
T-Type Ca2+ Enhancer SAK3 Activates CaMKII and Proteasome Activities in Lewy Body Dementia Mice Model



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
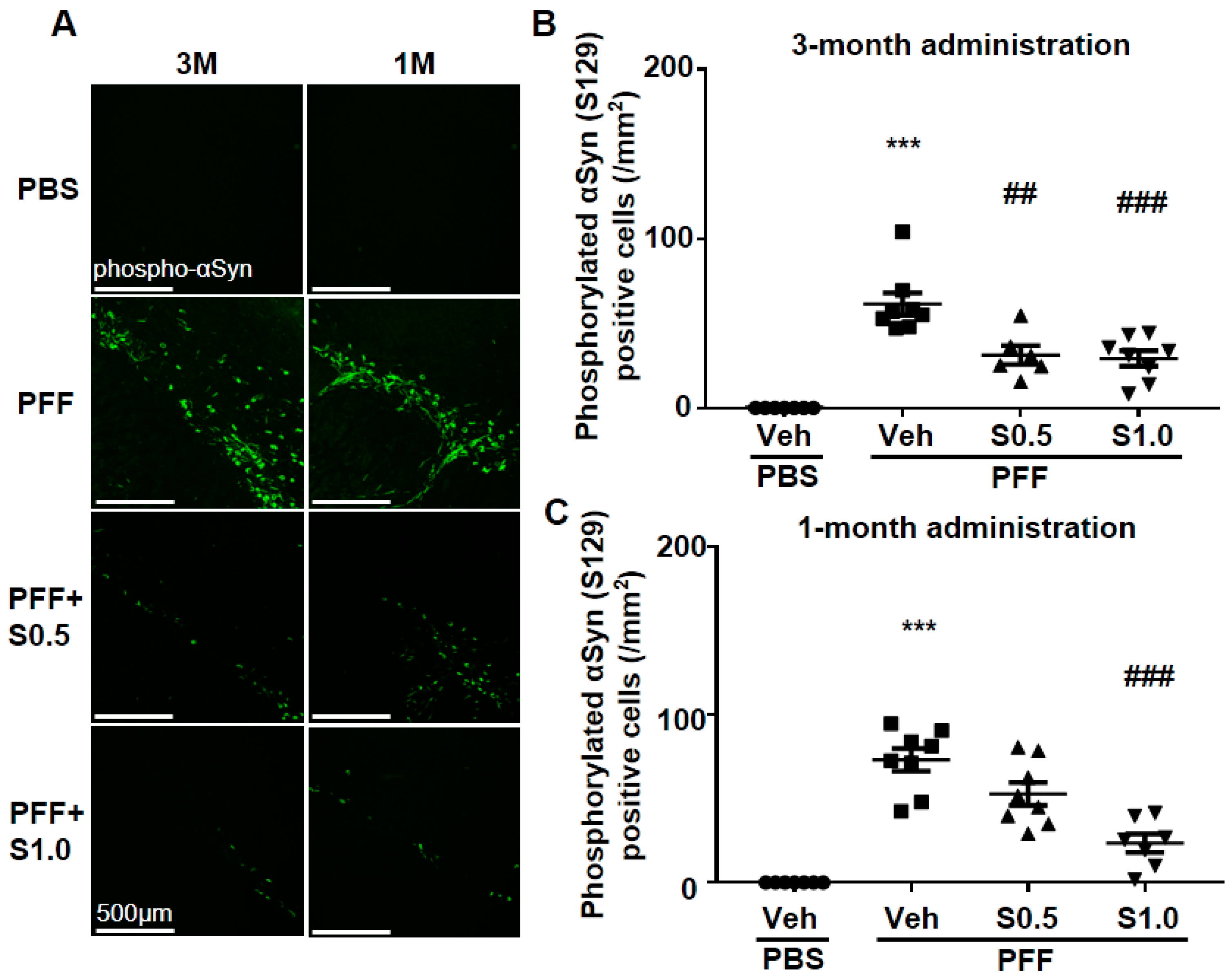
2.1. SAK3 Prevented the Development of Phosphorylated α-Syn (Ser129) of SNc in PF-Injected Mice
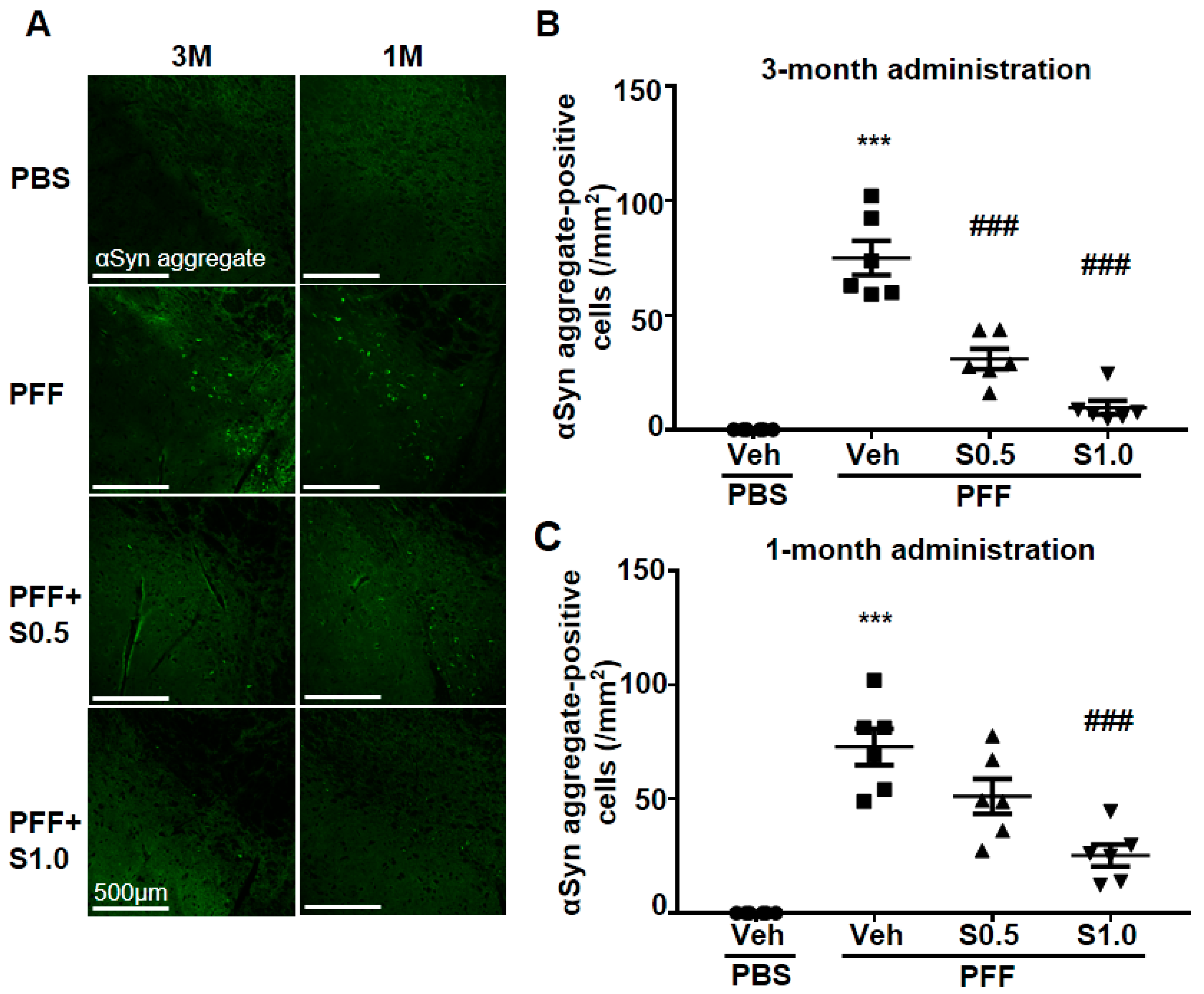
2.2. SAK3 Attenuated Neuronal Death and α-Syn Aggregation of Dopaminergic Neurons in PFF-Injected Mice
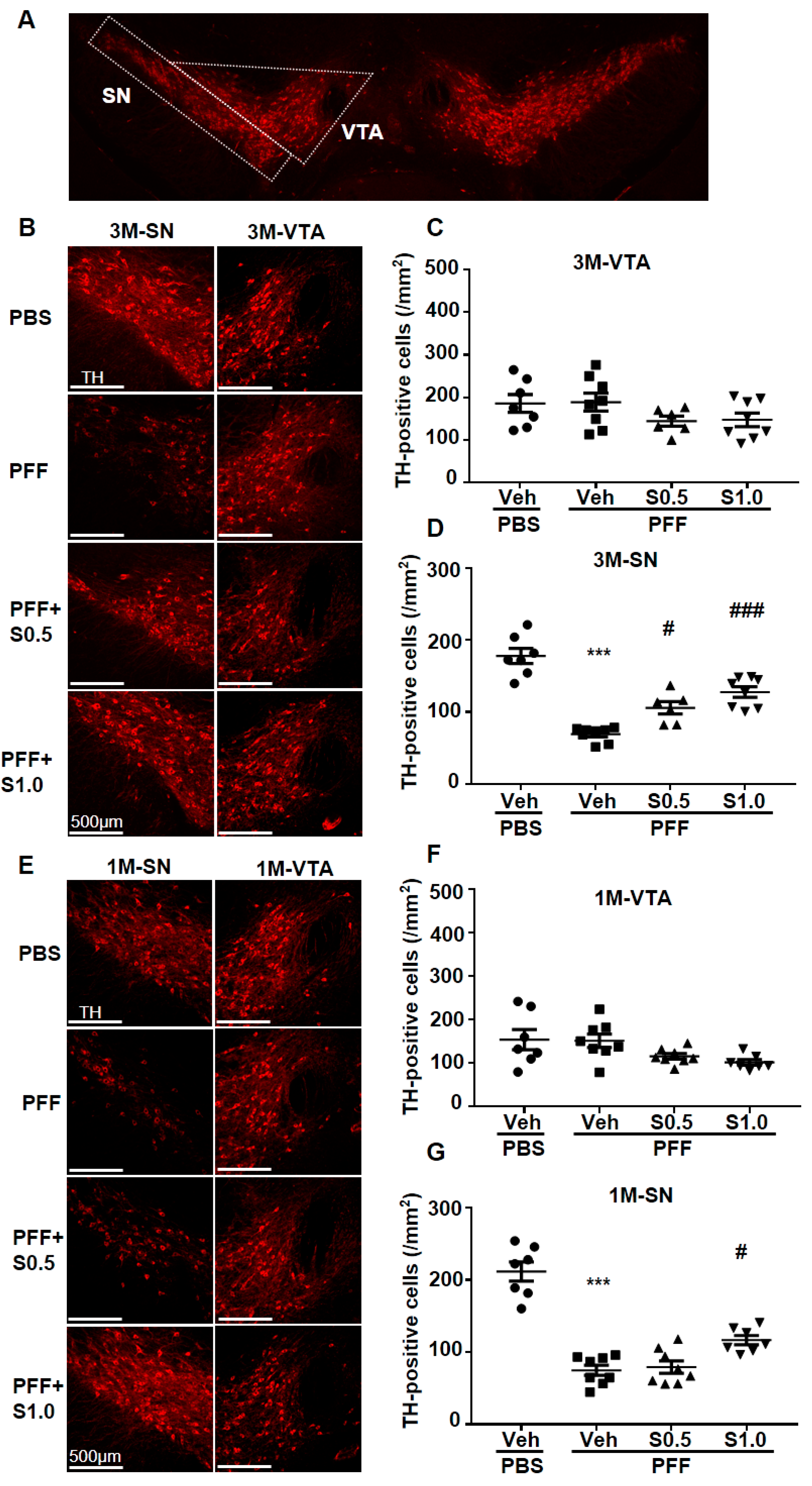
2.3. SAK3 Prevented the Dopaminergic Neuronal Death of SNc in PFF-Injected Mice
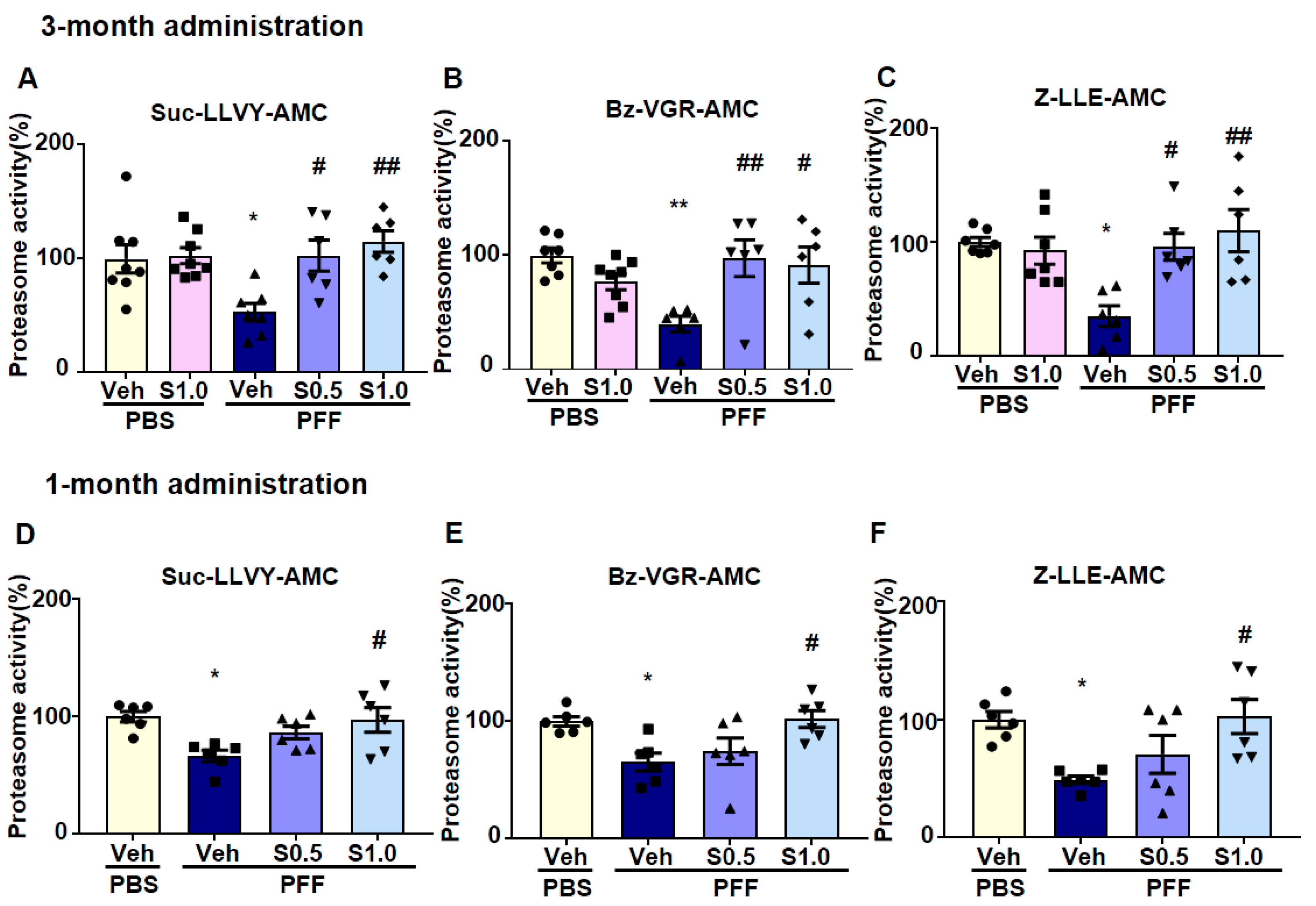
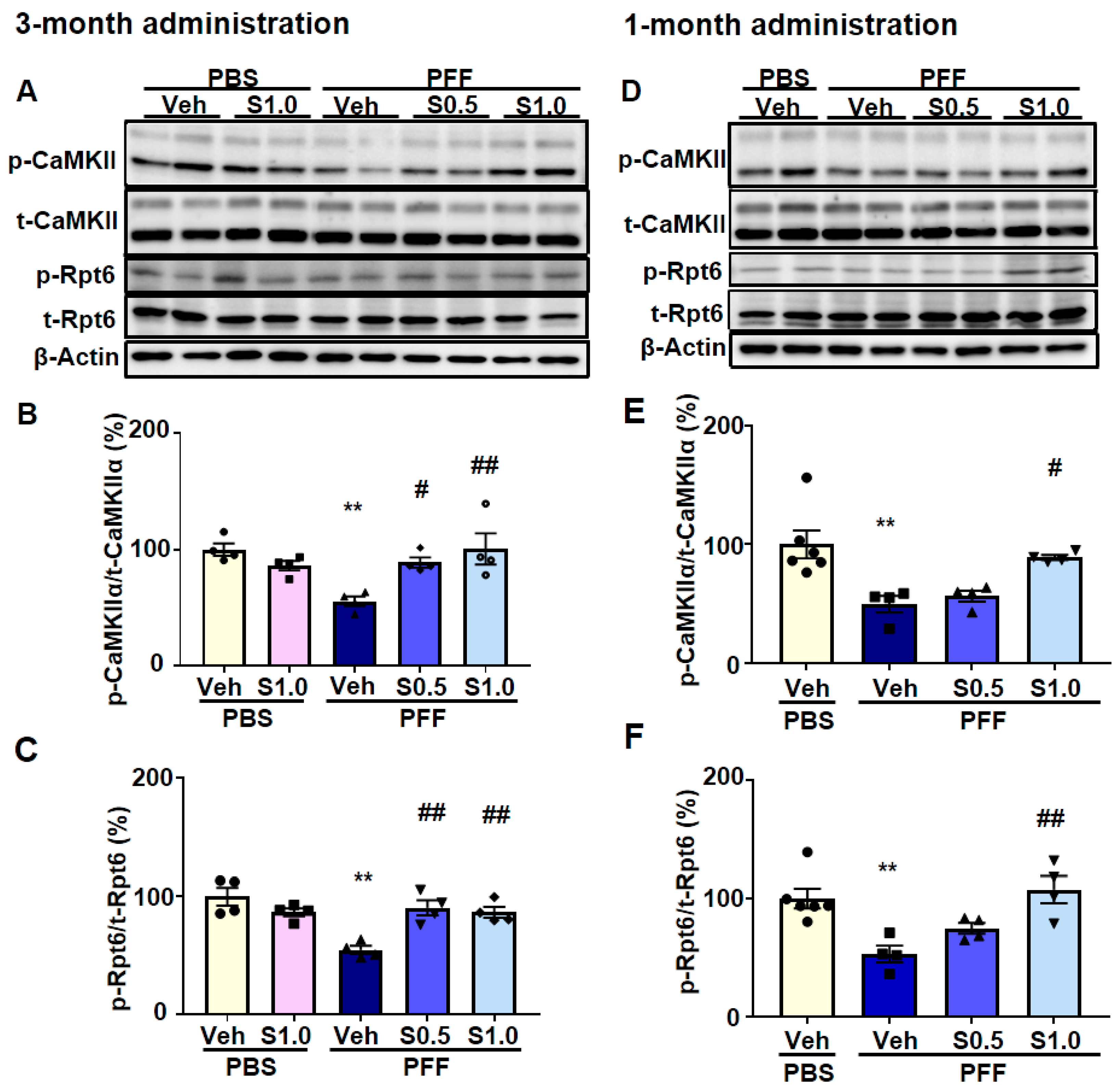
2.4. SAK3 Improved the Reduction of Proteasome Activity through the CaMKII-Rpt6 Signaling Pathway in PFF-Injected Mice
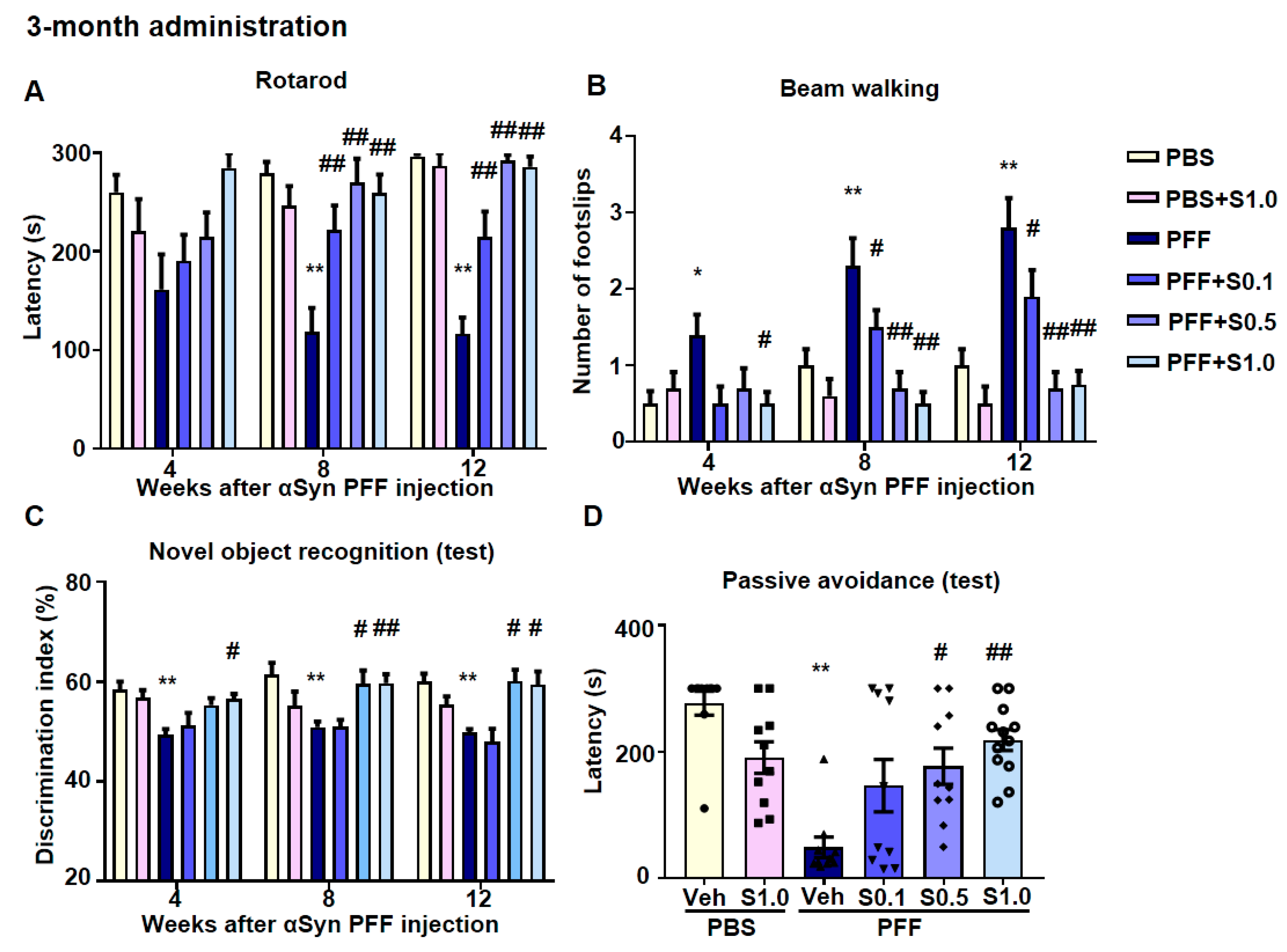
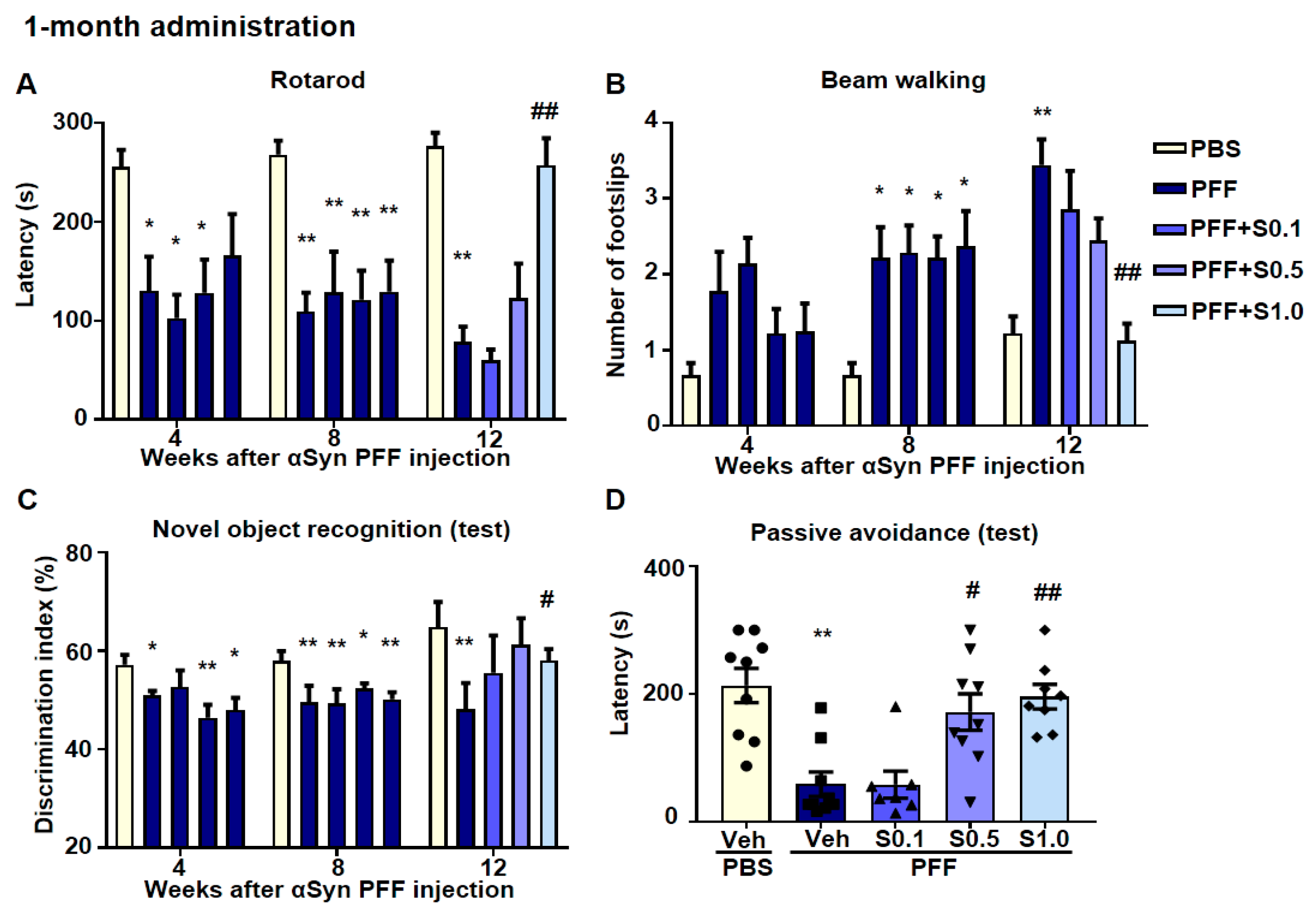
2.5. SAK3 Ameliorated Motor and Cognitive Impairments in PFF-Injected Mice
3. Discussion
4. Materials and Methods
4.1. Animals and Murine Model
4.2. Preformed Fibrillization of α-Syn
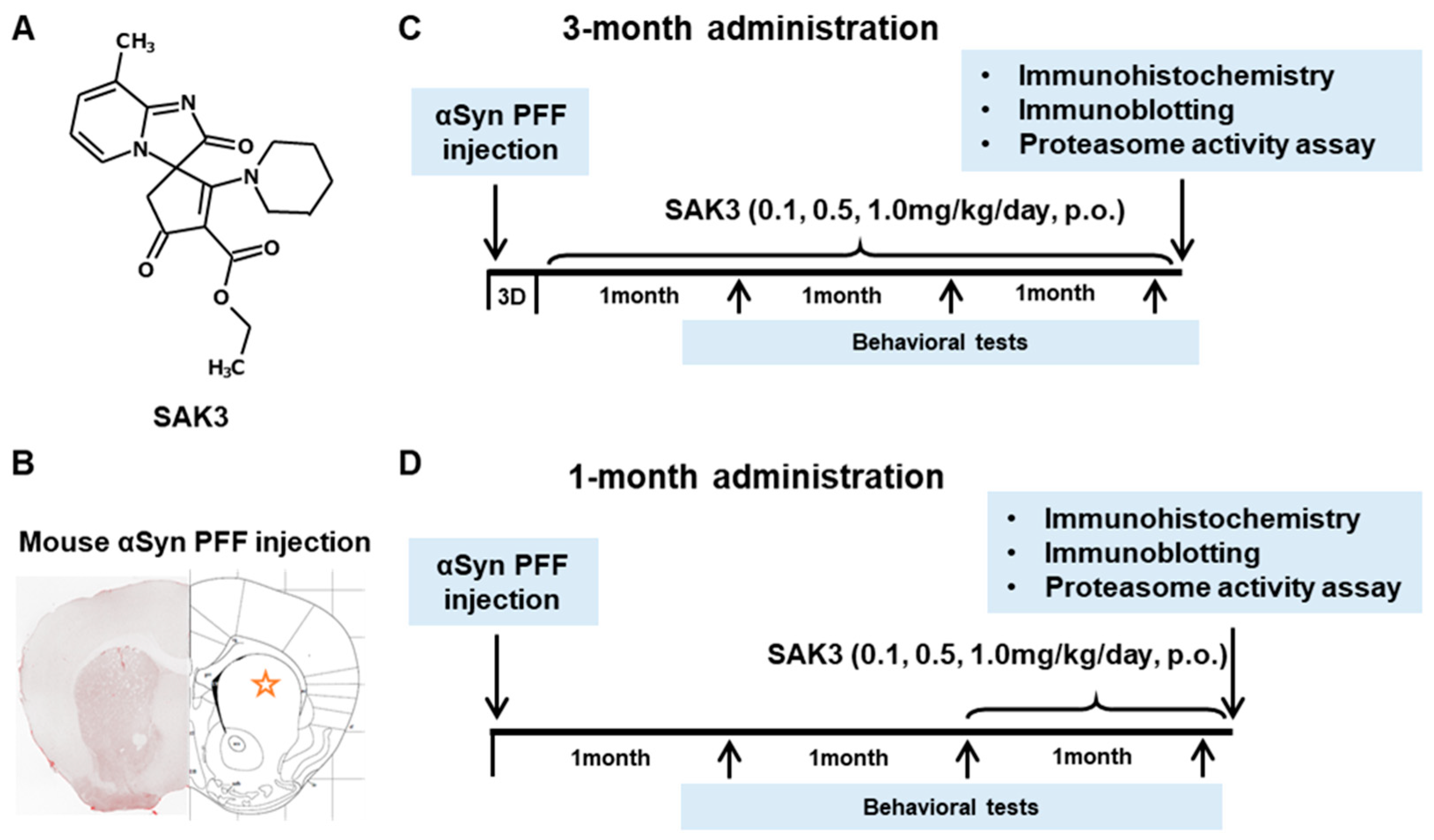
4.3. Drug Administration and Experimental Design
4.4. Behavioral Analyses
4.4.1. Rotarod Task
4.4.2. Beam-Walking Task
4.4.3. Y-Maze Task
4.4.4. Novel Object Recognition Task
4.4.5. Step-through Passive Avoidance Task
4.5. Immunohistochemistry
4.6. Western Blotting Analysis
4.7. Proteasome Activity Assay
4.8. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| α-Syn | alpha-synuclein |
| Aβ | amyloid-beta |
| AD | Alzheimer’s disease |
| APP-KI | amyloid precursor protein-knock-in mice |
| CaMKII | Ca2+/calmodulin-dependent protein kinase II |
| Cav3.1 | CACNA1G |
| Cav3.2 | CACNA1H |
| Cav3.3 | CACNA1I |
| LBD | Lewy body dementia |
| LBs | Lewy bodies |
| NK-G-F | App NL-F/NL-F mice |
| PD | Parkinson’s disease |
| PFF | α-Syn preformed fibril |
| SAK3 | ethyl-8′-methyl-2′,4-dioxo-2-(piperidin-1-yl)-2′H-spiro[cyclopentane-1,3′-imidazo[1,2-a]pyridin]-2-ene-3-carboxylate |
| SN | substantia nigra |
| SNc | substantia nigra pars compacta |
| T-VGCC | T-type voltage-gated calcium channels |
| UPS | ubiquitin–proteasome system |
| VTA | ventral tegmental area |
References
- Pillon, B.; Dubois, B.; Cusimano, G.; Bonnet, A.M.; Lhermitte, F.; Agid, Y. Does cognitive impairment in Parkinson’s disease result from non-dopaminergic lesions? J. Neurol. Neurosurg Psychiatry 1989, 52, 201–206. [Google Scholar] [CrossRef]
- Walker, Z.; Possin, K.L.; Boeve, B.F.; Aarsland, D. Lewy body dementias. Lancet 2015, 386, 1683–1697. [Google Scholar] [CrossRef]
- Lippa, C.F.; Duda, J.E.; Grossman, M.; Hurtig, H.I.; Aarsland, D.; Boeve, B.F.; Brooks, D.J.; Dickson, D.W.; Dubois, B.; Emre, M.; et al. DLB and PDD boundary issues: Diagnosis, treatment, molecular pathology, and biomarkers. Neurology 2007, 68, 812–819. [Google Scholar] [CrossRef] [PubMed]
- Irwin, D.J.; Lee, V.M.; Trojanowski, J.Q. Parkinson’s disease dementia: Convergence of α-synuclein, tau and amyloid-β pathologies. Nat. Rev. Neurosci. 2013, 14, 626–636. [Google Scholar] [CrossRef]
- Giehm, L.; Svergun, D.I.; Otzen, D.E.; Vestergaard, B. Low-resolution structure of a vesicle disrupting α-synuclein oligomer that accumulates during fibrillation. Proc. Natl. Acad. Sci. USA 2011, 108, 3246–3251. [Google Scholar] [CrossRef] [PubMed]
- Colla, E.; Jensen, P.H.; Pletnikova, O.; Troncoso, J.C.; Glabe, C.; Lee, M.K. Accumulation of toxic α-synuclein oligomer within endoplasmic reticulum occurs in α-synucleinopathy in vivo. J. Neurosci. 2012, 32, 3301–3305. [Google Scholar] [CrossRef] [PubMed]
- Brundin, P.; Melki, R. Prying into the Prion Hypothesis for Parkinson’s Disease. J. Neurosci. 2017, 37, 9808–9818. [Google Scholar] [CrossRef]
- Luk, K.C.; Kehm, V.; Carroll, J.; Zhang, B.; O’Brien, P.; Trojanowski, J.Q.; Lee, V.M. Pathological α-synuclein transmission initiates Parkinson-like neurodegeneration in nontransgenic mice. Science 2012, 338, 949–953. [Google Scholar] [CrossRef]
- Schampel, A.; Kuerten, S. Danger: High Voltage-The Role of Voltage-Gated Calcium Channels in Central Nervous System Pathology. Cells 2017, 6, 43. [Google Scholar] [CrossRef]
- Jones, E.G. Calcium channels in higher-level brain function. Proc. Natl. Acad. Sci. USA 2007, 104, 17903–17904. [Google Scholar] [CrossRef]
- Blesneac, I.; Chemin, J.; Bidaud, I.; Huc-Brandt, S.; Vandermoere, F.; Lory, P. Phosphorylation of the Cav3.2 T-type calcium channel directly regulates its gating properties. Proc. Natl. Acad. Sci. USA 2015, 112, 13705–13710. [Google Scholar] [CrossRef] [PubMed]
- Powell, K.L.; Cain, S.M.; Snutch, T.P.; O’Brien, T.J. Low threshold T-type calcium channels as targets for novel epilepsy treatments. Br. J. Clin. Pharmacol. 2014, 77, 729–739. [Google Scholar] [CrossRef]
- Perez-Reyes, E.; Cribbs, L.L.; Daud, A.; Lacerda, A.E.; Barclay, J.; Williamson, M.P.; Fox, M.; Rees, M.; Lee, J.H. Molecular characterization of a neuronal low-voltage-activated T-type calcium channel. Nature 1998, 391, 896–900. [Google Scholar] [CrossRef]
- Talley, E.M.; Cribbs, L.L.; Lee, J.H.; Daud, A.; Perez-Reyes, E.; Bayliss, D.A. Differential distribution of three members of a gene family encoding low voltage-activated (T-type) calcium channels. J. Neurosci. 1999, 19, 1895–1911. [Google Scholar] [CrossRef] [PubMed]
- McCormick, D.A.; Bal, T. Sleep and arousal: Thalamocortical mechanisms. Annu. Rev. Neurosci. 1997, 20, 185–215. [Google Scholar] [CrossRef]
- Lambert, R.C.; Bessaïh, T.; Crunelli, V.; Leresche, N. The many faces of T-type calcium channels. Pflügers Arch. Eur. J. Physiol. 2014, 466, 415–423. [Google Scholar] [CrossRef] [PubMed]
- Nelson, M.T.; Todorovic, S.M.; Perez-Reyes, E. The role of T-type calcium channels in epilepsy and pain. Curr. Pharm. Des. 2006, 12, 2189–2197. [Google Scholar] [CrossRef] [PubMed]
- Yabuki, Y.; Matsuo, K.; Izumi, H.; Haga, H.; Yoshida, T.; Wakamori, M.; Kakei, A.; Sakimura, K.; Fukuda, T.; Fukunaga, K. Pharmacological properties of SAK3, a novel T-type voltage-gated Ca(2+) channel enhancer. Neuropharmacology 2017, 117, 1–13. [Google Scholar] [CrossRef]
- Xu, J.; Yabuki, Y.; Yu, M.; Fukunaga, K. T-type calcium channel enhancer SAK3 produces anti-depressant-like effects by promoting adult hippocampal neurogenesis in olfactory bulbectomized mice. J. Pharmacol. Sci. 2018, 137, 333–341. [Google Scholar] [CrossRef] [PubMed]
- Izumi, H.; Kawahata, I.; Shinoda, Y.; Helmstetter, F.J.; Fukunaga, K. SAK3 Administration Improves Spine Abnormalities and Cognitive Deficits in App(NL-G-F/NL-G-F) Knock-in Mice by Increasing Proteasome Activity through CaMKII/Rpt6 Signaling. Int. J. Mol. Sci. 2020, 21, 3833. [Google Scholar] [CrossRef]
- Palmqvist, S.; Schöll, M.; Strandberg, O.; Mattsson, N.; Stomrud, E.; Zetterberg, H.; Blennow, K.; Landau, S.; Jagust, W.; Hansson, O. Earliest accumulation of β-amyloid occurs within the default-mode network and concurrently affects brain connectivity. Nat. Commun. 2017, 8, 1214. [Google Scholar] [CrossRef]
- Bentea, E.; Verbruggen, L.; Massie, A. The Proteasome Inhibition Model of Parkinson’s Disease. J. Parkinsons Dis. 2017, 7, 31–63. [Google Scholar] [CrossRef]
- De Vrij, F.M.; Sluijs, J.A.; Gregori, L.; Fischer, D.F.; Hermens, W.T.; Goldgaber, D.; Verhaagen, J.; Van Leeuwen, F.W.; Hol, E.M. Mutant ubiquitin expressed in Alzheimer’s disease causes neuronal death. FASEB J. 2001, 15, 2680–2688. [Google Scholar] [CrossRef]
- Hope, A.D.; de Silva, R.; Fischer, D.F.; Hol, E.M.; van Leeuwen, F.W.; Lees, A.J. Alzheimer’s associated variant ubiquitin causes inhibition of the 26S proteasome and chaperone expression. J. Neurochem. 2003, 86, 394–404. [Google Scholar] [CrossRef] [PubMed]
- McNaught, K.S.; Jenner, P. Proteasomal function is impaired in substantia nigra in Parkinson’s disease. Neurosci. Lett. 2001, 297, 191–194. [Google Scholar] [CrossRef]
- Kawahata, I.; Tokuoka, H.; Parvez, H.; Ichinose, H. Accumulation of phosphorylated tyrosine hydroxylase into insoluble protein aggregates by inhibition of an ubiquitin-proteasome system in PC12D cells. J. Neural Transm. 2009, 116, 1571–1578. [Google Scholar] [CrossRef] [PubMed]
- Chu, Y.; Dodiya, H.; Aebischer, P.; Olanow, C.W.; Kordower, J.H. Alterations in lysosomal and proteasomal markers in Parkinson’s disease: Relationship to alpha-synuclein inclusions. Neurobiol. Dis. 2009, 35, 385–398. [Google Scholar] [CrossRef]
- Glickman, M.H.; Ciechanover, A. The ubiquitin-proteasome proteolytic pathway: Destruction for the sake of construction. Physiol. Rev. 2002, 82, 373–428. [Google Scholar] [CrossRef]
- Thibaudeau, T.A.; Anderson, R.T.; Smith, D.M. A common mechanism of proteasome impairment by neurodegenerative disease-associated oligomers. Nat. Commun. 2018, 9, 1097. [Google Scholar] [CrossRef]
- Pickart, C.M.; Cohen, R.E. Proteasomes and their kin: Proteases in the machine age. Nat. Rev. Mol. Cell Biol. 2004, 5, 177–187. [Google Scholar] [CrossRef]
- Snyder, H.; Mensah, K.; Theisler, C.; Lee, J.; Matouschek, A.; Wolozin, B. Aggregated and monomeric alpha-synuclein bind to the S6′ proteasomal protein and inhibit proteasomal function. J. Biol. Chem. 2003, 278, 11753–11759. [Google Scholar] [CrossRef]
- Ebrahimi-Fakhari, D.; Cantuti-Castelvetri, I.; Fan, Z.; Rockenstein, E.; Masliah, E.; Hyman, B.T.; McLean, P.J.; Unni, V.K. Distinct roles in vivo for the ubiquitin-proteasome system and the autophagy-lysosomal pathway in the degradation of α-synuclein. J. Neurosci. 2011, 31, 14508–14520. [Google Scholar] [CrossRef] [PubMed]
- Manecka, D.L.; Vanderperre, B.; Fon, E.A.; Durcan, T.M. The Neuroprotective Role of Protein Quality Control in Halting the Development of Alpha-Synuclein Pathology. Front. Mol. Neurosci. 2017, 10, 311. [Google Scholar] [CrossRef]
- McNaught, K.S.; Perl, D.P.; Brownell, A.L.; Olanow, C.W. Systemic exposure to proteasome inhibitors causes a progressive model of Parkinson’s disease. Ann. Neurol. 2004, 56, 149–162. [Google Scholar] [CrossRef] [PubMed]
- Alghamdi, A.; Vallortigara, J.; Howlett, D.R.; Broadstock, M.; Hortobágyi, T.; Ballard, C.G.; Thomas, A.J.; O’Brien, J.; Aarsland, D.; Attems, J.; et al. Reduction of RPT6/S8 (a Proteasome Component) and Proteasome Activity in the Cortex is Associated with Cognitive Impairment in Lewy Body Dementia. J. Alzheimers Dis. 2017, 57, 373–386. [Google Scholar] [CrossRef]
- Dulka, B.N.; Pullins, S.E.; Cullen, P.K.; Moyer, J.R., Jr.; Helmstetter, F.J. Age-related memory deficits are associated with changes in protein degradation in brain regions critical for trace fear conditioning. Neurobiol. Aging 2020, 91, 160–166. [Google Scholar] [CrossRef] [PubMed]
- Giannini, C.; Kloß, A.; Gohlke, S.; Mishto, M.; Nicholson, T.P.; Sheppard, P.W.; Kloetzel, P.M.; Dahlmann, B. Poly-Ub-substrate-degradative activity of 26S proteasome is not impaired in the aging rat brain. PLoS ONE 2013, 8, e64042. [Google Scholar] [CrossRef]
- Jarome, T.J.; Kwapis, J.L.; Ruenzel, W.L.; Helmstetter, F.J. CaMKII, but not protein kinase A, regulates Rpt6 phosphorylation and proteasome activity during the formation of long-term memories. Front. Behav. Neurosci. 2013, 7, 115. [Google Scholar] [CrossRef]
- Hamilton, A.M.; Oh, W.C.; Vega-Ramirez, H.; Stein, I.S.; Hell, J.W.; Patrick, G.N.; Zito, K. Activity-dependent growth of new dendritic spines is regulated by the proteasome. Neuron 2012, 74, 1023–1030. [Google Scholar] [CrossRef]
- Paumier, K.L.; Luk, K.C.; Manfredsson, F.P.; Kanaan, N.M.; Lipton, J.W.; Collier, T.J.; Steece-Collier, K.; Kemp, C.J.; Celano, S.; Schulz, E.; et al. Intrastriatal injection of preformed mouse alpha-synuclein fibrils into rats triggers alpha-synuclein pathology and bilateral nigrostriatal degeneration. Neurobiol. Dis. 2015, 82, 185–199. [Google Scholar] [CrossRef]
- Kawahata, I.; Fukunaga, K. Degradation of Tyrosine Hydroxylase by the Ubiquitin-Proteasome System in the Pathogenesis of Parkinson’s Disease and Dopa-Responsive Dystonia. Int. J. Mol. Sci. 2020, 21, 3779. [Google Scholar] [CrossRef]
- De Mattos, E.P.; Wentink, A.; Nussbaum-Krammer, C.; Hansen, C.; Bergink, S.; Melki, R.; Kampinga, H.H. Protein Quality Control Pathways at the Crossroad of Synucleinopathies. J. Parkinsons Dis. 2020, 10, 369–382. [Google Scholar] [CrossRef]
- Hsu, L.J.; Sagara, Y.; Arroyo, A.; Rockenstein, E.; Sisk, A.; Mallory, M.; Wong, J.; Takenouchi, T.; Hashimoto, M.; Masliah, E. Alpha-synuclein promotes mitochondrial deficit and oxidative stress. Am. J. Pathol. 2000, 157, 401–410. [Google Scholar] [CrossRef]
- Hashimoto, M.; Kawahara, K.; Bar-On, P.; Rockenstein, E.; Crews, L.; Masliah, E. The Role of alpha-synuclein assembly and metabolism in the pathogenesis of Lewy body disease. J. Mol. Neurosci. 2004, 24, 343–352. [Google Scholar] [CrossRef]
- Iwatsubo, T. Pathological biochemistry of alpha-synucleinopathy. Neuropathology 2007, 27, 474–478. [Google Scholar] [CrossRef]
- Hashimoto, M.; Hsu, L.J.; Xia, Y.; Takeda, A.; Sisk, A.; Sundsmo, M.; Masliah, E. Oxidative stress induces amyloid-like aggregate formation of NACP/alpha-synuclein in vitro. Neuroreport 1999, 10, 717–721. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, H.; Hasegawa, M.; Dohmae, N.; Kawashima, A.; Masliah, E.; Goldberg, M.S.; Shen, J.; Takio, K.; Iwatsubo, T. Alpha-Synuclein is phosphorylated in synucleinopathy lesions. Nat. Cell Biol. 2002, 4, 160–164. [Google Scholar] [CrossRef]
- Anderson, J.P.; Walker, D.E.; Goldstein, J.M.; de Laat, R.; Banducci, K.; Caccavello, R.J.; Barbour, R.; Huang, J.; Kling, K.; Lee, M.; et al. Phosphorylation of Ser-129 is the dominant pathological modification of alpha-synuclein in familial and sporadic Lewy body disease. J. Biol. Chem. 2006, 281, 29739–29752. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Feany, M.B. Alpha-synuclein phosphorylation controls neurotoxicity and inclusion formation in a Drosophila model of Parkinson disease. Nat. Neurosci. 2005, 8, 657–663. [Google Scholar] [CrossRef]
- Whitby, F.G.; Masters, E.I.; Kramer, L.; Knowlton, J.R.; Yao, Y.; Wang, C.C.; Hill, C.P. Structural basis for the activation of 20S proteasomes by 11S regulators. Nature 2000, 408, 115–120. [Google Scholar] [CrossRef]
- Baumeister, W.; Walz, J.; Zühl, F.; Seemüller, E. The proteasome: Paradigm of a self-compartmentalizing protease. Cell 1998, 92, 367–380. [Google Scholar] [CrossRef]
- Stefanis, L.; Larsen, K.E.; Rideout, H.J.; Sulzer, D.; Greene, L.A. Expression of A53T mutant but not wild-type alpha-synuclein in PC12 cells induces alterations of the ubiquitin-dependent degradation system, loss of dopamine release, and autophagic cell death. J. Neurosci. 2001, 21, 9549–9560. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.F.; Zhu, G.Q.; Wang, M.; Cheng, H.; Zhou, A.; Wang, N.; Fang, N.; Wang, X.C.; Xiao, X.Q.; Chen, Z.W.; et al. Involvement of ubiquitin proteasome system in protective mechanisms of Puerarin to MPP(+)-elicited apoptosis. Neurosci. Res. 2009, 63, 52–58. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Shi, Q.; Ma, S.; Feng, N.; Li, J.; Wang, L.; Wang, X. Striatal 19S Rpt6 deficit is related to alpha-synuclein accumulation in MPTP-treated mice. Biochem. Biophys. Res. Commun. 2008, 376, 277–282. [Google Scholar] [CrossRef] [PubMed]
- Buneeva, O.; Kopylov, A.; Kapitsa, I.; Ivanova, E.; Zgoda, V.; Medvedev, A. The Effect of Neurotoxin MPTP and Neuroprotector Isatin on the Profile of Ubiquitinated Brain Mitochondrial Proteins. Cells 2018, 7, 91. [Google Scholar] [CrossRef] [PubMed]
- Fukunaga, K.; Yamamoto, H.; Matsui, K.; Higashi, K.; Miyamoto, E. Purification and characterization of a Ca2+-and calmodulin-dependent protein kinase from rat brain. J. Neurochem. 1982, 39, 1607–1617. [Google Scholar] [CrossRef] [PubMed]
- Fukunaga, K.; Soderling, T.R.; Miyamoto, E. Activation of Ca2+/calmodulin-dependent protein kinase II and protein kinase C by glutamate in cultured rat hippocampal neurons. J. Biol. Chem. 1992, 267, 22527–22533. [Google Scholar] [CrossRef]
- Lisman, J.; Schulman, H.; Cline, H. The molecular basis of CaMKII function in synaptic and behavioural memory. Nat. Rev. Neurosci. 2002, 3, 175–190. [Google Scholar] [CrossRef]
- Izumi, H.; Shinoda, Y.; Saito, T.; Saido, T.C.; Sato, K.; Yabuki, Y.; Matsumoto, Y.; Kanemitsu, Y.; Tomioka, Y.; Abolhassani, N.; et al. The Disease-modifying Drug Candidate, SAK3 Improves Cognitive Impairment and Inhibits Amyloid beta Deposition in App Knock-in Mice. Neuroscience 2018, 377, 87–97. [Google Scholar] [CrossRef]
- Fahn, S. Parkinson disease, the effect of levodopa, and the ELLDOPA trial. Earlier vs Later L-DOPA. Arch. Neurol. 1999, 56, 529–535. [Google Scholar] [CrossRef]
- Haga, H.; Yamada, R.; Izumi, H.; Shnioda, Y.; Kawahata, I.; Miyachi, H.; Fukunaga, K. Novel fatty acid-binding protein 3 ligand inhibits dopaminergic neuronal death and improves motor and cognitive impairments in Parkinson’s disease model mice. Pharmcoll Biochem. Behav. 2020, 191, 172891. [Google Scholar] [CrossRef]
- Yabuki, Y.; Ohizumi, Y.; Yokosuka, A.; Mimaki, Y.; Fukunaga, K. Nobiletin treatment improves motor and cognitive deficits seen in MPTP-induced Parkinson model mice. Neuroscience 2014, 259, 126–141. [Google Scholar] [CrossRef] [PubMed]
- Yabuki, Y.; Matsuo, K.; Kawahata, I.; Fukui, N.; Mizobata, T.; Kawata, Y.; Owada, Y.; Shioda, N.; Fukunaga, K. Fatty Acid Binding Protein 3 Enhances the Spreading and Toxicity of alpha-Synuclein in Mouse Brain. Int. J. Mol. Sci. 2020, 21, 2230. [Google Scholar] [CrossRef] [PubMed]
- Volpicelli-Daley, L.A.; Luk, K.C.; Lee, V.M. Addition of exogenous α-synuclein preformed fibrils to primary neuronal cultures to seed recruitment of endogenous α-synuclein to Lewy body and Lewy neurite-like aggregates. Nat. Protoc. 2014, 9, 2135–2146. [Google Scholar] [CrossRef]
- Yabuki, Y.; Wu, L.; Fukunaga, K. Cognitive enhancer ST101 improves schizophrenia-like behaviors in neonatal ventral hippocampus-lesioned rats in association with improved CaMKII/PKC pathway. J. Pharmacol. Sci. 2019, 140, 263–272. [Google Scholar] [CrossRef] [PubMed]
- Fukunaga, K.; Goto, S.; Miyamoto, E. Immunohistochemical localization of Ca2+/calmodulin-dependent protein kinase II in rat brain and various tissues. J. Neurochem. 1988, 51, 1070–1078. [Google Scholar] [CrossRef] [PubMed]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, J.; Kawahata, I.; Izumi, H.; Fukunaga, K. T-Type Ca2+ Enhancer SAK3 Activates CaMKII and Proteasome Activities in Lewy Body Dementia Mice Model. Int. J. Mol. Sci. 2021, 22, 6185. https://doi.org/10.3390/ijms22126185
Xu J, Kawahata I, Izumi H, Fukunaga K. T-Type Ca2+ Enhancer SAK3 Activates CaMKII and Proteasome Activities in Lewy Body Dementia Mice Model. International Journal of Molecular Sciences. 2021; 22(12):6185. https://doi.org/10.3390/ijms22126185
Chicago/Turabian StyleXu, Jing, Ichiro Kawahata, Hisanao Izumi, and Kohji Fukunaga. 2021. "T-Type Ca2+ Enhancer SAK3 Activates CaMKII and Proteasome Activities in Lewy Body Dementia Mice Model" International Journal of Molecular Sciences 22, no. 12: 6185. https://doi.org/10.3390/ijms22126185
APA StyleXu, J., Kawahata, I., Izumi, H., & Fukunaga, K. (2021). T-Type Ca2+ Enhancer SAK3 Activates CaMKII and Proteasome Activities in Lewy Body Dementia Mice Model. International Journal of Molecular Sciences, 22(12), 6185. https://doi.org/10.3390/ijms22126185