
BET Protein-Mediated Transcriptional Regulation in Heart Failure


{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
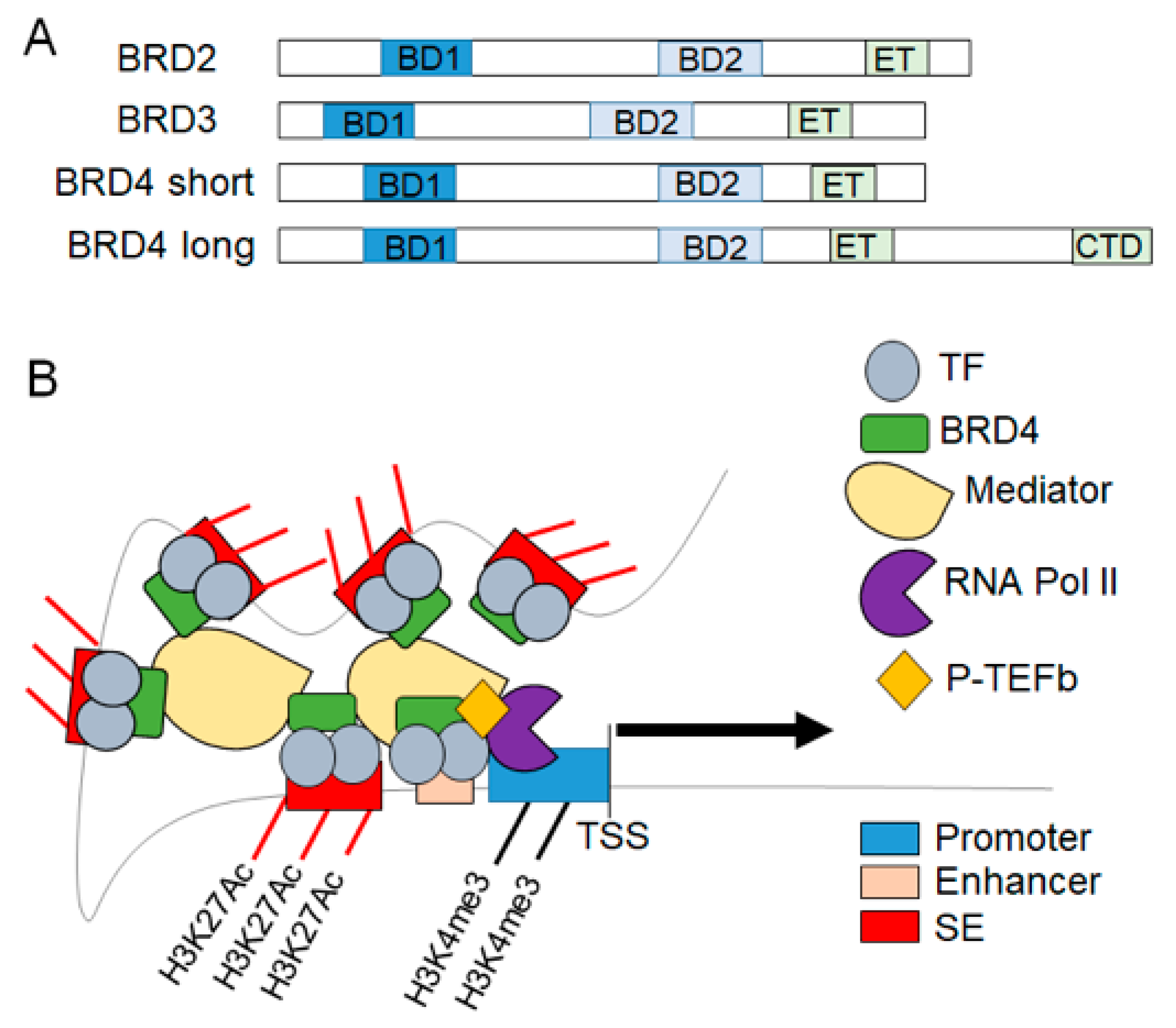
2. BET Proteins in Transcriptional Regulation
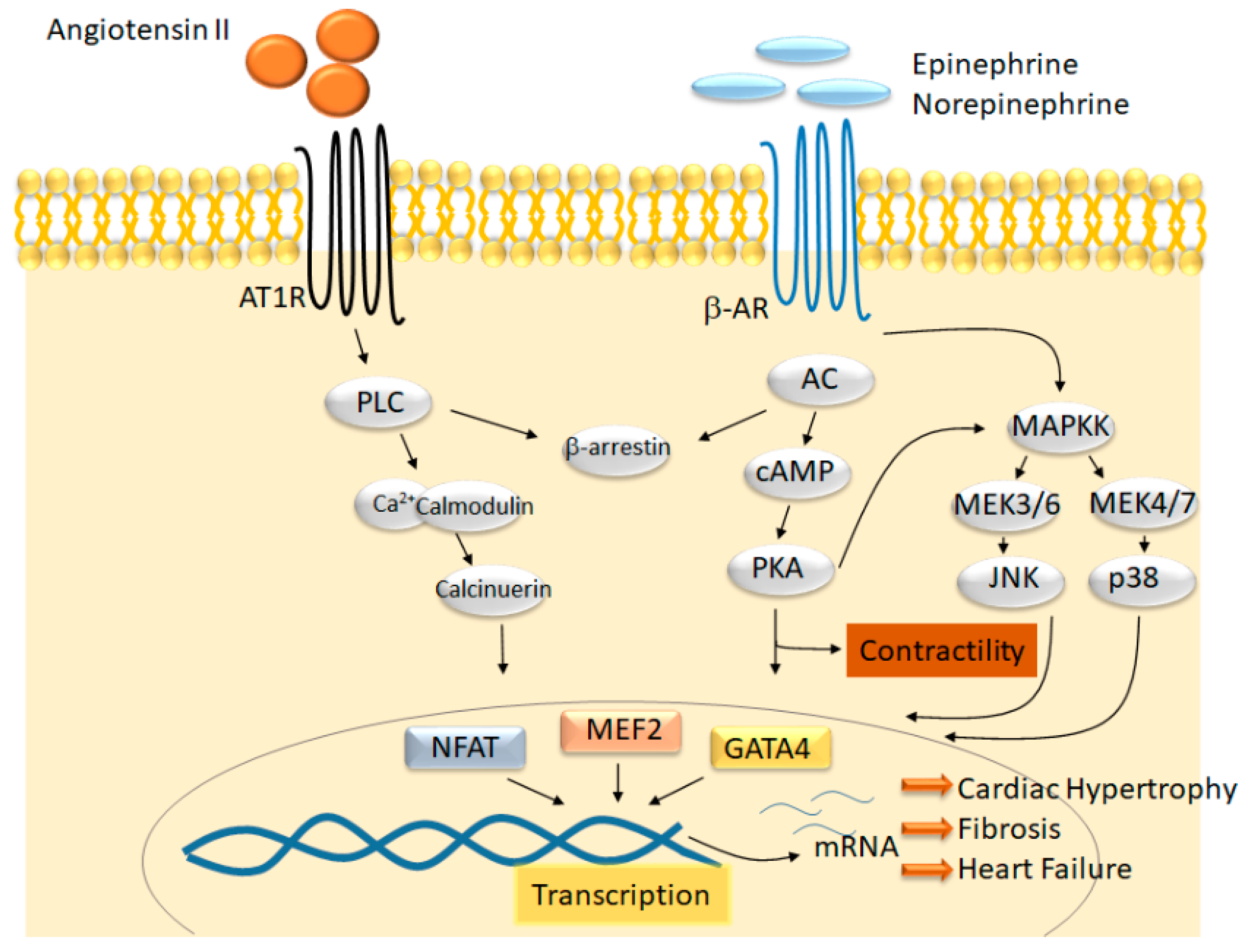
3. BET Protein Function in Cardiomyocytes In Vitro
4. BET Involvement in Pressure-Overload Cardiomyopathy
5. Role of BETs in Ischemic Cardiomyopathy
6. BETs in Genetic Dilated Cardiomyopathy
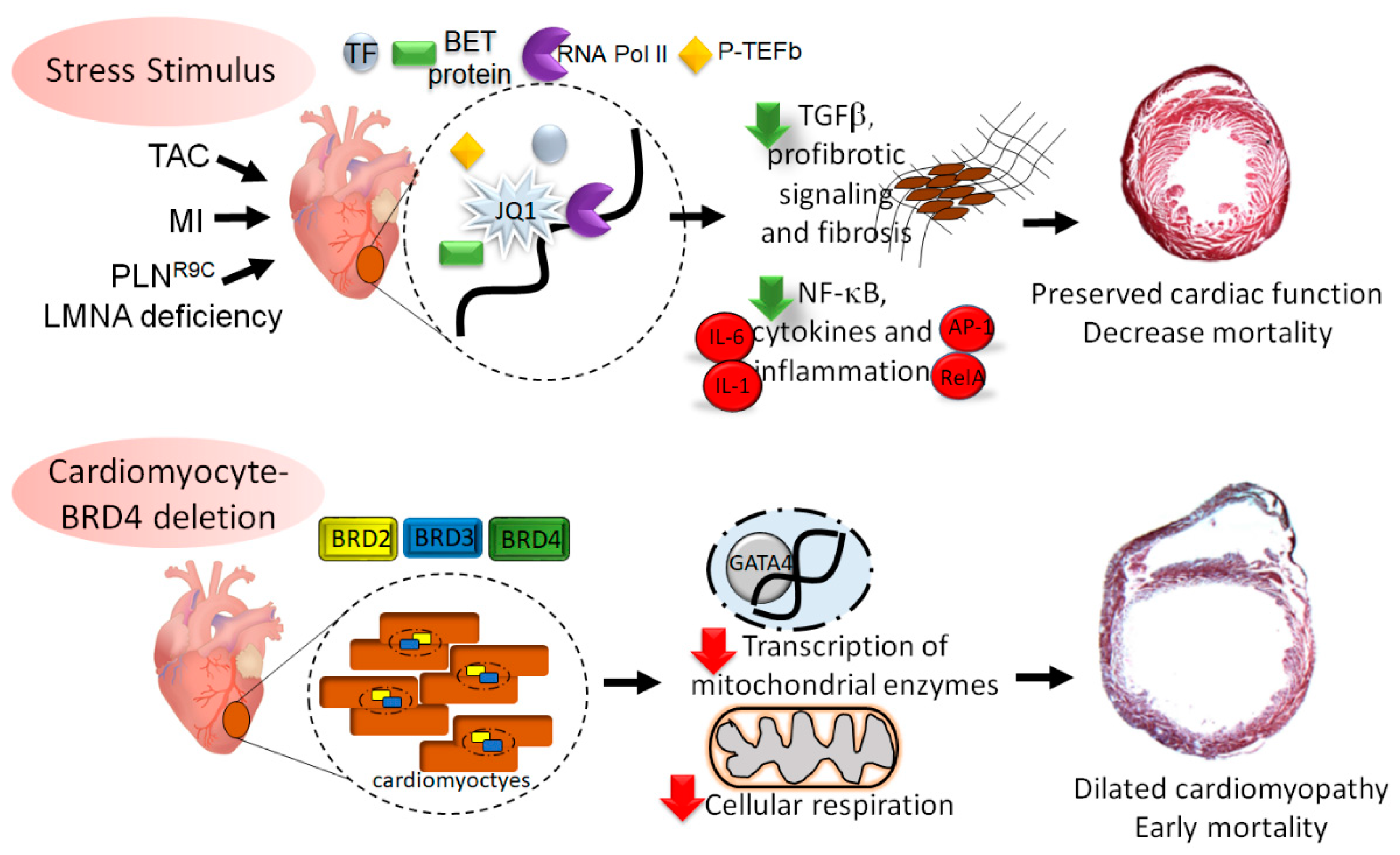
7. BRD4 Mediated Regulation of Cardiac Homeostasis
8. Summary and Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- van Berlo, J.H.; Maillet, M.; Molkentin, J.D. Signaling effectors underlying pathologic growth and remodeling of the heart. J. Clin. Investig. 2013, 123, 37–45. [Google Scholar] [CrossRef] [PubMed]
- Burke, M.A.; Cook, S.A.; Seidman, J.G.; Seidman, C.E. Clinical and Mechanistic Insights into the Genetics of Cardiomyopathy. J. Am. Coll. Cardiol. 2016, 68, 2871–2886. [Google Scholar] [CrossRef] [PubMed]
- Bacmeister, L.; Schwarzl, M.; Warnke, S.; Stoffers, B.; Blankenberg, S.; Westermann, D.; Lindner, D. Inflammation and fibrosis in murine models of heart failure. Basic Res. Cardiol. 2019, 114, 19. [Google Scholar] [CrossRef] [PubMed]
- Hartupee, J.; Mann, J.H.D.L. Neurohormonal activation in heart failure with reduced ejection fraction. Nat. Rev. Cardiol. 2017, 14, 30–38. [Google Scholar] [CrossRef]
- Spitz, F.; Furlong, E.E.M. Transcription factors: From enhancer binding to developmental control. Nat. Rev. Genet. 2012, 13, 613–626. [Google Scholar] [CrossRef] [PubMed]
- Creyghton, M.P.; Cheng, A.W.; Welstead, G.G.; Kooistra, T.; Carey, B.W.; Steine, E.J.; Hanna, J.; Lodato, M.A.; Frampton, G.M.; Sharp, P.A.; et al. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc. Natl. Acad. Sci. USA 2010, 107, 21931–21936. [Google Scholar] [CrossRef] [PubMed]
- Barski, A.; Cuddapah, S.; Cui, K.; Roh, T.-Y.; Schones, D.E.; Wang, Z.; Wei, G.; Chepelev, I.; Zhao, K. High-Resolution Profiling of Histone Methylations in the Human Genome. Cell 2007, 129, 823–837. [Google Scholar] [CrossRef]
- Rivera, C.M.; Ren, B. Mapping Human Epigenomes. Cell 2013, 155, 39–55. [Google Scholar] [CrossRef]
- Tang, F.; Yang, Z.; Tan, Y.; Li, Y. Super-enhancer function and its application in cancer targeted therapy. NPJ Precis. Oncol. 2020, 4, 2. [Google Scholar] [CrossRef]
- Jang, M.K.; Mochizuki, K.; Zhou, M.; Jeong, H.-S.; Brady, J.N.; Ozato, K. The Bromodomain Protein Brd4 Is a Positive Regulatory Component of P-TEFb and Stimulates RNA Polymerase II-Dependent Transcription. Mol. Cell 2005, 19, 523–534. [Google Scholar] [CrossRef]
- Tian, B.; Zhao, Y.; Sun, H.; Zhang, Y.; Yang, J.; Brasier, A.R. BRD4 mediates NF-κB-dependent epithelial-mesenchymal transition and pulmonary fibrosis via transcriptional elongation. Am. J. Physiol. Cell. Mol. Physiol. 2016, 311, L1183–L1201. [Google Scholar] [CrossRef] [PubMed]
- Ijaz, T.; Jamaluddin, M.; Zhao, Y.; Zhang, Y.; Jay, J.; Finnerty, C.C.; Herndon, D.N.; Tilton, R.G.; Brasier, A.R. Coordinate activities of BRD4 and CDK9 in the transcriptional elongation complex are required for TGFβ-induced Nox4 expression and myofibroblast transdifferentiation. Cell Death Dis. 2017, 8, e2606. [Google Scholar] [CrossRef] [PubMed]
- Papait, R.; Cattaneo, P.; Kunderfranco, P.; Greco, C.; Carullo, P.; Guffanti, A.; Viganò, V.; Stirparo, G.G.; Latronico, M.V.G.; Hasenfuss, G.; et al. Genome-wide analysis of histone marks identifying an epigenetic signature of promoters and enhancers underlying cardiac hypertrophy. Proc. Natl. Acad. Sci. USA 2013, 110, 20164–20169. [Google Scholar] [CrossRef] [PubMed]
- Anand, P.; Brown, J.D.; Lin, C.Y.; Qi, J.; Zhang, R.; Artero, P.C.; Alaiti, M.A.; Bullard, J.; Alazem, K.; Margulies, K.B.; et al. BET Bromodomains Mediate Transcriptional Pause Release in Heart Failure. Cell 2013, 154, 569–582. [Google Scholar] [CrossRef] [PubMed]
- Sayed, D.; He, M.; Yang, Z.; Lin, L.; Abdellatif, M. Transcriptional Regulation Patterns Revealed by High Resolution Chromatin Immunoprecipitation during Cardiac Hypertrophy. J. Biol. Chem. 2013, 288, 2546–2558. [Google Scholar] [CrossRef]
- Wei, J.Q.; Shehadeh, L.A.; Mitrani, J.M.; Pessanha, M.; Slepak, T.I.; Webster, K.A.; Bishopric, N.H. Quantitative Control of Adaptive Cardiac Hypertrophy by Acetyltransferase p300. Circulation 2008, 118, 934–946. [Google Scholar] [CrossRef]
- Kong, Y.; Tannous, P.; Lu, G.; Berenji, K.; Rothermel, B.A.; Olson, E.N.; Hill, J.A. Suppression of Class I and II Histone Deacetylases Blunts Pressure-Overload Cardiac Hypertrophy. Circulation 2006, 113, 2579–2588. [Google Scholar] [CrossRef]
- Antos, C.L.; McKinsey, T.A.; Dreitz, M.; Hollingsworth, L.M.; Zhang, C.-L.; Schreiber, K.; Rindt, H.; Gorczynski, R.J.; Olson, E.N. Dose-dependent Blockade to Cardiomyocyte Hypertrophy by Histone Deacetylase Inhibitors. J. Biol. Chem. 2003, 278, 28930–28937. [Google Scholar] [CrossRef]
- Kee, H.J. Inhibition of Histone Deacetylation Blocks Cardiac Hypertrophy Induced by Angiotensin II Infusion and Aortic Banding. Circulation 2006, 113, 51–59. [Google Scholar] [CrossRef]
- Di Salvo, T.G.; Haldar, S.M. Epigenetic mechanisms in heart failure pathogenesis. Circ. Heart Fail 2014, 7, 850–863. [Google Scholar] [CrossRef]
- Dhalluin, C.; Carlson, J.E.; Zeng, L.; He, C.; Aggarwal, A.K.; Zhou, M.-M. Structure and ligand of a histone acetyltransferase bromodomain. Nat. Cell Biol. 1999, 399, 491–496. [Google Scholar] [CrossRef]
- Filippakopoulos, P.; Qi, J.; Picaud, S.; Shen, Y.; Smith, M.C.; Fedorov, O.; Morse, E.M.; Keates, T.; Hickman, T.T.; Felletar, I.; et al. Selective inhibition of BET bromodomains. Nature 2010, 468, 1067–1073. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Vakoc, C.R. The Mechanisms behind the Therapeutic Activity of BET Bromodomain Inhibition. Mol. Cell 2014, 54, 728–736. [Google Scholar] [CrossRef] [PubMed]
- Haldar, S.M.; McKinsey, T.A. BET-ting on chromatin-based therapeutics for heart failure. J. Mol. Cell. Cardiol. 2014, 74, 98–102. [Google Scholar] [CrossRef]
- Antolic, A.; Wakimoto, H.; Jiao, Z.; Gorham, J.M.; DePalma, S.R.; Lemieux, M.E.; Conner, D.A.; Lee, D.Y.; Qi, J.; Seidman, J.G.; et al. BET bromodomain proteins regulate transcriptional reprogramming in genetic dilated cardiomyopathy. JCI Insight 2020, 5. [Google Scholar] [CrossRef] [PubMed]
- Cheung, K.L.; Zhang, F.; Jaganathan, A.; Sharma, R.; Zhang, Q.; Konuma, T.; Shen, T.; Lee, J.-Y.; Ren, C.; Chen, C.-H.; et al. Distinct Roles of Brd2 and Brd4 in Potentiating the Transcriptional Program for Th17 Cell Differentiation. Mol. Cell 2017, 65, 1068–1080.e5. [Google Scholar] [CrossRef]
- Lambert, J.-P.; Picaud, S.; Fujisawa, T.; Hou, H.; Savitsky, P.; Uusküla-Reimand, L.; Gupta, G.D.; Abdouni, H.; Lin, Z.-Y.; Tucholska, M.; et al. Interactome Rewiring Following Pharmacological Targeting of BET Bromodomains. Mol. Cell 2019, 73, 621–638.e17. [Google Scholar] [CrossRef]
- Winter, G.E.; Mayer, A.; Buckley, D.L.; Erb, M.A.; Roderick, J.E.; Vittori, S.; Reyes, J.M.; di Iulio, J.; Souza, A.; Ott, C.J.; et al. BET Bromodomain Proteins Function as Master Transcription Elongation Factors Independent of CDK9 Recruitment. Mol. Cell 2017, 67, 5–18.e19. [Google Scholar] [CrossRef]
- Devaiah, B.N.; Lewis, B.A.; Cherman, N.; Hewitt, M.C.; Albrecht, B.K.; Robey, P.G.; Ozato, K.; Sims, R.J.; Singer, D.S. BRD4 is an atypical kinase that phosphorylates Serine2 of the RNA Polymerase II carboxy-terminal domain. Proc. Natl. Acad. Sci. USA 2012, 109, 6927–6932. [Google Scholar] [CrossRef]
- Jiang, Y.W.; Veschambre, P.; Erdjument-Bromage, H.; Tempst, P.; Conaway, J.W.; Conaway, R.C.; Kornberg, R.D. Mammalian mediator of transcriptional regulation and its possible role as an end-point of signal transduction pathways. Proc. Natl. Acad. Sci. USA 1998, 95, 8538–8543. [Google Scholar] [CrossRef]
- Devaiah, B.N.; Case-Borden, C.; Gegonne, A.; Hsu, C.H.; Chen, Q.; Meerzaman, D.; Dey, A.; Ozato, K.; Singer, D.S. BRD4 is a histone acetyltransferase that evicts nucleosomes from chromatin. Nat. Struct. Mol. Biol. 2016, 23, 540–548. [Google Scholar] [CrossRef]
- Tian, B.; Liu, Z.; Yang, J.; Sun, H.; Zhao, Y.; Wakamiya, M.; Chen, H.; Rytting, E.; Zhou, J.; Brasier, A.R. Selective Antagonists of the Bronchiolar Epithelial NF-κB-Bromodomain-Containing Protein 4 Pathway in Viral-Induced Airway Inflammation. Cell Rep. 2018, 23, 1138–1151. [Google Scholar] [CrossRef] [PubMed]
- Uppal, S.; Gegonne, A.; Chen, Q.; Thompson, P.S.; Cheng, D.; Mu, J.; Meerzaman, D.; Misra, H.S.; Singer, D.S. The Bromodomain Protein 4 Contributes to the Regulation of Alternative Splicing. Cell Rep. 2019, 29, 2450–2460.e5. [Google Scholar] [CrossRef] [PubMed]
- Sabari, B.R.; Dall’Agnese, A.; Boija, A.; Klein, I.A.; Coffey, E.L.; Shrinivas, K.; Abraham, B.J.; Hannett, N.M.; Zamudio, A.V.; Manteiga, J.C.; et al. Coactivator condensation at super-enhancers links phase separation and gene control. Science 2018, 361, eaar3958. [Google Scholar] [CrossRef]
- Lee, J.-E.; Park, Y.-K.; Park, S.; Jang, Y.; Waring, N.; Dey, A.; Ozato, K.; Lai, B.; Peng, W.; Ge, K. Brd4 binds to active enhancers to control cell identity gene induction in adipogenesis and myogenesis. Nat. Commun. 2017, 8, 1–12. [Google Scholar] [CrossRef]
- Wu, T.; Kamikawa, Y.F.; Donohoe, M.E. Brd4′s Bromodomains Mediate Histone H3 Acetylation and Chromatin Remodeling in Pluripotent Cells through P300 and Brg1. Cell Rep. 2018, 25, 1756–1771. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.-P.; Yang, J.; Han, P.; Cheng, H.-L.; Shang, C.; Ashley, E.; Zhou, B. Chromatin regulation by Brg1 underlies heart muscle development and disease. Nat. Cell Biol. 2010, 466, 62–67. [Google Scholar] [CrossRef]
- Spiltoir, J.I.; Stratton, M.S.; Cavasin, M.A.; Demos-Davies, K.; Reid, B.G.; Qi, J.; Bradner, J.E.; McKinsey, T.A. BET acetyl-lysine binding proteins control pathological cardiac hypertrophy. J. Mol. Cell. Cardiol. 2013, 63, 175–179. [Google Scholar] [CrossRef] [PubMed]
- Stratton, M.S.; Lin, C.Y.; Anand, P.; Tatman, P.D.; Ferguson, B.S.; Wickers, S.T.; Ambardekar, A.V.; Sucharov, C.C.; Bradner, J.E.; Haldar, S.M.; et al. Signal-Dependent Recruitment of BRD4 to Cardiomyocyte Super-Enhancers Is Suppressed by a MicroRNA. Cell Rep. 2016, 16, 1366–1378. [Google Scholar] [CrossRef]
- Duan, Q.; McMahon, S.; Anand, P.; Shah, H.; Thomas, S.; Salunga, H.T.; Huang, Y.; Zhang, R.; Sahadevan, A.; Lemieux, M.E.; et al. BET bromodomain inhibition suppresses innate inflammatory and profibrotic transcriptional networks in heart failure. Sci. Transl. Med. 2017, 9, eaah5084. [Google Scholar] [CrossRef]
- Burke, M.A.; Chang, S.; Wakimoto, H.; Gorham, J.M.; Conner, D.A.; Christodoulou, D.C.; Parfenov, M.G.; DePalma, S.R.; Eminaga, S.; Konno, T.; et al. Molecular Profiling of Dilated Cardiomyopathy That Progresses to Heart Failure. Available online: https://insight.jci.org/articles/view/86898/pdf (accessed on 22 August 2020).
- Auguste, G.; Rouhi, L.; Matkovich, S.J.; Coarfa, C.; Robertson, M.J.; Czernuszewicz, G.; Gurha, P.; Marian, A.J. BET bromodomain inhibition attenuates cardiac phenotype in myocyte-specific lamin A/C–deficient mice. J. Clin. Investig. 2020, 130, 4740–4758. [Google Scholar] [CrossRef]
- Padmanabhan, A.; Alexanian, M.; Linares-Saldana, R.; González-Terán, B.; Andreoletti, G.; Huang, Y.; Connolly, A.J.; Kim, W.; Hsu, A.; Duan, Q.; et al. BRD4 (Bromodomain-Containing Protein 4) Interacts with GATA4 (GATA Binding Protein 4) to Govern Mitochondrial Homeostasis in Adult Cardiomyocytes. Circulation 2020, 142, 2338–2355. [Google Scholar] [CrossRef]
- Kim, S.Y.; Zhang, X.; Schiattarella, G.G.; Altamirano, F.; Ramos, T.A.; French, K.M.; Jiang, N.; Szweda, P.A.; Evers, B.M.; May, H.I.; et al. Epigenetic Reader BRD4 (Bromodomain-Containing Protein 4) Governs Nucleus-Encoded Mitochondrial Transcriptome to Regulate Cardiac Function. Circulation 2020, 142, 2356–2370. [Google Scholar] [CrossRef]
- Bolden, J.E.; Tasdemir, N.; Dow, L.E.; van Es, J.H.; Wilkinson, J.E.; Zhao, Z.; Clevers, H.; Lowe, S.W. Inducible in vivo Silencing of Brd4 Identifies Potential Toxicities of Sustained BET Protein Inhibition. Cell Rep. 2014, 8, 1919–1929. [Google Scholar] [CrossRef]
- Bechter, O.; Schöffski, P. Make Your Best BET: The Emerging Role of BET Inhibitor Treatment in Malignant Tumors. Pharmacol. Ther. 2020, 208, 107479. [Google Scholar] [CrossRef]
- Nicholls, S.J.; Puri, R.; Wolski, K.; Ballantyne, C.M.; Barter, P.J.; Brewer, H.B.; Kastelein, J.J.P.; Hu, B.; Uno, K.; Kataoka, Y.; et al. Effect of the BET Protein Inhibitor, RVX-208, on Progression of Coronary Atherosclerosis: Results of the Phase 2b, Randomized, Double-Blind, Multicenter, ASSURE Trial. Am. J. Cardiovasc. Drugs 2016, 16, 55–65. [Google Scholar] [CrossRef]
- Shishikura, D.; Kataoka, Y.; Honda, S.; Takata, K.; Kim, S.W.; Andrews, J.; Psaltis, P.J.; Sweeney, M.; Kulikowski, E.; Johansson, J.; et al. The Effect of Bromodomain and Extra-Terminal Inhibitor Apabetalone on Attenuated Coronary Atherosclerotic Plaque: Insights from the ASSURE Trial. Am. J. Cardiovasc. Drugs 2019, 19, 49–57. [Google Scholar] [CrossRef] [PubMed]
- Ray, K.K.; Nicholls, S.J.; Buhr, K.A.; Ginsberg, H.N.; Johansson, J.O.; Kalantar-Zadeh, K.; Kulikowski, E.; Toth, P.P.; Wong, N.; Sweeney, M.; et al. Effect of Apabetalone Added to Standard Therapy on Major Adverse Cardiovascular Events in Patients With Recent Acute Coronary Syndrome and Type 2 Diabetes: A Randomized Clinical Trial. JAMA 2020, 323, 1565. [Google Scholar] [CrossRef] [PubMed]
- Nicholls, S.J.; Schwartz, G.G.; Buhr, K.A.; Ginsberg, H.N.; Johansson, J.O.; Kalantar-Zadeh, K.; Kulikowski, E.; Toth, P.P.; Wong, N.; Sweeney, M.; et al. Apabetalone and Hospitalization for Heart Failure in Patients Following an Acute Coronary Syndrome: A Prespecified Analysis of the BETonMACE Study. Cardiovasc. Diabetol. 2021, 20, 13. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ijaz, T.; Burke, M.A. BET Protein-Mediated Transcriptional Regulation in Heart Failure. Int. J. Mol. Sci. 2021, 22, 6059. https://doi.org/10.3390/ijms22116059
Ijaz T, Burke MA. BET Protein-Mediated Transcriptional Regulation in Heart Failure. International Journal of Molecular Sciences. 2021; 22(11):6059. https://doi.org/10.3390/ijms22116059
Chicago/Turabian StyleIjaz, Talha, and Michael A. Burke. 2021. "BET Protein-Mediated Transcriptional Regulation in Heart Failure" International Journal of Molecular Sciences 22, no. 11: 6059. https://doi.org/10.3390/ijms22116059
APA StyleIjaz, T., & Burke, M. A. (2021). BET Protein-Mediated Transcriptional Regulation in Heart Failure. International Journal of Molecular Sciences, 22(11), 6059. https://doi.org/10.3390/ijms22116059