
Interaction between Lipopolysaccharide and Gut Microbiota in Inflammatory Bowel Diseases

 ,
,  ,
, 


 ,
, 
Abstract
1. Introduction
2. IBD: More Than One Cause
2.1. Immunologic Pathways
2.2. Microbiota Influence
3. LPS and IBD, a Complex Crosstalk
3.1. LPS and the Immune System
3.2. LPS and Microbiota
3.3. LPS and IBD, What Do We Know
4. Materials and Methods
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Bertani, B.; Ruiz, N. Function and Biogenesis of Lipopolysaccharides. EcoSal Plus 2018, 8. [Google Scholar] [CrossRef] [PubMed]
- Diks, S.H.; Richel, D.J.; Peppelenbosch, M.P. LPS signal transduction: The picture is becoming more complex. Curr. Top. Med. Chem. 2004, 4, 1115–1126. [Google Scholar] [CrossRef]
- Yang, J.; Zhao, Y.; Shao, F. Non-canonical activation of inflammatory caspases by cytosolic LPS in innate immunity. Curr. Opin. Immunol. 2015, 32, 78–83. [Google Scholar] [CrossRef] [PubMed]
- Mohammad, S.; Thiemermann, C. Role of Metabolic Endotoxemia in Systemic Inflammation and Potential Interventions. Front. Immunol. 2021, 11, 594150. [Google Scholar] [CrossRef]
- Ramendra, R.; Isnard, S.; Mehraj, V.; Chen, J.; Zhang, Y.; Finkelman, M.; Routy, J.P. Circulating LPS and (1→3)-β-D-Glucan: A Folie à Deux Contributing to HIV-Associated Immune Activation. Front. Immunol. 2019, 10, 465. [Google Scholar] [CrossRef]
- Tulkens, J.; Vergauwen, G.; Van Deun, J.; Geeurickx, E.; Dhondt, B. Increased levels of systemic LPS-positive bacterial extracellular vesicles in patients with intestinal barrier dysfunction. Gut 2020, 69, 191–193. [Google Scholar] [CrossRef]
- Roberti, R.; Iannone, L.F.; Palleria, C.; De Sarro, C.; Spagnuolo, R.; Barbieri, M.A.; Vero, A.; Manti, A.; Pisana, V.; Fries, W.; et al. Safety profiles of biologic agents for inflammatory bowel diseases: A prospective pharmacovigilance study in Southern Italy. Curr. Med. Res. Opin. 2020, 36, 1457–1463. [Google Scholar] [CrossRef]
- Bazzazi, H.; Aghaei, M.; Memarian, A.; Asgarian-Omran, H.; Behnampour, N.; Yazdani, Y. Th1-Th17 Ratio as a New Insight in Rheumatoid Arthritis Disease. Iran. J. Allergy Asthma Immunol. 2018, 17, 68–77. [Google Scholar]
- Rostami, A.; Ciric, B. Role of Th17 cells in the pathogenesis of CNS inflammatory demyelination. J. Neurol. Sci. 2013, 333, 76–87. [Google Scholar] [CrossRef]
- Lee, S.H.; Kwon, J.E.; Cho, M.-L. Immunological pathogenesis of inflammatory bowel disease. Intest. Res. 2018, 16, 26–42. [Google Scholar] [CrossRef]
- Cosnes, J.; Gower-Rousseau, C.; Seksik, P.; Cortot, A. Epidemiology and natural history of inflammatory bowel diseases. Gastroenterology 2011, 140, 1785–1794. [Google Scholar] [CrossRef]
- Nemeth, Z.H.; Bogdanovski, D.A.; Barratt-Stopper, P.; Paglinco, S.R.; Antonioli, L.; Rolandelli, R.H. Crohn’s Disease and Ulcerative Colitis Show Unique Cytokine Profiles. Cureus 2017, 9, e1177. [Google Scholar] [CrossRef]
- Coufal, S.; Galanova, N.; Bajer, L.; Gajdarova, Z.; Schierova, D.; Jiraskova Zakostelska, Z.; Kostovcikova, K.; Jackova, Z.; Stehlikova, Z.; Drastich, P.; et al. Inflammatory Bowel Disease Types Differ in Markers of Inflammation, Gut Barrier and in Specific Anti-Bacterial Response. Cells 2019, 8, 719. [Google Scholar] [CrossRef] [PubMed]
- Rizzello, F.; Spisni, E.; Giovanardi, E.; Imbesi, V.; Salice, M.; Alvisi, P.; Valerii, M.C.; Gionchetti, P. Implications of the Westernized Diet in the Onset and Progression of IBD. Nutrients 2019, 11, 1033. [Google Scholar] [CrossRef] [PubMed]
- Raman, M.; Ghosh, S. Diet and Nutrition in IBD-Progress and Gaps. Nutrients 2019, 11, 1740. [Google Scholar] [CrossRef]
- Sands, B.E. Biomarkers of Inflammation in Inflammatory Bowel Disease. Gastroenterology 2015, 149, 1275–1285.e2. [Google Scholar] [CrossRef] [PubMed]
- Fagarasan, S.; Kawamoto, S.; Kanagawa, O.; Suzuki, K. Adaptive immune regulation in the gut: T cell-dependent and T cell-independent IgA synthesis. Annu. Rev. Immunol. 2010, 28, 243–273. [Google Scholar] [CrossRef] [PubMed]
- Pararasa, C.; Zhang, N.; Tull, T.J.; Chong, M.H.A.; Siu, J.H.Y.; Guesdon, W.; Chavele, K.M.; Sanderson, J.D.; Langmead, L.; Kok, K.; et al. Reduced CD27(-)IgD(-) B Cells in Blood and Raised CD27(-)IgD(-) B Cells in Gut-Associated Lymphoid Tissue in Inflammatory Bowel Disease. Front. Immunol. 2019, 10, 361. [Google Scholar] [CrossRef]
- Iweala, O.I.; Nagler, C.R. The Microbiome and Food Allergy. Annu. Rev. Immunol. 2019, 37, 377–403. [Google Scholar] [CrossRef]
- Calder, P.C. Feeding the immune system. Proc. Nutr. Soc. 2013, 72, 299–309. [Google Scholar] [CrossRef]
- Tindemans, I.; Joosse, M.E.; Samsom, J.N. Dissecting the Heterogeneity in T-Cell Mediated Inflammation in IBD. Cells 2020, 9, 110. [Google Scholar] [CrossRef] [PubMed]
- Zundler, S.; Becker, E.; Spocinska, M.; Slawik, M.; Parga-Vidal, L.; Stark, R.; Wiendl, M.; Atreya, R.; Rath, T.; Leppkes, M.; et al. Hobit- and Blimp-1-driven CD4(+) tissue-resident memory T cells control chronic intestinal inflammation. Nat. Immunol. 2019, 20, 288–300. [Google Scholar] [CrossRef] [PubMed]
- Chang, S.; Malter, L.; Hudesman, D. Disease monitoring in inflammatory bowel disease. World J. Gastroenterol. 2015, 21, 11246–11259. [Google Scholar] [CrossRef] [PubMed]
- Park, S.C.; Jeen, Y.T. Anti-integrin therapy for inflammatory bowel disease. World J. Gastroenterol. 2018, 24, 1868–1880. [Google Scholar] [CrossRef] [PubMed]
- Pandolfi, F.; Franza, L.; Altamura, S.; Mandolini, C.; Cianci, R.; Ansari, A.; Kurnick, J.T. Integrins: Integrating the Biology and Therapy of Cell-cell Interactions. Clin. Ther. 2017, 39, 2420–2436. [Google Scholar] [CrossRef] [PubMed]
- Friedrich, M.; Pohin, M.; Powrie, F. Cytokine Networks in the Pathophysiology of Inflammatory Bowel Disease. Immunity 2019, 50, 992–1006. [Google Scholar] [CrossRef]
- Moschen, A.R.; Tilg, H.; Raine, T. IL-12, IL-23 and IL-17 in IBD: Immunobiology and therapeutic targeting. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 185–196. [Google Scholar] [CrossRef]
- Hohenberger, M.; Cardwell, L.A.; Oussedik, E.; Feldman, S.R. Interleukin-17 inhibition: Role in psoriasis and inflammatory bowel disease. J. Dermatol. Treat. 2018, 29, 13–18. [Google Scholar] [CrossRef] [PubMed]
- Lavelle, A.; Sokol, H. Gut microbiota-derived metabolites as key actors in inflammatory bowel disease. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 223–237. [Google Scholar] [CrossRef]
- Tomkovich, S.; Jobin, C. Microbiota and host immune responses: A love-hate relationship. Immunology 2016, 147, 1–10. [Google Scholar] [CrossRef]
- Fattorusso, A.; Di Genova, L.; Dell’Isola, G.B.; Mencaroni, E.; Esposito, S. Autism Spectrum Disorders and the Gut Microbiota. Nutrients 2019, 11, 521. [Google Scholar] [CrossRef]
- Federico, A.; Dallio, M.; Di Sarno, R.; Giorgio, V.; Miele, L. Gut microbiota, obesity and metabolic disorders. Minerva Gastroenterol. Dietol. 2017, 63, 337–344. [Google Scholar]
- Tang, W.H.; Kitai, T.; Hazen, S.L. Gut Microbiota in Cardiovascular Health and Disease. Circ. Res. 2017, 120, 1183–1196. [Google Scholar] [CrossRef] [PubMed]
- Matsuoka, K.; Kanai, T. The gut microbiota and inflammatory bowel disease. Semin. Immunopathol. 2015, 37, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Ni, J.; Wu, G.D.; Albenberg, L.; Tomov, V.T. Gut microbiota and IBD: Causation or correlation? Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 573–584. [Google Scholar] [CrossRef] [PubMed]
- Feller, M.; Huwiler, K.; Stephan, R.; Altpeter, E.; Shang, A.; Furrer, H.; Pfyffer, G.E.; Jemmi, T.; Baumgartner, A.; Egger, M. Mycobacterium avium subspecies paratuberculosis and Crohn’s disease: A systematic review and meta-analysis. Lancet Infect. Dis. 2007, 7, 607–613. [Google Scholar] [CrossRef]
- Ohkusa, T.; Okayasu, I.; Ogihara, T.; Morita, K.; Ogawa, M.; Sato, N. Induction of experimental ulcerative colitis by Fusobacterium varium isolated from colonic mucosa of patients with ulcerative colitis. Gut 2003, 52, 79–83. [Google Scholar] [CrossRef]
- Varela, E.; Manichanh, C.; Gallart, M.; Torrejón, A.; Borruel, N.; Casellas, F.; Guarner, F.; Antolin, M. Colonisation by Faecalibacterium prausnitzii and maintenance of clinical remission in patients with ulcerative colitis. Aliment. Pharmacol. Ther. 2013, 38, 151–161. [Google Scholar] [CrossRef]
- Ott, S.J.; Plamondon, S.; Hart, A.; Begun, A.; Rehman, A.; Kamm, M.A.; Schreiber, S. Dynamics of the mucosa-associated flora in ulcerative colitis patients during remission and clinical relapse. J. Clin. Microbiol. 2008, 46, 3510–3513. [Google Scholar] [CrossRef]
- Liguori, G.; Lamas, B.; Richard, M.L.; Brandi, G.; da Costa, G.; Hoffmann, T.W.; Di Simone, M.P.; Calabrese, C.; Poggioli, G.; Langella, P.; et al. Fungal Dysbiosis in Mucosa-associated Microbiota of Crohn’s Disease Patients. J. Crohn’s Colitis 2016, 10, 296–305. [Google Scholar] [CrossRef]
- Sokol, H.; Leducq, V.; Aschard, H.; Pham, H.P.; Jegou, S.; Landman, C.; Cohen, D.; Liguori, G.; Bourrier, A.; Nion-Larmurier, I.; et al. Fungal microbiota dysbiosis in IBD. Gut 2017, 66, 1039–1048. [Google Scholar] [CrossRef] [PubMed]
- Candelli, M.; Nista, E.C.; Nestola, M.; Armuzzi, A.; Silveri, N.G.; Gasbarrini, G.; Gasbarrini, A. Saccharomyces cerevisiae-associated diarrhea in an immunocompetent patient with ulcerative colitis. J. Clin. Gastroenterol. 2003, 36, 39–40. [Google Scholar] [CrossRef] [PubMed]
- Candelli, M.; Papa, A.; Nista, E.C.; Danese, S.; Armuzzi, A.; Bartolozzi, F.; Tondi, P.; Ojetti, V.; Gasbarrini, G.; Gasbarrini, A. Antibodies to Saccharomyces cerevisiae: Are they useful in clinical practice? Hepato-Gastroenterology 2003, 50, 718–720. [Google Scholar] [PubMed]
- Nahar, S.; Hokama, A.; Fujita, J. Clinical significance of cytomegalovirus and other herpes virus infections in ulcerative colitis. Pol. Arch. Intern. Med. 2019, 129, 620–626. [Google Scholar]
- Wang, W.; Jovel, J.; Halloran, B.; Wine, E.; Patterson, J.; Ford, G.; OʼKeefe, S.; Meng, B.; Song, D.; Zhang, Y.; et al. Metagenomic analysis of microbiome in colon tissue from subjects with inflammatory bowel diseases reveals interplay of viruses and bacteria. Inflamm. Bowel Dis. 2015, 21, 1419–1427. [Google Scholar] [CrossRef]
- Smits, L.P.; Bouter, K.E.; de Vos, W.M.; Borody, T.J.; Nieuwdorp, M. Therapeutic potential of fecal microbiota transplantation. Gastroenterology 2013, 145, 946–953. [Google Scholar] [CrossRef]
- Zocco, M.A.; dal Verme, L.Z.; Cremonini, F.; Piscaglia, A.C.; Nista, E.C.; Candelli, M.; Novi, M.; Rigante, D.; Cazzato, I.A.; Ojetti, V.; et al. Efficacy of Lactobacillus GG in maintaining remission of ulcerative colitis. Aliment. Pharmacol. Ther. 2006, 23, 1567–1574. [Google Scholar] [CrossRef]
- Liu, Y.; Yin, F.; Huang, L.; Teng, H.; Shen, T.; Qin, H. Long-term and continuous administration of Bacillus subtilis during remission effectively maintains the remission of inflammatory bowel disease by protecting intestinal integrity, regulating epithelial proliferation, and reshaping microbial structure and function. Food Funct. 2021, 12, 2201–2210. [Google Scholar] [PubMed]
- Hurley, J.C.; Guidet, B.; Offenstadt, G.; Maury, E. Endotoxemia and mortality prediction in ICU and other settings: Underlying risk and co-detection of gram negative bacteremia are confounders. Crit. Care 2012, 16, R148. [Google Scholar] [CrossRef]
- Napier, B.A.; Andres-Terre, M.; Massis, L.M.; Hryckowian, A.J.; Higginbottom, S.K.; Cumnock, K.; Casey, K.M.; Haileselassie, B.; Lugo, K.A.; Schneider, D.S.; et al. Western diet regulates immune status and the response to LPS-driven sepsis independent of diet-associated microbiome. Proc. Natl. Acad. Sci. USA 2019, 116, 3688–3694. [Google Scholar] [CrossRef]
- Suzuki, K. Chronic Inflammation as an Immunological Abnormality and Effectiveness of Exercise. Biomolecules 2019, 9, 223. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, X.; Luo, Z.; Ma, L.; Zhu, S.; Wang, Z.; Wen, J.; Cheng, S.; Gu, W.; Lian, Q.; et al. ECM1 is an essential factor for the determination of M1 macrophage polarization in IBD in response to LPS stimulation. Proc. Natl. Acad. Sci. USA 2020, 117, 3083–3092. [Google Scholar] [CrossRef] [PubMed]
- Bian, Y.; Dong, Y.; Sun, J.; Sun, M.; Hou, Q.; Lai, Y.; Zhang, B. Protective Effect of Kaempferol on LPS-Induced Inflammation and Barrier Dysfunction in a Coculture Model of Intestinal Epithelial Cells and Intestinal Microvascular Endothelial Cells. J. Agric. Food Chem. 2020, 68, 160–167. [Google Scholar] [CrossRef] [PubMed]
- Heinbockel, L.; Weindl, G.; Martinez-de-Tejada, G.; Correa, W.; Sanchez-Gomez, S.; Bárcena-Varela, S.; Goldmann, T.; Garidel, P.; Gutsmann, T.; Brandenburg, K. Inhibition of Lipopolysaccharide- and Lipoprotein-Induced Inflammation by Antitoxin Peptide Pep19-2.5. Front. Immunol. 2018, 9, 1704. [Google Scholar] [CrossRef] [PubMed]
- Jaffer, U.; Wade, R.G.; Gourlay, T. Cytokines in the systemic inflammatory response syndrome: A review. HSR Proc. Intensive Care Cardiovasc. Anesth. 2010, 2, 161–175. [Google Scholar]
- Vaure, C.; Liu, Y. A comparative review of toll-like receptor 4 expression and functionality in different animal species. Front. Immunol. 2014, 5, 316. [Google Scholar] [CrossRef]
- Rahhal, R.M.; Vanden Bush, T.J.; McLendon, M.K.; Apicella, M.A.; Bishop, G.A. Differential effects of Francisella tularensis lipopolysaccharide on B lymphocytes. J. Leukoc. Biol. 2007, 82, 813–820. [Google Scholar] [CrossRef]
- Vazquez-Torres, A.; Vallance, B.A.; Bergman, M.A.; Finlay, B.B.; Cookson, B.T.; Jones-Carson, J.; Fang, F.C. Toll-Like Receptor 4 Dependence of Innate and Adaptive Immunity to Salmonella: Importance of the Kupffer Cell Network. J. Immunol. 2004, 172, 6202–6208. [Google Scholar] [CrossRef]
- Fenini, G.; Contassot, E.; French, L.E. Potential of IL-1, IL-18 and Inflammasome Inhibition for the Treatment of Inflammatory Skin Diseases. Front. Pharmacol. 2017, 8, 278. [Google Scholar] [CrossRef]
- Ding, F.; Fu, Z.; Liu, B. Lipopolysaccharide Exposure Alleviates Asthma in Mice by Regulating Th1/Th2 and Treg/Th17 Balance. Med. Sci. Monit. Int. Med. J. Exp. Clin. Res. 2018, 24, 3220–3229. [Google Scholar] [CrossRef]
- Straub, T.; Freudenberg, M.A.; Schleicher, U.; Bogdan, C.; Gasteiger, G.; Pircher, H. Bacterial coinfection restrains antiviral CD8 T-cell response via LPS-induced inhibitory NK cells. Nat. Commun. 2018, 9, 4117. [Google Scholar] [CrossRef] [PubMed]
- Bannerman, D.D.; Goldblum, S.E. Mechanisms of bacterial lipopolysaccharide-induced endothelial apoptosis. Am. J. Physiol. Lung Cell. Mol. Physiol. 2003, 284, L899–L914. [Google Scholar] [CrossRef] [PubMed]
- Vatanen, T.; Kostic, A.D.; d’Hennezel, E.; Siljander, H.; Franzosa, E.A.; Yassour, M.; Kolde, R.; Vlamakis, H.; Arthur, T.D.; Hämäläinen, A.-M.; et al. Variation in Microbiome LPS Immunogenicity Contributes to Autoimmunity in Humans. Cell 2016, 165, 842–853. [Google Scholar] [CrossRef] [PubMed]
- Orivuori, L.; Mustonen, K.; de Goffau, M.C.; Hakala, S.; Paasela, M.; Roduit, C.; Dalphin, J.C.; Genuneit, J.; Lauener, R.; Riedler, J.; et al. High level of fecal calprotectin at age 2 months as a marker of intestinal inflammation predicts atopic dermatitis and asthma by age 6. Clin. Exp. Allergy J. Br. Soc. Allergy Clin. Immunol. 2015, 45, 928–939. [Google Scholar] [CrossRef] [PubMed]
- Fuke, N.; Nagata, N.; Suganuma, H.; Ota, T. Regulation of Gut Microbiota and Metabolic Endotoxemia with Dietary Factors. Nutrients 2019, 11, 2277. [Google Scholar] [CrossRef] [PubMed]
- Wassenaar, T.M.; Zimmermann, K. Lipopolysaccharides in Food, Food Supplements, and Probiotics: Should We be Worried? Eur. J. Microbiol. Immunol. 2018, 8, 63–69. [Google Scholar] [CrossRef]
- Ahola, A.J.; Lassenius, M.I.; Forsblom, C.; Harjutsalo, V.; Lehto, M.; Groop, P.H. Dietary patterns reflecting healthy food choices are associated with lower serum LPS activity. Sci. Rep. 2017, 7, 6511. [Google Scholar] [CrossRef]
- Lindenberg, F.C.B.; Ellekilde, M.; Thörn, A.C.; Kihl, P.; Larsen, C.S.; Hansen, C.H.F.; Metzdorff, S.B.; Aalbæk, B.; Hansen, A.K. Dietary LPS traces influences disease expression of the diet-induced obese mouse. Res. Vet. Sci. 2019, 123, 195–203. [Google Scholar] [CrossRef]
- Netto Candido, T.L.; Bressan, J.; Alfenas, R.C.G. Dysbiosis and metabolic endotoxemia induced by high-fat diet. Nutr. Hosp. 2018, 35, 1432–1440. [Google Scholar] [CrossRef]
- Wang, J.; Chen, W.-D.; Wang, Y.-D. The Relationship Between Gut Microbiota and Inflammatory Diseases: The Role of Macrophages. Front. Microbiol. 2020, 11, 1065. [Google Scholar] [CrossRef]
- Franza, L.; Carusi, V.; Altamura, S.; Caraffa, A.; Gallenga, C.E.; Kritas, S.K.; Ronconi, G.; Conti, P.; Pandolfi, F. Interrelationship between inflammatory cytokines (IL-1, IL-6, IL-33, IL-37) and acquired immunity. J. Biol. Regul. Homeost. Agents 2019, 33, 1321–1326. [Google Scholar]
- Pandolfi, F.; Franza, L.; Carusi, V.; Altamura, S.; Andriollo, G.; Nucera, E. Interleukin-6 in Rheumatoid Arthritis. Int. J. Mol. Sci. 2020, 21, 5238. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.; Xu, X.-H.; Jin, L. Macrophage Polarization in Physiological and Pathological Pregnancy. Front. Immunol. 2019, 10, 792. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.W.; Pan, Y.B.; Cao, Y.Q.; Wang, C.; Jiang, W.D.; Zhai, W.F.; Lu, J.G. Loganin alleviates LPS-activated intestinal epithelial inflammation by regulating TLR4/NF-κB and JAK/STAT3 signaling pathways. Kaohsiung J. Med. Sci. 2020, 36, 257–264. [Google Scholar] [CrossRef]
- Larabi, A.; Barnich, N.; Nguyen, H.T.T. New insights into the interplay between autophagy, gut microbiota and inflammatory responses in IBD. Autophagy 2020, 16, 38–51. [Google Scholar] [CrossRef]
- Førland, D.T.; Johnson, E.; Saetre, L.; Lyberg, T.; Lygren, I.; Hetland, G. Effect of an extract based on the medicinal mushroom Agaricus blazei Murill on expression of cytokines and calprotectin in patients with ulcerative colitis and Crohn’s disease. Scand. J. Immunol. 2011, 73, 66–75. [Google Scholar] [CrossRef]
- D’Amico, F.; Rubin, D.T.; Kotze, P.G.; Magro, F. International consensus on methodological issues in standardization of fecal calprotectin measurement in inflammatory bowel diseases. UEG J. 2021, 9, 451–460. [Google Scholar]
- Jaworska, K.; Konop, M.; Bielinska, K.; Hutsch, T.; Dziekiewicz, M.; Banaszkiewicz, A.; Ufnal, M. Inflammatory bowel disease is associated with increased gut-to-blood penetration of short-chain fatty acids: A new, non-invasive marker of a functional intestinal lesion. Exp. Physiol. 2019, 104, 1226–1236. [Google Scholar] [CrossRef]
- Kiecolt-Glaser, J.K.; Wilson, S.J.; Bailey, M.L.; Andridge, R.; Peng, J.; Jaremka, L.M.; Fagundes, C.P.; Malarkey, W.B.; Laskowski, B.; Belury, M.A. Marital distress, depression, and a leaky gut: Translocation of bacterial endotoxin as a pathway to inflammation. Psychoneuroendocrinology 2018, 98, 52–60. [Google Scholar] [CrossRef] [PubMed]
- Dickson, K.; Lehmann, C. Inflammatory Response to Different Toxins in Experimental Sepsis Models. Int. J. Mol. Sci. 2019, 20, 4341. [Google Scholar] [CrossRef]
- Wu, X.X.; Huang, X.L.; Chen, R.R.; Li, T.; Ye, H.J.; Xie, W.; Huang, Z.M.; Cao, G.Z. Paeoniflorin Prevents Intestinal Barrier Disruption and Inhibits Lipopolysaccharide (LPS)-Induced Inflammation in Caco-2 Cell Monolayers. Inflammation 2019, 42, 2215–2225. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Wan, G.; Han, B.; Zhang, Z. Echinacoside alleviated LPS-induced cell apoptosis and inflammation in rat intestine epithelial cells by inhibiting the mTOR/STAT3 pathway. Biomed. Pharmacother. 2018, 104, 622–628. [Google Scholar] [CrossRef] [PubMed]


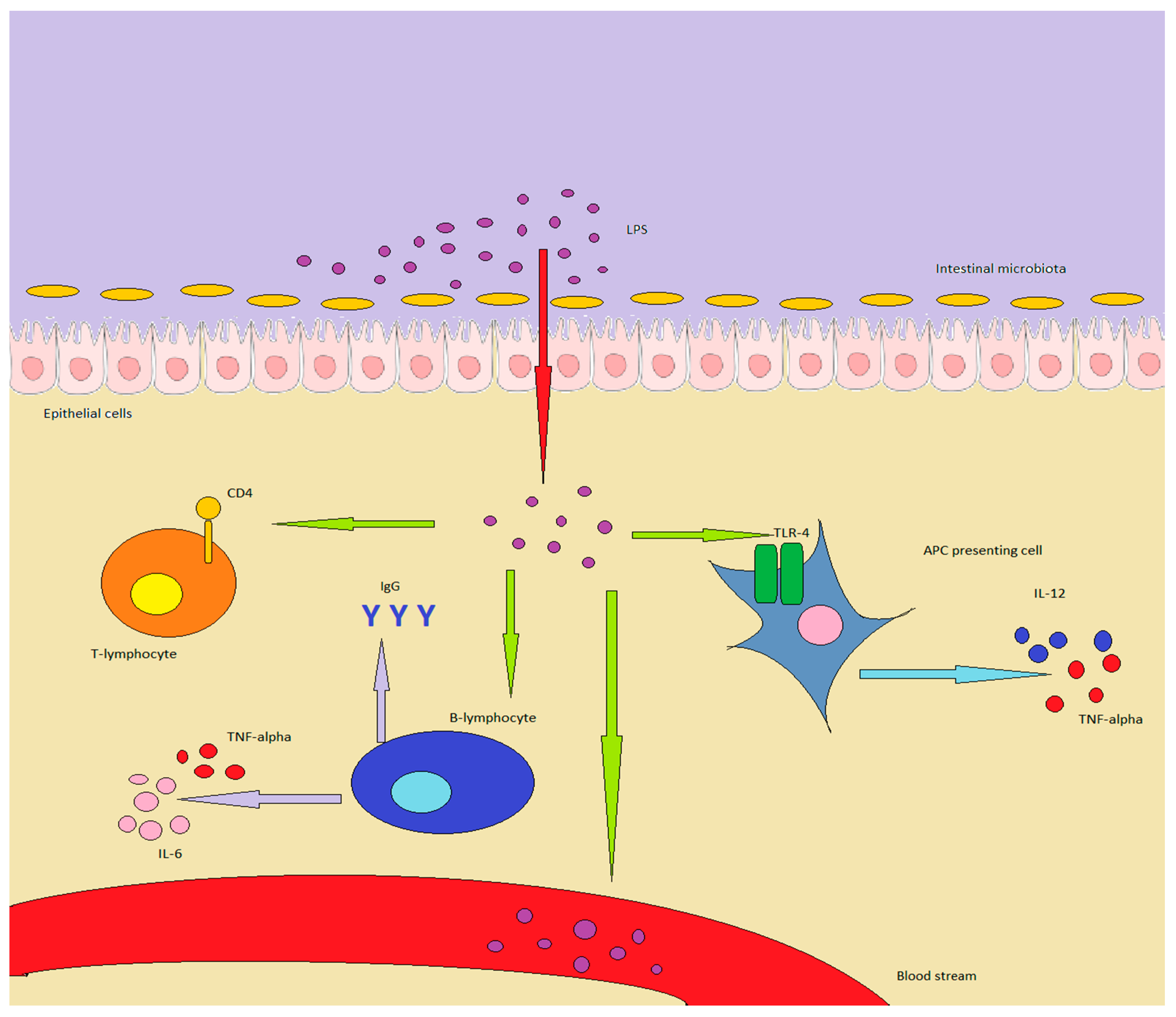{kind=link}
| Microbiota | Effect | Reference |
|---|---|---|
| B. fragilis enterotoxigenic variant | T-reg stimulation; Th17 stimulation. | [30] |
| M. avium subspecies paratuberculosis | Linked to the development of Crohn’s disease. | [36] |
| F. varium | Associated to IBD development. | [37] |
| F. prausnitzii | A reduced number is associated with ulcerative colitis. | [38] |
| C. albicans | Associated with the development of Crohn’s disease. | [40] |
| A. clavatus | Associated with the development of Crohn’s disease. | [40] |
| S. cerevisiae | Reduced in patients who develop IBD; antibodies present in patients with Crohn’s disease. | [40,41,42] |
| Lactobacillus GG | Maintains remission in ulcerative colitis. | [47] |
| B. subtilis | Regulates epithelial integrity, possibly preventing relapses. | [48] |
| E. coli | Increased in patients with the disease; decreases before relapse. | [34,39] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Candelli, M.; Franza, L.; Pignataro, G.; Ojetti, V.; Covino, M.; Piccioni, A.; Gasbarrini, A.; Franceschi, F. Interaction between Lipopolysaccharide and Gut Microbiota in Inflammatory Bowel Diseases. Int. J. Mol. Sci. 2021, 22, 6242. https://doi.org/10.3390/ijms22126242
Candelli M, Franza L, Pignataro G, Ojetti V, Covino M, Piccioni A, Gasbarrini A, Franceschi F. Interaction between Lipopolysaccharide and Gut Microbiota in Inflammatory Bowel Diseases. International Journal of Molecular Sciences. 2021; 22(12):6242. https://doi.org/10.3390/ijms22126242
Chicago/Turabian StyleCandelli, Marcello, Laura Franza, Giulia Pignataro, Veronica Ojetti, Marcello Covino, Andrea Piccioni, Antonio Gasbarrini, and Francesco Franceschi. 2021. "Interaction between Lipopolysaccharide and Gut Microbiota in Inflammatory Bowel Diseases" International Journal of Molecular Sciences 22, no. 12: 6242. https://doi.org/10.3390/ijms22126242
APA StyleCandelli, M., Franza, L., Pignataro, G., Ojetti, V., Covino, M., Piccioni, A., Gasbarrini, A., & Franceschi, F. (2021). Interaction between Lipopolysaccharide and Gut Microbiota in Inflammatory Bowel Diseases. International Journal of Molecular Sciences, 22(12), 6242. https://doi.org/10.3390/ijms22126242