
Ginsenoside Re Protects against Serotonergic Behaviors Evoked by 2,5-Dimethoxy-4-iodo-amphetamine in Mice via Inhibition of PKCδ-Mediated Mitochondrial Dysfunction

 ,
,  ,
,
Abstract
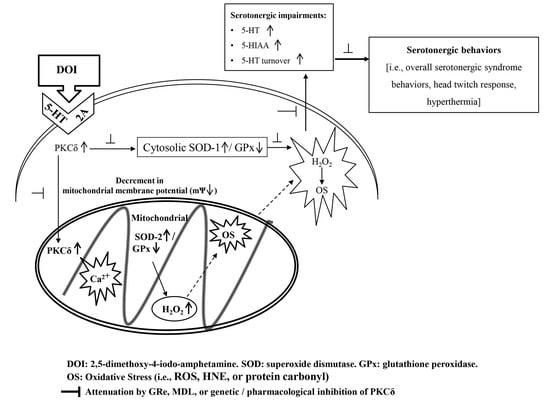
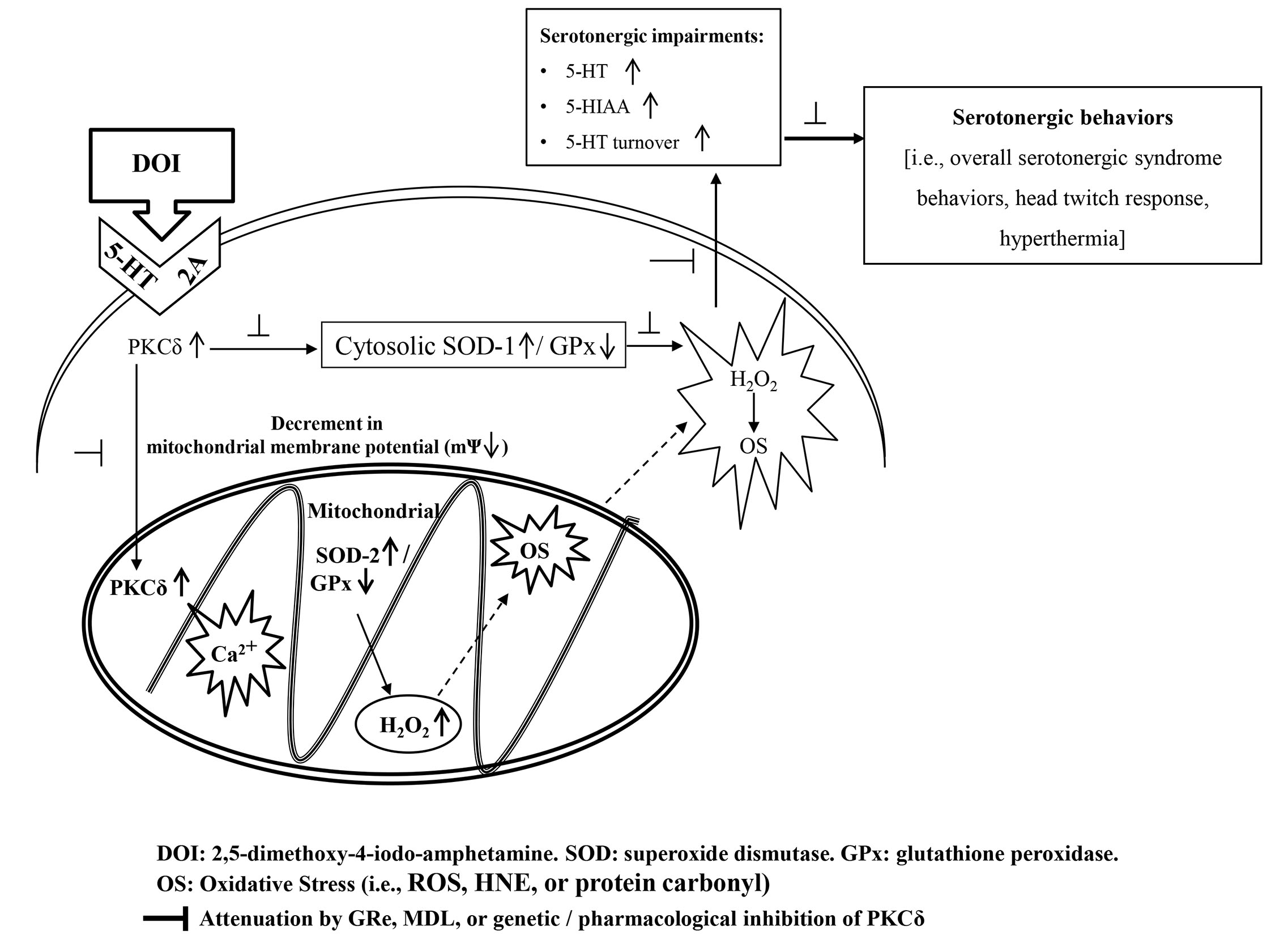{kind=link}
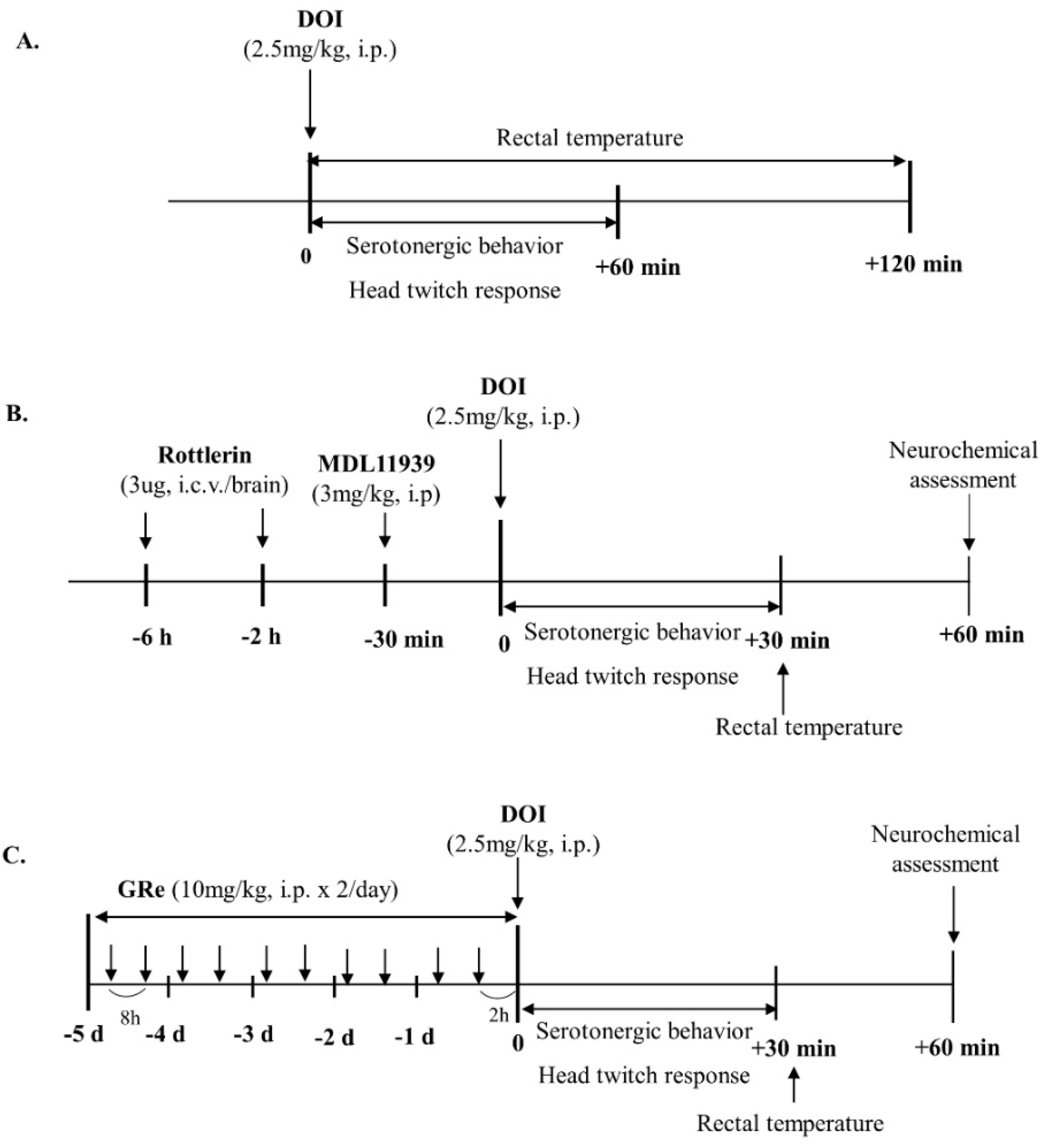{kind=link}
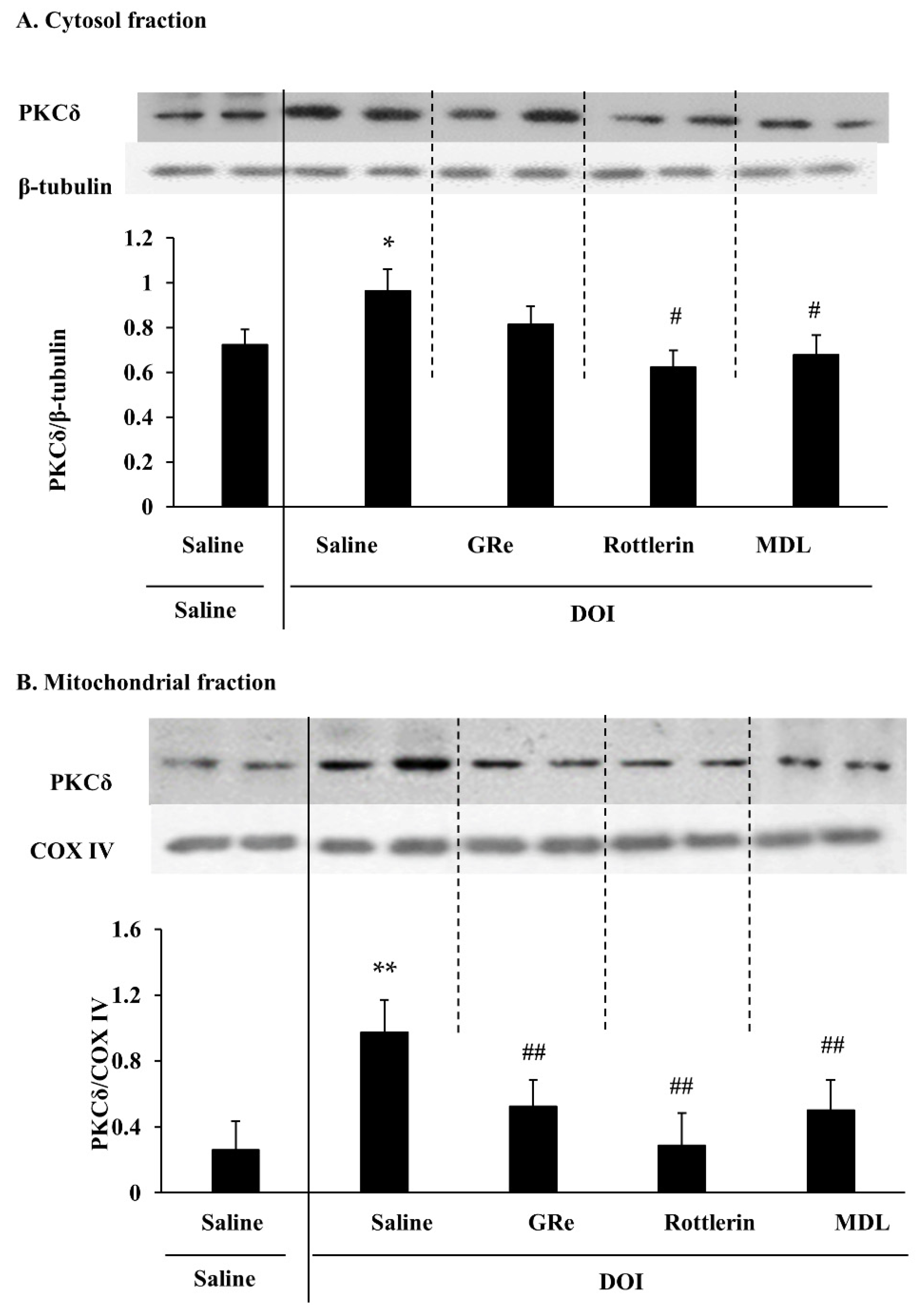{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. Effect of GRe, Rottlerin, or MDL11939 (MDL) on the Mitochondrial Translocation of PKCδ Induced by DOI in the Wild-Type Mice
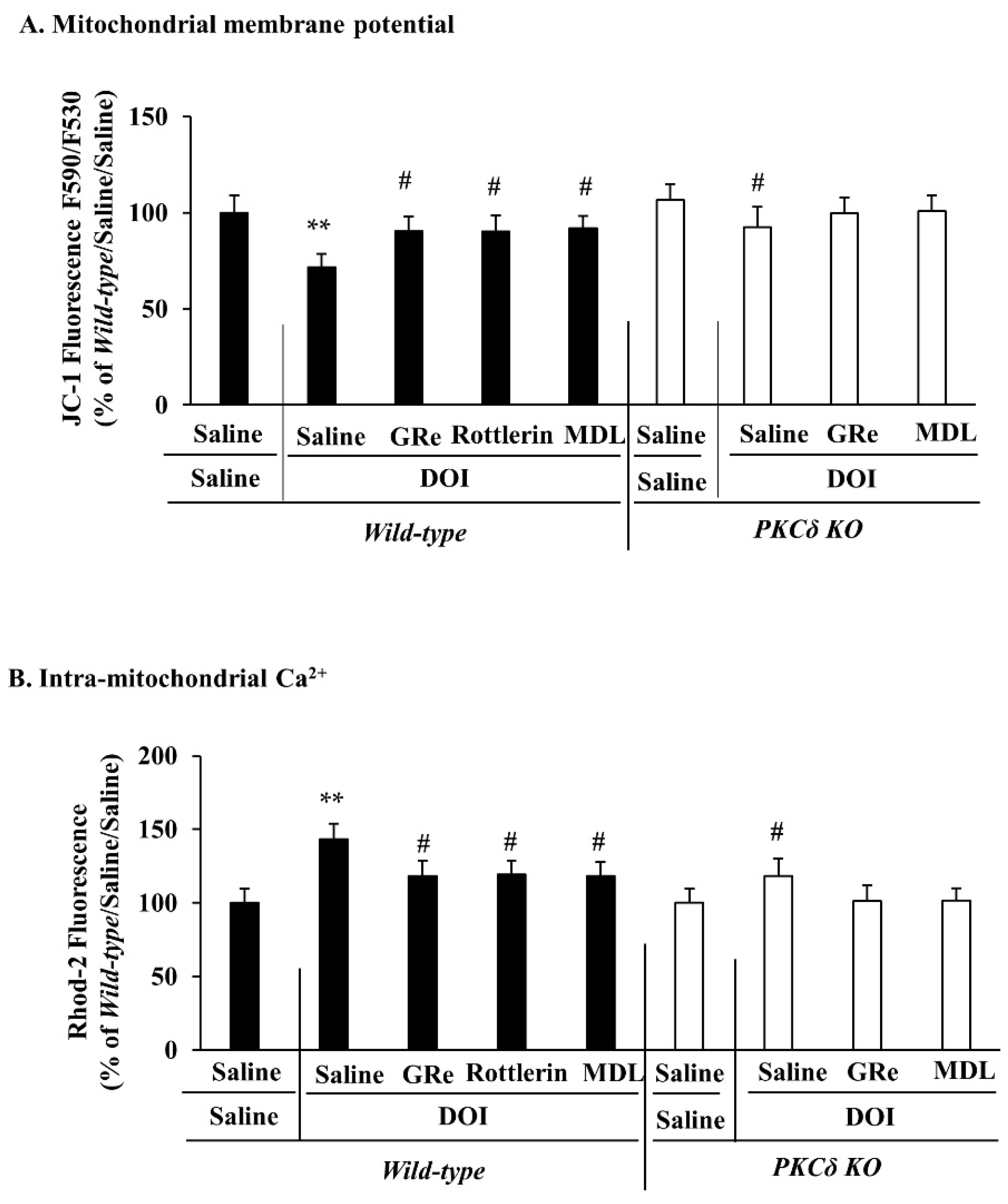
2.2. Effect of GRe or MDL11939 (MDL) on the Alterations in Mitochondrial Membrane Potential and Intra-Mitochondrial Ca2+ Level Elicited by DOI in the Wild-Type and PKCδ KO Mice
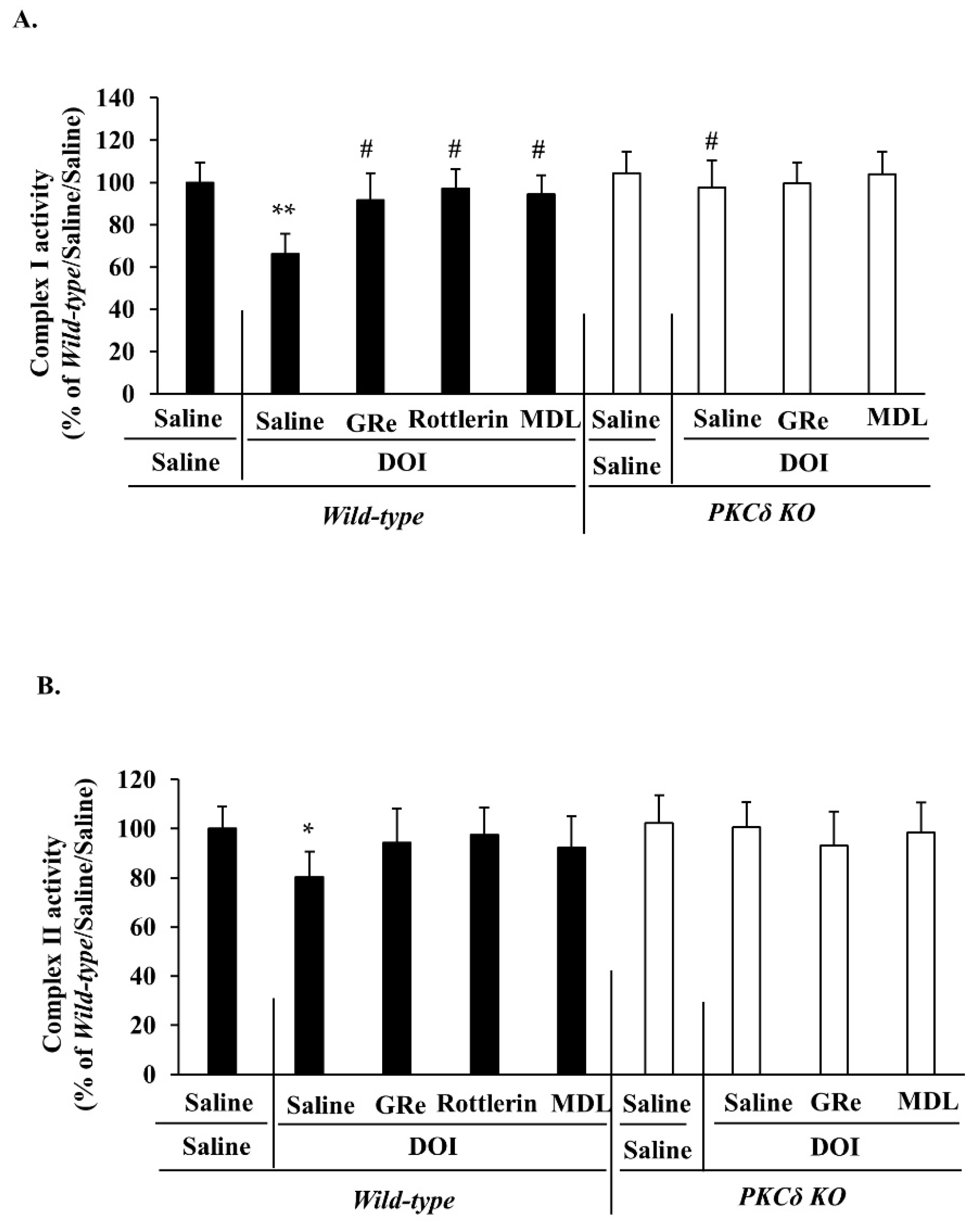
2.3. Effect of GRe or MDL11939 (MDL) on the Alterations in the Mitochondrial Complex I and Complex II Activities Caused by DOI in Wild-Type and PKCδ KO Mice
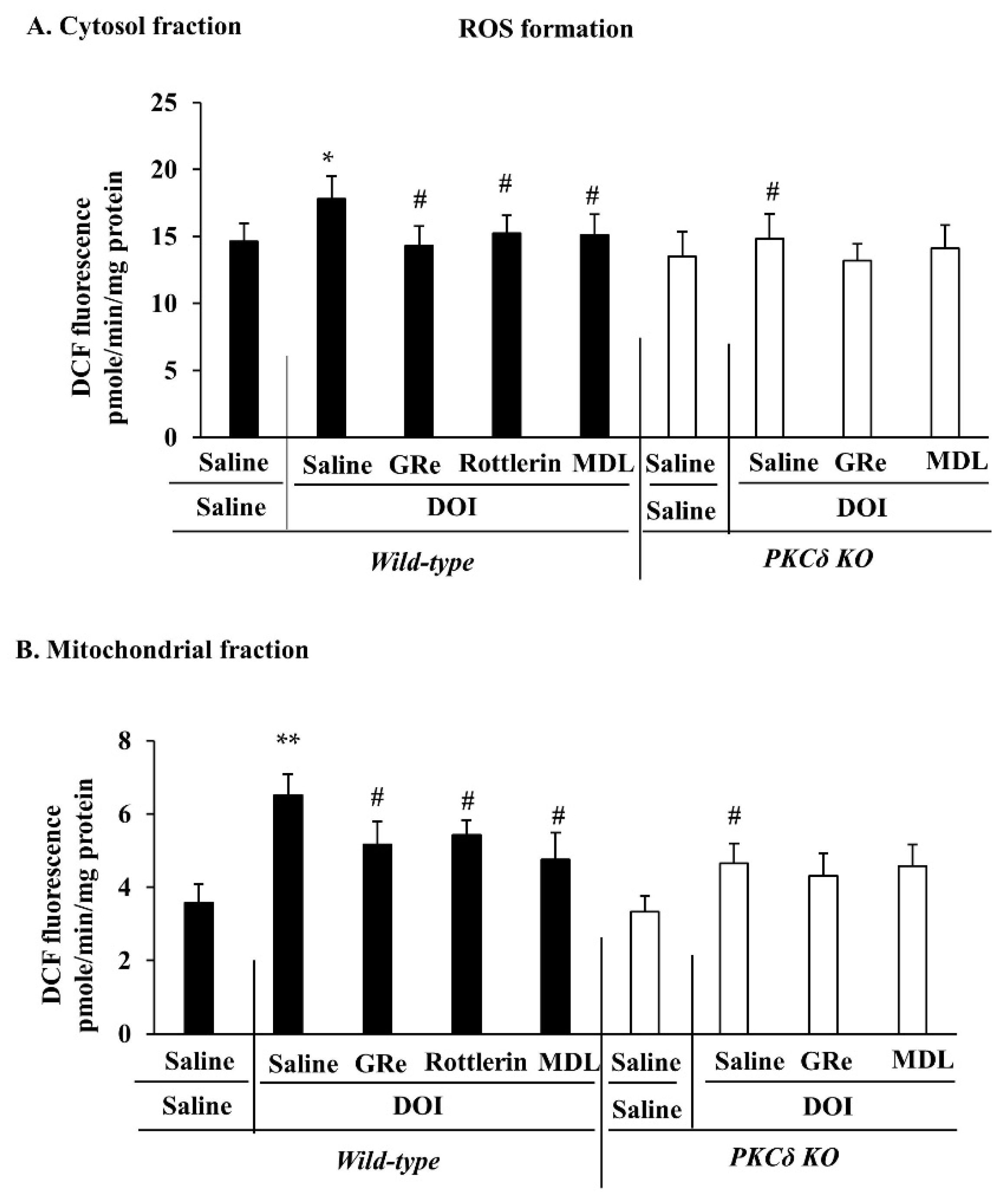
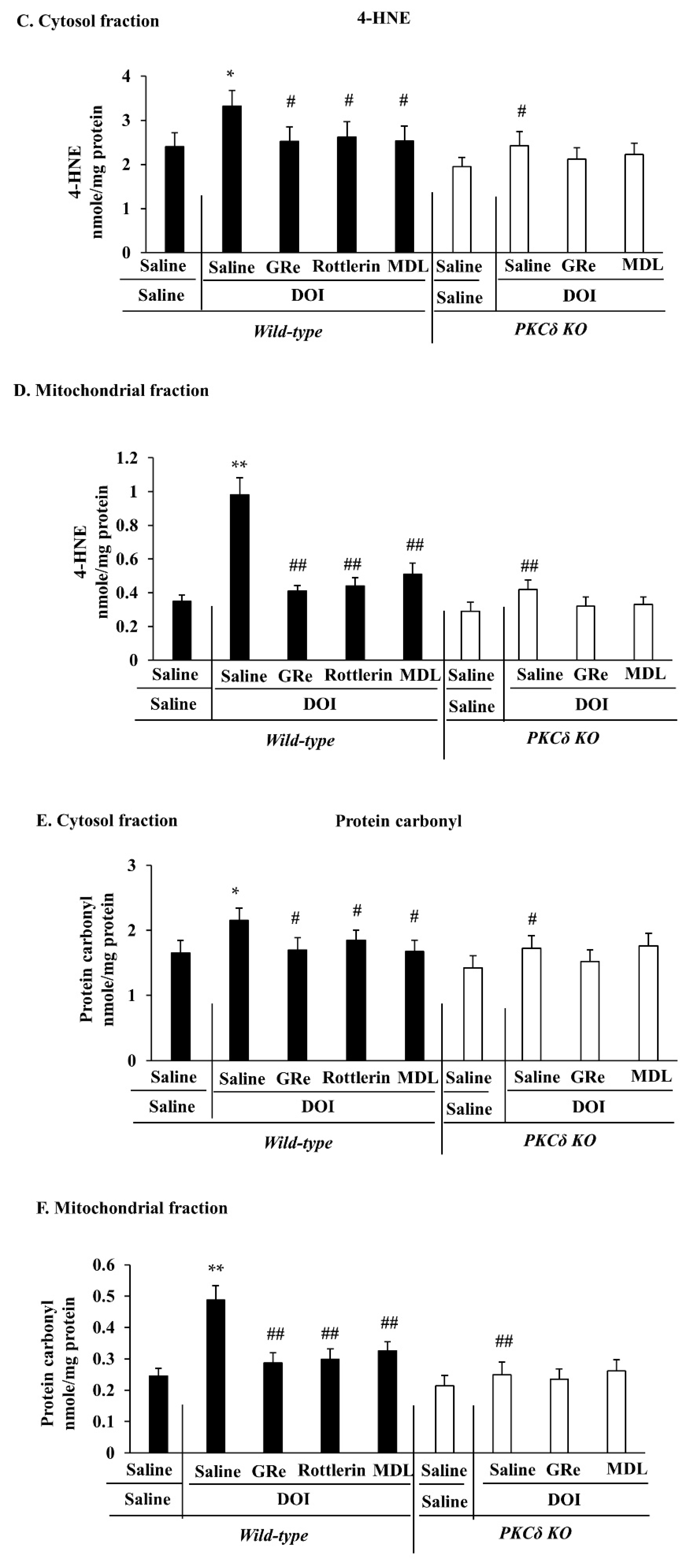
2.4. Effect of GRe or MDL11939 (MDL) on DOI-Induced Oxidative Stress in the Mitochondrial and Cytosolic Fractions of the Hypothalamus of Wild-Type and PKCδ KO Mice
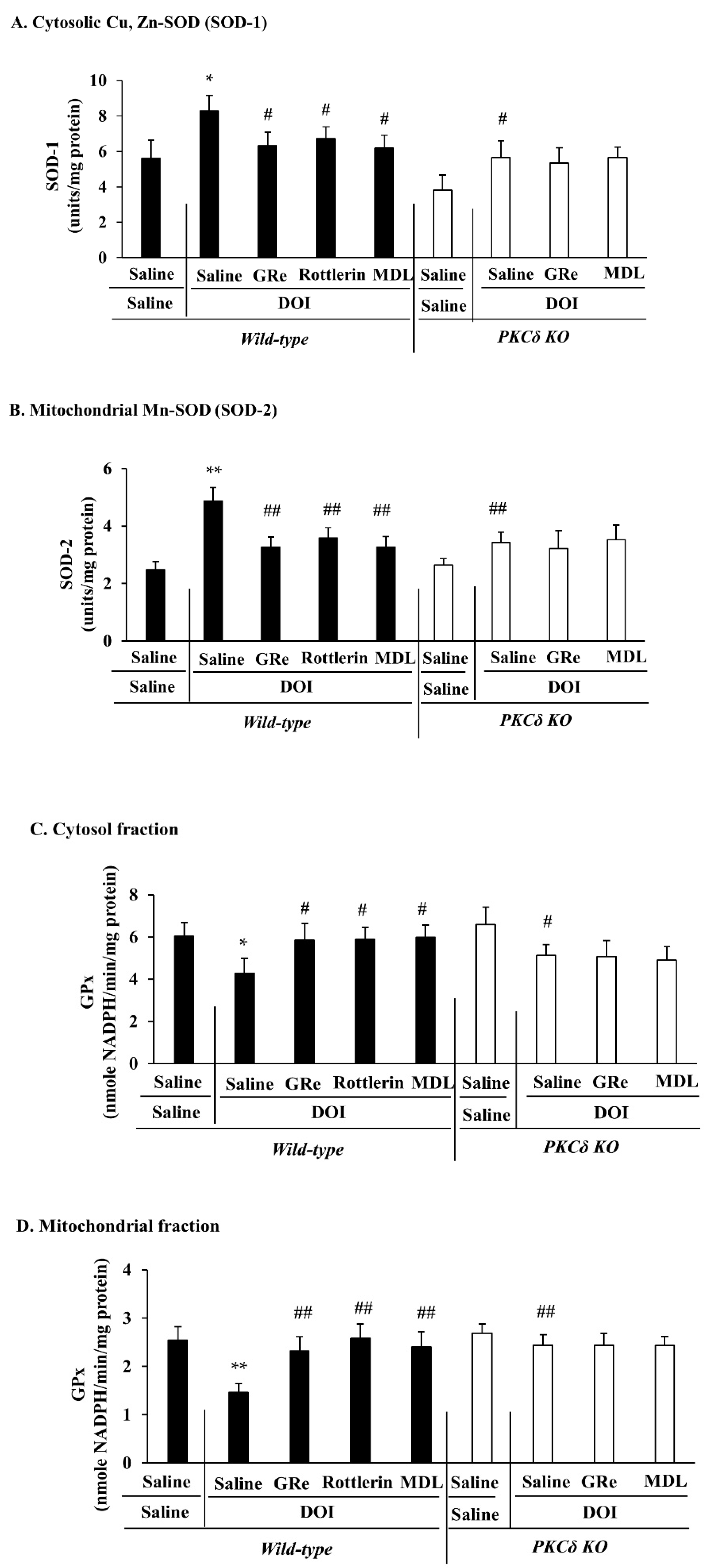
2.5. Effect of GRe or MDL11939 (MDL) on Changes in the Mitochondrial and Cytosolic Activities of Superoxide Dismutase (SOD) and Glutathione Peroxidase (GPx) Induced by DOI in Wild-Type and PKCδ KO Mice
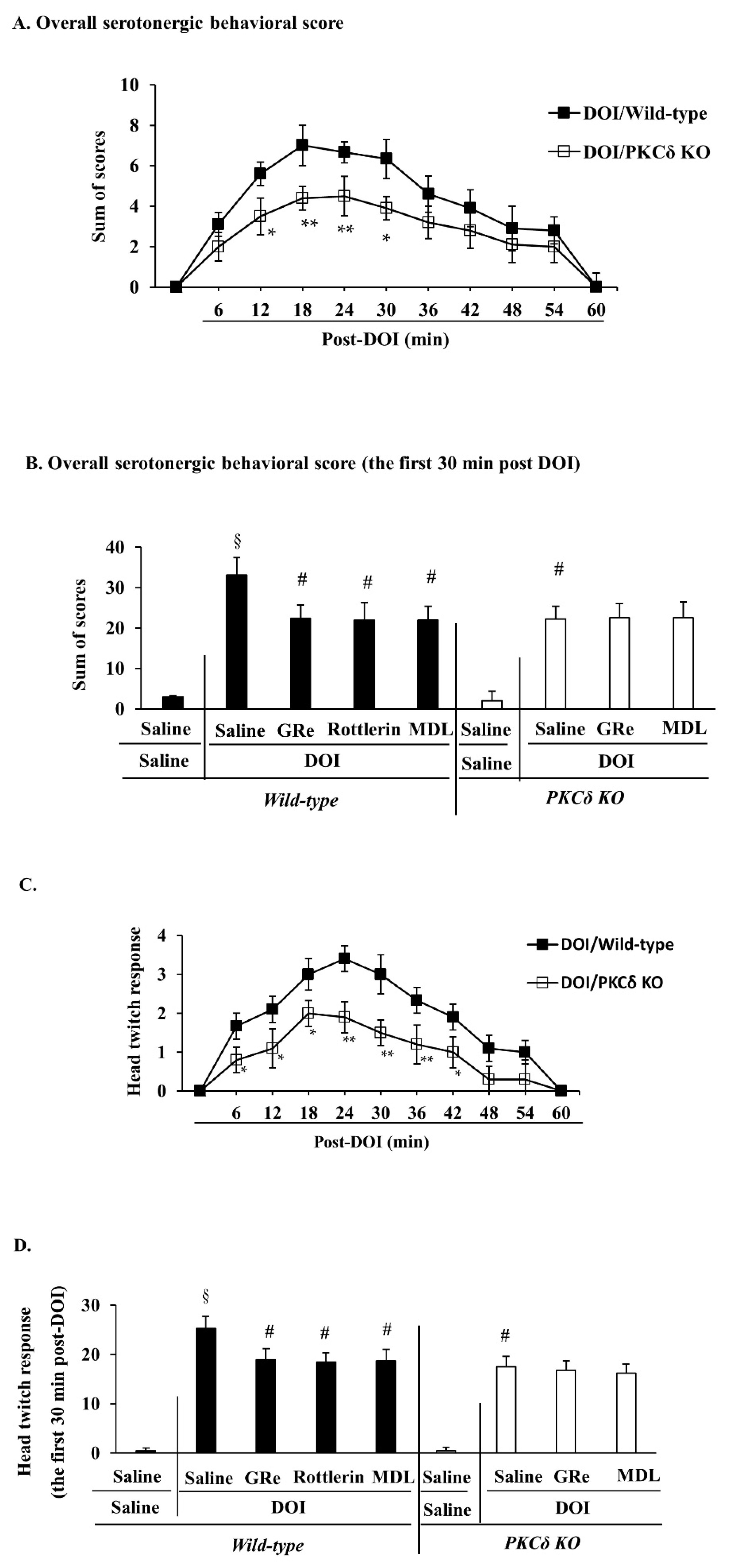
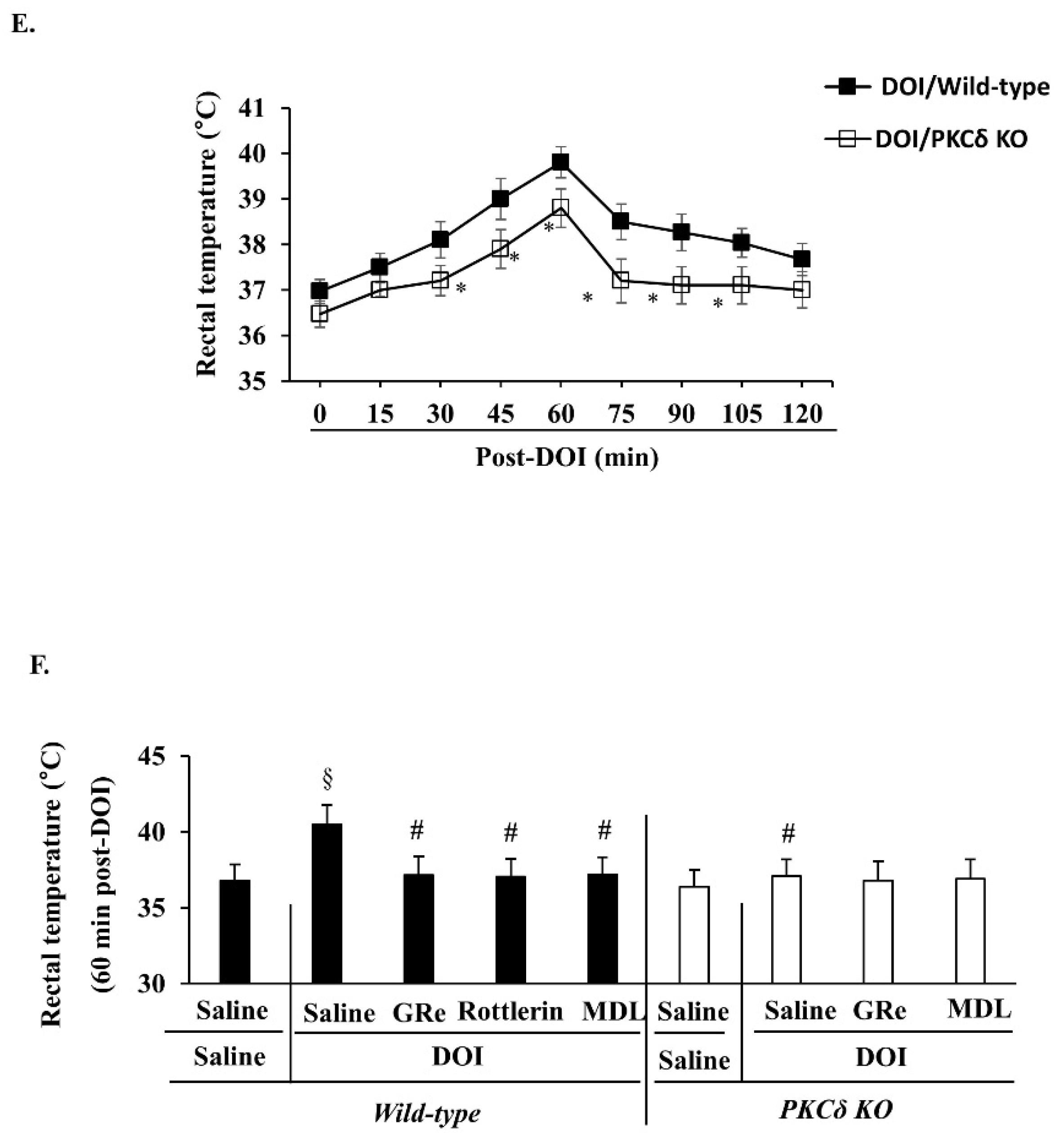
2.6. Effect of GRe or MDL11939 (MDL) against Serotonergic Behaaviors, Head Twitch Response, and Hyperthermia Caused by DOI in Wild-Type and PKCΔ KO Mice
3. Discussion
4. Materials and Methods
4.1. Preparation of GRe
4.2. Animals
4.3. Drug Treatment
4.4. Serotonergic Behaviors
4.5. Head Twitch Response
4.6. Rectal Temperature
4.7. Preparation of Cytosolic and Mitochondrial Fraction for Neurochemical and Western Blot Analyses
4.8. Mitochondrial Preparation for the Measurement of Mitochondrial Membrane Potential and Intramitochondrial Ca2+ Level
4.9. Western Blot Analysis
4.10. Measurement of Mitochondrial Transmembrane Potential
4.11. Measurement of Intramitochondrial Ca2+ Levels
4.12. Measurement of Complex I Activity
4.13. Measurement of Mitochondrial Complex II Activity
4.14. Determination of ROS
4.15. Determination of HNE
4.16. Determination of Protein Carbonyl
4.17. Determination of SOD
4.18. Determination of GPx
4.19. Statistical Analyses
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lü, J.M.; Yao, Q.; Chen, C. Ginseng compounds: An update on their molecular mechanisms and medical applications. Curr. Vasc. Pharmacol. 2009, 7, 293–302. [Google Scholar] [CrossRef] [PubMed]
- Christensen, L.P. Ginsenosides chemistry, biosynthesis, analysis, and potential health effects. Adv. Food Nutr. Res. 2009, 55, 1–99. [Google Scholar] [CrossRef]
- Attele, A.S.; Wu, J.A.; Yuan, C.S. Ginseng pharmacology: Multiple constituents and multiple actions. Biochem. Pharmacol. 1999, 58, 1685–1693. [Google Scholar] [CrossRef]
- Ko, S.K.; Bae, H.M.; Cho, O.S.; Im, B.O.; Chung, S.H.; Lee, B.Y. Analysis of ginsenoside composition of ginseng berry and seed. Food Sci. Biotechnol. 2008, 17, 1379–1382. [Google Scholar]
- Ko, S.K.; Cho, O.S.; Bae, H.M.; Im, B.O.; Lee, O.H.; Lee, B.Y. Quantitative analysis of ginsenosides composition in flower buds of various ginseng plants. J. Korean Soc. Appl. Biol. Chem. 2011, 54, 154–157. [Google Scholar] [CrossRef]
- Joo, K.M.; Lee, J.H.; Jeon, H.Y.; Park, C.W.; Hong, D.K.; Jeong, H.J.; Lee, S.J.; Lee, S.Y.; Lim, K.M. Pharmacokinetic study of ginsenoside Re with pure ginsenoside Re and ginseng berry extracts in mouse using ultra performance liquid chromatography/mass spectrometric method. J. Pharm. Biomed. Anal. 2010, 51, 278–283. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.T.; Shao, Z.H.; Vanden Hoek, T.L.; Chang, W.T.; Li, J.; Mehendale, S.; Wang, C.Z.; Hsu, C.W.; Becker, L.B.; Yin, J.J.; et al. Antioxidant effects of ginsenoside Re in cardiomyocytes. Eur. J. Pharmacol. 2006, 532, 201–207. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.K.; Yoo, D.S.; Xu, H.; Park, N.I.; Kim, H.H.; Choi, J.E.; Park, S.U. Ginsenoside content of berries and roots of three typical Korean ginseng (Panax ginseng) cultivars. Nat. Prod. Commun. 2009, 4, 903–906. [Google Scholar] [CrossRef]
- Dang, D.K.; Shin, E.J.; Kim, D.J.; Tran, H.Q.; Jeong, J.H.; Jang, C.G.; Nah, S.Y.; Jeong, J.H.; Byun, J.K.; Ko, S.K.; et al. Ginsenoside Re protects methamphetamine-induced dopaminergic neurotoxicity in mice via upregulation of dynorphin-mediated κ-opioid receptor and downregulation of substance P-mediated neurokinin1 receptor. J. Neuroinflammation 2018, 15, 52. [Google Scholar] [CrossRef]
- Tran, T.V.; Shin, E.J.; Dang, D.K.; Ko, S.K.; Jeong, J.H.; Nah, S.Y.; Jang, C.G.; Lee, Y.J.; Toriumi, K.; Nabeshima, T.; et al. Ginsenoside Re protects against phencyclidine-induced behavioral changes and mitochondrial dysfunction via interactive modulation of glutathione peroxidase-1 and NADPH oxidase in the dorsolateral cortex of mice. Food Chem. Toxicol. 2017, 110, 300–315. [Google Scholar] [CrossRef]
- Tu, T.T.; Sharma, N.; Shin, E.J.; Tran, H.Q.; Lee, Y.J.; Jeong, J.H.; Jeong, J.H.; Nah, S.Y.; Tran, H.P.; Byun, J.K.; et al. Ginsenoside Re Protects Trimethyltin-Induced Neurotoxicity via Activation of IL-6-Mediated Phosphoinositol 3-Kinase/Akt Signaling in Mice. Neurochem. Res. 2017, 42, 3125–3139. [Google Scholar] [CrossRef] [PubMed]
- Nam, Y.; Wie, M.B.; Shin, E.J.; Nguyen, T.T.; Nah, S.Y.; Ko, S.K.; Jeong, J.H.; Jang, C.G.; Kim, H.C. Ginsenoside Re protects methamphetamine-induced mitochondrial burdens and proapoptosis via genetic inhibition of protein kinase C delta in human neuroblastoma dopaminergic SH-SY5Y cell lines. J. Appl. Toxicol. 2015, 35, 927–944. [Google Scholar] [CrossRef] [PubMed]
- Shin, E.J.; Shin, S.W.; Nguyen, T.T.; Park, D.H.; Wie, M.B.; Jang, C.G.; Nah, S.Y.; Yang, B.W.; Ko, S.K.; Nabeshima, T.; et al. Ginsenoside Re rescues methamphetamine-induced oxidative damage, mitochondrial dysfunction, microglial activation, and dopaminergic degeneration by inhibiting the protein kinase Cδ gene. Mol. Neurobiol. 2014, 49, 1400–1421. [Google Scholar] [CrossRef] [PubMed]
- Boyer, E.W.; Shannon, M. The serotonin syndrome. N. Engl. J. Med. 2005, 352, 1112–1120. [Google Scholar] [CrossRef]
- Birmes, P.; Coppin, D.; Schmitt, L.; Lauque, D. Serotonin syndrome: A brief review. CMAJ 2003, 168, 1439–1442. [Google Scholar]
- Ener, R.A.; Meglathery, S.B.; Van Decker, W.A.; Gallagher, R.M. Serotonin syndrome and other serotonergic disorders. Pain Med. 2003, 4, 63–74. [Google Scholar] [CrossRef] [PubMed]
- Voizeux, P.; Lewandowski, R.; Daily, T.; Ellouze, O.; Bouchot, O.; Bouhemad, B.; Guinot, P.G. Case of Cardiac Arrest Treated with Extra-Corporeal Life Support after MDMA Intoxication. Case Rep. Crit. Care 2019, 2019, 7825915. [Google Scholar] [CrossRef] [PubMed]
- Nadkarni, G.N.; Hoskote, S.S.; Piotrkowski, J.; Annapureddy, N. Serotonin syndrome, disseminated intravascular coagulation, and hepatitis after a single ingestion of MDMA in an Asian woman. Am. J. Ther. 2014, 21, e117–e119. [Google Scholar] [CrossRef] [PubMed]
- Davies, O.; Batajoo-Shrestha, B.; Sosa-Popoteur, J.; Olibrice, M. Full recovery after severe serotonin syndrome, severe rhabdomyolysis, multi-organ failure and disseminated intravascular coagulopathy from MDMA. Heart Lung. 2014, 43, 117–119. [Google Scholar] [CrossRef] [PubMed]
- Dobry, Y.; Rice, T.; Sher, L. Ecstasy use and serotonin syndrome: A neglected danger to adolescents and young adults prescribed selective serotonin reuptake inhibitors. Int. J. Adolesc. Med. Health 2013, 25, 193–199. [Google Scholar] [CrossRef]
- Vuori, E.; Henry, J.A.; Ojanperä, I.; Nieminen, R.; Savolainen, T.; Wahlsten, P.; Jäntti, M. Death following ingestion of MDMA (ecstasy) and moclobemide. Addiction 2003, 98, 365–368. [Google Scholar] [CrossRef] [PubMed]
- Parrott, A.C. Recreational Ecstasy/MDMA, the serotonin syndrome, and serotonergic neurotoxicity. Pharmacol. Biochem. Behav. 2002, 71, 837–844. [Google Scholar] [CrossRef]
- Brush, D.E.; Bird, S.B.; Boyer, E.W. Monoamine oxidase inhibitor poisoning resulting from Internet misinformation on illicit substances. J. Toxicol. Clin. Toxicol. 2004, 42, 191–195. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, K. Three cases of acute serotonin syndrome due to psilocybin mushroom poisoning. Chudoku Kenkyu 2016, 29, 33–35. [Google Scholar] [PubMed]
- Kinoshita, H.; Ohkubo, T.; Yasuda, M.; Yakushiji, F. Serotonin syndrome induced by dextromethorphan (Medicon) administrated at the conventional dose. Geriatr. Gerontol. Int. 2011, 11, 121–122. [Google Scholar] [CrossRef]
- Schwartz, A.R.; Pizon, A.F.; Brooks, D.E. Dextromethorphan-induced serotonin syndrome. Clin. Toxicol. 2008, 46, 771–773. [Google Scholar] [CrossRef]
- Ganetsky, M.; Babu, K.M.; Boyer, E.W. Serotonin syndrome in dextromethorphan ingestion responsive to propofol therapy. Pediatr. Emerg. Care 2007, 23, 829–831. [Google Scholar] [CrossRef] [PubMed]
- Navarro, A.; Perry, C.; Bobo, W.V. A case of serotonin syndrome precipitated by abuse of the anticough remedy dextromethorphan in a bipolar patient treated with fluoxetine and lithium. Gen. Hosp. Psychiatry 2006, 28, 78–80. [Google Scholar] [CrossRef]
- Isbister, G.K.; Buckley, N.A. The pathophysiology of serotonin toxicity in animals and humans: Implications for diagnosis and treatment. Clin. Neuropharmacol. 2005, 28, 205–214. [Google Scholar] [CrossRef]
- Souza, M.E.; Polizello, A.C.; Uyemura, S.A.; Castro-Silva, O.; Curti, C. Effect of fluoxetine on rat liver mitochondria. Biochem. Pharmacol. 1994, 48, 535–541. [Google Scholar] [CrossRef]
- Nahon, E.; Israelson, A.; Abu-Hamad, S.; Varda, S.B. Fluoxetine (Prozac) interaction with the mitochondrial voltage-dependent anion channel and protection against apoptotic cell death. FEBS Lett. 2005, 579, 5105–5110. [Google Scholar] [CrossRef] [PubMed]
- Curti, C.; Mingatto, F.E.; Polizello, A.C.; Galastri, L.O.; Uyemura, S.A.; Santos, A.C. Fluoxetine interacts with the lipid bilayer of the inner membrane in isolated rat brain mitochondria, inhibiting electron transport and F1F0-ATPase activity. Mol. Cell. Biochem. 1999, 199, 103–109. [Google Scholar] [CrossRef]
- Abdel-Razaq, W.; Kendall, D.A.; Bates, T.E. The effects of antidepressants on mitochondrial function in a model cell system and isolated mitochondria. Neurochem. Res. 2011, 36, 327–338. [Google Scholar] [CrossRef] [PubMed]
- Scotton, W.J.; Hill, L.J.; Williams, A.C.; Barnes, N.M. Serotonin Syndrome: Pathophysiology, Clinical Features, Management, and Potential Future Directions. Int. J. Tryptophan Res. 2019, 12, 1178646919873925. [Google Scholar] [CrossRef]
- Tran, H.Q.; Shin, E.J.; Hoai Nguyen, B.C.; Phan, D.H.; Kang, M.J.; Jang, C.G.; Jeong, J.H.; Nah, S.Y.; Mouri, A.; Saito, K.; et al. 5-HT1A receptor agonist 8-OH-DPAT induces serotonergic behaviors in mice via interaction between PKCδ and p47phox. Food Chem. Toxicol. 2019, 123, 125–141. [Google Scholar] [CrossRef]
- Fox, M.A.; French, H.T.; LaPorte, J.L.; Blackler, A.R.; Murphy, D.L. The serotonin 5-HT(2A) receptor agonist TCB-2: A behavioral and neurophysiological analysis. Psychopharmacology 2010, 212, 13–23. [Google Scholar] [CrossRef]
- Haberzettl, R.; Fink, H.; Bert, B. Role of 5-HT(1A)- and 5-HT(2A) receptors for the murine model of the serotonin syndrome. J. Pharmacol. Toxicol. Methods 2014, 70, 129–133. [Google Scholar] [CrossRef] [PubMed]
- Takuwa, N.; Ganz, M.; Takuwa, Y.; Sterzel, R.B.; Rasmussen, H. Studies of the mitogenic effect of serotonin in rat renal mesangial cells. Am. J. Physiol. 1989, 257 Pt 2, F431–F439. [Google Scholar] [CrossRef]
- Steinberg, S.F. Distinctive activation mechanisms and functions for protein kinase Cdelta. Biochem. J. 2004, 384 Pt 3, 449–459. [Google Scholar] [CrossRef]
- Yoshida, K. PKCdelta signaling: Mechanisms of DNA damage response and apoptosis. Cell Signal. 2007, 19, 892–901. [Google Scholar] [CrossRef] [PubMed]
- Shin, E.J.; Duong, C.X.; Nguyen, X.T.; Li, Z.; Bing, G.; Bach, J.H.; Park, D.H.; Nakayama, K.; Ali, S.F.; Kanthasamy, A.G.; et al. Role of oxidative stress in methamphetamine-induced dopaminergic toxicity mediated by protein kinase Cδ. Behav. Brain Res. 2012, 232, 98–113. [Google Scholar] [CrossRef]
- Shin, E.J.; Nam, Y.; Tu, T.H.; Lim, Y.K.; Wie, M.B.; Kim, D.J.; Jeong, J.H.; Kim, H.C. Protein kinase Cδ mediates trimethyltin-induced neurotoxicity in mice in vivo via inhibition of glutathione defense mechanism. Arch. Toxicol. 2016, 90, 937–953. [Google Scholar] [CrossRef] [PubMed]
- Shin, E.J.; Hwang, Y.G.; Sharma, N.; Tran, H.Q.; Dang, D.K.; Jang, C.G.; Jeong, J.H.; Nah, S.Y.; Nabeshima, T.; Kim, H.C. Role of protein kinase Cδ in dopaminergic neurotoxic events. Food Chem. Toxicol. 2018, 121, 254–261. [Google Scholar] [CrossRef] [PubMed]
- Shin, E.J.; Jeong, J.H.; Sharma, G.; Sharma, N.; Kim, D.J.; Pham, D.T.; Trinh, Q.D.; Dang, D.K.; Nah, S.Y.; Bing, G.; et al. Protein kinase Cδ mediates methamphetamine-induced dopaminergic neurotoxicity in mice via activation of microsomal epoxide hydrolase. Food Chem. Toxicol. 2019, 133, 110761. [Google Scholar] [CrossRef] [PubMed]
- Shin, E.J.; Dang, D.K.; Hwang, Y.G.; Tran, H.Q.; Sharma, N.; Jeong, J.H.; Jang, C.G.; Nah, S.Y.; Nabeshima, T.; Yoneda, Y.; et al. Significance of protein kinase C in the neuropsychotoxicity induced by methamphetamine-like psychostimulants. Neurochem. Int. 2019, 124, 162–170. [Google Scholar] [CrossRef] [PubMed]
- Dang, D.K.; Shin, E.J.; Kim, D.J.; Tran, H.Q.; Jeong, J.H.; Jang, C.G.; Ottersen, O.P.; Nah, S.Y.; Hong, J.S.; Nabeshima, T.; et al. PKCδ-dependent p47phox activation mediates methamphetamine-induced dopaminergic neurotoxicity. Free Radic. Biol. Med. 2018, 115, 318–337. [Google Scholar] [CrossRef]
- Mai, H.N.; Sharma, N.; Shin, E.J.; Nguyen, B.T.; Nguyen, P.T.; Jeong, J.H.; Cho, E.H.; Lee, Y.J.; Kim, N.H.; Jang, C.G.; et al. Exposure to far-infrared ray attenuates methamphetamine-induced impairment in recognition memory through inhibition of protein kinase C δ in male mice: Comparison with the antipsychotic clozapine. J. Neurosci. Res. 2018, 96, 1294–1310. [Google Scholar] [CrossRef]
- Mai, H.N.; Sharma, N.; Shin, E.J.; Nguyen, B.T.; Nguyen, P.T.; Jeong, J.H.; Jang, C.G.; Cho, E.H.; Nah, S.Y.; Kim, N.H.; et al. Exposure to far-infrared rays attenuates methamphetamine-induced recognition memory impairment via modulation of the muscarinic M1 receptor, Nrf2, and PKC. Neurochem. Int. 2018, 116, 63–76. [Google Scholar] [CrossRef]
- Shin, E.J.; Duong, C.X.; Nguyen, X.T.; Bing, G.; Bach, J.H.; Park, D.H.; Nakayama, K.; Ali, S.F.; Kanthasamy, A.G.; Cadet, J.L.; et al. PKCδ inhibition enhances tyrosine hydroxylase phosphorylation in mice after methamphetamine treatment. Neurochem. Int. 2011, 59, 39–50. [Google Scholar] [CrossRef]
- Kramer, H.K.; Poblete, J.C.; Azmitia, E.C. 3,4-Methylenedioxymethamphetamine (‘Ecstasy’) promotes the translocation of protein kinase C (PKC): Requirement of viable serotonin nerve terminals. Brain Res. 1995, 680, 1–8. [Google Scholar] [CrossRef]
- Kramer, H.K.; Poblete, J.C.; Azmitia, E.C. Activation of protein kinase C (PKC) by 3,4-methylenedioxymethamphetamine (MDMA) occurs through the stimulation of serotonin receptors and transporter. Neuropsychopharmacology 1997, 17, 117–129. [Google Scholar] [CrossRef]
- Wang, Q.; Bubula, N.; Brown, J.; Wang, Y.; Kondev, V.; Vezina, P. PKC phosphorylates residues in the N-terminal of the DA transporter to regulate amphetamine-induced DA efflux. Neurosci. Lett. 2016, 622, 78–82. [Google Scholar] [CrossRef]
- Loweth, J.A.; Svoboda, R.; Austin, J.D.; Guillory, A.M.; Vezina, P. The PKC inhibitor Ro31-8220 blocks acute amphetamine-induced dopamine overflow in the nucleus accumbens. Neurosci. Lett. 2009, 455, 88–92. [Google Scholar] [CrossRef]
- Tran, H.Q.; Lee, Y.; Shin, E.J.; Jang, C.G.; Jeong, J.H.; Mouri, A.; Saito, K.; Nabeshima, T.; Kim, H.C. PKCδ knockout mice are [rotected from dextromethorphan-induced serotonergic behaviors in mice: Involvements of downregulation of 5-HT1A receptor and upregulation of Nrf2-dependent GSH synthesis. Mol. Neurobiol. 2018, 55, 7802–7821. [Google Scholar] [CrossRef]
- Phan, D.H.; Shin, E.J.; Sharma, N.; Hoang Yen, T.P.; Dang, D.K.; Lee, Y.S.; Lee, Y.J.; Nah, S.Y.; Cheong, J.H.; Jeong, J.H.; et al. 5-HT2A receptor-mediated PKCδ phosphorylation is critical for serotonergic impairments induced by p-chloroamphetamine in mice. Food Chem. Toxicol. 2020, 141, 111395. [Google Scholar] [CrossRef]
- Capela, J.P.; da Costa Araújo, S.; Costa, V.M.; Ruscher, K.; Fernandes, E.; Bastos Mde, L.; Dirnagl, U.; Meisel, A.; Carvalho, F. The neurotoxicity of hallucinogenic amphetamines in primary cultures of hippocampal neurons. Neurotoxicology 2013, 34, 254–263. [Google Scholar] [CrossRef] [PubMed]
- Shin, E.J.; Jeong, J.H.; Kim, A.Y.; Koh, Y.H.; Nah, S.Y.; Kim, W.K.; Ko, K.H.; Kim, H.J.; Wie, M.B.; Kwon, Y.S.; et al. Protection against kainate neurotoxicity by ginsenosides: Attenuation of convulsive behavior, mitochondrial dysfunction, and oxidative stress. J. Neurosci. Res. 2009, 87, 710–722. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.M.; Cao, Y.L.; Dou, D.Q. Protective effect of ginsenoside-Re against cerebral ischemia/reperfusion damage in rats. Biol. Pharm. Bull. 2006, 29, 2502–2505. [Google Scholar] [CrossRef]
- González-Burgos, E.; Fernández-Moriano, C.; Lozano, R.; Iglesias, I.; Gómez- Serranillos, M.P. Ginsenosides Rd and Re co-treatments improve rotenone-induced oxidative stress and mitochondrial impairment in SH-SY5Y neuroblastoma cells. Food Chem. Toxicol. 2017, 109 Pt 1, 38–47. [Google Scholar] [CrossRef]
- Liu, M.; Bai, X.; Yu, S.; Zhao, W.; Qiao, J.; Liu, Y.; Zhao, D.; Wang, J.; Wang, S. Ginsenoside Re Inhibits ROS/ASK-1 dependent mitochondrial apoptosis pathway and activation of Nrf2-antioxidant response in Beta-amyloid-challenged SH-SY5Y cells. Molecules 2019, 24, 2687. [Google Scholar] [CrossRef]
- Halliwell, B. Reactive oxygen species and central nervous system. J. Neurochem. 1992, 59, 1609–1623. [Google Scholar] [CrossRef] [PubMed]
- Lismont, C.; Revenco, I.; Frabsen, M. Peroxisomal hydrogen peroxide metabolism and signaling in health and disease. Int. J. Mol. Sci. 2019, 20, 3673. [Google Scholar] [CrossRef]
- Sharma, G.; Shin, E.J.; Sharma, N.; Nah, S.Y.; Mai, H.N.; Nguyen, B.T.; Jeong, J.H.; Lei, X.G.; Kim, H.C. Glutathione peroxidase-1 and neuromodulation: Novel potentials of an old enzyme. Food Chem. Toxicol. 2021, 148, 111945. [Google Scholar] [CrossRef]
- Chance, B.; Sies, H.; Boveris, A. Hydroperoxide metabolism in mammalian organs. Physiol. Rev. 1979, 59, 527–605. [Google Scholar] [CrossRef]
- Xiong, Y.; Shie, F.S.; Zhang, J.; Lee, C.P.; Ho, Y.S. The protective role of cellular glutathione peroxidase against trauma-induced mitochondrial dysfunction in the mouse brain. J. Stroke Cereb. Dis. 2004, 13, 129–137. [Google Scholar] [CrossRef] [PubMed]
- Marí, M.; Morales, A.; Colell, A.; García-Ruiz, C.; Kaplowitz, N.; Fernández-Checa, J.C. Mitochondrial glutathione: Features, regulation and role in disease. Biochim. Biophys. Acta. 2013, 1830, 3317–3328. [Google Scholar] [CrossRef]
- Ramamoorthy, S.; Giovanetti, E.; Qian, Y.; Blakely, R.D. Phosphorylation and regulation of antidepressant-sensitive serotonin transporters. J. Biol. Chem. 1998, 273, 2458–2466. [Google Scholar] [CrossRef] [PubMed]
- Anji, A.; Sullivan Hanley, N.R.; Kumari, M.; Hensler, J.G. The role of protein kinase C in the regulation of serotonin-2A receptor expression. J. Neurochem. 2001, 77, 589–597. [Google Scholar] [CrossRef]
- Mizutani, K.; Sonoda, S.; Wakita, H. Ritanserin, a serotonin-2 receptor antagonist, inhibits functional recovery after cerebral infarction. Neuroreport 2018, 29, 54–58. [Google Scholar] [CrossRef]
- Bhattacharyya, S.; Puri, S.; Miledi, R.; Panicker, M.M. Internalization and recycling of 5-HT2A receptors activated by serotonin and protein kinase C-mediated mechanisms. Proc. Natl. Acad. Sci. USA 2002, 99, 14470–14475. [Google Scholar] [CrossRef]
- Bhattacharyya, S.; Raote, I.; Bhattacharya, A.; Miledi, R.; Panicker, M.M. Activation, internalization, and recycling of the serotonin 2A receptor by dopamine. Proc. Natl. Acad. Sci. USA 2006, 103, 15248–15253. [Google Scholar] [CrossRef]
- Haberzettl, R.; Bert, B.; Fink, H.; Fox, M.A. Animal models of the serotonin syndrome: A systematic review. Behav. Brain Res. 2013, 256, 328–345. [Google Scholar] [CrossRef] [PubMed]
- Nisijima, K.; Yoshino, T.; Yui, K.; Katoh, S. Potent serotonin 5-HT2A receptor antagonists completely prevent the development of hyperthermia in an animal model of the 5-HT syndrome. Brain Res. 2001, 890, 23–31. [Google Scholar] [CrossRef]
- Shin, E.J.; Koh, Y.H.; Kim, A.Y.; Nah, S.Y.; Jeong, J.H.; Chae, J.S.; Kim, S.C.; Yen, T.P.; Yoon, H.J.; Kim, W.K.; et al. Ginsenosides attenuate kainic acid-induced synaptosomal oxidative stress via stimulation of adenosine A(2A) receptors in rat hippocampus. Behav. Brain Res. 2009, 197, 239–245. [Google Scholar] [CrossRef]
- Wu, C.W.; Ping, Y.H.; Yen, J.C.; Chang, C.Y.; Wang, S.F.; Yeh, C.L.; Chi, C.W.; Lee, H.C. Enhanced oxidative stress and aberrant mitochondrial biogenesis in human neuroblastoma SH-SY5Y cells during methamphetamine induced apoptosis. Toxicol. Appl. Pharmacol. 2007, 220, 243–251. [Google Scholar] [CrossRef] [PubMed]
- Beauvais, G.; Atwell, K.; Jayanthi, S.; Ladenheim, B.; Cadet, J.L. Involvement of dopamine receptors in binge methamphetamine-induced activation of endoplasmic reticulum and mitochondrial stress pathways. PLoS ONE 2011, 6, e28946. [Google Scholar] [CrossRef]
- Basu, A.; Pal, D. Two faces of protein kinase Cδ: The contrasting roles of PKCδ in cell survival and cell death. Sci. World J. 2010, 10, 2272–2284. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.F.; Chen, Y.C.; Liu, C.Y.; Wei, Y.H. Involvement of protein kinase C delta in the alteration of mitochondrial mass in human cells under oxidative stress. Free Radic. Biol. Med. 2006, 40, 2136–2146. [Google Scholar] [CrossRef] [PubMed]
- Kanthasamy, A.G.; Kitazawa, M.; Kanthasamy, A.; Anantharam, V. Role of proteolytic activation of protein kinase Cdelta in oxidative stress-induced apoptosis. Antioxid. Redox Signal. 2003, 5, 609–620. [Google Scholar] [CrossRef]
- Handy, D.E.; Lubos, E.; Yang, Y.; Galbraith, J.D.; Kelly, N.; Zhang, Y.Y.; Leopold, J.A.; Loscalzo, J. Glutathione peroxidase-1 regulates mitochondrial function to modulate redox-dependent cellular responses. J. Biol. Chem. 2009, 284, 11913–11921. [Google Scholar] [CrossRef]
- Esposito, L.A.; Kokoszka, J.E.; Waymire, K.G.; Cottrell, B.; MacGregor, G.R.; Wallace, D.C. Mitochondrial oxidative stress in mice lacking the glutathione peroxidase-1 gene. Free Radic. Biol. Med. 2000, 28, 754–766. [Google Scholar] [CrossRef][Green Version]
- Nicholls, D.G. Mitochondrial calcium function and dysfunction in the central nervous system. Biochim. Biophys. Acta 2009, 1787, 1416–1424. [Google Scholar] [CrossRef] [PubMed]
- Garratt, J.C.; Kidd, E.J.; Wright, I.K.; Marsden, C.A. Inhibition of 5-hydroxytryptamine neuronal activity by the 5-HT agonist, DOI. Eur. J. Pharmacol. 1991, 199, 349–355. [Google Scholar] [CrossRef]
- Kidd, E.J.; Garratt, J.C.; Marsden, C.A. Effects of repeated treatment with 1-(2,5-dimethoxy-4-iodophenyl)-2-aminopropane (DOI) on the autoregulatory control of dorsal raphe 5-HT neuronal firing and cortical 5-HT release. Eur. J. Pharmacol. 1991, 200, 131–139. [Google Scholar] [CrossRef]
- Martín-Ruiz, R.; Puig, M.V.; Celada, P.; Shapiro, D.A.; Roth, B.L.; Mengod, G.; Artigas, F. Control of serotonergic function in medial prefrontal cortex by serotonin-2A receptors through a glutamate-dependent mechanism. J. Neurosci. 2001, 21, 9856–9866. [Google Scholar] [CrossRef]
- Bortolozzi, A.; Díaz-Mataix, L.; Toth, M.; Celada, P.; Artigas, F. In vivo actions of aripiprazole on serotonergic and dopaminergic systems in rodent brain. Psychopharmacology 2007, 191, 745–758. [Google Scholar] [CrossRef][Green Version]
- Bortolozzi, A.; Amargós-Bosch, M.; Adell, A.; Díaz-Mataix, L.; Serrats, J.; Pons, S.; Artigas, F. In vivo modulation of 5-hydroxytryptamine release in mouse prefrontal cortex by local 5-HT(2A) receptors: Effect of antipsychotic drugs. Eur. J. Neurosci. 2003, 18, 1235–1246. [Google Scholar] [CrossRef]
- Sanders-Bush, E.; Steranka, L.R. Immediate and long-term effects of p-chloroamphetamine on brain amines. Ann. N. Y. Acad. Sci. 1978, 305, 208–221. [Google Scholar] [CrossRef]
- Perry, K.W.; Kostrzewa, R.M.; Fuller, R.W. Persistence of long-lasting serotonin depletion by p-chloroamphetamine in rat brain after 6-hydroxydopamine lesioning of dopamine neurons. Biochem. Pharmacol. 1995, 50, 1305–1307. [Google Scholar] [CrossRef]
- Van de Kar, L.D.; Javed, A.; Zhang, Y.; Serres, F.; Raap, D.K.; Gray, T.S. 5-HT2A receptors stimulate ACTH, corticosterone, oxytocin, renin, and prolactin release and activate hypothalamic CRF and oxytocin-expressing cells. J. Neurosci. 2001, 21, 3572–3579. [Google Scholar] [CrossRef]
- Zhang, Y.; Damjanoska, K.J.; Carrasco, G.A.; Dudas, B.; D’Souza, D.N.; Tetzlaff, J.; Garcia, F.; Hanley, N.R.; Scripathirathan, K.; Petersen, B.R.; et al. Evidence that 5-HT2A receptors in the hypothalamic paraventricular nucleus mediate neuroendocrine responses to (-) DOI. J. Neurosci. 2002, 22, 9635–9642. [Google Scholar] [CrossRef]
- Gudelsky, G.A.; Koenig, J.I.; Meltzer, H.Y. Thermoregulatory responses to serotonin (5-HT) receptor stimulation in the rat. Evidence for opposing roles of 5-HT2 and 5-HT1A receptors. Neuropharmacology 1986, 25, 1307–1313. [Google Scholar] [CrossRef]
- Mazzola-Pomietto, P.; Aulakh, C.S.; Wozniak, K.M.; Hill, J.L.; Murphy, D.L. Evidence that 1-(2,5-dimethoxy-4-iodophenyl)-2-aminopropane (DOI)-induced hyperthermia in rats is mediated by stimulation of 5-HT2A receptors. Psychopharmacology 1995, 117, 193–199. [Google Scholar] [CrossRef]
- Hoyer, D.; Hannon, J.P.; Martin, G.R. Molecular, pharmacological and functional diversity of 5-HT receptors. Pharmacol. Biochem. Behav. 2002, 71, 533–554. [Google Scholar] [CrossRef]
- Willins, D.L.; Meltzer, H.Y. Direct injection of 5-HT2A receptor agonists into the medial prefrontal cortex produces a head-twitch response in rats. J. Pharmacol. Exp. Ther. 1997, 282, 699–706. [Google Scholar] [PubMed]
- González-Maeso, J.; Yuen, T.; Ebersole, B.J.; Wurmbach, E.; Lira, A.; Zhou, M.; Weisstaub, N.; Hen, R.; Gingrich, J.A.; Sealfon, S.C. Transcriptome fingerprints distinguish hallucinogenic and nonhallucinogenic 5-hydroxytryptamine 2A receptor agonist effects in mouse somatosensory cortex. J. Neurosci. 2003, 23, 8836–8843. [Google Scholar] [CrossRef]
- González-Maeso, J.; Weisstaub, N.V.; Zhou, M.; Chan, P.; Ivic, L.; Ang, R.; Lira, A.; Bradley-Moore, M.; Ge, Y.; Zhou, Q.; et al. Hallucinogens recruit specific cortical 5-HT(2A) receptor-mediated signaling pathways to affect behavior. Neuron 2007, 53, 439–452. [Google Scholar] [CrossRef]
- Tran, T.V.; Shin, E.J.; Ko, S.K.; Nam, Y.; Chung, Y.H.; Jeong, J.H.; Jang, C.G.; Nah, S.Y.; Yamada, K.; Nabeshima, T.; et al. Mountain-cultivated ginseng attenuates phencyclidine-induced abnormal behaviors in mice by positive modulation of glutathione in the prefrontal cortex of mice. J. Med. Food 2016, 19, 961–969. [Google Scholar] [CrossRef]
- Miyamoto, A.; Nakayama, K.; Imaki, H.; Hirose, S.; Jiang, Y.; Abe, M.; Tsukiyama, T.; Nagahama, H.; Ohno, S.; Hatakeyama, S.; et al. Increased proliferation of B cells and auto-immunity in mice lacking protein kinase Cdelta. Nature 2002, 416, 865–869. [Google Scholar] [CrossRef]
- Yamada, J.; Sugimoto, Y.; Ohkura, M.; Inoue, K.; Shinozuka, K.; Kunitomo, M. Role of 5-HT(2) receptor subtypes in depletion of 5-HT induced by p-chloroamphetamine in the mouse frontal cortex. Brain Res. 2001, 911, 141–145. [Google Scholar] [CrossRef]
- Sugimoto, Y.; Ohkura, M.; Inoue, K.; Yamada, J. Involvement of serotonergic and dopaminergic mechanisms in hyperthermia induced by a serotonin-releasing drug, p-chloroamphetamine in mice. Eur. J. Pharmacol. 2001, 430, 265–268. [Google Scholar] [CrossRef]
- Fox, M.A.; Jensen, C.L.; Gallagher, P.S.; Murphy, D.L. Receptor mediation of exaggerated responses to serotonin-enhancing drugs in serotonin transporter (SERT)-deficient mice. Neuropharmacology 2007, 53, 643–656. [Google Scholar] [CrossRef] [PubMed]
- Gogvadze, V.; Orrenius, S.; Zhivotovsky, B. Analysis of mitochondrial dysfunction during cell death. Curr. Protoc. Cell Biol. 2003. [Google Scholar] [CrossRef] [PubMed]
- Shin, E.J.; Nam, Y.; Lee, J.W.; Nguyen, P.T.; Yoo, J.E.; Tran, T.V.; Jeong, J.H.; Jang, C.G.; Oh, Y.J.; Youdim, M.B.H.; et al. N-Methyl, N-propynyl-2-phenylethylamine (MPPE), a Selegiline Analog, Attenuates MPTP-induced dopaminergic toxicity with guaranteed behavioral safety: Involvement of inhibitions of mitochondrial oxidative burdens and p53 gene-elicited pro-apoptotic change. Mol. Neurobiol. 2016, 53, 6251–6269. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Y.; Gu, Q.; Peterson, P.L.; Muizelaar, J.P.; Lee, C.P. Mitochondrial dysfunction and calcium perturbation induced by traumatic brain injury. J. Neurotrauma 1997, 14, 23–34. [Google Scholar] [CrossRef]
- Bruce-Keller, A.J.; Geddes, J.W.; Knapp, P.E.; McFall, R.W.; Keller, J.N.; Holtsberg, F.W.; Parthasarathy, S.; Steiner, S.M.; Mattson, M.P. Anti-death properties of TNF against metabolic poisoning: Mitochondrial stabilization by MnSOD. J. Neuroimmunol. 1999, 93, 53–71. [Google Scholar] [CrossRef]
- Qu, M.; Zhou, Z.; Xu, S.; Chen, C.; Yu, Z.; Wang, D. Mortalin overexpression attenuates beta-amyloid-induced neurotoxicity in SH-SY5Y cells. Brain Res. 2011, 1368, 336–345. [Google Scholar] [CrossRef]
- Tran, H.Q.; Shin, E.J.; Saito, K.; Tran, T.V.; Phan, D.H.; Sharma, N.; Kim, D.W.; Choi, S.Y.; Jeong, J.H.; Jang, C.G.; et al. Indoleamine-2,3-dioxygenase-1 is a molecular target for the protective activity of mood stabilizers against mania-like behavior induced by d-amphetamine. Food Chem. Toxicol. 2020, 136, 110986. [Google Scholar] [CrossRef]
- Mattson, M.P.; Keller, J.N.; Begley, J.G. Evidence for synaptic apoptosis. Exp. Neurol. 1998, 153, 35–48. [Google Scholar] [CrossRef]
- Xu, S.; Pi, H.; Chen, Y.; Zhang, N.; Guo, P.; Lu, Y.; He, M.; Xie, J.; Zhong, M.; Zhang, Y.; et al. Cadmium induced Drp1-dependent mitochondrial fragmentation by disturbing calcium homeostasis in its hepatotoxicity. Cell Death Dis. 2013, 4, e540. [Google Scholar] [CrossRef]
- Janssen, A.J.; Trijbels, F.J.; Sengers, R.C.; Smeitink, J.A.; van den Heuvel, L.P.; Wintjes, L.T.; Stoltenborg-Hogenkamp, B.J.; Rodenburg, R.J. Spectrophotometric assay for complex I of the respiratory chain in tissue samples and cultured fibroblasts. Clin. Chem. 2007, 53, 729–734. [Google Scholar] [CrossRef]
- Lebel, C.P.; Bondy, S.C. Sensitive and rapid quantitation of oxygen reactive species formation in rat synaptosomes. Neurochem. Int. 1990, 17, 435–440. [Google Scholar] [CrossRef]
- Oliver, C.N.; Ahn, B.W.; Moerman, E.J.; Goldstein, S.; Stadtman, E.R. Age-related changes in oxidized proteins. J. Biol. Chem. 1987, 262, 5488–5491. [Google Scholar] [CrossRef]
- Lawrence, R.A.; Burk, R.F. Glutathione peroxidase activity in selenium-deficient rat liver. Biochem. Biophys. Res. Commun. 1976, 71, 952–958. [Google Scholar] [CrossRef]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shin, E.-J.; Jeong, J.H.; Nguyen, B.-T.; Sharma, N.; Nah, S.-Y.; Chung, Y.H.; Lee, Y.; Byun, J.K.; Nabeshima, T.; Ko, S.K.; et al. Ginsenoside Re Protects against Serotonergic Behaviors Evoked by 2,5-Dimethoxy-4-iodo-amphetamine in Mice via Inhibition of PKCδ-Mediated Mitochondrial Dysfunction. Int. J. Mol. Sci. 2021, 22, 7219. https://doi.org/10.3390/ijms22137219
Shin E-J, Jeong JH, Nguyen B-T, Sharma N, Nah S-Y, Chung YH, Lee Y, Byun JK, Nabeshima T, Ko SK, et al. Ginsenoside Re Protects against Serotonergic Behaviors Evoked by 2,5-Dimethoxy-4-iodo-amphetamine in Mice via Inhibition of PKCδ-Mediated Mitochondrial Dysfunction. International Journal of Molecular Sciences. 2021; 22(13):7219. https://doi.org/10.3390/ijms22137219
Chicago/Turabian StyleShin, Eun-Joo, Ji Hoon Jeong, Bao-Trong Nguyen, Naveen Sharma, Seung-Yeol Nah, Yoon Hee Chung, Yi Lee, Jae Kyung Byun, Toshitaka Nabeshima, Sung Kwon Ko, and et al. 2021. "Ginsenoside Re Protects against Serotonergic Behaviors Evoked by 2,5-Dimethoxy-4-iodo-amphetamine in Mice via Inhibition of PKCδ-Mediated Mitochondrial Dysfunction" International Journal of Molecular Sciences 22, no. 13: 7219. https://doi.org/10.3390/ijms22137219
APA StyleShin, E.-J., Jeong, J. H., Nguyen, B.-T., Sharma, N., Nah, S.-Y., Chung, Y. H., Lee, Y., Byun, J. K., Nabeshima, T., Ko, S. K., & Kim, H.-C. (2021). Ginsenoside Re Protects against Serotonergic Behaviors Evoked by 2,5-Dimethoxy-4-iodo-amphetamine in Mice via Inhibition of PKCδ-Mediated Mitochondrial Dysfunction. International Journal of Molecular Sciences, 22(13), 7219. https://doi.org/10.3390/ijms22137219