
3′ Tth Endonuclease Cleavage Polymerase Chain Reaction (3TEC-PCR) Technology for Single-Base-Specific Multiplex Pathogen Detection using a Two-Oligonucleotide System


Abstract
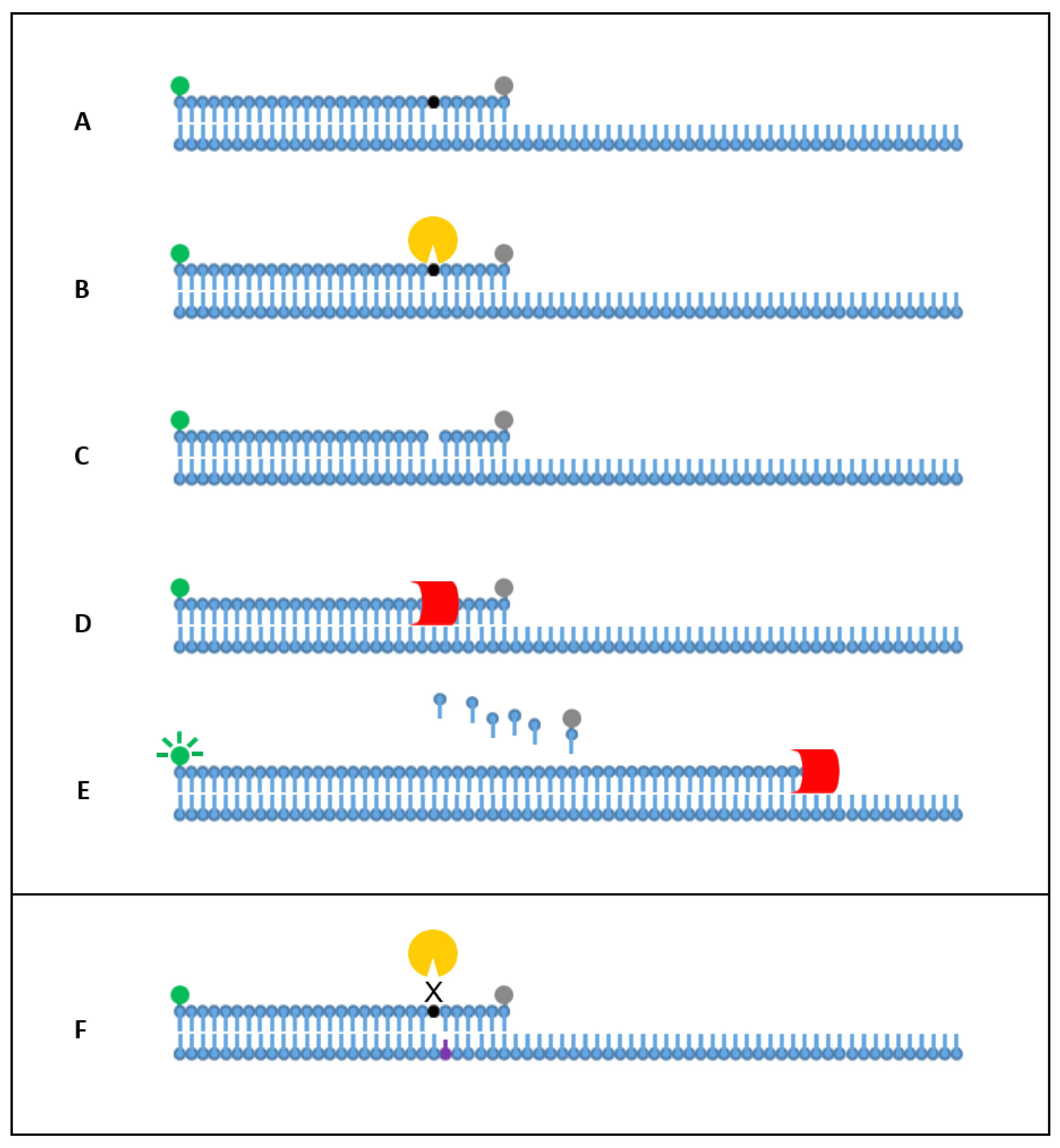
1. Introduction
2. Results
2.1. Singleplex Detection
2.2. Analytical Specificity and Sensitivity
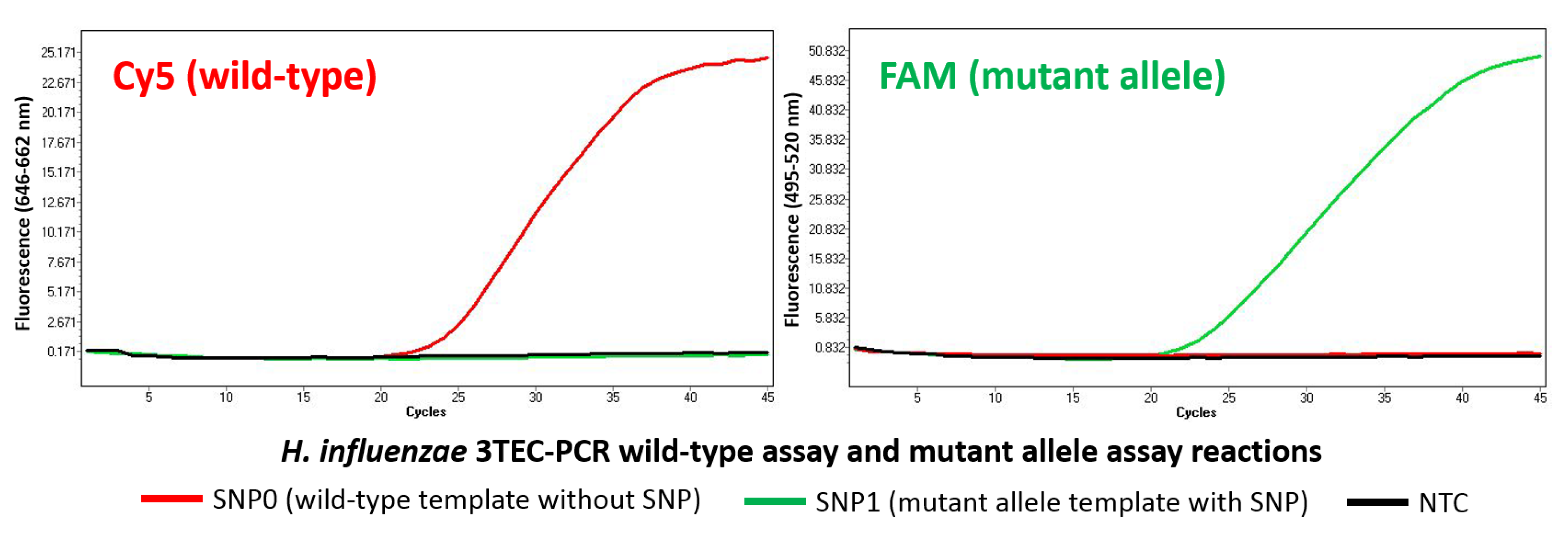
2.3. Allele-Specific Detection
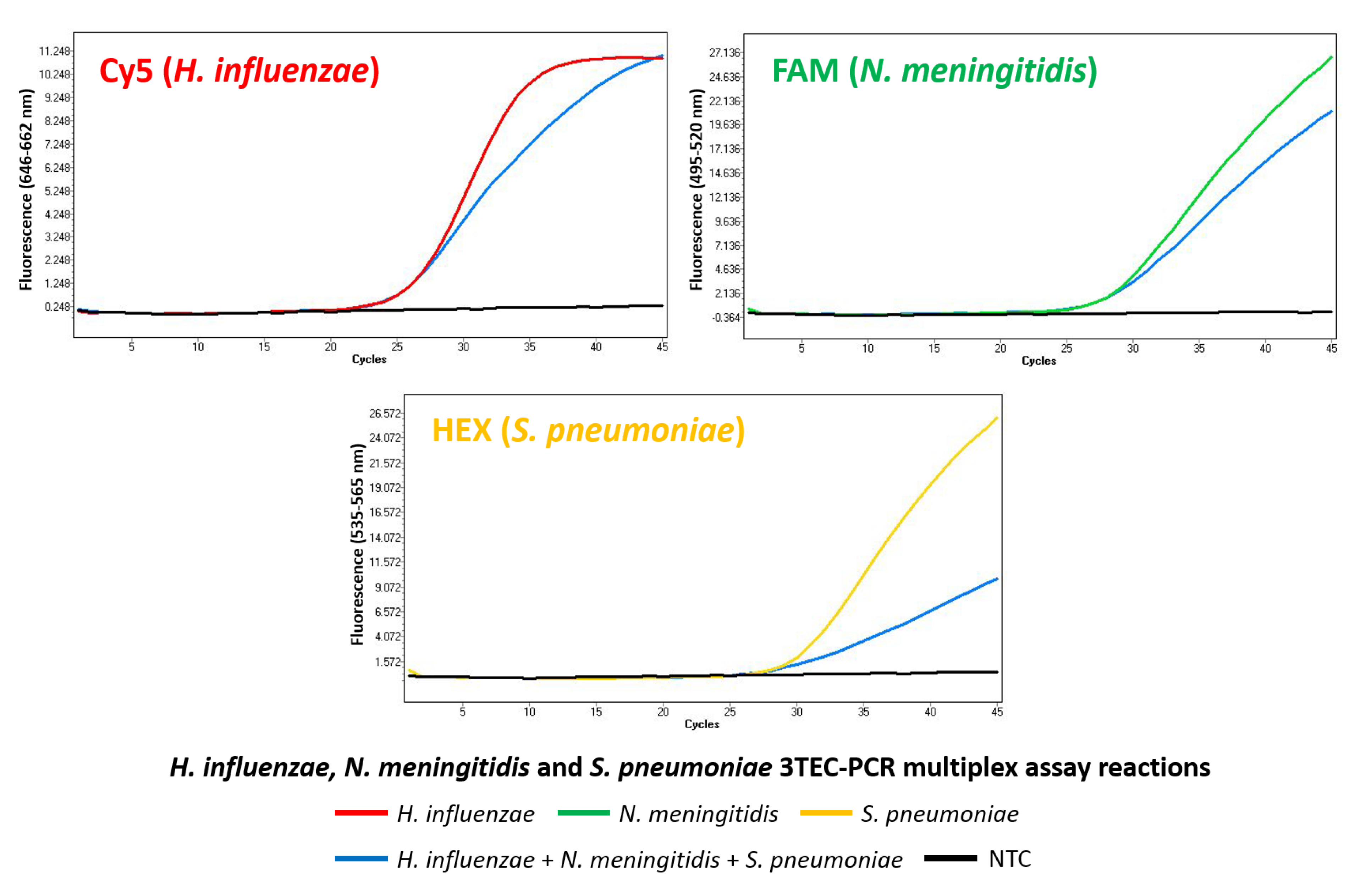
2.4. Multiplex Detection
3. Discussion
4. Materials and Methods
4.1. Bacterial Strains, DNA Extraction, and Quantification
4.2. Diagnostic Targets, Oligonucleotides, and Synthetic Templates
4.3. Singleplex Detection
4.4. Analytical Specificity and Sensitivity
4.5. Allele-Specific Detection
4.6. Multiplex Detection
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Belmont, P.; Constant, J.-F.; Demeunynck, M. Nucleic acid conformation diversity: From structure to function and regulation. Chem. Soc. Rev. 2001, 30, 70–81. [Google Scholar] [CrossRef]
- Minchin, S.; Lodge, J. Understanding biochemistry: Structure and function of nucleic acids. Essays Biochem. 2019, 63, 433–456. [Google Scholar] [CrossRef]
- Scheler, O.; Glynn, B.; Kurg, A. Nucleic acid detection technologies and marker molecules in bacterial diagnostics. Expert Rev. Mol. Diagn. 2014, 14, 489–500. [Google Scholar] [CrossRef]
- Lazcka, O.; Del Campo, F.J.; Munoz, F.X. Pathogen detection: A perspective of traditional methods and biosensors. Biosens. Bioelectron. 2007, 22, 1205–1217. [Google Scholar] [CrossRef]
- Saiki, R.K.; Scharf, S.; Faloona, F.; Mullis, K.B.; Horn, G.T.; Erlich, H.A.; Arnheim, N. Enzymatic amplification of beta-globin genomic sequences and restriction site analysis for diagnosis of sickle cell anemia. Science 1985, 230, 1350–1354. [Google Scholar] [CrossRef]
- Mullis, K.; Faloona, F.; Scharf, S.; Saiki, R.; Horn, G.; Erlich, H. Specific enzymatic amplification of DNA in vitro: The polymerase chain reaction. In Cold Spring Harbor Symposia on Quantitative Biology; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 1986; pp. 263–273. [Google Scholar]
- Mullis, K.B. Process for Amplifying Nucleic Acid Sequences. U.S. Patent US4683202A, 28 July 1987. [Google Scholar]
- Saiki, R.K.; Gelfand, D.H.; Stoffel, S.; Scharf, S.J.; Higuchi, R.; Horn, G.T.; Mullis, K.B.; Erlich, H.A. Primer-directed enzymatic amplification of DNA with a thermostable DNA polymerase. Science 1988, 239, 487–491. [Google Scholar] [CrossRef]
- Yu, A.C.-H.; Vatcher, G.; Yue, X.; Dong, Y.; Li, M.H.; Tam, P.H.; Tsang, P.Y.; Wong, A.K.; Hui, M.H.; Yang, B. Nucleic acid-based diagnostics for infectious diseases in public health affairs. Front. Med. 2012, 6, 173–186. [Google Scholar] [CrossRef]
- Amawi, H.; Abu Deiab, G.A.I.; AAljabali, A.A.; Dua, K.; Tambuwala, M.M. COVID-19 pandemic: An overview of epidemiology, pathogenesis, diagnostics and potential vaccines and therapeutics. Ther. Deliv. 2020, 11, 245–268. [Google Scholar] [CrossRef] [PubMed]
- Weissleder, R.; Lee, H.; Ko, J.; Pittet, M.J. COVID-19 diagnostics in context. Sci. Transl. Med. 2020, 12, eabc1931. [Google Scholar] [CrossRef] [PubMed]
- Kubista, M.; Andrade, J.M.; Bengtsson, M.; Forootan, A.; Jonák, J.; Lind, K.; Sindelka, R.; Sjöback, R.; Sjögreen, B.; Strömbom, L. The real-time polymerase chain reaction. Mol. Asp. Med. 2006, 27, 95–125. [Google Scholar] [CrossRef] [PubMed]
- Tajadini, M.; Panjehpour, M.; Javanmard, S.H. Comparison of SYBR Green and TaqMan methods in quantitative real-time polymerase chain reaction analysis of four adenosine receptor subtypes. Adv. Biomed. Res. 2014, 3, 85. [Google Scholar] [CrossRef]
- Navarro, E.; Serrano-Heras, G.; Castaño, M.; Solera, J. Real-time PCR detection chemistry. Clin. Chim. Acta 2015, 439, 231–250. [Google Scholar] [CrossRef] [PubMed]
- Hoorfar, J.; Malorny, B.; Abdulmawjood, A.; Cook, N.; Wagner, M.; Fach, P. Practical Considerations in Design of Internal Amplification Controls for Diagnostic PCR Assays. J. Clin. Microbiol. 2004, 42, 1863–1868. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Theodore, M.J.; Mair, R.; Trujillo-Lopez, E.; du Plessis, M.; Wolter, N.; Baughman, A.L.; Hatcher, C.; Vuong, J.; Lott, L.; et al. Clinical validation of multiplex real-time PCR assays for detection of bacterial meningitis pathogens. J. Clin. Microbiol. 2012, 50, 702–708. [Google Scholar] [CrossRef] [PubMed]
- Heid, C.A.; Stevens, J.; Livak, K.J.; Williams, P.M. Real time quantitative PCR. Genome Res. 1996, 6, 986–994. [Google Scholar] [CrossRef]
- Brookes, A.J. The essence of SNPs. Gene 1999, 234, 177–186. [Google Scholar] [CrossRef]
- Kim, S.; Misra, A. SNP genotyping: Technologies and biomedical applications. Annu. Rev. Biomed. Eng. 2007, 9, 289–320. [Google Scholar] [CrossRef] [PubMed]
- Fareed, M.; Afzal, M. Review Single nucleotide polymorphism in genome-wide association of human population: A tool for broad spectrum service. Egypt. J. Med. Hum. Genet. 2013, 14, 123–134. [Google Scholar] [CrossRef]
- Shen, W.; Tian, Y.; Ran, T.; Gao, Z. Genotyping and quantification techniques for single-nucleotide polymorphisms. TrAC Trends Anal. Chem. 2015, 69, 1–13. [Google Scholar] [CrossRef]
- Ugozzoli, L.; Wallace, R.B. Allele-specific polymerase chain reaction. Methods 1991, 2, 42–48. [Google Scholar] [CrossRef]
- Cha, R.S.; Zarbl, H.; Keohavong, P.; Thilly, W.G. Mismatch amplification mutation assay (MAMA): Application to the cH-ras gene. Genome Res. 1992, 2, 14–20. [Google Scholar] [CrossRef]
- Livak, K.J. Allelic discrimination using fluorogenic probes and the 5′ nuclease assay. Genet. Anal. Biomol. Eng. 1999, 14, 143–149. [Google Scholar] [CrossRef]
- McGuigan, F.E.; Ralston, S.H. Single nucleotide polymorphism detection: Allelic discrimination using TaqMan. Psychiatr. Genet. 2002, 12, 133–136. [Google Scholar] [CrossRef]
- Imyanitov, E.; Buslov, K.; Suspitsin, E.; Kuligina, E.S.; Belogubova, E.; Grigoriev, M.Y.; Togo, A.; Hanson, K. Improved reliability of allele-specific PCR. Biotechniques 2002, 33, 484–490. [Google Scholar] [CrossRef]
- Ugozzoli, L.A.; Latorra, D.; Pucket, R.; Arar, K.; Hamby, K. Real-time genotyping with oligonucleotide probes containing locked nucleic acids. Anal. Biochem. 2004, 324, 143–152. [Google Scholar] [CrossRef]
- Letertre, C.; Perelle, S.; Dilasser, F.; Arar, K.; Fach, P. Evaluation of the performance of LNA and MGB probes in 5′-nuclease PCR assays. Mol. Cell. Probes 2003, 17, 307–311. [Google Scholar] [CrossRef]
- Karlen, Y.; McNair, A.; Perseguers, S.; Mazza, C.; Mermod, N. Statistical significance of quantitative PCR. BMC Bioinform. 2007, 8, 131. [Google Scholar] [CrossRef]
- Medhurst, A.D.; Harrison, D.C.; Read, S.J.; Campbell, C.A.; Robbins, M.J.; Pangalos, M.N. The use of TaqMan RT-PCR assays for semiquantitative analysis of gene expression in CNS tissues and disease models. J. Neurosci. Methods 2000, 98, 9–20. [Google Scholar] [CrossRef]
- Bustin, S.A.; Benes, V.; Garson, J.A.; Hellemans, J.; Huggett, J.; Kubista, M.; Mueller, R.; Nolan, T.; Pfaffl, M.W.; Shipley, G.L. The MIQE guidelines: Minimum information for publication of quantitative real-time PCR experiments. Clin. Chem. 2009, 55, 611–622. [Google Scholar] [CrossRef]
- Kralik, P.; Ricchi, M. A basic guide to real time PCR in microbial diagnostics: Definitions, parameters, and everything. Front. Microbiol. 2017, 8, 108. [Google Scholar] [CrossRef]
- Corless, C.E.; Guiver, M.; Borrow, R.; Edwards-Jones, V.; Fox, A.J.; Kaczmarski, E.B. Simultaneous detection of Neisseria meningitidis, Haemophilus influenzae, and Streptococcus pneumoniae in suspected cases of meningitis and septicemia using real-time PCR. J. Clin. Microbiol. 2001, 39, 1553–1558. [Google Scholar] [CrossRef] [PubMed]
- Henegariu, O.; Heerema, N.; Dlouhy, S.; Vance, G.; Vogt, P. Multiplex PCR: Critical parameters and step-by-step protocol. Biotechniques 1997, 23, 504–511. [Google Scholar] [CrossRef] [PubMed]
- Elnifro, E.M.; Ashshi, A.M.; Cooper, R.J.; Klapper, P.E. Multiplex PCR: Optimization and application in diagnostic virology. Clin. Microbiol. Rev. 2000, 13, 559–570. [Google Scholar] [CrossRef]
- Whitcombe, D.; Theaker, J.; Guy, S.P.; Brown, T.; Little, S. Detection of PCR products using self-probing amplicons and fluorescence. Nat. Biotechnol. 1999, 17, 804–807. [Google Scholar] [CrossRef] [PubMed]
- Nazarenko, I.A.; Bhatnagar, S.; Hohman, R. A closed tube format for amplification and detection of DNA based on energy transfer. Nucleic Acids Res. 1997, 25, 2516–2521. [Google Scholar] [CrossRef]
- Nazarenko, I.; Lowe, B.; Darfler, M.; Ikonomi, P.; Schuster, D.; Rashtchian, A. Multiplex quantitative PCR using self-quenched primers labeled with a single fluorophore. Nucleic Acids Res. 2002, 30, e37. [Google Scholar] [CrossRef]
- Kandimalla, E.R.; Agrawal, S. ‘Cyclicons’ as hybridization-based fluorescent primer-probes: Synthesis, properties and application in real-time PCR. Bioorganic Med. Chem. 2000, 8, 1911–1916. [Google Scholar] [CrossRef]
- Lee, M.; Siddle, A.; Page, R. ResonSense®: Simple linear fluorescent probes for quantitative homogeneous rapid polymerase chain reaction. Anal. Chim. Acta 2002, 457, 61–70. [Google Scholar] [CrossRef]
- Sherrill, C.B.; Marshall, D.J.; Moser, M.J.; Larsen, C.A.; Daudé-Snow, L.; Prudent, J.R. Nucleic acid analysis using an expanded genetic alphabet to quench fluorescence. J. Am. Chem. Soc. 2004, 126, 4550–4556. [Google Scholar] [CrossRef]
- Chun, J.-Y.; Kim, K.-J.; Hwang, I.-T.; Kim, Y.-J.; Lee, D.-H.; Lee, I.-K.; Kim, J.-K. Dual priming oligonucleotide system for the multiplex detection of respiratory viruses and SNP genotyping of CYP2C19 gene. Nucleic Acids Res. 2007, 35, e40. [Google Scholar] [CrossRef]
- Dobosy, J.R.; Rose, S.D.; Beltz, K.R.; Rupp, S.M.; Powers, K.M.; Behlke, M.A.; Walder, J.A. RNase H-dependent PCR (rhPCR): Improved specificity and single nucleotide polymorphism detection using blocked cleavable primers. BMC Biotechnol. 2011, 11, 80. [Google Scholar] [CrossRef]
- Bekkaoui, F.; Poisson, I.; Crosby, W.; Cloney, L.; Duck, P. Cycling probe technology with RNase H attached to an oligonucleotide. BioTechniques 1996, 20, 240–248. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, Y.; Saito, R.; Zaraket, H.; Dapat, C.; Caperig-Dapat, I.; Suzuki, H. Rapid and specific detection of amantadine-resistant influenza A viruses with a Ser31Asn mutation by the cycling probe method. J. Clin. Microbiol. 2010, 48, 57–63. [Google Scholar] [CrossRef][Green Version]
- Beggs, M.L.; Cave, M.D.; Marlowe, C.; Cloney, L.; Duck, P.; Eisenach, K.D. Characterization of Mycobacterium tuberculosis complex direct repeat sequence for use in cycling probe reaction. J. Clin. Microbiol. 1996, 34, 2985–2989. [Google Scholar] [CrossRef] [PubMed]
- Cairns, M.J.; Turner, R.; Sun, L.-Q. Homogeneous real-time detection and quantification of nucleic acid amplification using restriction enzyme digestion. Biochem. Biophys. Res. Commun. 2004, 318, 684–690. [Google Scholar] [CrossRef]
- Higgins, O.; Clancy, E.; Cormican, M.; Boo, T.W.; Cunney, R.; Smith, T.J. Evaluation of an Internally Controlled Multiplex Tth Endonuclease Cleavage Loop-Mediated Isothermal Amplification (TEC-LAMP) Assay for the Detection of Bacterial Meningitis Pathogens. Int. J. Mol. Sci. 2018, 19, 524. [Google Scholar] [CrossRef]
- Higgins, O.; Smith, T.J. Loop-Primer Endonuclease Cleavage–Loop-Mediated Isothermal Amplification Technology for Multiplex Pathogen Detection and Single-Nucleotide Polymorphism Identification. J. Mol. Diagn. 2020, 22, 640–651. [Google Scholar] [CrossRef] [PubMed]
- Xiang, D.; Li, F.; Wu, C.; Shi, B.; Zhai, K. The G-BHQ synergistic effect: Improved double quenching molecular beacons based on guanine and Black Hole Quencher for sensitive simultaneous detection of two DNAs. Talanta 2017, 174, 289–294. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type | Sequence (5′-3′) |
|---|---|
| H. influenzae | |
| Forward Primer/Probe (Wild-Type) | Cy5-TGCCGCAATGTTTACCTTTGCA(dSpacer)GCGTAG-BHQ2 |
| Forward Primer/Probe (Mutant Allele) | FAM-TGCCGCAATGTTTACCTTTGCA(dSpacer)CCGTAG-BHQ1 |
| Reverse Primer | CTTGCTGTGCCGCGTTTACATTTTCGTAA |
| N. meningitidis | |
| Forward Primer/Probe | FAM-TGTTGGTGGTGTCGCTGTTTGA(dSpacer)ATTGTG-BHQ1 |
| Reverse Primer | TGTGCAAACAGATACGTCCGCAAACCGCC |
| S. pneumoniae | |
| Forward Primer/Probe | HEX-TTTGGTGGTCGATGCGGCTCAA(dSpacer)GAATTG-BHQ1 |
| Reverse Primer | AAGCCAGATAAACGTTGGCAAGAGTTTGA |
| Primer/Probe | TGCCGCAATGTTTACCTTTGCA-GCGTAG |
| ||||||||||||||||||||||||||||| | |
| SNP0 | ACGGCGTTACAAATGGAAACGTCCGCATC |
| SNP1 | ACGGCGTTACAAATGGAAACGTCGGCATC |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Higgins, O.; Smith, T.J. 3′ Tth Endonuclease Cleavage Polymerase Chain Reaction (3TEC-PCR) Technology for Single-Base-Specific Multiplex Pathogen Detection using a Two-Oligonucleotide System. Int. J. Mol. Sci. 2021, 22, 6061. https://doi.org/10.3390/ijms22116061
Higgins O, Smith TJ. 3′ Tth Endonuclease Cleavage Polymerase Chain Reaction (3TEC-PCR) Technology for Single-Base-Specific Multiplex Pathogen Detection using a Two-Oligonucleotide System. International Journal of Molecular Sciences. 2021; 22(11):6061. https://doi.org/10.3390/ijms22116061
Chicago/Turabian StyleHiggins, Owen, and Terry J. Smith. 2021. "3′ Tth Endonuclease Cleavage Polymerase Chain Reaction (3TEC-PCR) Technology for Single-Base-Specific Multiplex Pathogen Detection using a Two-Oligonucleotide System" International Journal of Molecular Sciences 22, no. 11: 6061. https://doi.org/10.3390/ijms22116061
APA StyleHiggins, O., & Smith, T. J. (2021). 3′ Tth Endonuclease Cleavage Polymerase Chain Reaction (3TEC-PCR) Technology for Single-Base-Specific Multiplex Pathogen Detection using a Two-Oligonucleotide System. International Journal of Molecular Sciences, 22(11), 6061. https://doi.org/10.3390/ijms22116061