
LEDGF/p75 Is Required for an Efficient DNA Damage Response

 ,
,  , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. CRISPR/Cas9-Generated LEDGF Cell Models
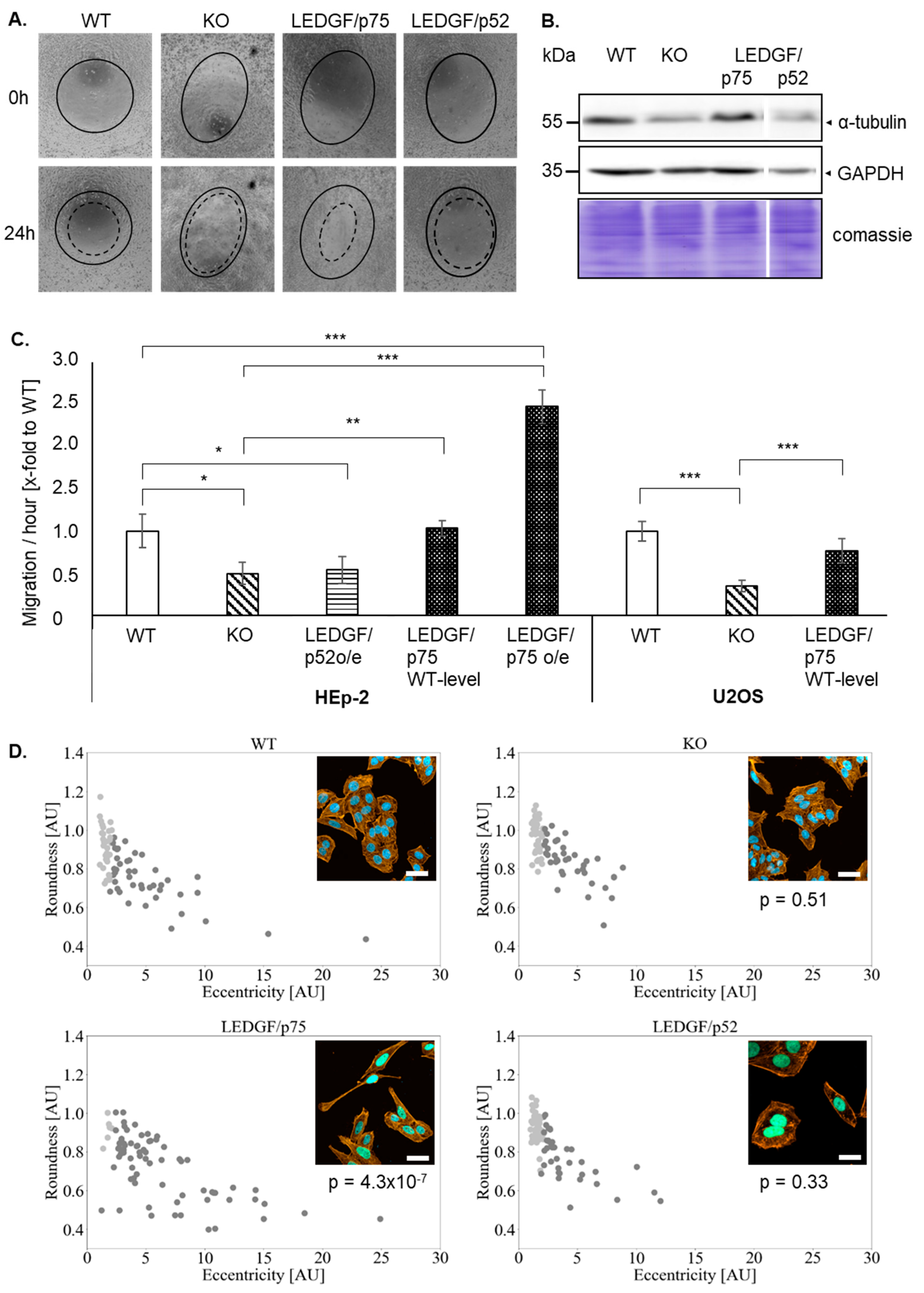
2.2. Depletion of LEDGF Decreases Cellular Migration
2.3. LEDGF Depletion Sensitizes Cancer Cells toward Etoposide
2.4. LEDGF Depletion Impairs DNA Damage Response via Homology-Directed Repair
2.5. LEDGF Depletion Results in Dysfunctional DNA Damage Response
3. Discussion
4. Materials and Methods
4.1. Antibodies
4.2. Cell Lines and Culture
4.3. Cloning
4.4. Generation of LEDGF-Modified Cell Clones
4.5. Proliferation Analysis
4.6. Exposure to Etoposide
4.7. Digital Image Analysis and Bioimage Informatics
4.8. Scratch Assay
4.9. Immunofluorescence
4.10. Immunoblotting
4.11. Pulsed Field Gel Electrophoresis (PFGE)
4.12. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Basu, A.; Woods-Burnham, L.; Ortiz, G.; Rios-Colon, L.; Figueroa, J.; Albesa, R.; Andrade, L.E.; Mahler, M.; Casiano, C.A. Specificity of Antinuclear Autoantibodies Recognizing the Dense Fine Speckled Nuclear Pattern: Preferential Targeting of DFS70/LEDGFp75 over Its Interacting Partner MeCP2. Clin. Immunol. 2015, 161, 241–250. [Google Scholar] [CrossRef][Green Version]
- Debyser, Z.; Christ, F.; De Rijck, J.; Gijsbers, R. Host Factors for Retroviral Integration Site Selection. Trends Biochem. Sci. 2015, 40, 108–116. [Google Scholar] [CrossRef]
- Leitz, J.; Reuschenbach, M.; Lohrey, C.; Honegger, A.; Accardi, R.; Tommasino, M.; Llano, M.; von Knebel Doeberitz, M.; Hoppe-Seyler, K.; Hoppe-Seyler, F. Oncogenic Human Papillomaviruses Activate the Tumor-Associated Lens Epithelial-Derived Growth Factor (LEDGF) Gene. PLoS Pathog. 2014, 10, e1003957. [Google Scholar] [CrossRef] [PubMed]
- Basu, A.; Rojas, H.; Banerjee, H.; Cabrera, I.B.; Perez, K.Y.; De León, M.; Casiano, C.A. Expression of the Stress Response Oncoprotein LEDGF/P75 in Human Cancer: A Study of 21 Tumor Types. PLoS ONE 2012, 7, e30132. [Google Scholar] [CrossRef] [PubMed]
- Daniels, T.; Zhang, J.; Gutierrez, I.; Elliot, M.L.; Yamada, B.; Heeb, M.J.; Sheets, S.M.; Wu, X.; Casiano, C.A. Antinuclear Autoantibodies in Prostate Cancer: Immunity to LEDGF/P75, a Survival Protein Highly Expressed in Prostate Tumors and Cleaved during Apoptosis. Prostate 2005, 62, 14–26. [Google Scholar] [CrossRef] [PubMed]
- Brown-Bryan, T.A.; Leoh, L.S.; Ganapathy, V.; Pacheco, F.J.; Mediavilla-Varela, M.; Filippova, M.; Linkhart, T.A.; Gijsbers, R.; Debyser, Z.; Casiano, C.A. Alternative Splicing and Caspase-Mediated Cleavage Generate Antagonistic Variants of the Stress Oncoprotein LEDGF/P75. Mol. Cancer Res. 2008, 6, 1293–1307. [Google Scholar] [CrossRef]
- Pradeepa, M.M.; Sutherland, H.G.; Ule, J.; Grimes, G.R.; Bickmore, W.A. Psip1/Ledgf P52 Binds Methylated Histone H3K36 and Splicing Factors and Contributes to the Regulation of Alternative Splicing. PLoS Genet. 2012, 8, e1002717. [Google Scholar] [CrossRef]
- Ochs, R.L.; Mahler, M.; Basu, A.; Rios-Colon, L.; Sanchez, T.W.; Andrade, L.E.; Fritzler, M.J.; Casiano, C.A. The Significance of Autoantibodies to DFS70/LEDGFp75 in Health and Disease: Integrating Basic Science with Clinical Understanding. Clin. Exp. Med. 2016, 16, 273–293. [Google Scholar] [CrossRef]
- Ishihara, K.; Fatma, N.; Bhargavan, B.; Chhunchha, B.; Kubo, E.; Dey, S.; Takamura, Y.; Kumar, A.; Singh, D.P. Lens Epithelium-Derived Growth Factor DeSumoylation by Sumo-Specific Protease-1 Regulates Its Transcriptional Activation of Small Heat Shock Protein and the Cellular Response: Senp-1 Regulates LEDGF Transcriptional Activity. FEBS J. 2012, 279, 3048–3070. [Google Scholar] [CrossRef]
- Bhargavan, B.; Fatma, N.; Chhunchha, B.; Singh, V.; Kubo, E.; Singh, D.P. LEDGF Gene Silencing Impairs the Tumorigenicity of Prostate Cancer DU145 Cells by Abating the Expression of Hsp27 and Activation of the Akt/ERK Signaling Pathway. Cell Death Dis. 2012, 3, e316. [Google Scholar] [CrossRef]
- Fatma, N.; Singh, D.P.; Shinohara, T.; Chylack, L.T. Transcriptional Regulation of the Antioxidant Protein 2 Gene, a Thiol-Specific Antioxidant, by Lens Epithelium-Derived Growth Factor to Protect Cells from Oxidative Stress. J. Biol. Chem. 2001, 276, 48899–48907. [Google Scholar] [CrossRef]
- Ríos-Colón, L.; Ross, C.K.C.-D.; Basu, A.; Elix, C.; Alicea-Polanco, I.; Sanchez, T.W.; Radhakrishnan, V.; Chen, C.-S.; Casiano, C.A. Targeting the Stress Oncoprotein LEDGF/P75 to Sensitize Chemoresistant Prostate Cancer Cells to Taxanes. Oncotarget 2017, 8, 24915. [Google Scholar] [CrossRef]
- Li, L.; Wang, Y. Cross-Talk between the H3K36me3 and H4K16ac Histone Epigenetic Marks in DNA Double-Strand Break Repair. J. Biol. Chem. 2017, 292, 11951–11959. [Google Scholar] [CrossRef]
- Tang, J.; Cho, N.W.; Cui, G.; Manion, E.M.; Shanbhag, N.M.; Botuyan, M.V.; Mer, G.; Greenberg, R.A. Acetylation Limits 53BP1 Association with Damaged Chromatin to Promote Homologous Recombination. Nat. Struct. Mol. Biol. 2013, 20, 317–325. [Google Scholar] [CrossRef]
- Wang, Y.; Cortez, D.; Yazdi, P.; Neff, N.; Elledge, S.J.; Qin, J. BASC, a Super Complex of BRCA1-Associated Proteins Involved in the Recognition and Repair of Aberrant DNA Structures. Genes Dev. 2000, 14, 927–939. [Google Scholar]
- Gruosso, T.; Mieulet, V.; Cardon, M.; Bourachot, B.; Kieffer, Y.; Devun, F.; Dubois, T.; Dutreix, M.; Vincent-Salomon, A.; Miller, K.M.; et al. Chronic Oxidative Stress Promotes H2 AX Protein Degradation and Enhances Chemosensitivity in Breast Cancer Patients. EMBO Mol. Med. 2016, 8, 527–549. [Google Scholar] [CrossRef]
- Kim, S.; Jin, H.; Seo, H.-R.; Lee, H.J.; Lee, Y.-S. Regulating BRCA1 Protein Stability by Cathepsin S-Mediated Ubiquitin Degradation. Cell Death Differ. 2019, 26, 812–825. [Google Scholar] [CrossRef]
- Densham, R.M.; Garvin, A.J.; Stone, H.R.; Strachan, J.; Baldock, R.A.; Daza-Martin, M.; Fletcher, A.; Blair-Reid, S.; Beesley, J.; Johal, B.; et al. Human BRCA1–BARD1 Ubiquitin Ligase Activity Counteracts Chromatin Barriers to DNA Resection. Nat. Struct. Mol. Biol. 2016, 23, 647–655. [Google Scholar] [CrossRef]
- Mallery, D.L. Activation of the E3 Ligase Function of the BRCA1/BARD1 Complex by Polyubiquitin Chains. EMBO J. 2002, 21, 6755–6762. [Google Scholar] [CrossRef]
- Levy-Barda, A.; Lerentha, Y.; Davis, A.J.; Chung, Y.M.; Essers, J.; Shao, Z.; van Vliet, N.; Che, D.J.; Hu, M.C.-T.; Kanaar, R.; et al. Involvement of the Nuclear Proteasome Activator PA28γ in the Cellular Response to DNA Doublestrand Break. Cell Cycle 2011. [Google Scholar] [CrossRef]
- Daugaard, M.; Baude, A.; Fugger, K.; Povlsen, L.K.; Beck, H.; Sørensen, C.S.; Petersen, N.H.T.; Sorensen, P.H.B.; Lukas, C.; Bartek, J.; et al. LEDGF (P75) Promotes DNA-End Resection and Homologous Recombination. Nat. Struct. Mol. Biol. 2012, 19, 803–810. [Google Scholar] [CrossRef]
- Singh, D.K.; Gholamalamdari, O.; Jadaliha, M.; Ling Li, X.; Lin, Y.-C.; Zhang, Y.; Guang, S.; Hashemikhabir, S.; Tiwari, S.; Zhu, Y.J.; et al. PSIP1/P75 Promotes Tumorigenicity in Breast Cancer Cells by Promoting the Transcription of Cell Cycle Genes. Carcinogenesis 2017, 38, 966–975. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.-H.; Tee, L.Y.; Wang, X.-G.; Huang, Q.-S.; Yang, S.-H. Off-Target Effects in CRISPR/Cas9-Mediated Genome Engineering. Mol. Ther. Nucleic Acids 2015, 4, e264. [Google Scholar] [CrossRef]
- Mediavilla-Varela, M.; Pacheco, F.J.; Almaguel, F.; Perez, J.; Sahakian, E.; Daniels, T.R.; Leoh, L.; Padilla, A.; Wall, N.R.; Lilly, M.B.; et al. Docetaxel-Induced Prostate Cancer Cell Death Involves Concomitant Activation of Caspase and Lysosomal Pathways and Is Attenuated by LEDGF/P75. Mol. Cancer 2009, 8, 68. [Google Scholar] [CrossRef] [PubMed]
- SilMon. SilMon/Cell-Morphology-Notebook: V. 1.1 (Version v1.1). Zenodo. Available online: http://doi.org/10.5281/zenodo.4271745 (accessed on 13 November 2020).
- Boggs, A.E.; Vitolo, M.I.; Whipple, R.A.; Charpentier, M.S.; Goloubeva, O.G.; Ioffe, O.B.; Tuttle, K.C.; Slovic, J.; Lu, Y.; Mills, G.B.; et al. α-Tubulin Acetylation Elevated in Metastatic and Basal-like Breast Cancer Cells Promotes Microtentacle Formation, Adhesion, and Invasive Migration. Cancer Res. 2015, 75, 203–215. [Google Scholar] [CrossRef]
- Lee, C.-C.; Cheng, Y.-C.; Chang, C.-Y.; Lin, C.-M.; Chang, J.-Y. Alpha-Tubulin Acetyltransferase/MEC-17 Regulates Cancer Cell Migration and Invasion through Epithelial–Mesenchymal Transition Suppression and Cell Polarity Disruption. Sci. Rep. 2018, 8. [Google Scholar] [CrossRef]
- Lebok, P.; Öztürk, M.; Heilenkötter, U.; Jaenicke, F.; Müller, V.; Paluchowski, P.; Geist, S.; Wilke, C.; Burandt, E.; Lebeau, A.; et al. High Levels of Class III β-Tubulin Expression Are Associated with Aggressive Tumor Features in Breast Cancer. Oncol. Lett. 2016, 11, 1987–1994. [Google Scholar] [CrossRef]
- Huang, T.; Myklebust, L.M.; Kjarland, E.; Gjertsen, B.; Pendino, F.; Bruserud, Ø.; Døskeland, S.; Lillehaug, J.R. LEDGF/P75 Has Increased Expression in Blasts from Chemotherapy-Resistant Human Acute Myelogenic Leukemia Patients and Protects Leukemia Cells from Apoptosis in Vitro. Mol. Cancer 2007, 6, 31. [Google Scholar] [CrossRef]
- Lin, S.; Staahl, B.T.; Alla, R.K.; Doudna, J.A. Enhanced Homology-Directed Human Genome Engineering by Controlled Timing of CRISPR/Cas9 Delivery. eLife 2014, 3. [Google Scholar] [CrossRef]
- Liu, S.; Opiyo, S.O.; Manthey, K.; Glanzer, J.G.; Ashley, A.K.; Amerin, C.; Troksa, K.; Shrivastav, M.; Nickoloff, J.A.; Oakley, G.G. Distinct Roles for DNA-PK, ATM and ATR in RPA Phosphorylation and Checkpoint Activation in Response to Replication Stress. Nucleic Acids Res. 2012, 40, 10780–10794. [Google Scholar] [CrossRef] [PubMed]
- Kanu, N.; Grönroos, E.; Martinez, P.; Burrell, R.A.; Goh, X.Y.; Bartkova, J.; Maya-Mendoza, A.; Mistrík, M.; Rowan, A.J.; Patel, H.; et al. SETD2 Loss-of-Function Promotes Renal Cancer Branched Evolution through Replication Stress and Impaired DNA Repair. Oncogene 2015, 34, 5699–5708. [Google Scholar] [CrossRef]
- Bartkova, J.; Hořejší, Z.; Koed, K.; Krämer, A.; Tort, F.; Zieger, K.; Guldberg, P.; Sehested, M.; Nesland, J.M.; Lukas, C.; et al. DNA Damage Response as a Candidate Anti-Cancer Barrier in Early Human Tumorigenesis. Nature 2005, 434, 864–870. [Google Scholar] [CrossRef] [PubMed]
- Vargas-Rondón, N.; Villegas, V.; Rondón-Lagos, M. The Role of Chromosomal Instability in Cancer and Therapeutic Responses. Cancers 2017, 10, 4. [Google Scholar] [CrossRef]
- Paull, T.T.; Rogakou, E.P.; Yamazaki, V.; Kirchgessner, C.U.; Gellert, M.; Bonner, W.M. A Critical Role for Histone H2AX in Recruitment of Repair Factors to Nuclear Foci after DNA Damage. Curr. Biol. 2000, 10, 886–895. [Google Scholar] [CrossRef]
- An, J.; Huang, Y.-C.; Xu, Q.-Z.; Zhou, L.-J.; Shang, Z.-F.; Huang, B.; Wang, Y.; Liu, X.-D.; Wu, D.-C.; Zhou, P.-K. DNA-PKcs Plays a Dominant Role in the Regulation of H2AX Phosphorylation in Response to DNA Damage and Cell Cycle Progression. BMC Mol. Biol. 2010, 11, 18. [Google Scholar] [CrossRef]
- Zeng, S.; Shen, W.; Liu, L. Senescence and Cancer. Cancer Transl. Med. 2018, 4, 70. [Google Scholar] [CrossRef]
- González-Gualda, E.; Baker, A.G.; Fruk, L.; Muñoz-Espín, D. A Guide to Assessing Cellular Senescence in vitro and in vivo. FEBS J. 2021, 288, 56–80. [Google Scholar] [CrossRef]
- Brinkmann, K.; Schell, M.; Hoppe, T.; Kashkar, H. Regulation of the DNA Damage Response by Ubiquitin Conjugation. Front. Genet. 2015, 6. [Google Scholar] [CrossRef] [PubMed]
- Krum, S.A.; de la Dalugdugan, E.R.; Miranda-Carboni, G.A.; Lane, T.F. BRCA1 Forms a Functional Complex with γ-H2AX as a Late Response to Genotoxic Stress. J. Nucleic Acids 2010, 2010, 1–9. [Google Scholar] [CrossRef]
- Ramadan, K.; Meerang, M. Degradation-Linked Ubiquitin Signal and Proteasome Are Integral Components of DNA Double Strand Break Repair: New Perspectives for Anti-Cancer Therapy. FEBS Lett. 2011, 585, 2868–2875. [Google Scholar] [CrossRef]
- Stone, H.R.; Morris, J.R. DNA Damage Emergency: Cellular Garbage Disposal to the Rescue? Oncogene 2014, 33, 805–813. [Google Scholar] [CrossRef]
- Vissers, J.H.; Nicassio, F.; van Lohuizen, M.; Di Fiore, P.; Citterio, E. The Many Faces of Ubiquitinated Histone H2A: Insights from the DUBs. Cell Div. 2008, 3, 8. [Google Scholar] [CrossRef]
- Ran, F.A.; Hsu, P.D.; Wright, J.; Agarwala, V.; Scott, D.A.; Zhang, F. Genome Engineering Using the CRISPR-Cas9 System. Nat. Protoc. 2013, 8, 2281–2308. [Google Scholar] [CrossRef]
- Oceguera-Yanez, F. Engineering the AAVS1 Locus for Consistent and Scalable Transgene Expression in Human IPSCs and Their Differentiated Derivatives. Methods 2016, 15, 43–55. [Google Scholar] [CrossRef]
- Vichai, V.; Kirtikara, K. Sulforhodamine B Colorimetric Assay for Cytotoxicity Screening. Nat. Protoc. 2006, 1, 1112–1116. [Google Scholar] [CrossRef] [PubMed]
- Rödiger, S.; Schierack, P.; Böhm, A.; Nitschke, J.; Berger, I.; Frömmel, U.; Schmidt, C.; Ruhland, M.; Schimke, I.; Roggenbuck, D.; et al. A Highly Versatile Microscope Imaging Technology Platform for the Multiplex Real-Time Detection of Biomolecules and Autoimmune Antibodies. In Molecular Diagnostics; Advances in Biochemical Engineering/Biotechnology; Seitz, H., Schumacher, S., Eds.; Springer: Berlin/Heidelberg, Germany, 2012; Volume 133, pp. 35–74. ISBN 978-3-642-37690-0. [Google Scholar]
- Willitzki, A.; Lorenz, S.; Hiemann, R.; Guttek, K.; Goihl, A.; Hartig, R.; Conrad, K.; Feist, E.; Sack, U.; Schierack, P.; et al. Fully Automated Analysis of Chemically Induced ΓH2AX Foci in Human Peripheral Blood Mononuclear Cells by Indirect Immunofluorescence: Fully Automated ΓH2AX Foci Analysis in PBMCs. Cytometry A 2013, 83, 1017–1026. [Google Scholar] [CrossRef]
- Schneider, J.; Weiss, R.; Ruhe, M.; Jung, T.; Roggenbuck, D.; Stohwasser, R.; Schierack, P.; Rödiger, S. Open Source Bioimage Informatics Tools for the Analysis of DNA Damage and Associated Biomarkers. J. Lab. Precis. Med. 2019, 4, 1–27. [Google Scholar] [CrossRef]
- Ni, D.; Xu, P.; Gallagher, S. Immunoblotting and Immunodetection. Curr. Protoc. Cell Biol. 2008, 83, 10–18. [Google Scholar] [CrossRef]
- R: The R Project for Statistical Computing. Available online: https://www.r-project.org/ (accessed on 3 June 2020).
- Ritz, C.; Baty, F.; Streibig, J.C.; Gerhard, D. Dose-Response Analysis Using R. PLoS ONE 2015, 10, e0146021. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liedtke, V.; Schröder, C.; Roggenbuck, D.; Weiss, R.; Stohwasser, R.; Schierack, P.; Rödiger, S.; Schenk, L. LEDGF/p75 Is Required for an Efficient DNA Damage Response. Int. J. Mol. Sci. 2021, 22, 5866. https://doi.org/10.3390/ijms22115866
Liedtke V, Schröder C, Roggenbuck D, Weiss R, Stohwasser R, Schierack P, Rödiger S, Schenk L. LEDGF/p75 Is Required for an Efficient DNA Damage Response. International Journal of Molecular Sciences. 2021; 22(11):5866. https://doi.org/10.3390/ijms22115866
Chicago/Turabian StyleLiedtke, Victoria, Christian Schröder, Dirk Roggenbuck, Romano Weiss, Ralf Stohwasser, Peter Schierack, Stefan Rödiger, and Lysann Schenk. 2021. "LEDGF/p75 Is Required for an Efficient DNA Damage Response" International Journal of Molecular Sciences 22, no. 11: 5866. https://doi.org/10.3390/ijms22115866
APA StyleLiedtke, V., Schröder, C., Roggenbuck, D., Weiss, R., Stohwasser, R., Schierack, P., Rödiger, S., & Schenk, L. (2021). LEDGF/p75 Is Required for an Efficient DNA Damage Response. International Journal of Molecular Sciences, 22(11), 5866. https://doi.org/10.3390/ijms22115866