSNP Discovery
3. Modern Array of Methods for Studying Individual rSNPs
4. Recent Comprehensive Examples
4.1. Allele C of rs36115365 from chr5p15.33 Multi-Cancer Risk Locus Enhances ZNF148 Binding and Telomerase Reverse Transcriptase (TERT) Expression
4.2. Allele G of rs11672691 from Chr19q13.2, Associated with Aggressive Prostate Cancer, Creates a HOXA2 Binding Site and Raises the Transcription Levels of PCAT19 and CEACAM21 Genes, Implicated in Prostate Cancer Cell Growth and Tumor Progression
4.3. Atherosclerosis Risk Variant A of rs2107595 from Chr7p21.1 Interferes with E2F3 in Putative Enhancer Region, Which Leads to HDAC9 Activation
4.4. Allele A of rs12411216 from Chr1q22 Decreases E2F4 Binding, Which Results in a Decreased GBA Expression and an Increased Cognitive Damage in Parkinson’s Disease
4.5. Allele A of rs13239597, Associated with Two Systemic Autoimmune Diseases, Enhances the Binding of EVI1, Which Promotes Formation of a Long-Range Chromatin Loop and an Increased Expression of IRF5, Located 118 kb Away
4.6. Allele T of rs17079281 Decreases Lung Cancer Risk through Creating an YY1 Binding Site to Suppress Proto-Oncogene DCBLD1 Expression
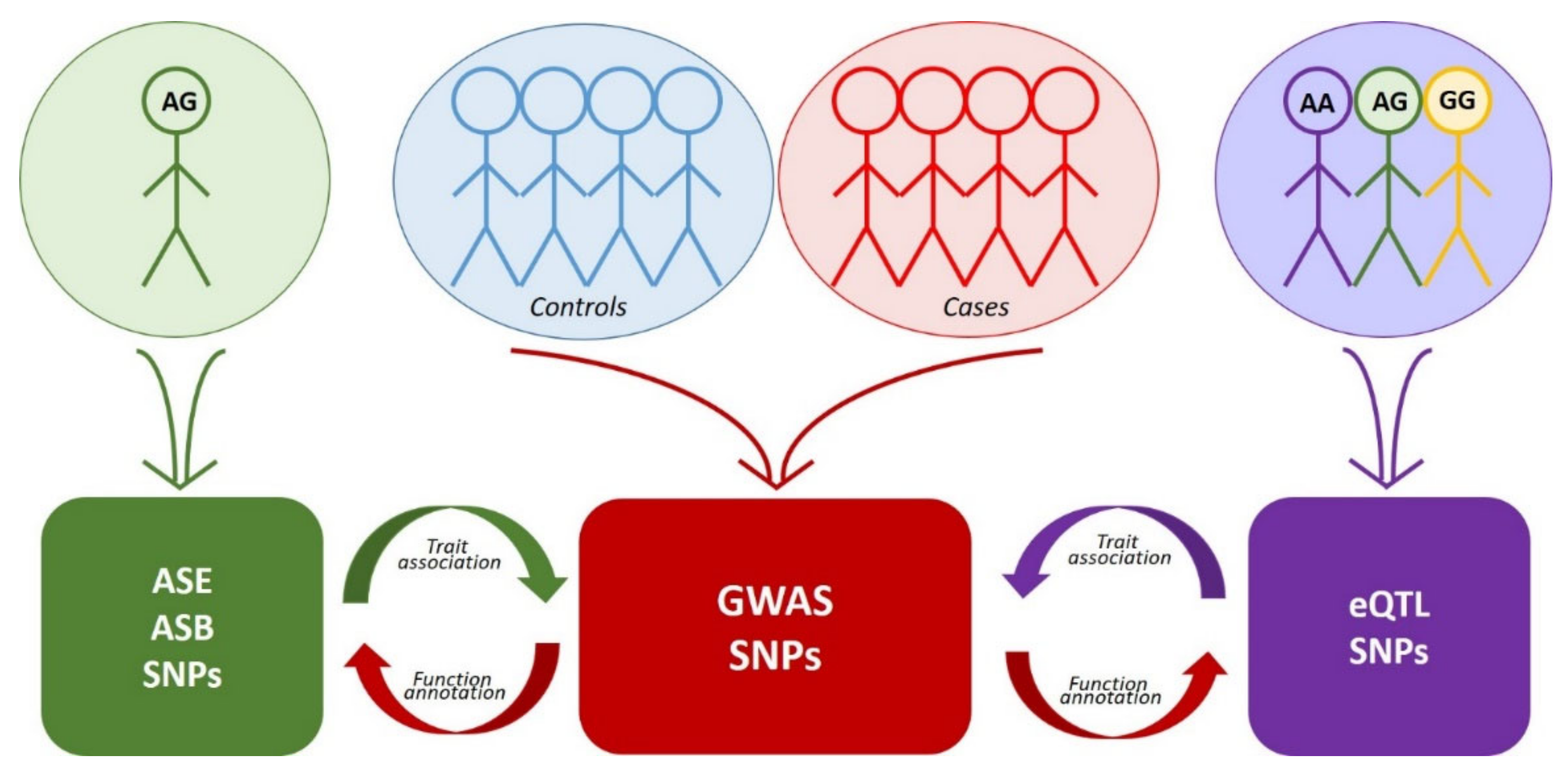
5. rSNPs on a Genome-Wide Scale
5.1. Making Molecular Sense of GWAS
5.2. eQTL Analysis
5.3. Allele-Specific Expression (ASE) Analysis
5.4. Allele-Specific Binding (ASB) Analysis
6. Conclusions
Funding
Conflicts of Interest
References
- Buniello, A.; MacArthur, J.A.L.; Cerezo, M.; Harris, L.W.; Hayhurst, J.; Malangone, C.; McMahon, A.; Morales, J.; Mountjoy, E.; Sollis, E.; et al. The NHGRI-EBI GWAS Catalog of published genome-wide association studies, targeted arrays and summary statistics 2019. Nucleic Acids Res. 2019, 47, D1005–D1012. [Google Scholar] [CrossRef] [PubMed]
- Claussnitzer, M.; Cho, J.H.; Collins, R.; Cox, N.J.; Dermitzakis, E.T.; Hurles, M.E.; Kathiresan, S.; Kenny, E.E.; Lindgren, C.M.; MacArthur, D.G.; et al. A brief history of human disease genetics. Nature 2020, 577, 179–189. [Google Scholar] [CrossRef] [PubMed]
- Bryzgalov, L.O.; Antontseva, E.V.; Matveeva, M.Y.; Shilov, A.G.; Kashina, E.V.; Mordvinov, V.A.; Merkulova, T.I. Detection of Regulatory SNPs in Human Genome Using ChIP-seq ENCODE Data. PLoS ONE 2013, 8, e78833. [Google Scholar] [CrossRef] [PubMed]
- Farh, K.K.-H.; Marson, A.; Zhu, J.; Kleinewietfeld, M.; Housley, W.J.; Beik, S.; Shoresh, N.; Whitton, H.; Ryan, R.J.H.; Shishkin, A.A.; et al. Genetic and epigenetic fine mapping of causal autoimmune disease variants. Nature 2015, 518, 337–343. [Google Scholar] [CrossRef] [PubMed]
- Maurano, M.T.; Humbert, R.; Rynes, E.; Thurman, R.E.; Haugen, E.; Wang, H.; Reynolds, A.P.; Sandstrom, R.; Qu, H.; Brody, J.; et al. Systematic Localization of Common Disease-Associated Variation in Regulatory DNA. Science 2012, 337, 1190–1195. [Google Scholar] [CrossRef] [PubMed]
- GTEx Consortium. Genetic effects on gene expression across human tissues. Nature 2017, 550, 204–213. [Google Scholar] [CrossRef]
- Levo, M.; Segal, E. In pursuit of design principles of regulatory sequences. Nat. Rev. Genet. 2014, 15, 453–468. [Google Scholar] [CrossRef]
- Andersson, R. Promoter or enhancer, what’s the difference? Deconstruction of established distinctions and presentation of a unifying model. BioEssays 2015, 37, 314–323. [Google Scholar] [CrossRef]
- Erokhin, M.; Vassetzky, Y.; Georgiev, P.; Chetverina, D. Eukaryotic enhancers: Common features, regulation, and participation in diseases. Cell. Mol. Life Sci. 2015, 72, 2361–2375. [Google Scholar] [CrossRef]
- Chen, H.; Pugh, B.F. What Do Transcription Factors Interact with? J. Mol. Biol. 2021, 166883. [Google Scholar] [CrossRef]
- Tobias, I.C.; Abatti, L.E.; Moorthy, S.D.; Mullany, S.; Taylor, T.; Khader, N.; Filice, M.A.; Mitchell, J.A. Transcriptional enhancers: From prediction to functional assessment on a genome-wide scale. Genome 2021, 64, 426–448. [Google Scholar] [CrossRef]
- Singh, G.; Mullany, S.; Moorthy, S.D.; Zhang, R.; Mehdi, T.; Tian, R.; Duncan, A.G.; Moses, A.M.; Mitchell, J.A. A flexible repertoire of transcription factor binding sites and a diversity threshold determines enhancer activity in embryonic stem cells. Genome Res. 2021, 31, 564–575. [Google Scholar] [CrossRef]
- Lambert, S.A.; Jolma, A.; Campitelli, L.F.; Das, P.K.; Yin, Y.; Albu, M.; Chen, X.; Taipale, J.; Hughes, T.R.; Weirauch, M.T. The Human Transcription Factors. Cell 2018, 175, 598–599. [Google Scholar] [CrossRef]
- Lelli, K.M.; Slattery, M.; Mann, R.S. Disentangling the Many Layers of Eukaryotic Transcriptional Regulation. Annu. Rev. Genet. 2012, 46, 43–68. [Google Scholar] [CrossRef]
- Merkulova, T.I.; Ananko, E.A.; Ignat’eva, E.V.; Kolchanov, N.A. Regulatory transcription codes in eukaryotic genomes. Genetika 2013, 49, 37–54. [Google Scholar] [CrossRef]
- Wang, Y.; Ma, R.; Liu, B.; Kong, J.; Lin, H.; Yu, X.; Wang, R.; Li, L.; Gao, M.; Zhou, B.; et al. SNP rs17079281 decreases lung cancer risk through creating an YY1-binding site to suppress DCBLD1 expression. Oncogene 2020, 39, 4092–4102. [Google Scholar] [CrossRef]
- Padhy, B.; Hayat, B.; Nanda, G.G.; Mohanty, P.P.; Alone, D.P. Pseudoexfoliation and Alzheimer’s associated CLU risk variant, rs2279590, lies within an enhancer element and regulates CLU, EPHX2 and PTK2B gene expression. Hum. Mol. Genet. 2017, 26, 4519–4529. [Google Scholar] [CrossRef]
- Krause, M.D.; Huang, R.-T.; Wu, D.; Shentu, T.-P.; Harrison, D.L.; Whalen, M.B.; Stolze, L.K.; Di Rienzo, A.; Moskowitz, I.P.; Civelek, M.; et al. Genetic variant at coronary artery disease and ischemic stroke locus 1p32.2 regulates endothelial responses to hemodynamics. Proc. Natl. Acad. Sci. USA 2018, 115, e11349–e11358. [Google Scholar] [CrossRef]
- Hazelett, D.J.; Rhie, S.K.; Gaddis, M.; Yan, C.; Lakeland, D.L.; Coetzee, S.G.; Henderson, B.E.; Noushmehr, H.; Cozen, W.; Kote-Jarai, Z.; et al. Comprehensive Functional Annotation of 77 Prostate Cancer Risk Loci. PLoS Genet. 2014, 10, e1004102. [Google Scholar] [CrossRef]
- Gao, P.; Xia, J.-H.; Sipeky, C.; Dong, X.-M.; Zhang, Q.; Yang, Y.; Zhang, P.; Cruz, S.P.; Zhang, K.; Zhu, J.; et al. Biology and Clinical Implications of the 19q13 Aggressive Prostate Cancer Susceptibility Locus. Cell 2018, 174, 576–589.e18. [Google Scholar] [CrossRef]
- Afanasyeva, M.A.; Putlyaeva, L.V.; Demin, D.E.; Kulakovskiy, I.V.; Vorontsov, I.E.; Fridman, M.V.; Makeev, V.J.; Kuprash, D.V.; Schwartz, A.M. The single nucleotide variant rs12722489 determines differential estrogen receptor binding and enhancer properties of an IL2RA intronic region. PLoS ONE 2017, 12, e0172681. [Google Scholar] [CrossRef]
- Korneev, K.V.; Sviriaeva, E.N.; Mitkin, N.A.; Gorbacheva, A.M.; Uvarova, A.N.; Ustiugova, A.S.; Polanovsky, O.L.; Kulakovskiy, I.V.; Afanasyeva, M.A.; Schwartz, A.M.; et al. Minor C allele of the SNP rs7873784 associated with rheumatoid arthritis and type-2 diabetes mellitus binds PU.1 and enhances TLR4 expression. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165626. [Google Scholar] [CrossRef]
- Fang, J.; Jia, J.; Makowski, M.; Xu, M.; Wang, Z.; Zhang, T.; Hoskins, J.W.; Choi, J.; Han, Y.; Zhang, M.; et al. Functional characterization of a multi-cancer risk locus on chr5p15.33 reveals regulation of TERT by ZNF148. Nat. Commun. 2017, 8, 15034. [Google Scholar] [CrossRef]
- Choi, J.; Zhang, T.; Vu, A.; Ablain, J.; Makowski, M.M.; Colli, L.M.; Xu, M.; Hennessey, R.C.; Yin, J.; Rothschild, H.; et al. Massively parallel reporter assays of melanoma risk variants identify MX2 as a gene promoting melanoma. Nat. Commun. 2020, 11, 2718. [Google Scholar] [CrossRef]
- Zhao, Y.; Wu, D.; Jiang, D.; Zhang, X.; Wu, T.; Cui, J.; Qian, M.; Zhao, J.; Oesterreich, S.; Sun, W.; et al. A sequential methodology for the rapid identification and characterization of breast cancer-associated functional SNPs. Nat. Commun. 2020, 11, 3340. [Google Scholar] [CrossRef]
- Prestel, M.; Prell-Schicker, C.; Webb, T.; Malik, R.; Lindner, B.; Ziesch, N.; Rex-Haffner, M.; Röh, S.; Viturawong, T.; Lehm, M.; et al. The Atherosclerosis Risk Variant rs2107595 Mediates Allele-Specific Transcriptional Regulation of HDAC9 via E2F3 and Rb1. Stroke 2019, 50, 2651–2660. [Google Scholar] [CrossRef] [PubMed]
- Thomas, R.; Trapani, D.; Goodyer-Sait, L.; Tomkova, M.; Fernandez-Rozadilla, C.; Sahnane, N.; Woolley, C.; Davis, H.; Chegwidden, L.; Kriaucionis, S.; et al. The polymorphic variant rs1800734 influences methylation acquisition and allele-specific TFAP4 binding in the MLH1 promoter leading to differential mRNA expression. Sci. Rep. 2019, 9, 13463. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Z.; Huang, Y.; Zhang, P.; Han, C.; Lu, Y.; Mo, Z.; Zhang, Z.; Li, X.; Zhao, S.; Cai, F.; et al. Characterization of a pathogenic variant in GBA for Parkinson’s disease with mild cognitive impairment patients. Mol. Brain 2020, 13, 102. [Google Scholar] [CrossRef] [PubMed]
- Allen, E.K.; Randolph, A.G.; Bhangale, T.; Dogra, P.; Ohlson, M.; Oshansky, C.M.; Zamora, A.E.; Shannon, J.P.; Finkelstein, D.; Dressen, A.; et al. SNP-mediated disruption of CTCF binding at the IFITM3 promoter is associated with risk of severe influenza in humans. Nat. Med. 2017, 23, 975–983. [Google Scholar] [CrossRef]
- Vasiliev, G.V.; Merkulov, V.M.; Kobzev, V.F.; Merkulova, T.I.; Ponomarenko, M.P.; Kolchanov, N.A. Point mutations within 663–666 bp of intron 6 of the human TDO2 gene, associated with a number of psychiatric disorders, damage the YY-1 transcription factor binding site. FEBS Lett. 1999, 462, 85–88. [Google Scholar] [CrossRef]
- Cooper, D. The human gene mutation database. Nucleic Acids Res. 1998, 26, 285–287. [Google Scholar] [CrossRef]
- Deplancke, B.; Alpern, D.; Gardeux, V. The Genetics of Transcription Factor DNA Binding Variation. Cell 2016, 166, 538–554. [Google Scholar] [CrossRef]
- Ponomarenko, J.V. rSNP_Guide, a database system for analysis of transcription factor binding to target sequences: Application to SNPs and site-directed mutations. Nucleic Acids Res. 2001, 29, 312–316. [Google Scholar] [CrossRef][Green Version]
- Bienvenu, T.; Lacronique, V.; Raymondjean, M.; Cazeneuve, C.; Hubert, D.; Kaplan, J.-C.; Beldjord, C. Three novel sequence variations in the 5? upstream region of the cystic fibrosis transmembrane conductance regulator (CFTR) gene: Two polymorphisms and one putative molecular defect. Hum. Genet. 1995, 95, 698–702. [Google Scholar] [CrossRef]
- Ludlow, L.B.; Schick, B.P.; Budarf, M.L.; Driscoll, D.A.; Zackai, E.H.; Cohen, A.; Konkle, B.A. Identification of a Mutation in a GATA Binding Site of the Platelet Glycoprotein Ibβ Promoter Resulting in the Bernard-Soulier Syndrome. J. Biol. Chem. 1996, 271, 22076–22080. [Google Scholar] [CrossRef]
- Comings, D.E.; Gade, R.; Muhleman, D.; Chiu, C.; Wu, S.; To, M.; Spence, M.; Dietz, G.; Winn-Deen, E.; Rosenthal, R.J.; et al. Exon and intron variants in the human tryptophan 2,3-dioxygenase gene: Potential association with Tourette syndrome, substance abuse and other disorders. Pharmacogenetics 1996, 6, 307–318. [Google Scholar] [CrossRef]
- Merkulov, V.M.; Merkulova, T.I. Nucleotide sequence of a fragment of the rat tryptophan oxygenase gene showing high affinity to glucocorticoid receptor in vitro. Biochim. Biophys. Acta Gene Struct. Expr. 1992, 1132, 100–102. [Google Scholar] [CrossRef]
- Ponomarenko, J.V.; Ponomarenko, M.P.; Frolov, A.S.; Vorobyev, D.G.; Overton, G.C.; Kolchanov, N.A. Conformational and physicochemical DNA features specific for transcription factor binding sites. Bioinformatics 1999, 15, 654–668. [Google Scholar] [CrossRef]
- Verheul, T.C.J.; van Hijfte, L.; Perenthaler, E.; Barakat, T.S. The Why of YY1: Mechanisms of Transcriptional Regulation by Yin Yang 1. Front. Cell Dev. Biol. 2020, 8. [Google Scholar] [CrossRef]
- Knight, J.C.; Udalova, I.; Hill, A.V.S.; Greenwood, B.M.; Peshu, N.; Marsh, K.; Kwiatkowski, D. A polymorphism that affects OCT-1 binding to the TNF promoter region is associated with severe malaria. Nat. Genet. 1999, 22, 145–150. [Google Scholar] [CrossRef]
- Piedrafita, F.J.; Molander, R.B.; Vansant, G.; Orlova, E.A.; Pfahl, M.; Reynolds, W.F. An Alu Element in the Myeloperoxidase Promoter Contains a Composite SP1-Thyroid Hormone-Retinoic Acid Response Element. J. Biol. Chem. 1996, 271, 14412–14420. [Google Scholar] [CrossRef] [PubMed]
- Moi, P.; Loudianos, G.; Lavinha, J.; Murru, S.; Cossu, P.; Casu, R.; Oggiano, L.; Longinotti, M.; Cao, A.; Pirastu, M. Delta-thalassemia due to a mutation in an erythroid-specific binding protein sequence 3’ to the delta-globin gene. Blood 1992, 79, 512–516. [Google Scholar] [CrossRef] [PubMed]
- Wingender, E. TRANSFAC: A database on transcription factors and their DNA binding sites. Nucleic Acids Res. 1996, 24, 238–241. [Google Scholar] [CrossRef]
- Nishizaki, S.S.; Boyle, A.P. Mining the Unknown: Assigning Function to Noncoding Single Nucleotide Polymorphisms. Trends Genet. 2017, 33, 34–45. [Google Scholar] [CrossRef]
- Liu, B.; Montgomery, S.B. Identifying causal variants and genes using functional genomics in specialized cell types and contexts. Hum. Genet. 2020, 139, 95–102. [Google Scholar] [CrossRef]
- Moyerbrailean, G.A.; Richards, A.L.; Kurtz, D.; Kalita, C.A.; Davis, G.O.; Harvey, C.T.; Alazizi, A.; Watza, D.; Sorokin, Y.; Hauff, N.; et al. High-throughput allele-specific expression across 250 environmental conditions. Genome Res. 2016, 26, 1627–1638. [Google Scholar] [CrossRef]
- Chen, Q.; Deng, X.; Hu, X.; Guan, S.; He, M.; Wang, Y.; Wei, B.; Zhang, J.; Zhao, H.; Yao, W.; et al. Breast Cancer Risk–Associated SNPs in the mTOR Promoter Form De Novo KLF5- and ZEB1-Binding Sites that Influence the Cellular Response to Paclitaxel. Mol. Cancer Res. 2019, 17, 2244–2256. [Google Scholar] [CrossRef] [PubMed]
- Matana, A.; Ziros, P.G.; Chartoumpekis, D.V.; Renaud, C.O.; Polašek, O.; Hayward, C.; Zemunik, T.; Sykiotis, G.P. Rare and common genetic variations in the Keap1/Nrf2 antioxidant response pathway impact thyroglobulin gene expression and circulating levels, respectively. Biochem. Pharmacol. 2020, 173, 113605. [Google Scholar] [CrossRef]
- Levings, D.; Shaw, K.E.; Lacher, S.E. Genomic resources for dissecting the role of non-protein coding variation in gene-environment interactions. Toxicology 2020, 441, 152505. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.B.; McCarthy, M.; Ren, H.; Carrillo-Roa, T.; Shekhtman, T.; DeModena, A.; Liu, J.J.; Leckband, S.G.; Mors, O.; Rietschel, M.; et al. A functional variant in the serotonin receptor 7 gene (HTR7), rs7905446, is associated with good response to SSRIs in bipolar and unipolar depression. Mol. Psychiatry 2020, 25, 1312–1322. [Google Scholar] [CrossRef] [PubMed]
- Boldes, T.; Merenbakh-Lamin, K.; Journo, S.; Shachar, E.; Lipson, D.; Yeheskel, A.; Pasmanik-Chor, M.; Rubinek, T.; Wolf, I. R269C variant of ESR1: High prevalence and differential function in a subset of pancreatic cancers. BMC Cancer 2020, 20, 531. [Google Scholar] [CrossRef]
- Zhao, Y.Y.; Zhou, J.; Narayanan, C.S.; Cui, Y.; Kumar, A. Role of C/A Polymorphism at −20 on the Expression of Human Angiotensinogen Gene. Hypertension 1999, 33, 108–115. [Google Scholar] [CrossRef]
- López Rodríguez, M.; Kaminska, D.; Lappalainen, K.; Pihlajamäki, J.; Kaikkonen, M.U.; Laakso, M. Identification and characterization of a FOXA2-regulated transcriptional enhancer at a type 2 diabetes intronic locus that controls GCKR expression in liver cells. Genome Med. 2017, 9, 63. [Google Scholar] [CrossRef]
- Boulling, A.; Masson, E.; Zou, W.; Paliwal, S.; Wu, H.; Issarapu, P.; Bhaskar, S.; Génin, E.; Cooper, D.N.; Li, Z.; et al. Identification of a functional enhancer variant within the chronic pancreatitis-associated SPINK1 c.101A>G (p.Asn34Ser)-containing haplotype. Hum. Mutat. 2017, 38, 1014–1024. [Google Scholar] [CrossRef]
- Li, X.-X.; Peng, T.; Gao, J.; Feng, J.-G.; Wu, D.-D.; Yang, T.; Zhong, L.; Fu, W.-P.; Sun, C. Allele-specific expression identified rs2509956 as a novel long-distance cis -regulatory SNP for SCGB1A1, an important gene for multiple pulmonary diseases. Am. J. Physiol. Cell. Mol. Physiol. 2019, 317, L456–L463. [Google Scholar] [CrossRef]
- Peng, T.; Zhong, L.; Gao, J.; Wan, Z.; Fu, W.-P.; Sun, C. Identification of rs11615992 as a novel regulatory SNP for human P2RX7 by allele-specific expression. Mol. Genet. Genom. 2020, 295, 23–30. [Google Scholar] [CrossRef]
- Kuang, X.; Zhou, Q.; Li, Z.; Hu, Y.; Kang, Y.; Huang, W. −254C>G SNP in the TRPC6 Gene Promoter Influences Its Expression via Interaction with the NF- κ B Subunit RELA in Steroid-Resistant Nephrotic Syndrome Children. Int. J. Genom. 2019, 2019, 1–7. [Google Scholar] [CrossRef]
- Pan, G.; Cavalli, M.; Carlsson, B.; Skrtic, S.; Kumar, C.; Wadelius, C. rs953413 Regulates Polyunsaturated Fatty Acid Metabolism by Modulating ELOVL2 Expression. iScience 2020, 23, 100808. [Google Scholar] [CrossRef]
- Thynn, H.N.; Chen, X.-F.; Hu, W.-X.; Duan, Y.-Y.; Zhu, D.-L.; Chen, H.; Wang, N.-N.; Chen, H.-H.; Rong, Y.; Lu, B.-J.; et al. An Allele-Specific Functional SNP Associated with Two Systemic Autoimmune Diseases Modulates IRF5 Expression by Long-Range Chromatin Loop Formation. J. Investig. Dermatol. 2020, 140, 348–360.e11. [Google Scholar] [CrossRef]
- Coetzee, S.G.; Coetzee, G.A.; Hazelett, D.J. motifbreakR: An R/Bioconductor package for predicting variant effects at transcription factor binding sites: Fig. 1. Bioinformatics 2015, btv470. [Google Scholar] [CrossRef]
- Kumar, S.; Ambrosini, G.; Bucher, P. SNP2TFBS—A database of regulatory SNPs affecting predicted transcription factor binding site affinity. Nucleic Acids Res. 2017, 45, D139–D144. [Google Scholar] [CrossRef]
- Kulakovskiy, I.V.; Vorontsov, I.E.; Yevshin, I.S.; Sharipov, R.N.; Fedorova, A.D.; Rumynskiy, E.I.; Medvedeva, Y.A.; Magana-Mora, A.; Bajic, V.B.; Papatsenko, D.A.; et al. HOCOMOCO: Towards a complete collection of transcription factor binding models for human and mouse via large-scale ChIP-Seq analysis. Nucleic Acids Res. 2018, 46, D252–D259. [Google Scholar] [CrossRef]
- Fornes, O.; Castro-Mondragon, J.A.; Khan, A.; van der Lee, R.; Zhang, X.; Richmond, P.A.; Modi, B.P.; Correard, S.; Gheorghe, M.; Baranašić, D.; et al. JASPAR 2020: Update of the open-access database of transcription factor binding profiles. Nucleic Acids Res. 2019. [Google Scholar] [CrossRef]
- Nishizaki, S.S.; Ng, N.; Dong, S.; Porter, R.S.; Morterud, C.; Williams, C.; Asman, C.; Switzenberg, J.A.; Boyle, A.P. Predicting the effects of SNPs on transcription factor binding affinity. Bioinformatics 2020, 36, 364–372. [Google Scholar] [CrossRef]
- Shin, S.; Hudson, R.; Harrison, C.; Craven, M.; Keleş, S. atSNP Search: A web resource for statistically evaluating influence of human genetic variation on transcription factor binding. Bioinformatics 2019, 35, 2657–2659. [Google Scholar] [CrossRef]
- Yan, J.; Qiu, Y.; Ribeiro dos Santos, A.M.; Yin, Y.; Li, Y.E.; Vinckier, N.; Nariai, N.; Benaglio, P.; Raman, A.; Li, X.; et al. Systematic analysis of binding of transcription factors to noncoding variants. Nature 2021, 591, 147–151. [Google Scholar] [CrossRef]
- Stormo, G.D. DNA binding sites: Representation and discovery. Bioinformatics 2000, 16, 16–23. [Google Scholar] [CrossRef]
- Slattery, M.; Zhou, T.; Yang, L.; Dantas Machado, A.C.; Gordân, R.; Rohs, R. Absence of a simple code: How transcription factors read the genome. Trends Biochem. Sci. 2014, 39, 381–399. [Google Scholar] [CrossRef]
- Srivastava, D.; Mahony, S. Sequence and chromatin determinants of transcription factor binding and the establishment of cell type-specific binding patterns. Biochim. Biophys. Acta Gene Regul. Mech. 2020, 1863, 194443. [Google Scholar] [CrossRef]
- Inukai, S.; Kock, K.H.; Bulyk, M.L. Transcription factor–DNA binding: Beyond binding site motifs. Curr. Opin. Genet. Dev. 2017, 43, 110–119. [Google Scholar] [CrossRef] [PubMed]
- Fagny, M.; Paulson, J.N.; Kuijjer, M.L.; Sonawane, A.R.; Chen, C.-Y.; Lopes-Ramos, C.M.; Glass, K.; Quackenbush, J.; Platig, J. Exploring regulation in tissues with eQTL networks. Proc. Natl. Acad. Sci. USA 2017, 114, e7841–e7850. [Google Scholar] [CrossRef] [PubMed]
- Syddall, C.M.; Reynard, L.N.; Young, D.A.; Loughlin, J. The Identification of Trans-acting Factors That Regulate the Expression of GDF5 via the Osteoarthritis Susceptibility SNP rs143383. PLoS Genet. 2013, 9, e1003557. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Qin, S.; Ray, B.; Kalari, K.R.; Wang, L.; Weinshilboum, R.M. Single Nucleotide Polymorphisms (SNPs) Distant from Xenobiotic Response Elements Can Modulate Aryl Hydrocarbon Receptor Function: SNP-Dependent CYP1A1 Induction. Drug Metab. Dispos. 2018, 46, 1372–1381. [Google Scholar] [CrossRef] [PubMed]
- Tian, J.; Lou, J.; Cai, Y.; Rao, M.; Lu, Z.; Zhu, Y.; Zou, D.; Peng, X.; Wang, H.; Zhang, M.; et al. Risk SNP-Mediated Enhancer–Promoter Interaction Drives Colorectal Cancer through Both FADS2 and AP002754.2. Cancer Res. 2020, 80, 1804–1818. [Google Scholar] [CrossRef]
- Merkulov, V.M.; Leberfarb, E.Y.; Merkulova, T.I. Regulatory SNPs and their widespread effects on the transcriptome. J. Biosci. 2018, 43, 1069–1075. [Google Scholar] [CrossRef]
- ENCODE Project Consortium. Expanded encyclopaedias of DNA elements in the human and mouse genomes. Nature 2020, 583, 699–710. [Google Scholar] [CrossRef]
- Berner, D.; Hoja, U.; Zenkel, M.; Ross, J.J.; Uebe, S.; Paoli, D.; Frezzotti, P.; Rautenbach, R.M.; Ziskind, A.; Williams, S.E.; et al. The protective variant rs7173049 at LOXL1 locus impacts on retinoic acid signaling pathway in pseudoexfoliation syndrome. Hum. Mol. Genet. 2019, 28, 2531–2548. [Google Scholar] [CrossRef]
- Ali, M.W.; Patro, C.P.K.; Zhu, J.J.; Dampier, C.H.; Plummer, S.J.; Kuscu, C.; Adli, M.; Lau, C.; Lai, R.K.; Casey, G. A functional variant on 20q13.33 related to glioma risk alters enhancer activity and modulates expression of multiple genes. Hum. Mutat. 2021, 42, 77–88. [Google Scholar] [CrossRef]
- Liu, S.; Liu, Y.; Zhang, Q.; Wu, J.; Liang, J.; Yu, S.; Wei, G.-H.; White, K.P.; Wang, X. Systematic identification of regulatory variants associated with cancer risk. Genome Biol. 2017, 18, 194. [Google Scholar] [CrossRef]
- Gupta, R.M.; Hadaya, J.; Trehan, A.; Zekavat, S.M.; Roselli, C.; Klarin, D.; Emdin, C.A.; Hilvering, C.R.E.; Bianchi, V.; Mueller, C.; et al. A Genetic Variant Associated with Five Vascular Diseases Is a Distal Regulator of Endothelin-1 Gene Expression. Cell 2017, 170, 522–533. [Google Scholar] [CrossRef]
- Zhu, D.-L.; Chen, X.-F.; Hu, W.-X.; Dong, S.-S.; Lu, B.-J.; Rong, Y.; Chen, Y.-X.; Chen, H.; Thynn, H.N.; Wang, N.-N.; et al. Multiple Functional Variants at 13q14 Risk Locus for Osteoporosis Regulate RANKL Expression through Long-Range Super-Enhancer. J. Bone Miner. Res. 2018, 33, 1335–1346. [Google Scholar] [CrossRef]
- Wang, X.; Hayes, J.E.; Xu, X.; Gao, X.; Mehta, D.; Lilja, H.G.; Klein, R.J. Validation of prostate cancer risk variants rs10993994 and rs7098889 by CRISPR/Cas9 mediated genome editing. Gene 2021, 768, 145265. [Google Scholar] [CrossRef]
- Gutierrez-Arcelus, M.; Baglaenko, Y.; Arora, J.; Hannes, S.; Luo, Y.; Amariuta, T.; Teslovich, N.; Rao, D.A.; Ermann, J.; Jonsson, A.H.; et al. Allele-specific expression changes dynamically during T cell activation in HLA and other autoimmune loci. Nat. Genet. 2020, 52, 247–253. [Google Scholar] [CrossRef]
- Klein, J.C.; Keith, A.; Rice, S.J.; Shepherd, C.; Agarwal, V.; Loughlin, J.; Shendure, J. Functional testing of thousands of osteoarthritis-associated variants for regulatory activity. Nat. Commun. 2019, 10, 2434. [Google Scholar] [CrossRef]
- Azghandi, S.; Prell, C.; van der Laan, S.W.; Schneider, M.; Malik, R.; Berer, K.; Gerdes, N.; Pasterkamp, G.; Weber, C.; Haffner, C.; et al. Deficiency of the Stroke Relevant HDAC9 Gene Attenuates Atherosclerosis in Accord with Allele-Specific Effects at 7p21.1. Stroke 2015, 46, 197–202. [Google Scholar] [CrossRef]
- Roca-Ayats, N.; Martínez-Gil, N.; Cozar, M.; Gerousi, M.; Garcia-Giralt, N.; Ovejero, D.; Mellibovsky, L.; Nogués, X.; Díez-Pérez, A.; Grinberg, D.; et al. Functional characterization of the C7ORF76 genomic region, a prominent GWAS signal for osteoporosis in 7q21.3. Bone 2019, 123, 39–47. [Google Scholar] [CrossRef]
- Malecová, B.; Morris, K.V. Transcriptional gene silencing through epigenetic changes mediated by non-coding RNAs. Curr. Opin. Mol. Ther. 2010, 12, 214–222. [Google Scholar]
- Butter, F.; Davison, L.; Viturawong, T.; Scheibe, M.; Vermeulen, M.; Todd, J.A.; Mann, M. Proteome-Wide Analysis of Disease-Associated SNPs That Show Allele-Specific Transcription Factor Binding. PLoS Genet. 2012, 8, e1002982. [Google Scholar] [CrossRef]
- Amin Al Olama, A.; Kote-Jarai, Z.; Schumacher, F.R.; Wiklund, F.; Berndt, S.I.; Benlloch, S.; Giles, G.G.; Severi, G.; Neal, D.E.; Hamdy, F.C.; et al. A meta-analysis of genome-wide association studies to identify prostate cancer susceptibility loci associated with aggressive and non-aggressive disease. Hum. Mol. Genet. 2013, 22, 408–415. [Google Scholar] [CrossRef]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio Cancer Genomics Portal: An Open Platform for Exploring Multidimensional Cancer Genomics Data: Figure 1. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef]
- Ran, F.A.; Hsu, P.D.; Wright, J.; Agarwala, V.; Scott, D.A.; Zhang, F. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013, 8, 2281–2308. [Google Scholar] [CrossRef]
- Malik, R.; Chauhan, G.; Traylor, M.; Sargurupremraj, M.; Okada, Y.; Mishra, A.; Rutten-Jacobs, L.; Giese, A.-K.; van der Laan, S.W.; Gretarsdottir, S.; et al. Multiancestry genome-wide association study of 520,000 subjects identifies 32 loci associated with stroke and stroke subtypes. Nat. Genet. 2018, 50, 524–537. [Google Scholar] [CrossRef]
- Nelson, C.P.; Goel, A.; Butterworth, A.S.; Kanoni, S.; Webb, T.R.; Marouli, E.; Zeng, L.; Ntalla, I.; Lai, F.Y.; Hopewell, J.C.; et al. Association analyses based on false discovery rate implicate new loci for coronary artery disease. Nat. Genet. 2017, 49, 1385–1391. [Google Scholar] [CrossRef]
- Takeuchi, F.; Akiyama, M.; Matoba, N.; Katsuya, T.; Nakatochi, M.; Tabara, Y.; Narita, A.; Saw, W.-Y.; Moon, S.; Spracklen, C.N.; et al. Interethnic analyses of blood pressure loci in populations of East Asian and European descent. Nat. Commun. 2018, 9, 5052. [Google Scholar] [CrossRef]
- Hoffmann, T.J.; Ehret, G.B.; Nandakumar, P.; Ranatunga, D.; Schaefer, C.; Kwok, P.-Y.; Iribarren, C.; Chakravarti, A.; Risch, N. Genome-wide association analyses using electronic health records identify new loci influencing blood pressure variation. Nat. Genet. 2017, 49, 54–64. [Google Scholar] [CrossRef]
- Giri, A.; Hellwege, J.N.; Keaton, J.M.; Park, J.; Qiu, C.; Warren, H.R.; Torstenson, E.S.; Kovesdy, C.P.; Sun, Y.V.; Wilson, O.D.; et al. Trans-ethnic association study of blood pressure determinants in over 750,000 individuals. Nat. Genet. 2019, 51, 51–62. [Google Scholar] [CrossRef]
- Karasu, N.; Sexton, T. 4C-Seq: Interrogating Chromatin Looping with Circular Chromosome Conformation Capture. In Capturing Chromosome Conformation; Humana: New York, NY, USA, 2021; pp. 19–34. [Google Scholar]
- Sardi, S.P.; Clarke, J.; Viel, C.; Chan, M.; Tamsett, T.J.; Treleaven, C.M.; Bu, J.; Sweet, L.; Passini, M.A.; Dodge, J.C.; et al. Augmenting CNS glucocerebrosidase activity as a therapeutic strategy for parkinsonism and other Gaucher-related synucleinopathies. Proc. Natl. Acad. Sci. USA 2013, 110, 3537–3542. [Google Scholar] [CrossRef]
- Mata, I.F.; Leverenz, J.B.; Weintraub, D.; Trojanowski, J.Q.; Chen-Plotkin, A.; Van Deerlin, V.M.; Ritz, B.; Rausch, R.; Factor, S.A.; Wood-Siverio, C.; et al. GBA Variants are associated with a distinct pattern of cognitive deficits in Parkinson’s disease. Mov. Disord. 2016, 31, 95–102. [Google Scholar] [CrossRef]
- van der Lee, S.J.; Knol, M.J.; Chauhan, G.; Satizabal, C.L.; Smith, A.V.; Hofer, E.; Bis, J.C.; Hibar, D.P.; Hilal, S.; van den Akker, E.B.; et al. A genome-wide association study identifies genetic loci associated with specific lobar brain volumes. Commun. Biol. 2019, 2, 285. [Google Scholar] [CrossRef]
- Lan, Q.; Hsiung, C.A.; Matsuo, K.; Hong, Y.-C.; Seow, A.; Wang, Z.; Hosgood, H.D.; Chen, K.; Wang, J.-C.; Chatterjee, N.; et al. Genome-wide association analysis identifies new lung cancer susceptibility loci in never-smoking women in Asia. Nat. Genet. 2012, 44, 1330–1335. [Google Scholar] [CrossRef]
- McKay, J.D.; Hung, R.J.; Han, Y.; Zong, X.; Carreras-Torres, R.; Christiani, D.C.; Caporaso, N.E.; Johansson, M.; Xiao, X.; Li, Y.; et al. Large-scale association analysis identifies new lung cancer susceptibility loci and heterogeneity in genetic susceptibility across histological subtypes. Nat. Genet. 2017, 49, 1126–1132. [Google Scholar] [CrossRef] [PubMed]
- Lappalainen, T. Functional genomics bridges the gap between quantitative genetics and molecular biology. Genome Res. 2015, 25, 1427–1431. [Google Scholar] [CrossRef] [PubMed]
- Qian, Y.; Zhang, L.; Cai, M.; Li, H.; Xu, H.; Yang, H.; Zhao, Z.; Rhie, S.K.; Farnham, P.J.; Shi, J.; et al. The prostate cancer risk variant rs55958994 regulates multiple gene expression through extreme long-range chromatin interaction to control tumor progression. Sci. Adv. 2019, 5, eaaw6710. [Google Scholar] [CrossRef]
- Ulirsch, J.C.; Nandakumar, S.K.; Wang, L.; Giani, F.C.; Zhang, X.; Rogov, P.; Melnikov, A.; McDonel, P.; Do, R.; Mikkelsen, T.S.; et al. Systematic Functional Dissection of Common Genetic Variation Affecting Red Blood Cell Traits. Cell 2016, 165, 1530–1545. [Google Scholar] [CrossRef] [PubMed]
- Kalita, C.A.; Brown, C.D.; Freiman, A.; Isherwood, J.; Wen, X.; Pique-Regi, R.; Luca, F. High-throughput characterization of genetic effects on DNA–protein binding and gene transcription. Genome Res. 2018, 28, 1701–1708. [Google Scholar] [CrossRef] [PubMed]
- Patwardhan, R.P.; Lee, C.; Litvin, O.; Young, D.L.; Pe’er, D.; Shendure, J. High-resolution analysis of DNA regulatory elements by synthetic saturation mutagenesis. Nat. Biotechnol. 2009, 27, 1173–1175. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Xia, J.-H.; Zhu, J.; Gao, P.; Tian, Y.-J.; Du, M.; Guo, Y.-C.; Suleman, S.; Zhang, Q.; Kohli, M.; et al. High-throughput screening of prostate cancer risk loci by single nucleotide polymorphisms sequencing. Nat. Commun. 2018, 9, 2022. [Google Scholar] [CrossRef]
- Kolchanov, N.A.; Merkulova, T.I.; Ignatieva, E.V.; Ananko, E.A.; Oshchepkov, D.Y.; Levitsky, V.G.; Vasiliev, G.V.; Klimova, N.V.; Merkulov, V.M.; Charles Hodgman, T. Combined experimental and computational approaches to study the regulatory elements in eukaryotic genes. Brief. Bioinform. 2007, 8, 266–274. [Google Scholar] [CrossRef]
- Li, S.; Li, Y.; Li, X.; Liu, J.; Huo, Y.; Wang, J.; Liu, Z.; Li, M.; Luo, X.-J. Regulatory mechanisms of major depressive disorder risk variants. Mol. Psychiatry 2020, 25, 1926–1945. [Google Scholar] [CrossRef]
- Wray, N.R.; Ripke, S.; Mattheisen, M.; Trzaskowski, M.; Byrne, E.M.; Abdellaoui, A.; Adams, M.J.; Agerbo, E.; Air, T.M.; Andlauer, T.M.F.; et al. Genome-wide association analyses identify 44 risk variants and refine the genetic architecture of major depression. Nat. Genet. 2018, 50, 668–681. [Google Scholar] [CrossRef]
- Sun, W.; Yao, S.; Tang, J.; Liu, S.; Chen, J.; Deng, D.; Zeng, C. Integrative analysis of super enhancer SNPs for type 2 diabetes. PLoS ONE 2018, 13, e0192105. [Google Scholar] [CrossRef] [PubMed]
- Gong, J.; Qiu, C.; Huang, D.; Zhang, Y.; Yu, S.; Zeng, C. Integrative functional analysis of super enhancer SNPs for coronary artery disease. J. Hum. Genet. 2018, 63, 627–638. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Du, Y.; Qu, S.; Wang, J. rVarBase: An updated database for regulatory features of human variants. Nucleic Acids Res. 2016, 44, D888–D893. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Jones, M.R.; Peng, P.-C.; Coetzee, S.G.; Tyrer, J.; Reyes, A.L.P.; Corona, R.I.; Davis, B.; Chen, S.; Dezem, F.; Seo, J.-H.; et al. Ovarian Cancer Risk Variants Are Enriched in Histotype-Specific Enhancers and Disrupt Transcription Factor Binding Sites. Am. J. Hum. Genet. 2020, 107, 622–635. [Google Scholar] [CrossRef]
- Guo, Y.; Perez, A.A.; Hazelett, D.J.; Coetzee, G.A.; Rhie, S.K.; Farnham, P.J. CRISPR-mediated deletion of prostate cancer risk-associated CTCF loop anchors identifies repressive chromatin loops. Genome Biol. 2018, 19, 160. [Google Scholar] [CrossRef]
- Gamazon, E.R.; Segrè, A.V.; van de Bunt, M.; Wen, X.; Xi, H.S.; Hormozdiari, F.; Ongen, H.; Konkashbaev, A.; Derks, E.M.; Aguet, F.; et al. Using an atlas of gene regulation across 44 human tissues to inform complex disease- and trait-associated variation. Nat. Genet. 2018, 50, 956–967. [Google Scholar] [CrossRef]
- Barbeira, A.N.; Bonazzola, R.; Gamazon, E.R.; Liang, Y.; Park, Y.; Kim-Hellmuth, S.; Wang, G.; Jiang, Z.; Zhou, D.; Hormozdiari, F.; et al. Exploiting the GTEx resources to decipher the mechanisms at GWAS loci. Genome Biol. 2021, 22, 49. [Google Scholar] [CrossRef]
- Corces, M.R.; Shcherbina, A.; Kundu, S.; Gloudemans, M.J.; Frésard, L.; Granja, J.M.; Louie, B.H.; Eulalio, T.; Shams, S.; Bagdatli, S.T.; et al. Single-cell epigenomic analyses implicate candidate causal variants at inherited risk loci for Alzheimer’s and Parkinson’s diseases. Nat. Genet. 2020, 52, 1158–1168. [Google Scholar] [CrossRef]
- Ray, J.P.; de Boer, C.G.; Fulco, C.P.; Lareau, C.A.; Kanai, M.; Ulirsch, J.C.; Tewhey, R.; Ludwig, L.S.; Reilly, S.K.; Bergman, D.T.; et al. Prioritizing disease and trait causal variants at the TNFAIP3 locus using functional and genomic features. Nat. Commun. 2020, 11, 1237. [Google Scholar] [CrossRef]
- Zeng, B.; Lloyd-Jones, L.R.; Montgomery, G.W.; Metspalu, A.; Esko, T.; Franke, L.; Vosa, U.; Claringbould, A.; Brigham, K.L.; Quyyumi, A.A.; et al. Comprehensive Multiple eQTL Detection and Its Application to GWAS Interpretation. Genetics 2019, 212, 905–918. [Google Scholar] [CrossRef]
- Zhao, J.; Cheng, F.; Jia, P.; Cox, N.; Denny, J.C.; Zhao, Z. An integrative functional genomics framework for effective identification of novel regulatory variants in genome–phenome studies. Genome Med. 2018, 10, 7. [Google Scholar] [CrossRef]
- Gerring, Z.F.; Vargas, A.M.; Gamazon, E.R.; Derks, E.M. An integrative systems-based analysis of substance use: eQTL-informed gene-based tests, gene networks, and biological mechanisms. Am. J. Med. Genet. Part B Neuropsychiatr. Genet. 2020. [Google Scholar] [CrossRef]
- GTEx Consortium. The GTEx Consortium atlas of genetic regulatory effects across human tissues. Science 2020, 369, 1318–1330. [Google Scholar] [CrossRef]
- Fairfax, B.P.; Humburg, P.; Makino, S.; Naranbhai, V.; Wong, D.; Lau, E.; Jostins, L.; Plant, K.; Andrews, R.; McGee, C.; et al. Innate Immune Activity Conditions the Effect of Regulatory Variants upon Monocyte Gene Expression. Science 2014, 343, 1246949. [Google Scholar] [CrossRef]
- Fan, J.; Hu, J.; Xue, C.; Zhang, H.; Susztak, K.; Reilly, M.P.; Xiao, R.; Li, M. ASEP: Gene-based detection of allele-specific expression across individuals in a population by RNA sequencing. PLoS Genet. 2020, 16, e1008786. [Google Scholar] [CrossRef]
- Kim-Hellmuth, S.; Bechheim, M.; Pütz, B.; Mohammadi, P.; Nédélec, Y.; Giangreco, N.; Becker, J.; Kaiser, V.; Fricker, N.; Beier, E.; et al. Genetic regulatory effects modified by immune activation contribute to autoimmune disease associations. Nat. Commun. 2017, 8, 266. [Google Scholar] [CrossRef]
- Werling, D.M.; Pochareddy, S.; Choi, J.; An, J.-Y.; Sheppard, B.; Peng, M.; Li, Z.; Dastmalchi, C.; Santpere, G.; Sousa, A.M.M.; et al. Whole-Genome and RNA Sequencing Reveal Variation and Transcriptomic Coordination in the Developing Human Prefrontal Cortex. Cell Rep. 2020, 31, 107489. [Google Scholar] [CrossRef]
- Göring, H.H.H.; Curran, J.E.; Johnson, M.P.; Dyer, T.D.; Charlesworth, J.; Cole, S.A.; Jowett, J.B.M.; Abraham, L.J.; Rainwater, D.L.; Comuzzie, A.G.; et al. Discovery of expression QTLs using large-scale transcriptional profiling in human lymphocytes. Nat. Genet. 2007, 39, 1208–1216. [Google Scholar] [CrossRef]
- Westra, H.-J.; Franke, L. From genome to function by studying eQTLs. Biochim. Biophys. Acta Mol. Basis Dis. 2014, 1842, 1896–1902. [Google Scholar] [CrossRef]
- GTEx Consortium. The Genotype-Tissue Expression (GTEx) project. Nat. Genet. 2013, 45, 580–585. [Google Scholar] [CrossRef]
- Umans, B.D.; Battle, A.; Gilad, Y. Where Are the Disease-Associated eQTLs? Trends Genet. 2021, 37, 109–124. [Google Scholar] [CrossRef] [PubMed]
- Zou, J.; Hormozdiari, F.; Jew, B.; Castel, S.E.; Lappalainen, T.; Ernst, J.; Sul, J.H.; Eskin, E. Leveraging allelic imbalance to refine fine-mapping for eQTL studies. PLoS Genet. 2019, 15, e1008481. [Google Scholar] [CrossRef] [PubMed]
- Gamazon, E.R.; Zwinderman, A.H.; Cox, N.J.; Denys, D.; Derks, E.M. Multi-tissue transcriptome analyses identify genetic mechanisms underlying neuropsychiatric traits. Nat. Genet. 2019, 51, 933–940. [Google Scholar] [CrossRef] [PubMed]
- Gerring, Z.F.; Lupton, M.K.; Edey, D.; Gamazon, E.R.; Derks, E.M. An analysis of genetically regulated gene expression across multiple tissues implicates novel gene candidates in Alzheimer’s disease. Alzheimers Res. Ther. 2020, 12, 43. [Google Scholar] [CrossRef] [PubMed]
- Hormozdiari, F.; van de Bunt, M.; Segrè, A.V.; Li, X.; Joo, J.W.J.; Bilow, M.; Sul, J.H.; Sankararaman, S.; Pasaniuc, B.; Eskin, E. Colocalization of GWAS and eQTL Signals Detects Target Genes. Am. J. Hum. Genet. 2016, 99, 1245–1260. [Google Scholar] [CrossRef]
- Fadason, T.; Ekblad, C.; Ingram, J.R.; Schierding, W.S.; O’Sullivan, J.M. Physical Interactions and Expression Quantitative Traits Loci Identify Regulatory Connections for Obesity and Type 2 Diabetes Associated SNPs. Front. Genet. 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Jaffe, A.E.; Hoeppner, D.J.; Saito, T.; Blanpain, L.; Ukaigwe, J.; Burke, E.E.; Collado-Torres, L.; Tao, R.; Tajinda, K.; Maynard, K.R.; et al. Profiling gene expression in the human dentate gyrus granule cell layer reveals insights into schizophrenia and its genetic risk. Nat. Neurosci. 2020, 23, 510–519. [Google Scholar] [CrossRef]
- Morrow, J.D.; Cho, M.H.; Platig, J.; Zhou, X.; DeMeo, D.L.; Qiu, W.; Celli, B.; Marchetti, N.; Criner, G.J.; Bueno, R.; et al. Ensemble genomic analysis in human lung tissue identifies novel genes for chronic obstructive pulmonary disease. Hum. Genom. 2018, 12, 1. [Google Scholar] [CrossRef]
- O’Brien, H.E.; Hannon, E.; Hill, M.J.; Toste, C.C.; Robertson, M.J.; Morgan, J.E.; McLaughlin, G.; Lewis, C.M.; Schalkwyk, L.C.; Hall, L.S.; et al. Expression quantitative trait loci in the developing human brain and their enrichment in neuropsychiatric disorders. Genome Biol. 2018, 19, 194. [Google Scholar] [CrossRef]
- Ratnapriya, R.; Sosina, O.A.; Starostik, M.R.; Kwicklis, M.; Kapphahn, R.J.; Fritsche, L.G.; Walton, A.; Arvanitis, M.; Gieser, L.; Pietraszkiewicz, A.; et al. Retinal transcriptome and eQTL analyses identify genes associated with age-related macular degeneration. Nat. Genet. 2019, 51, 606–610. [Google Scholar] [CrossRef]
- Zhang, T.; Choi, J.; Kovacs, M.A.; Shi, J.; Xu, M.; Goldstein, A.M.; Trower, A.J.; Bishop, D.T.; Iles, M.M.; Duffy, D.L.; et al. Cell-type–specific eQTL of primary melanocytes facilitates identification of melanoma susceptibility genes. Genome Res. 2018, 28, 1621–1635. [Google Scholar] [CrossRef]
- Zhu, Z.; Zhang, F.; Hu, H.; Bakshi, A.; Robinson, M.R.; Powell, J.E.; Montgomery, G.W.; Goddard, M.E.; Wray, N.R.; Visscher, P.M.; et al. Integration of summary data from GWAS and eQTL studies predicts complex trait gene targets. Nat. Genet. 2016, 48, 481–487. [Google Scholar] [CrossRef]
- Korbolina, E.E.; Bryzgalov, L.O.; Ustrokhanova, D.Z.; Postovalov, S.N.; Poverin, D.V.; Damarov, I.S.; Merkulova, T.I. A panel of rSNPs demonstrating allelic asymmetry in both ChIP-seq and RNA-seq data and the search for their phenotypic outcomes through analysis of DEGs. Int. J. Mol. Sci. 2021, (in press).
- Saha, A.; Kim, Y.; Gewirtz, A.D.H.; Jo, B.; Gao, C.; McDowell, I.C.; Engelhardt, B.E.; Battle, A. Co-expression networks reveal the tissue-specific regulation of transcription and splicing. Genome Res. 2017, 27, 1843–1858. [Google Scholar] [CrossRef]
- van der Wijst, M.; de Vries, D.; Groot, H.; Trynka, G.; Hon, C.; Bonder, M.; Stegle, O.; Nawijn, M.; Idaghdour, Y.; van der Harst, P.; et al. The single-cell eQTLGen consortium. Elife 2020, 9. [Google Scholar] [CrossRef]
- Tewhey, R.; Kotliar, D.; Park, D.S.; Liu, B.; Winnicki, S.; Reilly, S.K.; Andersen, K.G.; Mikkelsen, T.S.; Lander, E.S.; Schaffner, S.F.; et al. Direct Identification of Hundreds of Expression-Modulating Variants using a Multiplexed Reporter Assay. Cell 2016, 165, 1519–1529. [Google Scholar] [CrossRef]
- Richard, A.C.; Peters, J.E.; Lee, J.C.; Vahedi, G.; Schäffer, A.A.; Siegel, R.M.; Lyons, P.A.; Smith, K.G.C. Targeted genomic analysis reveals widespread autoimmune disease association with regulatory variants in the TNF superfamily cytokine signalling network. Genome Med. 2016, 8, 76. [Google Scholar] [CrossRef]
- Beer, M.A. Predicting enhancer activity and variant impact using gkm-SVM. Hum. Mutat. 2017, 38, 1251–1258. [Google Scholar] [CrossRef]
- Castel, S.E.; Aguet, F.; Mohammadi, P.; Ardlie, K.G.; Lappalainen, T. A vast resource of allelic expression data spanning human tissues. Genome Biol. 2020, 21, 234. [Google Scholar] [CrossRef]
- Kang, E.Y.; Martin, L.J.; Mangul, S.; Isvilanonda, W.; Zou, J.; Ben-David, E.; Han, B.; Lusis, A.J.; Shifman, S.; Eskin, E. Discovering Single Nucleotide Polymorphisms Regulating Human Gene Expression Using Allele Specific Expression from RNA-seq Data. Genetics 2016, 204, 1057–1064. [Google Scholar] [CrossRef]
- Liu, Y.; Li, C.; Shen, S.; Chen, X.; Szlachta, K.; Edmonson, M.N.; Shao, Y.; Ma, X.; Hyle, J.; Wright, S.; et al. Discovery of regulatory noncoding variants in individual cancer genomes by using cis-X. Nat. Genet. 2020, 52, 811–818. [Google Scholar] [CrossRef]
- Harvey, C.T.; Moyerbrailean, G.A.; Davis, G.O.; Wen, X.; Luca, F.; Pique-Regi, R. QuASAR: Quantitative allele-specific analysis of reads. Bioinformatics 2015, 31, 1235–1242. [Google Scholar] [CrossRef]
- Edsgärd, D.; Iglesias, M.J.; Reilly, S.-J.; Hamsten, A.; Tornvall, P.; Odeberg, J.; Emanuelsson, O. GeneiASE: Detection of condition-dependent and static allele-specific expression from RNA-seq data without haplotype information. Sci. Rep. 2016, 6, 21134. [Google Scholar] [CrossRef]
- Maurano, M.T.; Haugen, E.; Sandstrom, R.; Vierstra, J.; Shafer, A.; Kaul, R.; Stamatoyannopoulos, J.A. Large-scale identification of sequence variants influencing human transcription factor occupancy in vivo. Nat. Genet. 2015, 47, 1393–1401. [Google Scholar] [CrossRef]
- Cavalli, M.; Pan, G.; Nord, H.; Wallerman, O.; Wallén Arzt, E.; Berggren, O.; Elvers, I.; Eloranta, M.-L.; Rönnblom, L.; Lindblad Toh, K.; et al. Allele-specific transcription factor binding to common and rare variants associated with disease and gene expression. Hum. Genet. 2016, 135, 485–497. [Google Scholar] [CrossRef]
- Matys, V. TRANSFAC(R) and its module TRANSCompel(R): Transcriptional gene regulation in eukaryotes. Nucleic Acids Res. 2006, 34, D108–D110. [Google Scholar] [CrossRef]
- Portales-Casamar, E.; Thongjuea, S.; Kwon, A.T.; Arenillas, D.; Zhao, X.; Valen, E.; Yusuf, D.; Lenhard, B.; Wasserman, W.W.; Sandelin, A. JASPAR 2010: The greatly expanded open-access database of transcription factor binding profiles. Nucleic Acids Res. 2010, 38, D105–D110. [Google Scholar] [CrossRef] [PubMed]
- Newburger, D.E.; Bulyk, M.L. UniPROBE: An online database of protein binding microarray data on protein-DNA interactions. Nucleic Acids Res. 2009, 37, D77–D82. [Google Scholar] [CrossRef] [PubMed]
- Jolma, A.; Yan, J.; Whitington, T.; Toivonen, J.; Nitta, K.R.; Rastas, P.; Morgunova, E.; Enge, M.; Taipale, M.; Wei, G.; et al. DNA-Binding Specificities of Human Transcription Factors. Cell 2013, 152, 327–339. [Google Scholar] [CrossRef] [PubMed]
- Lappalainen, T.; Sammeth, M.; Friedländer, M.R.; ‘t Hoen, P.A.C.; Monlong, J.; Rivas, M.A.; Gonzàlez-Porta, M.; Kurbatova, N.; Griebel, T.; Ferreira, P.G.; et al. Transcriptome and genome sequencing uncovers functional variation in humans. Nature 2013, 501, 506–511. [Google Scholar] [CrossRef] [PubMed]
- Cavalli, M.; Pan, G.; Nord, H.; Wallén Arzt, E.; Wallerman, O.; Wadelius, C. Allele-specific transcription factor binding in liver and cervix cells unveils many likely drivers of GWAS signals. Genomics 2016, 107, 248–254. [Google Scholar] [CrossRef]
- Marinov, G.K.; Shipony, Z. Interrogating the Accessible Chromatin Landscape of Eukaryote Genomes Using ATAC-seq. In Deep Sequencing Data Analysis; Humana: New York, NY, USA, 2021; pp. 183–226. [Google Scholar] [CrossRef]
- Xu, S.; Feng, W.; Lu, Z.; Yu, C.Y.; Shao, W.; Nakshatri, H.; Reiter, J.L.; Gao, H.; Chu, X.; Wang, Y.; et al. regSNPs-ASB: A Computational Framework for Identifying Allele-Specific Transcription Factor Binding From ATAC-seq Data. Front. Bioeng. Biotechnol. 2020, 8. [Google Scholar] [CrossRef]
- Benaglio, P.; D’Antonio-Chronowska, A.; Ma, W.; Yang, F.; Young Greenwald, W.W.; Donovan, M.K.R.; DeBoever, C.; Li, H.; Drees, F.; Singhal, S.; et al. Allele-specific NKX2-5 binding underlies multiple genetic associations with human electrocardiographic traits. Nat. Genet. 2019, 51, 1506–1517. [Google Scholar] [CrossRef]
- van Setten, J.; Brody, J.A.; Jamshidi, Y.; Swenson, B.R.; Butler, A.M.; Campbell, H.; Del Greco, F.M.; Evans, D.S.; Gibson, Q.; Gudbjartsson, D.F.; et al. PR interval genome-wide association meta-analysis identifies 50 loci associated with atrial and atrioventricular electrical activity. Nat. Commun. 2018, 9, 2904. [Google Scholar] [CrossRef]
- D’Oliveira Albanus, R.; Kyono, Y.; Hensley, J.; Varshney, A.; Orchard, P.; Kitzman, J.O.; Parker, S.C.J. Chromatin information content landscapes inform transcription factor and DNA interactions. Nat. Commun. 2021, 12, 1307. [Google Scholar] [CrossRef]
- Li, M.; Huang, H.; Li, L.; He, C.; Zhu, L.; Guo, H.; Wang, L.; Liu, J.; Wu, S.; Liu, J.; et al. Core transcription regulatory circuitry orchestrates corneal epithelial homeostasis. Nat. Commun. 2021, 12, 420. [Google Scholar] [CrossRef]
- Liu, Y.; Guo, B.; Aguilera-Jimenez, E.; Chu, V.S.; Zhou, J.; Wu, Z.; Francis, J.M.; Yang, X.; Choi, P.S.; Bailey, S.D.; et al. Chromatin Looping Shapes KLF5-Dependent Transcriptional Programs in Human Epithelial Cancers. Cancer Res. 2020, 80, 5464–5477. [Google Scholar] [CrossRef]
- Sun, J.; Zhao, Y.; McGreal, R.; Cohen-Tayar, Y.; Rockowitz, S.; Wilczek, C.; Ashery-Padan, R.; Shechter, D.; Zheng, D.; Cvekl, A. Pax6 associates with H3K4-specific histone methyltransferases Mll1, Mll2, and Set1a and regulates H3K4 methylation at promoters and enhancers. Epigenet. Chromatin 2016, 9, 37. [Google Scholar] [CrossRef]
- Korbolina, E.E.; Brusentsov, I.I.; Bryzgalov, L.O.; Leberfarb, E.Y.; Degtyareva, A.O.; Merkulova, T.I. Novel approach to functional SNPs discovery from genome-wide data reveals promising variants for colon cancer risk. Hum. Mutat. 2018, 39, 851–859. [Google Scholar] [CrossRef]
- Bryzgalov, L.O.; Korbolina, E.E.; Brusentsov, I.I.; Leberfarb, E.Y.; Bondar, N.P.; Merkulova, T.I. Novel functional variants at the GWAS-implicated loci might confer risk to major depressive disorder, bipolar affective disorder and schizophrenia. BMC Neurosci. 2018, 19, 22. [Google Scholar] [CrossRef]
- Seshagiri, S.; Stawiski, E.W.; Durinck, S.; Modrusan, Z.; Storm, E.E.; Conboy, C.B.; Chaudhuri, S.; Guan, Y.; Janakiraman, V.; Jaiswal, B.S.; et al. Recurrent R-spondin fusions in colon cancer. Nature 2012, 488, 660–664. [Google Scholar] [CrossRef] [PubMed]
- Leberfarb, E.Y.; Degtyareva, A.O.; Brusentsov, I.I.; Maximov, V.N.; Voevoda, M.I.; Autenshlus, A.I.; Morozov, D.V.; Sokolov, A.V.; Merkulova, T.I. Potential regulatory SNPs in the ATXN7L3B and KRT15 genes are associated with gender-specific colorectal cancer risk. Per. Med. 2020, 17, 43–54. [Google Scholar] [CrossRef]
- Cavalli, M.; Baltzer, N.; Umer, H.M.; Grau, J.; Lemnian, I.; Pan, G.; Wallerman, O.; Spalinskas, R.; Sahlén, P.; Grosse, I.; et al. Allele specific chromatin signals, 3D interactions, and motif predictions for immune and B cell related diseases. Sci. Rep. 2019, 9, 2695. [Google Scholar] [CrossRef] [PubMed]
- Dubois-Chevalier, J.; Mazrooei, P.; Lupien, M.; Staels, B.; Lefebvre, P.; Eeckhoute, J. Organizing combinatorial transcription factor recruitment at cis -regulatory modules. Transcription 2018, 9, 233–239. [Google Scholar] [CrossRef] [PubMed]
- Gerstein, M.B.; Kundaje, A.; Hariharan, M.; Landt, S.G.; Yan, K.-K.; Cheng, C.; Mu, X.J.; Khurana, E.; Rozowsky, J.; Alexander, R.; et al. Architecture of the human regulatory network derived from ENCODE data. Nature 2012, 489, 91–100. [Google Scholar] [CrossRef]
- Lan, X.; Farnham, P.J.; Jin, V.X. Uncovering Transcription Factor Modules Using One- and Three-dimensional Analyses. J. Biol. Chem. 2012, 287, 30914–30921. [Google Scholar] [CrossRef]
- Gan, K.A.; Carrasco Pro, S.; Sewell, J.A.; Fuxman Bass, J.I. Identification of Single Nucleotide Non-coding Driver Mutations in Cancer. Front. Genet. 2018, 9. [Google Scholar] [CrossRef]
- Carrasco Pro, S.; Bulekova, K.; Gregor, B.; Labadorf, A.; Fuxman Bass, J.I. Prediction of genome-wide effects of single nucleotide variants on transcription factor binding. Sci. Rep. 2020, 10, 17632. [Google Scholar] [CrossRef]
- Badis, G.; Berger, M.F.; Philippakis, A.A.; Talukder, S.; Gehrke, A.R.; Jaeger, S.A.; Chan, E.T.; Metzler, G.; Vedenko, A.; Chen, X.; et al. Diversity and Complexity in DNA Recognition by Transcription Factors. Science 2009, 324, 1720–1723. [Google Scholar] [CrossRef]
- Nagy, G.; Nagy, L. Motif grammar: The basis of the language of gene expression. Comput. Struct. Biotechnol. J. 2020, 18, 2026–2032. [Google Scholar] [CrossRef]
- Crocker, J.; Preger-Ben Noon, E.; Stern, D.L. The Soft Touch: Low-affinity transcription factor binding sites in development and evolution. Curr. Top. Dev. Biol. 2016, 117, 455–469. [Google Scholar] [CrossRef]
- Levitsky, V.G.; Kulakovskiy, I.V.; Ershov, N.I.; Oshchepkov, D.; Makeev, V.J.; Hodgman, T.C.; Merkulova, T.I. Application of experimentally verified transcription factor binding sites models for computational analysis of ChIP-Seq data. BMC Genom. 2014, 15, 80. [Google Scholar] [CrossRef]
- Levitsky, V.G.; Oshchepkov, D.Y.; Klimova, N.V.; Ignatieva, E.; Vasiliev, G.V.; Merkulov, V.M.; Merkulova, T.I. Hidden heterogeneity of transcription factor binding sites: A case study of SF-1. Comput. Biol. Chem. 2016, 64, 19–32. [Google Scholar] [CrossRef]
- Osz, J.; McEwen, A.G.; Bourguet, M.; Przybilla, F.; Peluso-Iltis, C.; Poussin-Courmontagne, P.; Mély, Y.; Cianférani, S.; Jeffries, C.M.; Svergun, D.I.; et al. Structural basis for DNA recognition and allosteric control of the retinoic acid receptors RAR–RXR. Nucleic Acids Res. 2020, 48, 9969–9985. [Google Scholar] [CrossRef]
- Yin, M.; Wang, J.; Wang, M.; Li, X.; Zhang, M.; Wu, Q.; Wang, Y. Molecular mechanism of directional CTCF recognition of a diverse range of genomic sites. Cell Res. 2017, 27, 1365–1377. [Google Scholar] [CrossRef]
- Afek, A.; Cohen, H.; Barber-Zucker, S.; Gordân, R.; Lukatsky, D.B. Nonconsensus Protein Binding to Repetitive DNA Sequence Elements Significantly Affects Eukaryotic Genomes. PLoS Comput. Biol. 2015, 11, e1004429. [Google Scholar] [CrossRef]
- Teif, V.B. Soft Power of Nonconsensus Protein-DNA Binding. Biophys. J. 2020, 118, 1797–1798. [Google Scholar] [CrossRef]
- Zheng, A.; Lamkin, M.; Zhao, H.; Wu, C.; Su, H.; Gymrek, M. Deep neural networks identify sequence context features predictive of transcription factor binding. Nat. Mach. Intell. 2021, 3, 172–180. [Google Scholar] [CrossRef]
- Wang, S.; Zhang, Q.; Shen, Z.; He, Y.; Chen, Z.-H.; Li, J.; Huang, D.-S. Predicting transcription factor binding sites using DNA shape features based on shared hybrid deep learning architecture. Mol. Ther. Nucleic Acids 2021, 24, 154–163. [Google Scholar] [CrossRef]
- Wada, K.; Wada, Y.; Ikemura, T. Mb-level CpG and TFBS islands visualized by AI and their roles in the nuclear organization of the human genome. Genes Genet. Syst. 2020, 95, 29–41. [Google Scholar] [CrossRef]
- Pei, G.; Hu, R.; Dai, Y.; Manuel, A.M.; Zhao, Z.; Jia, P. Predicting regulatory variants using a dense epigenomic mapped CNN model elucidated the molecular basis of trait-tissue associations. Nucleic Acids Res. 2021, 49, 53–66. [Google Scholar] [CrossRef]
- Jing, F.; Zhang, S.-W.; Cao, Z.; Zhang, S. An Integrative Framework for Combining Sequence and Epigenomic Data to Predict Transcription Factor Binding Sites Using Deep Learning. IEEE/ACM Trans. Comput. Biol. Bioinform. 2021, 18, 355–364. [Google Scholar] [CrossRef]
- Chen, C.; Hou, J.; Shi, X.; Yang, H.; Birchler, J.A.; Cheng, J. DeepGRN: Prediction of transcription factor binding site across cell-types using attention-based deep neural networks. BMC Bioinform. 2021, 22, 38. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Aim | Method | Advantages | Shortcomings | Comments |
|---|---|---|---|---|
| Registration of the fact of an effect of nucleotide substitution on TF binding | EMSA with nuclear extract (cross-competition assay when necessary) | Simple procedure | In vitro; tissue-specific effects | Testing of several cell lines is desirable |
| Identification of TF the binding site of which is disrupted by a nucleotide substitution | EMSA with purified TF or specific antibody | Unambiguous result | In vitro; requires prior knowledge about TFBS, purified TF, specific antibody | Prescreening in competition assay with unlabeled oligonucleotides may be helpful |
| Confirmation of TF binding in vivo | ChIP-PCR | In vivo | Requires prior knowledge about TFBS and specific antibody | |
| Identification of TF the binding site of which is disrupted by a nucleotide substitution | ChIP-AS-qPCR | In vivo; unambiguous result | Requires prior knowledge about TFBS and specific antibody | Copy number variation must be taken into account when using cell lines |
| Identification of TF the binding site of which is disrupted by a nucleotide substitution | Pull-down assay followed by mass spectrometry analysis | Requires no prior knowledge about TFBS | In vitro | Confirmation by EMSA with purified TF or specific antibody is necessary in some cases |
| Registration of the fact of an effect of nucleotide substitution on the activity of regulatory element | Reporter assays | Simple procedure | Out of genome context | Testing of several cell lines is desirable |
| Registration of the fact of an effect of nucleotide substitution on the activity of regulatory element | CRISPR/Cas9-mediated single nucleotide editing | In genome context | Testing of several cell lines is desirable |
| ID | Location | Risk Allele | TFs with ASB | Genes with ASE | Risk Disease According to GWAS | Ref |
|---|---|---|---|---|---|---|
| rs36115365 | chr5p15.33 intergenic region, putative enhancer | C | ZNF148 (EMSA+AB, EMSA+ purified ZNF148) | TERT (ASE, siRNA-mediated knockdown of ZNF148) | Increased pancreatic and testicular cancer risk but a decreased lung cancer and melanoma risk | [23] |
| rs11672691 | Chr19q13.2 Intron 2 of lncRNA PCAT19 | G | HOXA2 (ChIP-AS-qPCR) | PCAT19 CEACAM21 (ASE, HOXA2 knockdown CRISPR/Cas9 | Aggressive prostate cancer | [20] |
| rs2107595 | Chr7p21 noncoding DNA 3’ to the HDAC, DHSs | A | E2F3 (ChIP-PCR) | HDAC9 (ASE) | Atherosclerosis, coronary artery disease, stroke | [26] |
| rs12411216 | Chr1q22 DHSs | A | E2F4 (EMSA+AB) | GBA (ASE, CRISPR/Cas9) | Parkinson’s disease, cognitive damage | [28] |
| rs13239597 | Chr7q32.1 TNPO3 promoter | A | EVI1 (ChIP-AS-qPCR) | IRF5 (ASE, shRNA-mediated knockdown of EVI1) | Systemic lupus erythematosus and systemic sclerosis | [59] |
| rs17079281 | Chr6q22.2 DCBLD1 promoter | C | YY1 (ChIP-qPCR) | DCBLD1 (ASE, CRISPR/Cas9) | Lung cancer | [16] |
| Approach | GWAS | eQTL Analysis | ASE | ASB | |
|---|---|---|---|---|---|
| 1 | Initial association with trait | + | − | − | − |
| 2 | Initial association with function | − | + | + | + |
| 3 | Causal or in LD | Both | + | ++ | +++ |
| 4 | Number of participants | Tens and hundreds of thousands (large cohorts) | Hundreds (modestly sized cohorts) | Few | Few |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Degtyareva, A.O.; Antontseva, E.V.; Merkulova, T.I. Regulatory SNPs: Altered Transcription Factor Binding Sites Implicated in Complex Traits and Diseases. Int. J. Mol. Sci. 2021, 22, 6454. https://doi.org/10.3390/ijms22126454
Degtyareva AO, Antontseva EV, Merkulova TI. Regulatory SNPs: Altered Transcription Factor Binding Sites Implicated in Complex Traits and Diseases. International Journal of Molecular Sciences. 2021; 22(12):6454. https://doi.org/10.3390/ijms22126454
Chicago/Turabian StyleDegtyareva, Arina O., Elena V. Antontseva, and Tatiana I. Merkulova. 2021. "Regulatory SNPs: Altered Transcription Factor Binding Sites Implicated in Complex Traits and Diseases" International Journal of Molecular Sciences 22, no. 12: 6454. https://doi.org/10.3390/ijms22126454
APA StyleDegtyareva, A. O., Antontseva, E. V., & Merkulova, T. I. (2021). Regulatory SNPs: Altered Transcription Factor Binding Sites Implicated in Complex Traits and Diseases. International Journal of Molecular Sciences, 22(12), 6454. https://doi.org/10.3390/ijms22126454