
Rapid Conversion of Amyloid-Beta 1-40 Oligomers to Mature Fibrils through a Self-Catalytic Bimolecular Process

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
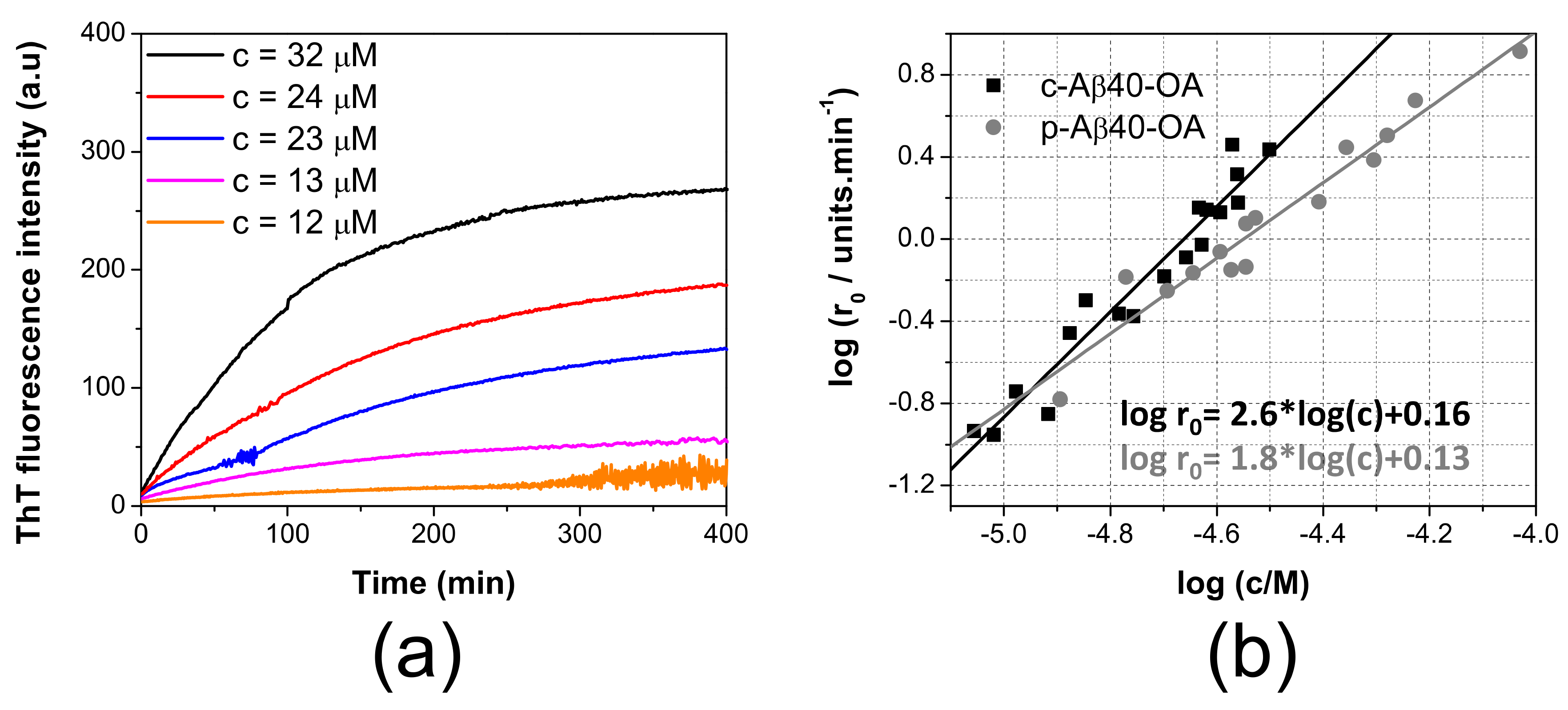
2.1. Effective Nucleation of Amyloid Structure from Disaggregated Aβ Depends on a Dynamic Exchange between Monomer and Oligomeric Species
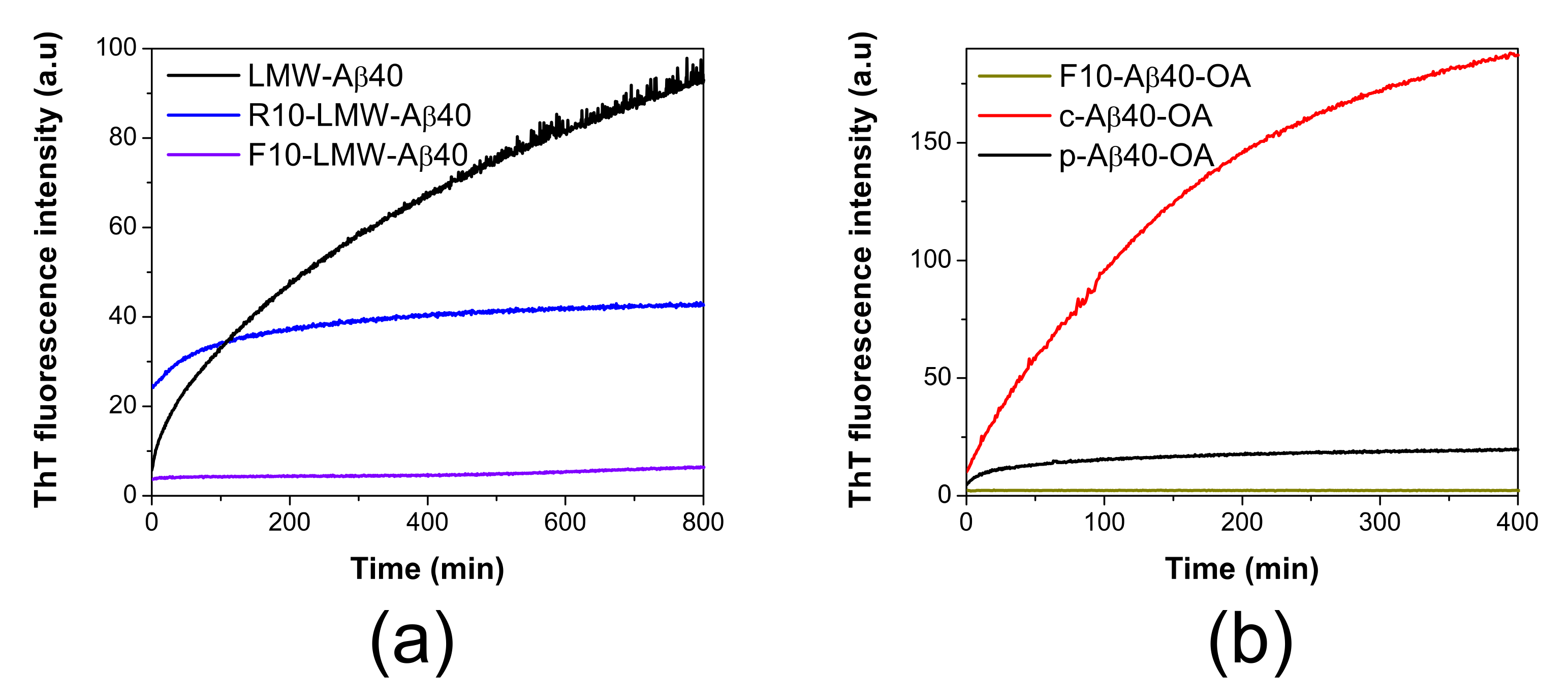
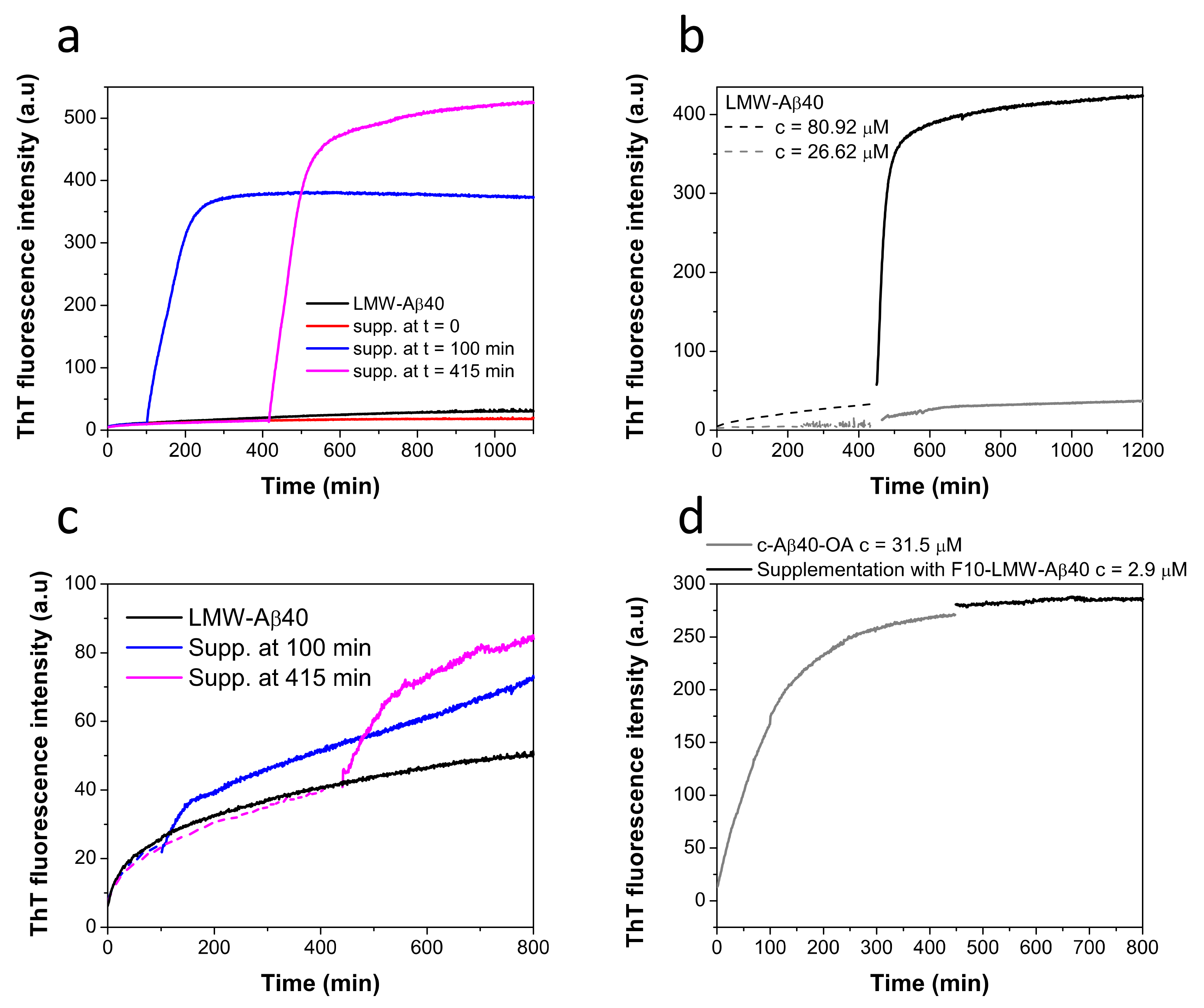
2.2. Conversion of Aβ40 Oligomers to Fibrils Is Dependent on the Presence of Low Molecular Weight Species
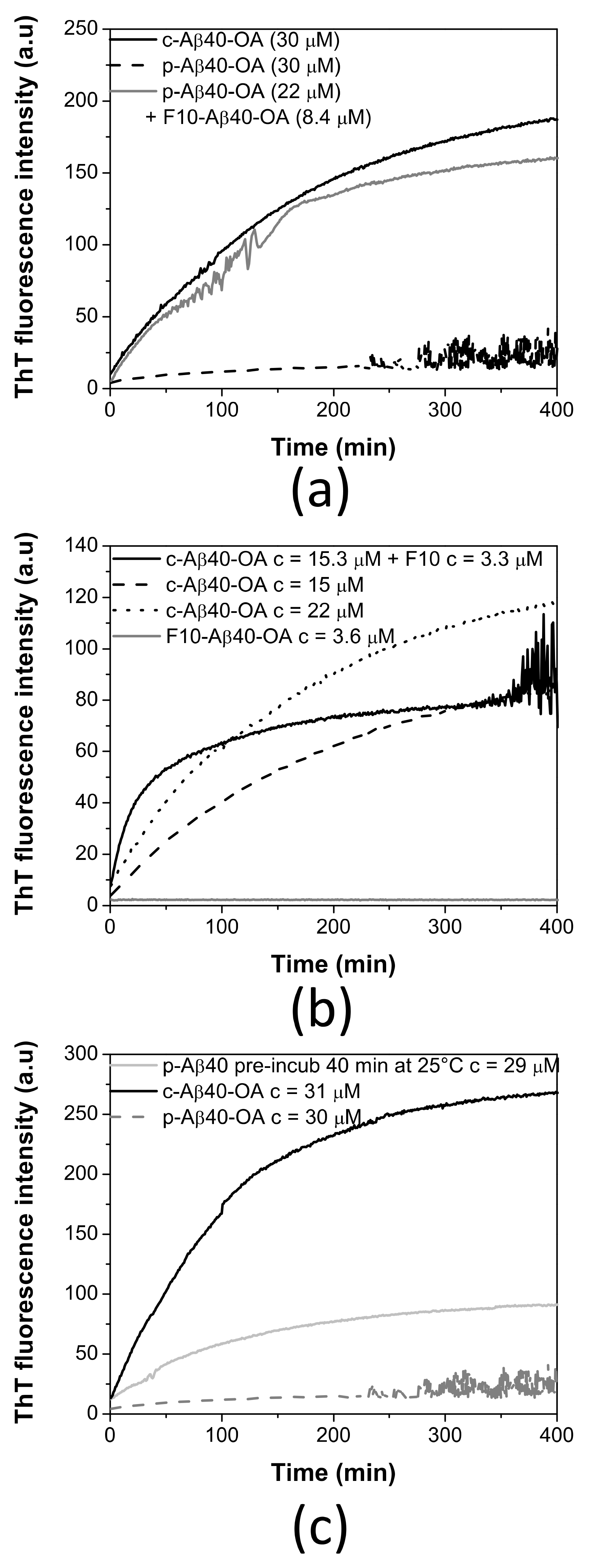
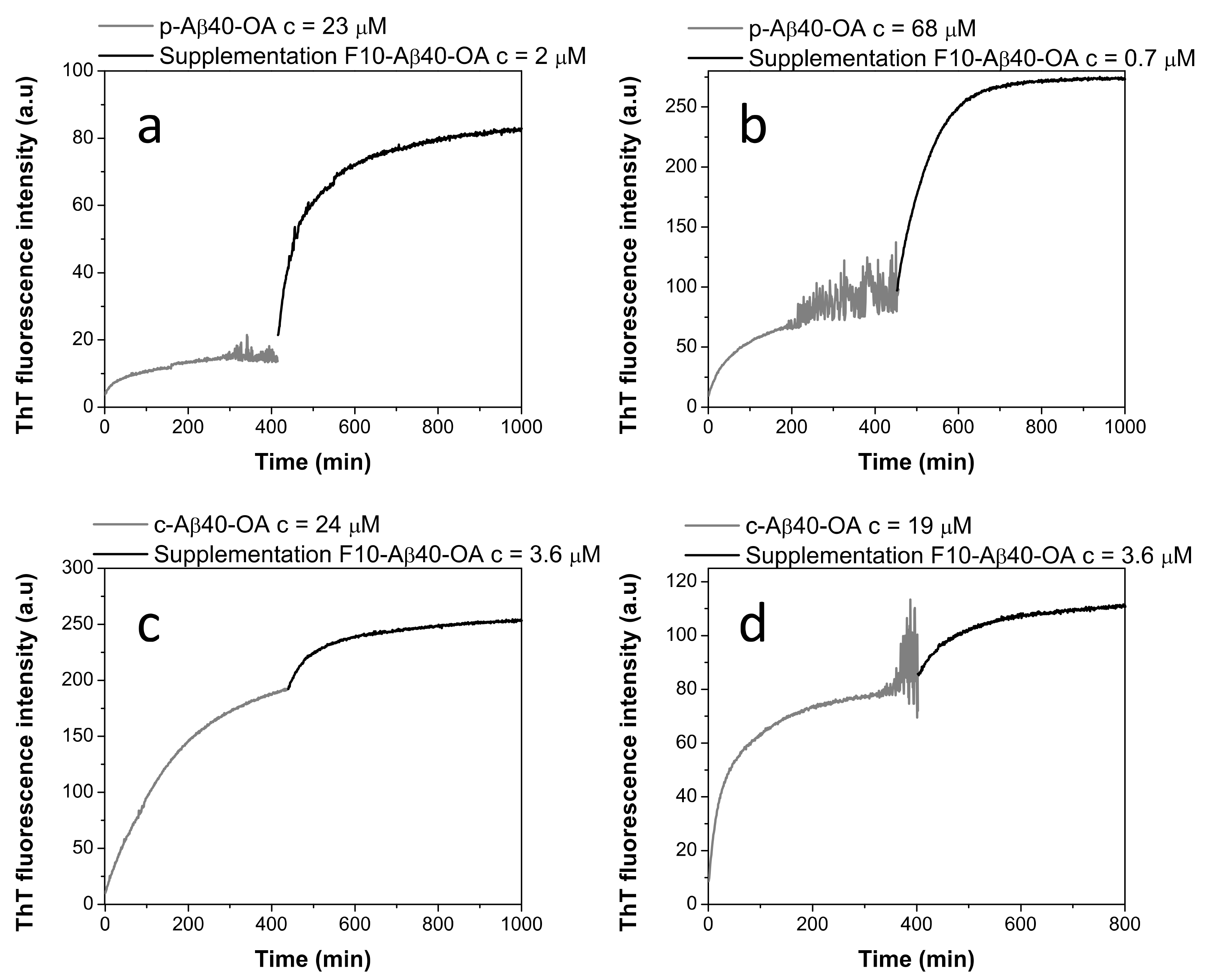
2.3. Pure Type A Oligomers (p-Aβ40-OA) Can Behave as c-Aβ40-OA under Certain Conditions
2.4. Low-Molecular Weight Species Act as Catalyst of Fibrillation
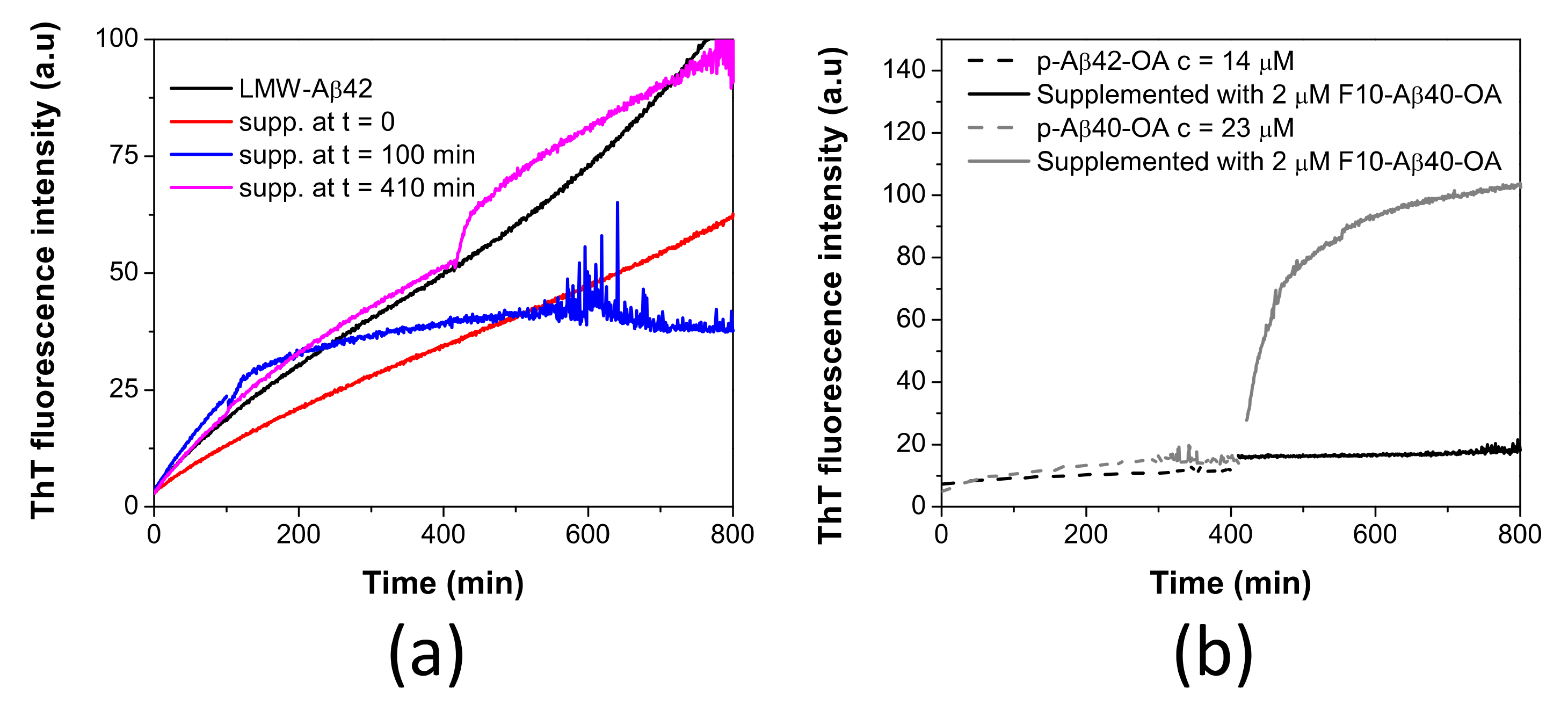
2.5. Aβ42 Spheroidal Oligomers Cannot Be Catalyzed to Fibrillate
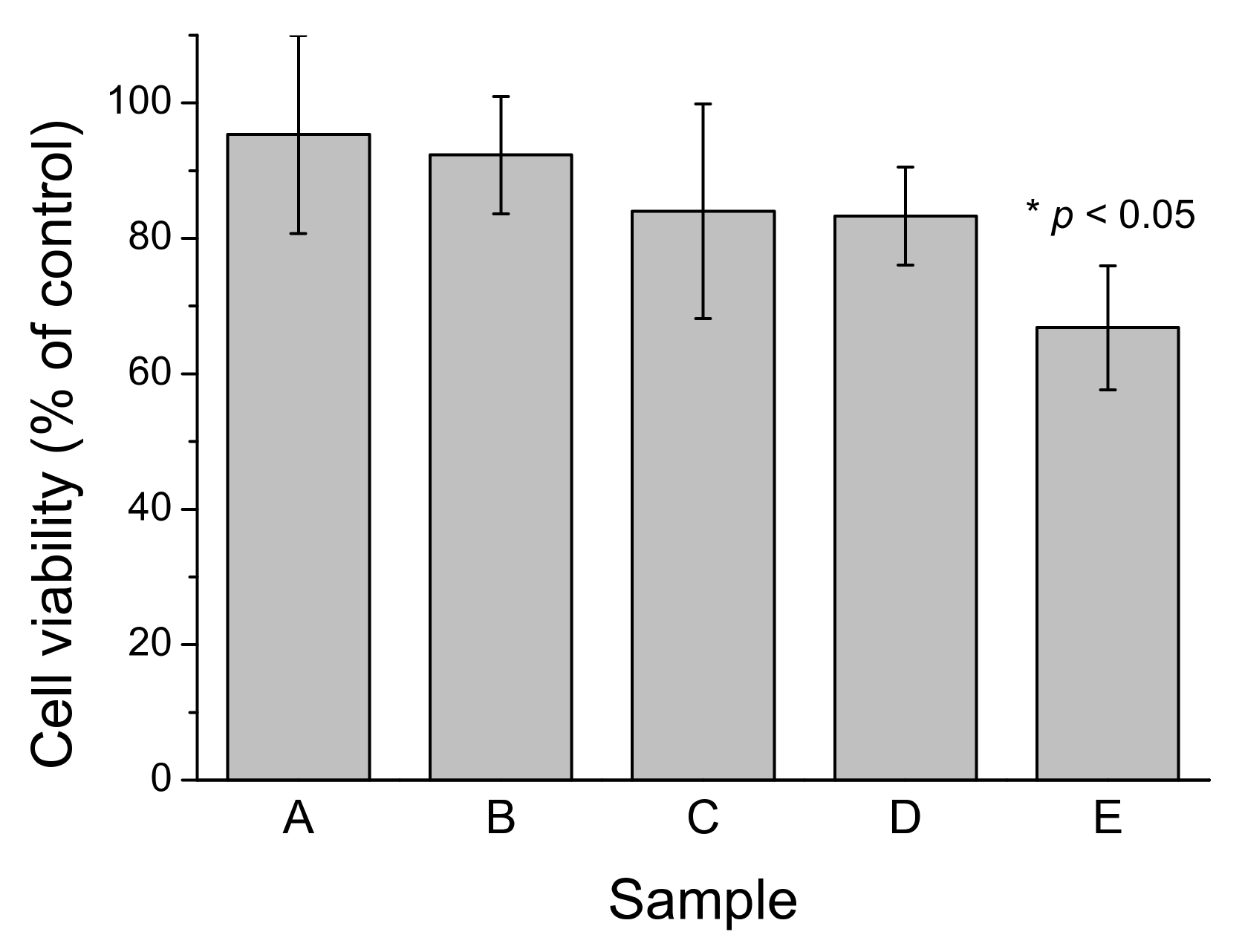
2.6. Cytotoxic Effects of Aβ40 Oligomers Are Related to Active Fibrillation
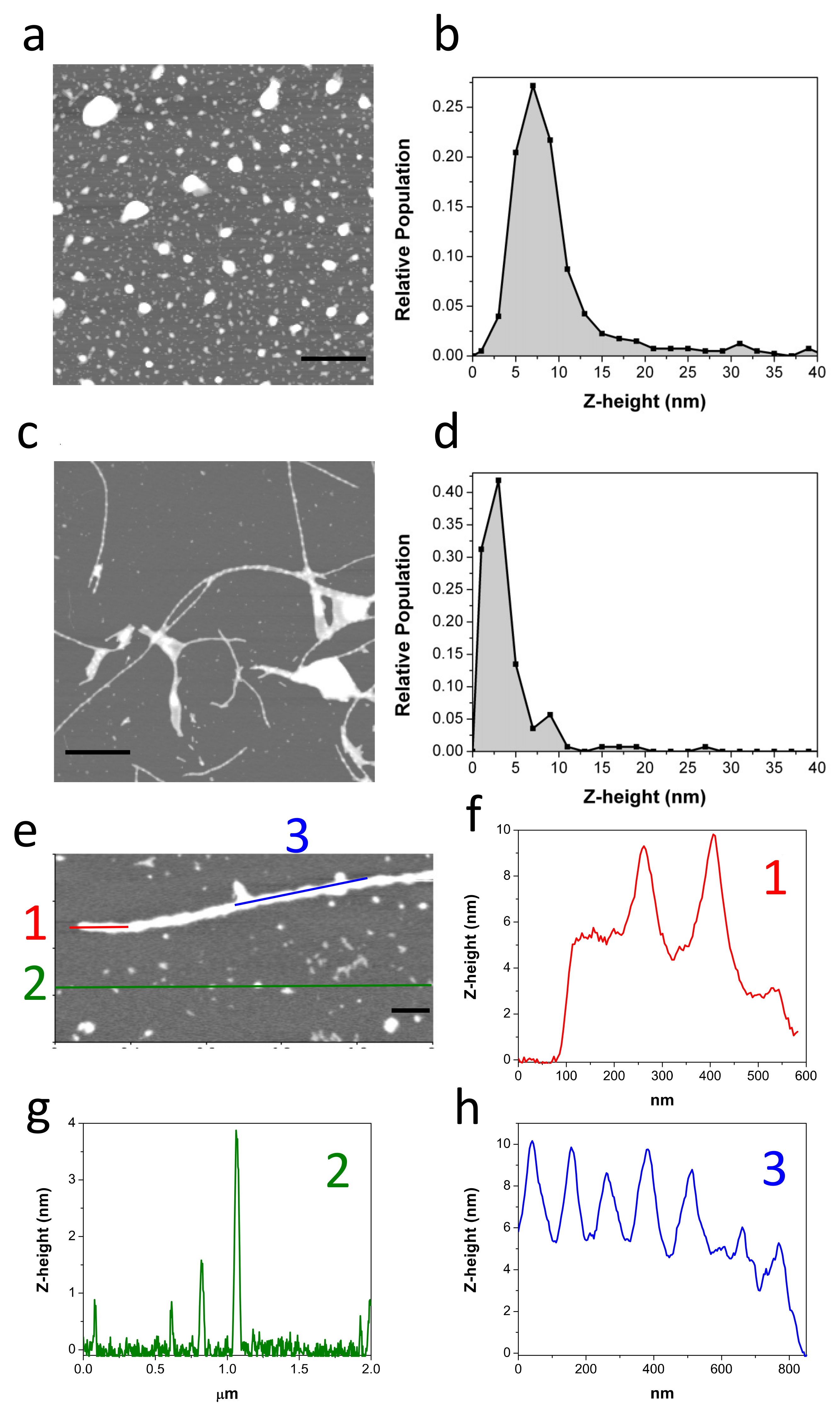
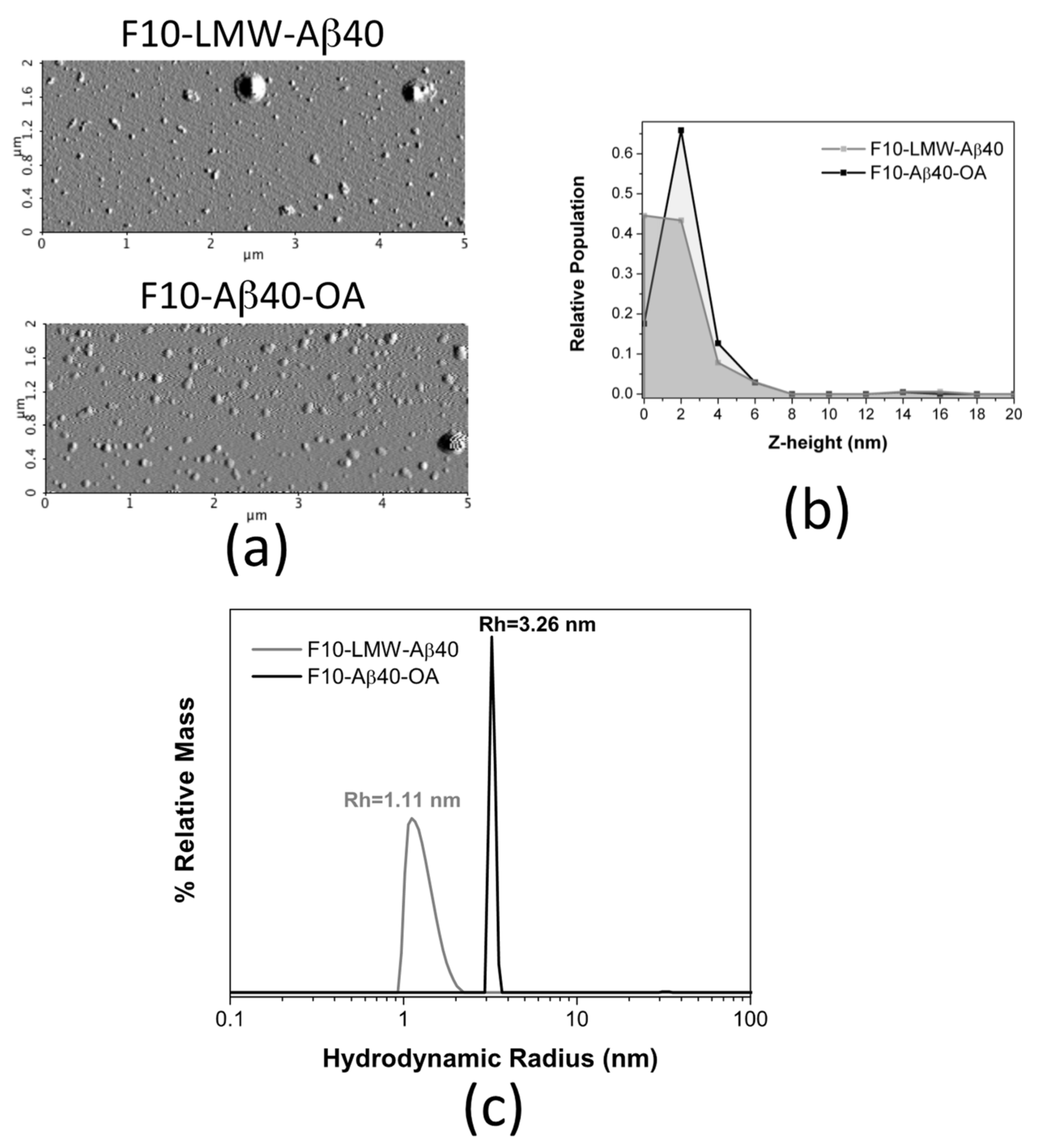
2.7. Biophysical Characterization of the Lowest Molecular Weight Species F10-Aβ40-OA and F10-LMW-Aβ40
3. Discussion
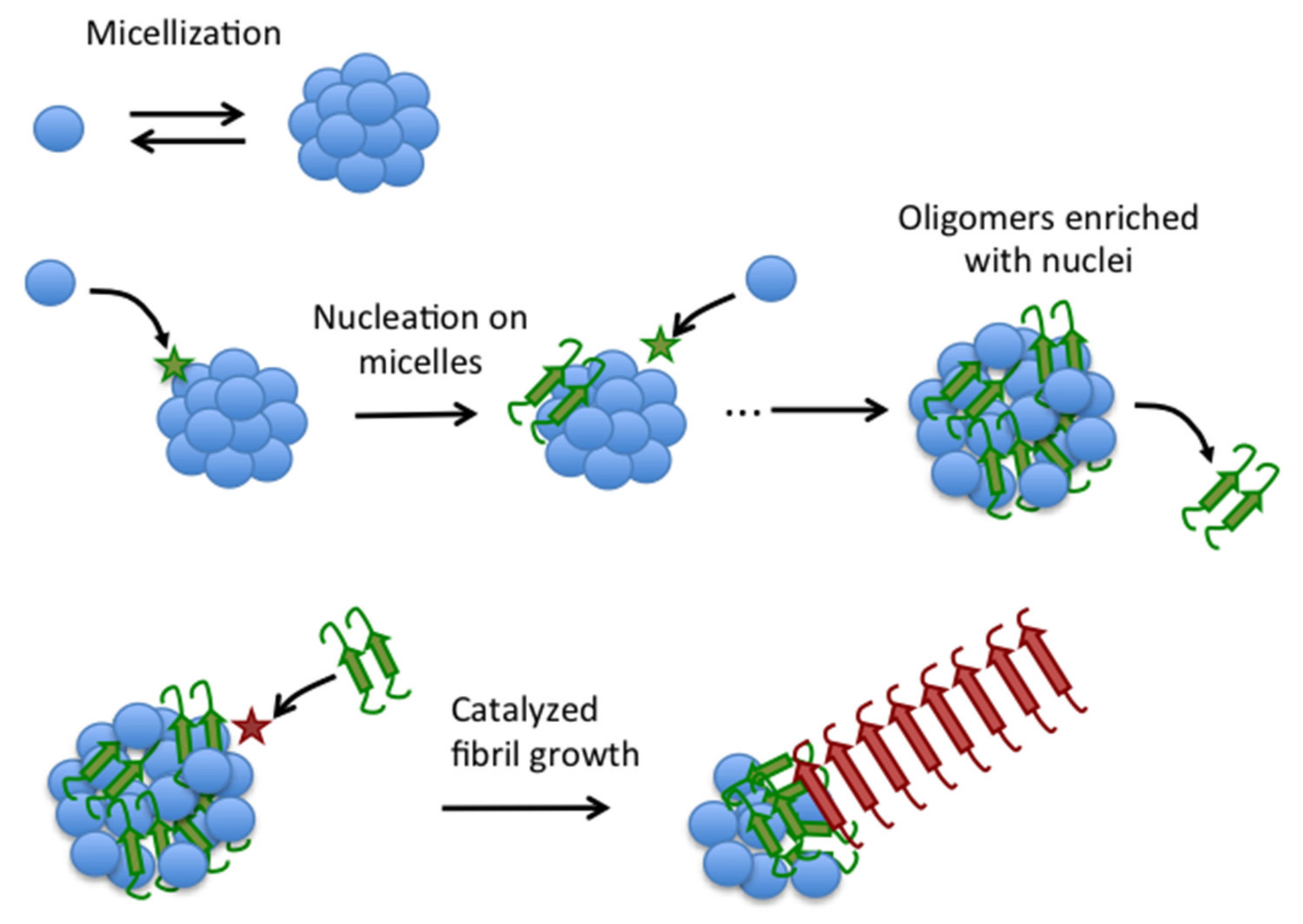
3.1. A Novel Mechanism of Aβ40 Fibrillation
3.2. Catalyzed Fibrillation Is a Source of Cytotoxicity of Aβ40
3.3. Aβ42 Oligomers Do Not Produce Fibrillation Active Species
3.4. Are the Fibrillation Catalytic Species Chemical or Conformational Isomers of Aβ40?
4. Materials and Methods
4.1. Preparation of Disaggregated Aβ Peptides
4.2. Preparation and Purification of Oligomer Samples
4.3. Formation of Amyloid Aggregates Determined by ThT Fluorescence
4.4. Atomic Force Microscopy (AFM)
4.5. Cell Viability Measurements
4.6. Particle Size Determination by Dynamic Light Scattering (DLS)
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chiti, F.; Dobson, C.M. Protein Misfolding, Amyloid Formation, and Human Disease: A Summary of Progress Over the Last Decade. Annu. Rev. Biochem. 2017, 86, 27–68. [Google Scholar] [CrossRef] [PubMed]
- Ross, C.A.; Poirier, M.A. Protein aggregation and neurodegenerative disease. Nat. Med. 2004, 10, S10–S17. [Google Scholar] [CrossRef] [PubMed]
- Westermark, P. Classification of amyloid fibril proteins and their precursors: An ongoing discussion. Amyloid 1997, 4, 216–218. [Google Scholar] [CrossRef]
- Klein, W.L.; Stine, W.B., Jr.; Teplow, D.B. Small assemblies of unmodified amyloid beta-protein are the proximate neurotoxin in Alzheimer’s disease. Neurobiol. Aging 2004, 25, 569–580. [Google Scholar] [CrossRef] [PubMed]
- Hsia, A.Y.; Masliah, E.; McConlogue, L.; Yu, G.Q.; Tatsuno, G.; Hu, K.; Kholodenko, D.; Malenka, R.C.; Nicoll, R.A.; Mucke, L. Plaque-independent disruption of neural circuits in Alzheimer’s disease mouse models. Proc. Natl. Acad. Sci. USA 1999, 96, 3228–3233. [Google Scholar] [CrossRef]
- Lambert, M.P.; Barlow, A.K.; Chromy, B.A.; Edwards, C.; Freed, R.; Liosatos, M.; Morgan, T.E.; Rozovsky, I.; Trommer, B.; Viola, K.L.; et al. Diffusible, nonfibrillar ligands derived from Abeta1-42 are potent central nervous system neurotoxins. Proc. Natl. Acad. Sci. USA 1998, 95, 6448–6453. [Google Scholar] [CrossRef] [PubMed]
- Benilova, I.; Karran, E.; De Strooper, B. The toxic Abeta oligomer and Alzheimer’s disease: An emperor in need of clothes. Nat. Neurosci. 2012, 15, 349–357. [Google Scholar] [CrossRef]
- Haass, C.; Selkoe, D.J. Soluble protein oligomers in neurodegeneration: Lessons from the Alzheimer’s amyloid beta-peptide. Nat. Rev. Mol. Cell. Biol. 2007, 8, 101–112. [Google Scholar] [CrossRef]
- Ono, K.; Condron, M.M.; Teplow, D.B. Structure-neurotoxicity relationships of amyloid beta-protein oligomers. Proc. Natl. Acad. Sci. USA 2009, 106, 14745–14750. [Google Scholar] [CrossRef]
- Limbocker, R.; Chia, S.; Ruggeri, F.S.; Perni, M.; Cascella, R.; Heller, G.T.; Meisl, G.; Mannini, B.; Habchi, J.; Michaels, T.C.T.; et al. Trodusquemine enhances Abeta42 aggregation but suppresses its toxicity by displacing oligomers from cell membranes. Nat. Commun. 2019, 10, 225. [Google Scholar] [CrossRef]
- Cohen, S.I.A.; Cukalevski, R.; Michaels, T.C.T.; Saric, A.; Tornquist, M.; Vendruscolo, M.; Dobson, C.M.; Buell, A.K.; Knowles, T.P.J.; Linse, S. Distinct thermodynamic signatures of oligomer generation in the aggregation of the amyloid-beta peptide. Nat. Chem. 2018, 10, 523–531. [Google Scholar] [CrossRef] [PubMed]
- Meisl, G.; Yang, X.; Dobson, C.M.; Linse, S.; Knowles, T.P.J. Modulation of electrostatic interactions to reveal a reaction network unifying the aggregation behaviour of the Abeta42 peptide and its variants. Chem. Sci. 2017, 8, 4352–4362. [Google Scholar] [CrossRef]
- Habchi, J.; Chia, S.; Limbocker, R.; Mannini, B.; Ahn, M.; Perni, M.; Hansson, O.; Arosio, P.; Kumita, J.R.; Challa, P.K.; et al. Systematic development of small molecules to inhibit specific microscopic steps of Abeta42 aggregation in Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2017, 114, E200–E208. [Google Scholar] [CrossRef]
- Zhang, J.; Mao, X.; Xu, W. Fibril Nucleation Kinetics of a Pharmaceutical Peptide: The Role of Conformation Stability, Formulation Factors, and Temperature Effect. Mol. Pharm. 2018. [Google Scholar] [CrossRef] [PubMed]
- Zurdo, J.; Guijarro, J.I.; Jimenez, J.L.; Saibil, H.R.; Dobson, C.M. Dependence on solution conditions of aggregation and amyloid formation by an SH3 domain. J. Mol. Biol. 2001, 311, 325–340. [Google Scholar] [CrossRef]
- Ruzafa, D.; Conejero-Lara, F.; Morel, B. Modulation of the stability of amyloidogenic precursors by anion binding strongly influences the rate of amyloid nucleation. Phys. Chem. Chem. Phys. 2013, 15, 15508–15517. [Google Scholar] [CrossRef] [PubMed]
- Ruzafa, D.; Hernandez-Gomez, Y.S.; Bisello, G.; Broersen, K.; Morel, B.; Conejero-Lara, F. The influence of N-terminal acetylation on micelle-induced conformational changes and aggregation of alpha-Synuclein. PLoS ONE 2017, 12, e0178576. [Google Scholar] [CrossRef] [PubMed]
- Knowles, T.P.; Waudby, C.A.; Devlin, G.L.; Cohen, S.I.; Aguzzi, A.; Vendruscolo, M.; Terentjev, E.M.; Welland, M.E.; Dobson, C.M. An analytical solution to the kinetics of breakable filament assembly. Science 2009, 326, 1533–1537. [Google Scholar] [CrossRef] [PubMed]
- Morris, A.M.; Watzky, M.A.; Finke, R.G. Protein aggregation kinetics, mechanism, and curve-fitting: A review of the literature. Biochim. Biophys. Acta 2009, 1794, 375–397. [Google Scholar] [CrossRef]
- Ruzafa, D.; Morel, B.; Varela, L.; Azuaga, A.I.; Conejero-Lara, F. Characterization of oligomers of heterogeneous size as precursors of amyloid fibril nucleation of an SH3 domain: An experimental kinetics study. PLoS ONE 2012, 7, e49690. [Google Scholar] [CrossRef] [PubMed]
- Morel, B.; Conejero-Lara, F. Early mechanisms of amyloid fibril nucleation in model and disease-related proteins. Biochim. Biophys Acta Proteins Proteom 2019, 1867, 140264. [Google Scholar] [CrossRef] [PubMed]
- Harper, J.D.; Lansbury, P.T., Jr. Models of amyloid seeding in Alzheimer’s disease and scrapie: Mechanistic truths and physiological consequences of the time-dependent solubility of amyloid proteins. Annu. Rev. Biochem. 1997, 66, 385–407. [Google Scholar] [CrossRef]
- Jarrett, J.T.; Lansbury, P.T., Jr. Seeding “one-dimensional crystallization” of amyloid: A pathogenic mechanism in Alzheimer’s disease and scrapie? Cell 1993, 73, 1055–1058. [Google Scholar] [CrossRef]
- Roychaudhuri, R.; Yang, M.; Hoshi, M.M.; Teplow, D.B. Amyloid beta-protein assembly and Alzheimer disease. J. Biol. Chem. 2009, 284, 4749–4753. [Google Scholar] [CrossRef]
- Wetzel, R. Kinetics and Thermodynamics of Amyloid Fibril Assembly. Acc. Chem. Res. 2006, 39, 671–679. [Google Scholar] [CrossRef]
- Morel, B.; Casares, S.; Conejero-Lara, F. A single mutation induces amyloid aggregation in the alpha-spectrin SH3 domain: Analysis of the early stages of fibril formation. J. Mol. Biol. 2006, 356, 453–468. [Google Scholar] [CrossRef] [PubMed]
- Morel, B.; Varela, L.; Azuaga, A.I.; Conejero-Lara, F. Environmental conditions affect the kinetics of nucleation of amyloid fibrils and determine their morphology. Biophys. J. 2010, 99, 3801–3810. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, J.S.; Christensen, G.; Otzen, D.E. Modulation of S6 Fibrillation by Unfolding Rates and Gatekeeper Residues. J. Mol. Biol. 2004, 341, 575–588. [Google Scholar] [CrossRef]
- Ruzafa, D.; Varela, L.; Azuaga, A.I.; Conejero-Lara, F.; Morel, B. Mapping the structure of amyloid nucleation precursors by protein engineering kinetic analysis. Phys. Chem. Chem. Phys. 2014, 16, 2989–3000. [Google Scholar] [CrossRef]
- Varela, L.; Morel, B.; Azuaga, A.I.; Conejero-Lara, F. A single mutation in an SH3 domain increases amyloid aggregation by accelerating nucleation, but not by destabilizing thermodynamically the native state. FEBS Lett. 2009, 583, 801–806. [Google Scholar] [CrossRef]
- Bernstein, S.L.; Dupuis, N.F.; Lazo, N.D.; Wyttenbach, T.; Condron, M.M.; Bitan, G.; Teplow, D.B.; Shea, J.E.; Ruotolo, B.T.; Robinson, C.V.; et al. Amyloid-beta protein oligomerization and the importance of tetramers and dodecamers in the aetiology of Alzheimer’s disease. Nat. Chem. 2009, 1, 326–331. [Google Scholar] [CrossRef]
- Kelly, J.W. The alternative conformations of amyloidogenic proteins and their multi-step assembly pathways. Curr. Opin. Struct. Biol. 1998, 8, 101–106. [Google Scholar] [CrossRef]
- Morel, B.; Carrasco, M.P.; Jurado, S.; Marco, C.; Conejero-Lara, F. Dynamic micellar oligomers of amyloid beta peptides play a crucial role in their aggregation mechanisms. Phys. Chem. Chem. Phys. 2018, 20, 20597–20614. [Google Scholar] [CrossRef] [PubMed]
- Sabate, R.; Estelrich, J. Evidence of the existence of micelles in the fibrillogenesis of beta-amyloid peptide. J. Phys. Chem. B 2005, 109, 11027–11032. [Google Scholar] [CrossRef] [PubMed]
- Serio, T.R.; Cashikar, A.G.; Kowal, A.S.; Sawicki, G.J.; Moslehi, J.J.; Serpell, L.; Arnsdorf, M.F.; Lindquist, S.L. Nucleated conformational conversion and the replication of conformational information by a prion determinant. Science 2000, 289, 1317–1321. [Google Scholar] [CrossRef]
- Lee, J.; Culyba, E.K.; Powers, E.T.; Kelly, J.W. Amyloid-beta forms fibrils by nucleated conformational conversion of oligomers. Nat. Chem. Biol. 2011, 7, 602–609. [Google Scholar] [CrossRef]
- Cohen, S.I.; Linse, S.; Luheshi, L.M.; Hellstrand, E.; White, D.A.; Rajah, L.; Otzen, D.E.; Vendruscolo, M.; Dobson, C.M.; Knowles, T.P. Proliferation of amyloid-beta42 aggregates occurs through a secondary nucleation mechanism. Proc. Natl. Acad. Sci. USA 2013, 110, 9758–9763. [Google Scholar] [CrossRef]
- Michaels, T.C.; Lazell, H.W.; Arosio, P.; Knowles, T.P. Dynamics of protein aggregation and oligomer formation governed by secondary nucleation. J. Chem. Phys. 2015, 143, 054901. [Google Scholar] [CrossRef]
- Close, W.; Neumann, M.; Schmidt, A.; Hora, M.; Annamalai, K.; Schmidt, M.; Reif, B.; Schmidt, V.; Grigorieff, N.; Fandrich, M. Physical basis of amyloid fibril polymorphism. Nat. Commun. 2018, 9, 699. [Google Scholar] [CrossRef] [PubMed]
- Paravastu, A.K.; Leapman, R.D.; Yau, W.M.; Tycko, R. Molecular structural basis for polymorphism in Alzheimer’s beta-amyloid fibrils. Proc. Natl. Acad. Sci. USA 2008, 105, 18349–18354. [Google Scholar] [CrossRef]
- Shekhawat, G.S.; Lambert, M.P.; Sharma, S.; Velasco, P.T.; Viola, K.L.; Klein, W.L.; Dravid, V.P. Soluble state high resolution atomic force microscopy study of Alzheimer’s β-amyloid oligomers. Appl. Phys. Lett. 2009, 95, 183701. [Google Scholar] [CrossRef] [PubMed]
- Brender, J.R.; Krishnamoorthy, J.; Sciacca, M.F.; Vivekanandan, S.; D’Urso, L.; Chen, J.; La Rosa, C.; Ramamoorthy, A. Probing the sources of the apparent irreproducibility of amyloid formation: Drastic changes in kinetics and a switch in mechanism due to micellelike oligomer formation at critical concentrations of IAPP. J. Phys. Chem. B 2015, 119, 2886–2896. [Google Scholar] [CrossRef]
- Galvagnion, C.; Buell, A.K.; Meisl, G.; Michaels, T.C.; Vendruscolo, M.; Knowles, T.P.; Dobson, C.M. Lipid vesicles trigger alpha-synuclein aggregation by stimulating primary nucleation. Nat. Chem. Biol. 2015, 11, 229–234. [Google Scholar] [CrossRef]
- Poma, A.B.; Chwastyk, M.; Cieplak, M. Elastic moduli of biological fibers in a coarse-grained model: Crystalline cellulose and Ab amyloids. Phys. Chem. Chem. Phys. 2017, 19, 28195–28206. [Google Scholar] [CrossRef]
- Ruggeri, F.S.; Adamcik, J.; Jeong, J.S.; Lashuel, H.A.; Mezzenga, R.; Dietler, G. Influence of the beta-sheet content on the mechanical properties of aggregates during amyloid fibrillization. Angew. Chem. Int. Ed. Engl. 2015, 54, 2462–2466. [Google Scholar] [CrossRef]
- Poma, A.B.; Guzman, H.V.; Li, M.S.; Theodorakis, P.E. Mechanical and thermodynamic properties of Ab42, Ab40, and a-synuclein fibrils: A coarse-grained method to complement experimental studies. Beilstein J. Nanotechnol. 2019, 10, 500–513. [Google Scholar] [CrossRef]
- Yuan, C.; Levin, A.; Chen, W.; Xing, R.; Zou, Q.; Herling, T.W.; Challa, P.K.; Knowles, T.P.J.; Yan, X. Nucleation and Growth of Amino Acid and Peptide Supramolecular Polymers through Liquid-Liquid Phase Separation. Angew. Chem. Int. Ed. Engl. 2019, 58, 18116–18123. [Google Scholar] [CrossRef]
- Dear, A.J.; Meisl, G.; Michaels, T.C.T.; Zimmermann, M.R.; Linse, S.; Knowles, T.P.J. The catalytic nature of protein aggregation. J. Chem. Phys. 2020, 152, 045101. [Google Scholar] [CrossRef] [PubMed]
- Saric, A.; Buell, A.K.; Meisl, G.; Michaels, T.C.T.; Dobson, C.M.; Linse, S.; Knowles, T.P.J.; Frenkel, D. Physical determinants of the self-replication of protein fibrils. Nat. Phys. 2016, 12, 874–880. [Google Scholar] [CrossRef]
- Jan, A.; Adolfsson, O.; Allaman, I.; Buccarello, A.L.; Magistretti, P.J.; Pfeifer, A.; Muhs, A.; Lashuel, H.A. Abeta42 neurotoxicity is mediated by ongoing nucleated polymerization process rather than by discrete Abeta42 species. J. Biol. Chem. 2011, 286, 8585–8596. [Google Scholar] [CrossRef] [PubMed]
- Krishtal, J.; Bragina, O.; Metsla, K.; Palumaa, P.; Tougu, V. In situ fibrillizing amyloid-beta 1-42 induces neurite degeneration and apoptosis of differentiated SH-SY5Y cells. PLoS ONE 2017, 12, e0186636. [Google Scholar] [CrossRef]
- Walker, L.C.; Jucker, M. Neurodegenerative diseases: Expanding the prion concept. Annu. Rev. Neurosci. 2015, 38, 87–103. [Google Scholar] [CrossRef]
- Katzmarski, N.; Ziegler-Waldkirch, S.; Scheffler, N.; Witt, C.; Abou-Ajram, C.; Nuscher, B.; Prinz, M.; Haass, C.; Meyer-Luehmann, M. Abeta oligomers trigger and accelerate Abeta seeding. Brain Pathol. 2020, 30, 36–45. [Google Scholar] [CrossRef]
- Cukalevski, R.; Yang, X.; Meisl, G.; Weininger, U.; Bernfur, K.; Frohm, B.; Knowles, T.P.J.; Linse, S. The Abeta40 and Abeta42 peptides self-assemble into separate homomolecular fibrils in binary mixtures but cross-react during primary nucleation. Chem. Sci. 2015, 6, 4215–4233. [Google Scholar] [CrossRef] [PubMed]
- Tornquist, M.; Michaels, T.C.T.; Sanagavarapu, K.; Yang, X.; Meisl, G.; Cohen, S.I.A.; Knowles, T.P.J.; Linse, S. Secondary nucleation in amyloid formation. Chem. Commun. 2018, 54, 8667–8684. [Google Scholar] [CrossRef]
- Yasumoto, T.; Takamura, Y.; Tsuji, M.; Watanabe-Nakayama, T.; Imamura, K.; Inoue, H.; Nakamura, S.; Inoue, T.; Kimura, A.; Yano, S.; et al. High molecular weight amyloid beta1-42 oligomers induce neurotoxicity via plasma membrane damage. FASEB J. 2019, 33, 9220–9234. [Google Scholar] [CrossRef]
- Shimizu, T.; Fukuda, H.; Murayama, S.; Izumiyama, N.; Shirasawa, T. Isoaspartate formation at position 23 of amyloid beta peptide enhanced fibril formation and deposited onto senile plaques and vascular amyloids in Alzheimer’s disease. J. Neurosci. Res. 2002, 70, 451–461. [Google Scholar] [CrossRef] [PubMed]
- Roher, A.E.; Lowenson, J.D.; Clarke, S.; Wolkow, C.; Wang, R.; Cotter, R.J.; Reardon, I.M.; Zurcher-Neely, H.A.; Heinrikson, R.L.; Ball, M.J.; et al. Structural alterations in the peptide backbone of beta-amyloid core protein may account for its deposition and stability in Alzheimer’ s disease. J. Biol. Chem. 1993, 268, 3072–3083. [Google Scholar] [CrossRef]
- Kamgar-Parsi, K.; Hong, L.; Naito, A.; Brooks, C.L., 3rd; Ramamoorthy, A. Growth-incompetent monomers of human calcitonin lead to a noncanonical direct relationship between peptide concentration and aggregation lag time. J. Biol. Chem. 2017, 292, 14963–14976. [Google Scholar] [CrossRef] [PubMed]
- Kashchiev, D. Modeling the Effect of Monomer Conformational Change on the Early Stage of Protein Self-Assembly into Fibrils. J. Phys. Chem. B 2017, 121, 35–46. [Google Scholar] [CrossRef]
- Jan, A.; Hartley, D.M.; Lashuel, H.A. Preparation and characterization of toxic Abeta aggregates for structural and functional studies in Alzheimer’s disease research. Nat. Protoc. 2010, 5, 1186–1209. [Google Scholar] [CrossRef] [PubMed]
- Morel, B.; Conejero-Lara, F. Formation of dynamic micellar oligomers of Aβ 40 and Aβ 42 in the early stages of aggregation. Methods Mol. Biol. 2021, in press. [Google Scholar]
- Tatulian, S.A. Structural Characterization of Membrane Proteins and Peptides by FTIR and ATR-FTIR Spectroscopy. Methods Mol. Biol. 2013, 974, 177–218. [Google Scholar] [CrossRef] [PubMed]










Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Morel, B.; Carrasco-Jiménez, M.P.; Jurado, S.; Conejero-Lara, F. Rapid Conversion of Amyloid-Beta 1-40 Oligomers to Mature Fibrils through a Self-Catalytic Bimolecular Process. Int. J. Mol. Sci. 2021, 22, 6370. https://doi.org/10.3390/ijms22126370
Morel B, Carrasco-Jiménez MP, Jurado S, Conejero-Lara F. Rapid Conversion of Amyloid-Beta 1-40 Oligomers to Mature Fibrils through a Self-Catalytic Bimolecular Process. International Journal of Molecular Sciences. 2021; 22(12):6370. https://doi.org/10.3390/ijms22126370
Chicago/Turabian StyleMorel, Bertrand, María P. Carrasco-Jiménez, Samuel Jurado, and Francisco Conejero-Lara. 2021. "Rapid Conversion of Amyloid-Beta 1-40 Oligomers to Mature Fibrils through a Self-Catalytic Bimolecular Process" International Journal of Molecular Sciences 22, no. 12: 6370. https://doi.org/10.3390/ijms22126370
APA StyleMorel, B., Carrasco-Jiménez, M. P., Jurado, S., & Conejero-Lara, F. (2021). Rapid Conversion of Amyloid-Beta 1-40 Oligomers to Mature Fibrils through a Self-Catalytic Bimolecular Process. International Journal of Molecular Sciences, 22(12), 6370. https://doi.org/10.3390/ijms22126370