
Genetic Dominant Variants in STUB1, Segregating in Families with SCA48, Display In Vitro Functional Impairments Indistinctive from Recessive Variants Associated with SCAR16


 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
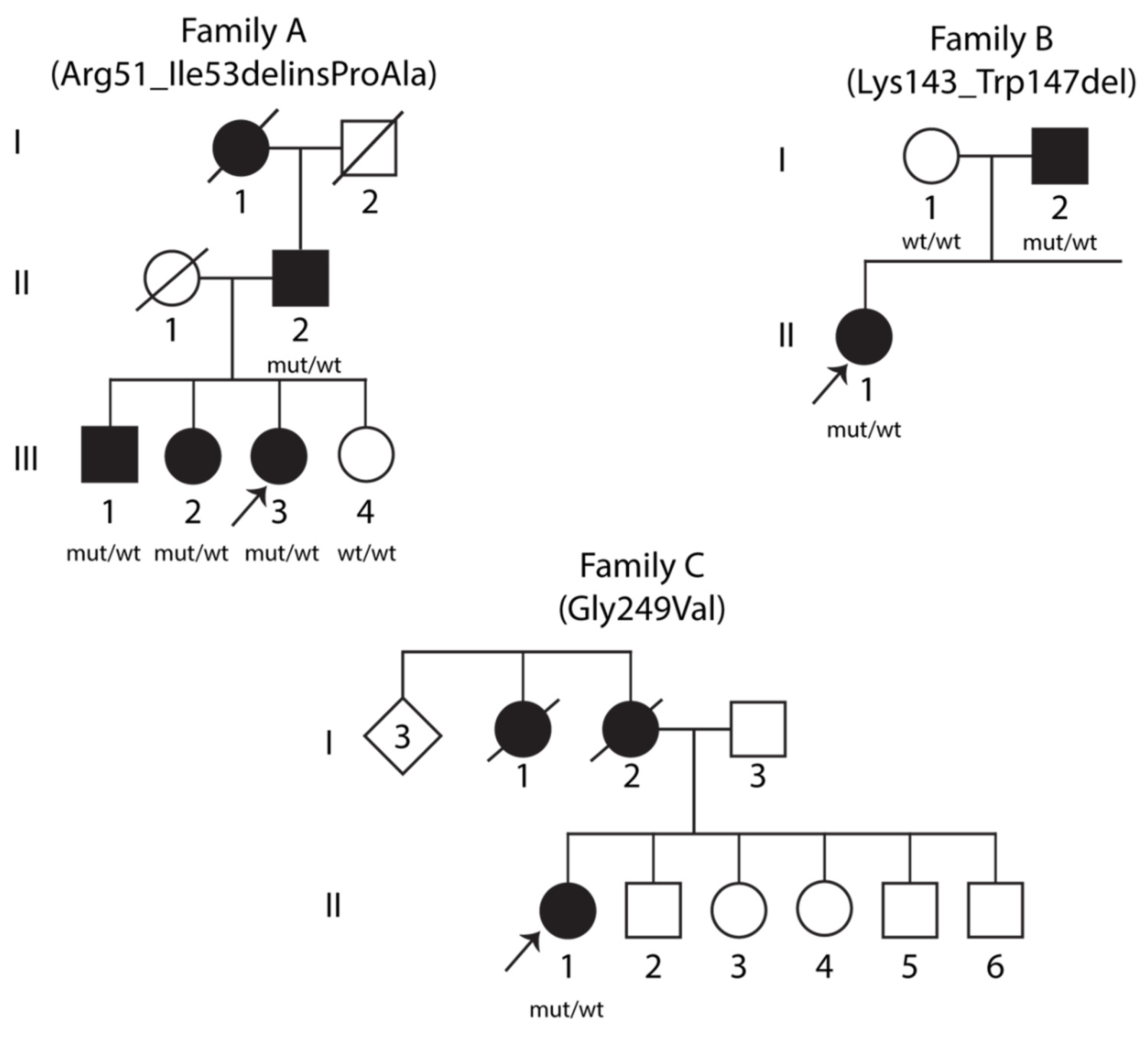
2.1. Genetic Findings
2.2. Clinical Features
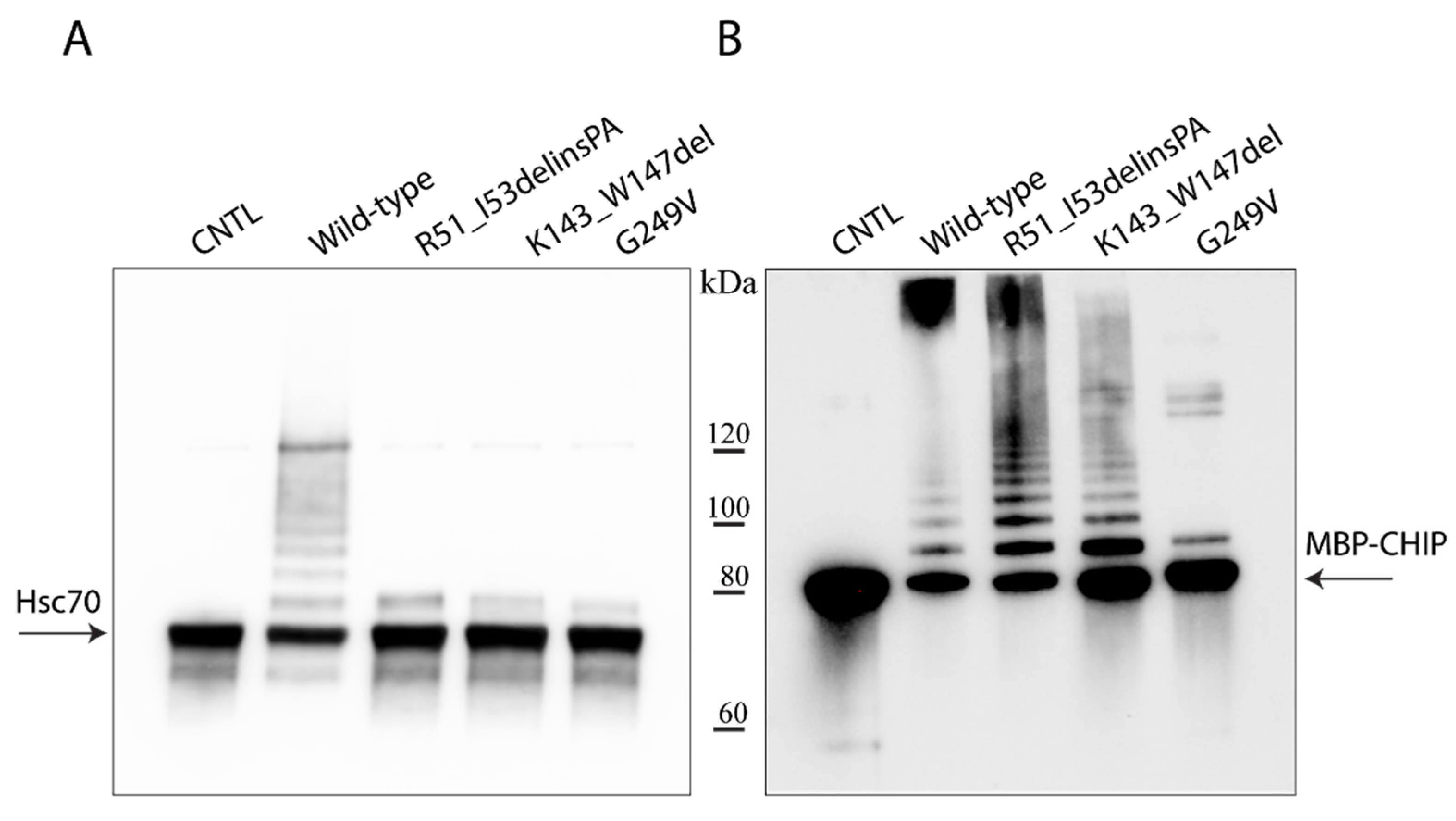
2.3. Hsc70- and Self-Ubiquitination Activity of the CHIP Variants
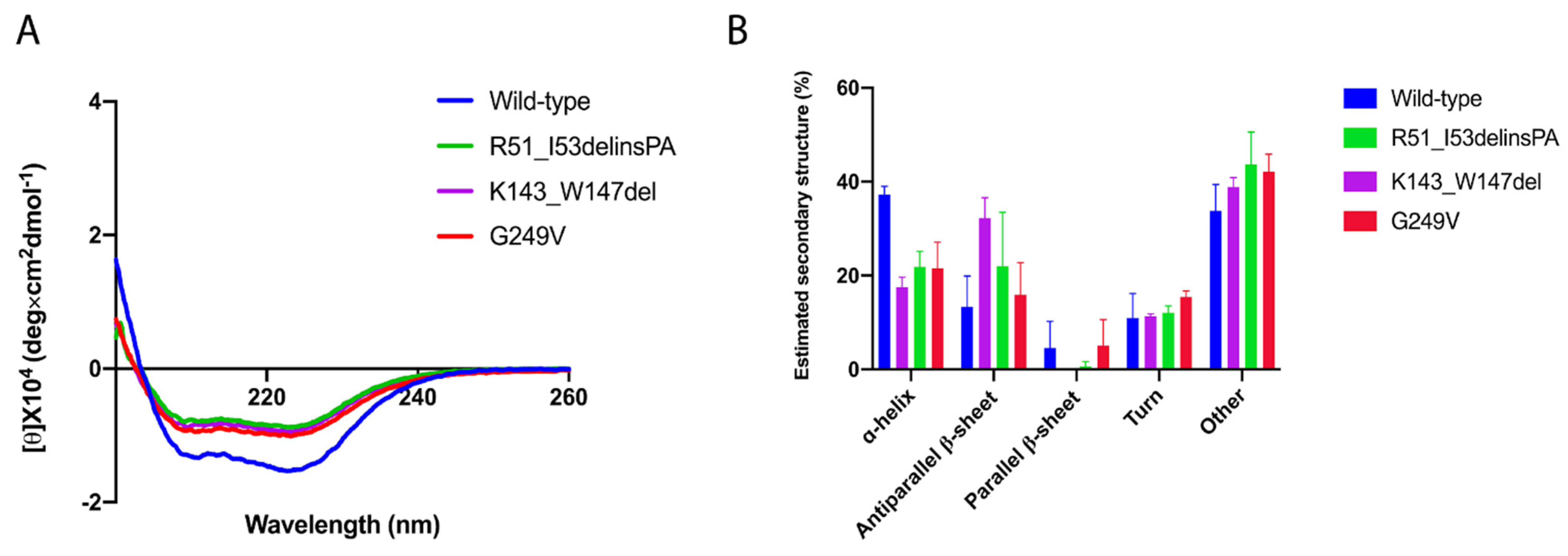
2.4. Thermal Unfolding of the CHIP Variants
2.5. Oligomerization States of the CHIP Variants
3. Discussion
4. Materials and Methods
4.1. Clinical Data
4.2. Genetic Analyses
4.3. In Vitro Expression of Mutant CHIP Proteins
4.4. In Vitro Ubiquitination Activity Assay
4.5. Circular Dichroism Spectroscopy
4.6. Native Polyacrylamide Gel Electrophoresis (Native–PAGE)
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A

References
- Durr, A. Autosomal dominant cerebellar ataxias: Polyglutamine expansions and beyond. Lancet Neurol. 2010, 9, 885–894. [Google Scholar] [CrossRef]
- Ruano, L.; Melo, C.; Silva, M.C.; Coutinho, P. The global epidemiology of hereditary ataxia and spastic paraplegia: A systematic review of prevalence studies. Neuroepidemiology 2014, 42, 174–183. [Google Scholar] [CrossRef] [PubMed]
- Bird, T.D. Hereditary Ataxia Overview. In GeneReviews(®); Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Mirzaa, G., Amemiya, A., Eds.; University of Washington: Washington, WA, USA, 1993. [Google Scholar]
- Genis, D.; Ortega-Cubero, S.; San Nicolás, H.; Corral, J.; Gardenyes, J.; de Jorge, L.; López, E.; Campos, B.; Lorenzo, E.; Tonda, R.; et al. Heterozygous STUB1 mutation causes familial ataxia with cognitive affective syndrome (SCA48). Neurology 2018, 91, e1988–e1998. [Google Scholar] [CrossRef] [PubMed]
- Lieto, M.; Riso, V.; Galatolo, D.; De Michele, G.; Rossi, S.; Barghigiani, M.; Cocozza, S.; Pontillo, G.; Trovato, R.; Saccà, F.; et al. The complex phenotype of spinocerebellar ataxia type 48 in eight unrelated Italian families. Eur. J. Neurol. 2020, 27, 498–505. [Google Scholar] [CrossRef]
- De Michele, G.; Lieto, M.; Galatolo, D.; Salvatore, E.; Cocozza, S.; Barghigiani, M.; Tessa, A.; Baldacci, J.; Pappatà, S.; Filla, A.; et al. Spinocerebellar ataxia 48 presenting with ataxia associated with cognitive, psychiatric, and extrapyramidal features: A report of two Italian families. Parkinsonism Relat. Disord. 2019, 65, 91–96. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.-H.; Latimer, C.; Yagi, M.; Ndugga-Kabuye, M.K.; Heigham, E.; Jayadev, S.; Meabon, J.S.; Gomez, C.M.; Keene, C.D.; Cook, D.G.; et al. Heterozygous STUB1 missense variants cause ataxia, cognitive decline, and STUB1 mislocalization. Neurol. Genet. 2020, 6, e397-13. [Google Scholar] [CrossRef]
- Ravel, J.-M.; Benkirane, M.; Calmels, N.; Marelli, C.; Ory-Magne, F.; Ewenczyk, C.; Halleb, Y.; Tison, F.; Lecocq, C.; Pische, G.; et al. Expanding the clinical spectrum of STIP1 homology and U-box containing protein 1-associated ataxia. J. Neurol. 2021, 268, 1927–1937. [Google Scholar] [CrossRef]
- De Michele, G.; Galatolo, D.; Barghigiani, M.; Iacovo, D.D.; Trovato, R.; Tessa, A.; Salvatore, E.; Filla, A.; De Michele, G.; Santorelli, F.M. Spinocerebellar ataxia type 48: Last but not least. Neurol. Sci. 2020, 41, 2423–2432. [Google Scholar] [CrossRef] [PubMed]
- Ballinger, C.A.; Connell, P.; Wu, Y.; Hu, Z.; Thompson, L.J.; Yin, L.-Y.; Patterson, C. Identification of CHIP, a Novel Tetratricopeptide Repeat-Containing Protein That Interacts with Heat Shock Proteins and Negatively Regulates Chaperone Functions. Mol. Cell. Biol. 1999, 19, 4535–4545. [Google Scholar] [CrossRef]
- Nikolay, R.; Wiederkehr, T.; Rist, W.; Kramer, G.; Mayer, M.P.; Bukau, B. Dimerization of the Human E3 Ligase CHIP via a Coiled-coil Domain Is Essential for Its Activity*. J. Biol. Chem. 2004, 279, 2673–2678. [Google Scholar] [CrossRef]
- Shi, Y.; Wang, J.; Li, J.-D.; Ren, H.; Guan, W.; He, M.; Yan, W.; Zhou, Y.; Hu, Z.; Zhang, J.; et al. Identification of CHIP as a novel causative gene for autosomal recessive cerebellar ataxia. PLoS ONE 2013, 8, e81884. [Google Scholar] [CrossRef] [PubMed]
- Pakdaman, Y.; Sanchez-Guixé, M.; Kleppe, R.; Erdal, S.; Bustad, H.J.; Bjørkhaug, L.; Haugarvoll, K.; Tzoulis, C.; Heimdal, K.; Knappskog, P.M.; et al. In vitro characterization of six STUB1 variants in spinocerebellar ataxia 16 reveals altered structural properties for the encoded CHIP proteins. Biosci. Rep. 2017, 37. [Google Scholar] [CrossRef] [PubMed]
- Kanack, A.J.; Newsom, O.J.; Scaglione, K.M. Most mutations that cause spinocerebellar ataxia autosomal recessive type 16 (SCAR16) destabilize the protein quality-control E3 ligase CHIP. J. Biol. Chem. 2018, 293, 2735–2743. [Google Scholar] [CrossRef] [PubMed]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [PubMed]
- Palvadeau, R.; Kaya-Güleç, Z.E.; Şimşir, G.; Vural, A.; Öztop-Çakmak, Ö.; Genç, G.; Aygün, M.S.; Falay, O.; Başak, A.N.; Ertan, S. Cerebellar cognitive-affective syndrome preceding ataxia associated with complex extrapyramidal features in a Turkish SCA48 family. Neurogenetics 2020, 21, 51–58. [Google Scholar] [CrossRef]
- Roux, T.; Barbier, M.; Papin, M.; Davoine, C.S.; Sayah, S.; Coarelli, G.; Charles, P.; Marelli, C.; Parodi, L.; Tranchant, C.; et al. Clinical, neuropathological, and genetic characterization of STUB1 variants in cerebellar ataxias: A frequent cause of predominant cognitive impairment. Genet. Med. 2020, 22, 1851–1862. [Google Scholar] [CrossRef]
- Park, J.; Deininger, N.; Rautenberg, M.; Saft, C.; Harmuth, F.; Sturm, M.; Riess, O.; Schöls, L.; Synofzik, M.; Haack, T.B. Correspondence on “Clinical, neuropathological, and genetic characterization of STUB1 variants in cerebellar ataxias: A frequent cause of predominant cognitive impairment” by Roux et al. Genet. Med. 2021, 1–2. [Google Scholar] [CrossRef]
- Zhang, M.; Windheim, M.; Roe, S.M.; Peggie, M.; Cohen, P.; Prodromou, C.; Pearl, L.H. Chaperoned Ubiquitylation—Crystal Structures of the CHIP U Box E3 Ubiquitin Ligase and a CHIP-Ubc13-Uev1a Complex. Mol. Cell 2005, 20, 525–538. [Google Scholar] [CrossRef]
- McLaughlin, B.; Buendia, M.A.; Saborido, T.P.; Palubinsky, A.M.; Stankowski, J.N.; Stanwood, G.D. Haploinsufficiency of the E3 ubiquitin ligase C-terminus of heat shock cognate 70 interacting protein (CHIP) produces specific behavioral impairments. PLoS ONE 2012, 7, e36340. [Google Scholar] [CrossRef]
- Shi, C.H.; Schisler, J.C.; Rubel, C.E.; Tan, S.; Song, B.; McDonough, H.; Xu, L.; Portbury, A.L.; Mao, C.Y.; True, C.; et al. Ataxia hypogonadism caused by the loss of ubiquitin ligase activity of the U box protein, C.H.I.P. Hum. Mol. Genet. 2014, 23, 1013–1024. [Google Scholar] [CrossRef]
- Heimdal, K.; Sanchez-Guixé, M.; Aukrust, I.; Bollerslev, J.; Bruland, O.; Jablonski, G.E.; Erichsen, A.K.; Gude, E.; A Koht, J.; Erdal, S.; et al. STUB1 mutations in autosomal recessive ataxias—Evidence for mutation-specific clinical heterogeneity. Orphanet J. Rare Dis. 2014, 9, 146. [Google Scholar] [CrossRef]
- Herrema, H.; Mikkelsen, T.; Robin, A.; LeWitt, P.; Sidiropoulos, C. SCA 17 phenotype with intermediate triplet repeat number. J. Neurol. Sci. 2014, 345, 269–270. [Google Scholar] [CrossRef] [PubMed]
- Doherty, K.M.; Warner, T.T.; Lees, A.J. Late onset ataxia: MSA-C or SCA 17? A gene penetrance dilemma. Mov. Disord. 2014, 29, 36–38. [Google Scholar] [CrossRef] [PubMed]
- Jih, K.-Y.; Lin, K.-P.; Tsai, P.-C.; Soong, B.-W.; Liao, Y.-C.; Lee, Y.-C. Investigating TBP CAG/CAA trinucleotide repeat expansions in a Taiwanese cohort with ALS. Amyotroph. Lateral Scler. Front. Degener. 2020, 2020, 1–6. [Google Scholar]
- Shin, J.H.; Park, H.; Ehm, G.H.; Lee, W.W.; Yun, J.Y.; Kim, Y.E.; Lee, J.-Y.; Kim, H.-J.; Kim, J.-M.; Jeon, B.S.; et al. The Pathogenic Role of Low Range Repeats in SCA17. PLoS ONE 2015, 10, e0135275. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Cai, Q.; You, H.; Zhou, X.; Chen, D.; Mo, G.; Li, X. Co-occurrence of ATXN3 and ATXN2 repeat expansions in Chinese ataxia patients with slow saccades. Mol. Genet. Genom. Med. 2019, 7, e663. [Google Scholar] [CrossRef]
- Holtan, J.P.; Aukrust, I.; Jansson, R.W.; Berland, S.; Bruland, O.; Gjerde, B.L.; Stokowy, T.; Bojovic, O.; Forsaa, V.; Austeng, D.; et al. Clinical features and molecular genetics of patients with ABCA4-retinal dystrophies. Acta Ophthalmol. 2020. [Google Scholar] [CrossRef]
- Bredrup, C.; Johansson, S.; Bindoff, L.A.; Sztromwasser, P.; Kråkenes, J.; Mellgren, A.E.; Brurås, K.R.; Lind, O.; Boman, H.; Knappskog, P.M.; et al. High Myopia–Excavated Optic Disc Anomaly Associated With a Frameshift Mutation in the MYC-Binding Protein 2 Gene (MYCBP2). Am. J. Ophthalmol. 2015, 159, 973–979e2. [Google Scholar] [CrossRef] [PubMed]
- Micsonai, A.; Wien, F.; Bulyáki, É.; Kun, J.; Moussong, É.; Lee, Y.-H.; Goto, Y.; Réfrégiers, M.; Kardos, J. BeStSel: A web server for accurate protein secondary structure prediction and fold recognition from the circular dichroism spectra. Nucleic Acids Res. 2018, 46, W315–W322. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pakdaman, Y.; Berland, S.; Bustad, H.J.; Erdal, S.; Thompson, B.A.; James, P.A.; Power, K.N.; Ellingsen, S.; Krooni, M.; Berge, L.I.; et al. Genetic Dominant Variants in STUB1, Segregating in Families with SCA48, Display In Vitro Functional Impairments Indistinctive from Recessive Variants Associated with SCAR16. Int. J. Mol. Sci. 2021, 22, 5870. https://doi.org/10.3390/ijms22115870
Pakdaman Y, Berland S, Bustad HJ, Erdal S, Thompson BA, James PA, Power KN, Ellingsen S, Krooni M, Berge LI, et al. Genetic Dominant Variants in STUB1, Segregating in Families with SCA48, Display In Vitro Functional Impairments Indistinctive from Recessive Variants Associated with SCAR16. International Journal of Molecular Sciences. 2021; 22(11):5870. https://doi.org/10.3390/ijms22115870
Chicago/Turabian StylePakdaman, Yasaman, Siren Berland, Helene J. Bustad, Sigrid Erdal, Bryony A. Thompson, Paul A. James, Kjersti N. Power, Ståle Ellingsen, Martin Krooni, Line I. Berge, and et al. 2021. "Genetic Dominant Variants in STUB1, Segregating in Families with SCA48, Display In Vitro Functional Impairments Indistinctive from Recessive Variants Associated with SCAR16" International Journal of Molecular Sciences 22, no. 11: 5870. https://doi.org/10.3390/ijms22115870
APA StylePakdaman, Y., Berland, S., Bustad, H. J., Erdal, S., Thompson, B. A., James, P. A., Power, K. N., Ellingsen, S., Krooni, M., Berge, L. I., Sexton, A., Bindoff, L. A., Knappskog, P. M., Johansson, S., & Aukrust, I. (2021). Genetic Dominant Variants in STUB1, Segregating in Families with SCA48, Display In Vitro Functional Impairments Indistinctive from Recessive Variants Associated with SCAR16. International Journal of Molecular Sciences, 22(11), 5870. https://doi.org/10.3390/ijms22115870