
Molecular Landscape and Therapeutic Strategies in Cholangiocarcinoma: An Integrated Translational Approach towards Precision Medicine

 , ,
, , 
Abstract
1. Introduction
2. CCA Molecular Landscape
3. Targeted Therapy: State-of-the-Art and Future Perspectives
3.1. Metabolic Regulators
3.2. Tyrosine Kinase Receptors
3.3. Epidermal Growth Factor Receptor
3.4. PI3k/AKT/mTOR Pathway
3.5. Proteasome Inhibitors
3.6. Immunotherapy
3.7. Future Perspectives
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Banales, J.M.; Cardinale, V.; Carpino, G.; Marzioni, M.; Andersen, J.B.; Invernizzi, P.; Lind, G.E.; Folseraas, T.; Forbes, S.J.; Fouassier, L.; et al. Cholangiocarcinoma: Current knowledge and future perspectives consensus statement from the European Network for the Study of Cholangiocarcinoma (ENS-CCA). Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 261–280. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.A.; Tavolari, S.; Brandi, G. Cholangiocarcinoma: Epidemiology and risk factors. Liver Int. 2019, 39, 19–31. [Google Scholar] [CrossRef] [PubMed]
- Cardinale, V.; Carpino, G.; Reid, L.; Gaudio, E.; Alvaro, D. Multiple cells of origin in cholangiocarcinoma underlie biological, epidemiological and clinical heterogeneity. World J. Gastrointest. Oncol. 2012, 4, 94–102. [Google Scholar] [CrossRef]
- Nakagawa, H.; Suzuki, N.; Hirata, Y.; Hikiba, Y.; Hayakawa, Y.; Kinoshita, H.; Ihara, S.; Uchino, K.; Nishikawa, Y.; Ijichi, H.; et al. Biliary epithelial injury-induced regenerative response by IL-33 promotes cholangiocarcinogenesis from peribiliary glands. Proc. Natl. Acad. Sci. USA 2017, 114, E3806–E3815. [Google Scholar] [CrossRef] [PubMed]
- Clements, O.; Eliahoo, J.; Kim, J.U.; Taylor-Robinson, S.D.; Khan, S.A. Risk factors for intrahepatic and extrahepatic cholangiocarcinoma: A systematic review and meta-analysis. J. Hepatol. 2020, 72, 95–103. [Google Scholar] [CrossRef]
- Banales, J.M.; Marin, J.J.G.; Lamarca, A.; Rodrigues, P.M.; Khan, S.A.; Roberts, L.R.; Cardinale, V.; Carpino, G.; Andersen, J.B.; Braconi, C.; et al. Cholangiocarcinoma 2020: The next horizon in mechanisms and management. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 577–588. [Google Scholar] [CrossRef]
- Alvaro, D.; Hassan, C.; Cardinale, V.; Carpino, G.; Fabris, L.; Gringeri, E.; Granata, V.; Mutignani, M.; Morement, H.; Giuliante, F.; et al. Italian Clinical Practice Guidelines on Cholangiocarcinoma—Part II: Treatment. Dig. Liver Dis. 2020, 52, 1430–1442. [Google Scholar] [CrossRef] [PubMed]
- Marin, J.J.G.; Prete, M.G.; Lamarca, A.; Tavolari, S.; Landa-Magdalena, A.; Brandi, G.; Segatto, O.; Vogel, A.; Macias, R.I.R.; Rodrigues, P.M.; et al. Current and novel therapeutic opportunities for systemic therapy in biliary cancer. Br. J. Cancer 2020, 123, 1–13. [Google Scholar] [CrossRef]
- Valle, J.W.; Kelley, R.K.; Nervi, B.; Oh, D.-Y.; Zhu, A.X. Biliary tract cancer. Lancet 2021, 397, 428–444. [Google Scholar] [CrossRef]
- O’Rourke, C.J.; Munoz-Garrido, P.; Andersen, J.B. Molecular targets in cholangiocarcinoma. Hepatology 2021, 73, 62–74. [Google Scholar] [CrossRef]
- Pellino, A.; Loupakis, F.; Cadamuro, M.; Dadduzio, V.; Fassan, M.; Guido, M.; Cillo, U.; Indraccolo, S.; Fabris, L. Precision medicine in cholangiocarcinoma. Transl. Gastroenterol. Hepatol. 2018, 3, 40. [Google Scholar] [CrossRef]
- Abou-Alfa, G.K.; Macarulla, T.; Javle, M.M.; Kelley, R.K.; Lubner, S.J.; Adeva, J.; Cleary, J.M.; Catenacci, D.V.; Borad, M.J.; Bridgewater, J.; et al. Ivosidenib in IDH1-mutant, chemotherapy-refractory cholangiocarcinoma (ClarIDHy): A multicentre, randomised, double-blind, placebo-controlled, phase 3 study. Lancet Oncol. 2020, 21, 796–807. [Google Scholar] [CrossRef]
- Verlingue, L.; Malka, D.; Allorant, A.; Massard, C.; Ferté, C.; Lacroix, L.; Rouleau, E.; Auger, N.; Ngo, M.; Nicotra, C.; et al. Precision medicine for patients with advanced biliary tract cancers: An effective strategy within the prospective MOSCATO-01 trial. Eur. J. Cancer 2017, 87, 122–130. [Google Scholar] [CrossRef]
- Angelakas, A.; Lamarca, A.; Hubner, R.A.; McNamara, M.G.; Valle, J.W. Ivosidenib: An investigational drug for the treatment of biliary tract cancers. Expert Opin. Investig. Drugs 2021, 30, 301–307. [Google Scholar] [CrossRef] [PubMed]
- Rizzo, A.; Ricci, A.D.; Brandi, G. Recent advances of immunotherapy for biliary tract cancer. Expert Rev. Gastroenterol. Hepatol. 2021, 1–10. [Google Scholar] [CrossRef]
- Piha-Paul, S.A.; Oh, D.; Ueno, M.; Malka, D.; Chung, H.C.; Nagrial, A.; Kelley, R.K.; Ros, W.; Italiano, A.; Nakagawa, K.; et al. Efficacy and safety of pembrolizumab for the treatment of advanced biliary cancer: Results from the KEYNOTE -158 and KEYNOTE -028 studies. Int. J. Cancer 2020, 147, 2190–2198. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Shi, J.; Wang, Y.; Zhou, H.; Zhang, Z.; Han, Z.; Li, G.; Yang, B.; Cao, G.; Ke, Y.; et al. Next-generation sequencing-guided molecular-targeted therapy and immunotherapy for biliary tract cancers. Cancer Immunol. Immunother. 2021, 70, 1001–1014. [Google Scholar] [CrossRef] [PubMed]
- Iyer, R.V.; Pokuri, V.K.; Groman, A.; Ma, W.W.; Malhotra, U.; Iancu, D.M.; Grande, C.; Saab, T.B. A Multicenter Phase II Study of Gemcitabine, Capecitabine, and Bevacizumab for Locally Advanced or Metastatic Biliary Tract Cancer. Am. J. Clin. Oncol. 2018, 41, 649–655. [Google Scholar] [CrossRef]
- Chen, J.S.; Hsu, C.; Chiang, N.J.; Tsai, C.S.; Tsou, H.H.; Huang, S.F.; Bai, L.Y.; Chang, I.C.; Shiah, H.S.; Ho, C.L.; et al. A KRAS mutation status-stratified randomized phase II trial of gemcitabine and oxaliplatin alone or in combination with cetuximab in advanced biliary tract cancer. Ann. Oncol. 2015, 26, 943–949. [Google Scholar] [CrossRef]
- Lee, J.K.; Capanu, M.; Oreilly, E.M.; Ma, J.; Chou, J.F.; Shia, J.; Katz, S.; Gansukh, B.; Reidylagunes, D.; Segal, N.H.; et al. A phase II study of gemcitabine and cisplatin plus sorafenib in patients with advanced biliary adenocarcinomas. Br. J. Cancer 2013, 109, 915–919. [Google Scholar] [CrossRef]
- Simbolo, M.; Fassan, M.; Ruzzenente, A.; Mafficini, A.; Wood, L.D.; Corbo, V.; Melisi, D.; Malleo, G.; Vicentini, C.; Malpeli, G.; et al. Multigene mutational profiling of cholangiocarcinomas identifies actionable molecular subgroups. Oncotarget 2014, 5, 2839–2852. [Google Scholar] [CrossRef] [PubMed]
- Chae, H.; Kim, D.; Yoo, C.; Kim, K.-P.; Jeong, J.H.; Chang, H.-M.; Lee, S.S.; Park, D.H.; Song, T.J.; Hwang, S.; et al. Therapeutic relevance of targeted sequencing in management of patients with advanced biliary tract cancer: DNA damage repair gene mutations as a predictive biomarker. Eur. J. Cancer 2019, 120, 31–39. [Google Scholar] [CrossRef]
- Nakamura, H.; Arai, Y.; Totoki, Y.; Shirota, T.; ElZawahry, A.; Kato, M.; Hama, N.; Hosoda, F.; Urushidate, T.; Ohashi, S.; et al. Genomic spectra of biliary tract cancer. Nat. Genet. 2015, 47, 1003–1010. [Google Scholar] [CrossRef]
- Wardell, C.P.; Fujita, M.; Yamada, T.; Simbolo, M.; Fassan, M.; Karlic, R.; Polak, P.; Kim, J.; Hatanaka, Y.; Maejima, K.; et al. Genomic characterization of biliary tract cancers identifies driver genes and predisposing mutations. J. Hepatol. 2018, 68, 959–969. [Google Scholar] [CrossRef] [PubMed]
- Tate, J.G.; Bamford, S.; Jubb, H.C.; Sondka, Z.; Beare, D.M.; Bindal, N.; Boutselakis, H.; Cole, C.G.; Creatore, C.; Dawson, E.; et al. COSMIC: The Catalogue of Somatic Mutations in Cancer. Nucleic Acids Res. 2018, 47, D941–D947. [Google Scholar] [CrossRef] [PubMed]
- Sia, D.; Hoshida, Y.; Villanueva, A.; Roayaie, S.; Ferrer, J.; Tabak, B.; Peix, J.; Sole, M.; Tovar, V.; Alsinet, C.; et al. Integrative Molecular Analysis of Intrahepatic Cholangiocarcinoma Reveals 2 Classes That Have Different Outcomes. Gastroenterology 2013, 144, 829–840. [Google Scholar] [CrossRef] [PubMed]
- Montal, R.; Sia, D.; Montironi, C.; Leow, W.Q.; Esteban-Fabró, R.; Pinyol, R.; Torres-Martin, M.; Bassaganyas, L.; Moeini, A.; Peix, J.; et al. Molecular classification and therapeutic targets in extrahepatic cholangiocarcinoma. J. Hepatol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Tomczak, K.; Czerwińska, P.; Wiznerowicz, M. Review The Cancer Genome Atlas (TCGA): An immeasurable source of knowledge. Współcz. Onkol. 2015, 1A, 68–77. [Google Scholar] [CrossRef]
- Nepal, C.; O’Rourke, C.J.; Oliveira, D.V.; Taranta, A.; Shema, S.; Gautam, P.; Calderaro, J.; Barbour, A.; Raggi, C.; Wennerberg, K.; et al. Genomic perturbations reveal distinct regulatory networks in intrahepatic cholangiocarcinoma. Hepatology 2018, 68, 949–963. [Google Scholar] [CrossRef]
- Farshidfar, F.; Zheng, S.; Gingras, M.-C.; Newton, Y.; Shih, J.; Robertson, A.G.; Hinoue, T.; Hoadley, K.A.; Gibb, E.A.; Roszik, J.; et al. Integrative Genomic Analysis of Cholangiocarcinoma Identifies Distinct IDH-Mutant Molecular Profiles. Cell Rep. 2017, 19, 2878–2880. [Google Scholar] [CrossRef]
- Zou, S.; Li, J.; Zhou, H.; Frech, C.; Jiang, X.; Chu, J.S.C.; Zhao, X.; Li, Y.; Li, Q.; Wang, H.; et al. Mutational landscape of intrahepatic cholangiocarcinoma. Nat. Commun. 2014, 5, 5696. [Google Scholar] [CrossRef]
- Goeppert, B.; Folseraas, T.; Roessler, S.; Kloor, M.; Volckmar, A.; Endris, V.; Buchhalter, I.; Stenzinger, A.; Grzyb, K.; Grimsrud, M.M.; et al. Genomic Characterization of Cholangiocarcinoma in Primary Sclerosing Cholangitis Reveals Therapeutic Opportunities. Hepatology 2020, 72, 1253–1266. [Google Scholar] [CrossRef] [PubMed]
- Carpino, G.; Cardinale, V.; Renzi, A.; Hov, J.R.; Berloco, P.B.; Rossi, M.; Karlsen, T.H.; Alvaro, D.; Gaudio, E. Activation of biliary tree stem cells within peribiliary glands in primary sclerosing cholangitis. J. Hepatol. 2015, 63, 1220–1228. [Google Scholar] [CrossRef] [PubMed]
- Ong, C.K.; Subimerb, C.; Pairojkul, C.; Wongkham, S.; Cutcutache, I.; Yu, W.; McPherson, J.R.; Allen, E.G.; Ng, C.C.Y.; Wong, B.H.; et al. Exome sequencing of liver fluke–associated cholangiocarcinoma. Nat. Genet. 2012, 44, 690–693. [Google Scholar] [CrossRef]
- Chan-On, W.; Nairismägi, M.-L.; Ong, C.K.; Lim, W.K.; Dima, S.; Pairojkul, C.; Lim, K.H.; McPherson, J.R.; Cutcutache, I.; Heng, H.L.; et al. Exome sequencing identifies distinct mutational patterns in liver fluke–related and non-infection-related bile duct cancers. Nat. Genet. 2013, 45, 1474–1478. [Google Scholar] [CrossRef] [PubMed]
- Jusakul, A.; Cutcutache, I.; Yong, C.H.; Lim, J.Q.; Ni Huang, M.; Padmanabhan, N.; Nellore, V.; Kongpetch, S.; Ng, A.W.T.; Ng, L.M.; et al. Whole-Genome and Epigenomic Landscapes of Etiologically Distinct Subtypes of Cholangiocarcinoma. Cancer Discov. 2017, 7, 1116–1135. [Google Scholar] [CrossRef]
- Lau, D.K.; Tay, R.Y.; Yeung, Y.H.; Chionh, F.; Mooi, J.; Murone, C.; Skrinos, E.; Price, T.J.; Mariadason, J.M.; Tebbutt, N.C. Phase II study of everolimus (RAD001) monotherapy as first-line treatment in advanced biliary tract cancer with biomarker exploration: The RADiChol Study. Br. J. Cancer 2018, 118, 966–971. [Google Scholar] [CrossRef]
- Zhang, Z.; Oyesanya, R.A.; Campbell, D.J.W.; Almenara, J.A.; DeWitt, J.L.; Sirica, A.E. Preclinical assessment of simultaneous targeting of epidermal growth factor receptor (ErbB1) and ErbB2 as a strategy for cholangiocarcinoma therapy. Hepatology 2010, 52, 975–986. [Google Scholar] [CrossRef]
- El-Khoueiry, A.B.; Rankin, C.J.; Ben-Josef, E.; Lenz, H.-J.; Gold, P.J.; Hamilton, R.D.; Govindarajan, R.; Eng, C.; Blanke, C.D. SWOG 0514: A phase II study of sorafenib in patients with unresectable or metastatic gallbladder carcinoma and cholangiocarcinoma. Investig. New Drugs 2012, 30, 1646–1651. [Google Scholar] [CrossRef]
- Buzzoni, R.; Pusceddu, S.; Bajetta, E.; De Braud, F.; Platania, M.; Iannacone, C.; Cantore, M.; Mambrini, A.; Bertolini, A.; Alabiso, O.; et al. Activity and safety of RAD001 (everolimus) in patients affected by biliary tract cancer progressing after prior chemotherapy: A phase II ITMO study. Ann. Oncol. 2014, 25, 1597–1603. [Google Scholar] [CrossRef]
- Sun, W.; Patel, A.; Normolle, D.; Patel, K.; Ohr, J.; Lee, J.J.; Bahary, N.; Chu, E.; Streeter, N.; Drummond, S. A phase 2 trial of regorafenib as a single agent in patients with chemotherapy-refractory, advanced, and metastatic biliary tract adenocarcinoma. Cancer 2019, 125, 902–909. [Google Scholar] [CrossRef] [PubMed]
- Sohal, D.P.S.; Mykulowycz, K.; Uehara, T.; Teitelbaum, U.R.; Damjanov, N.; Giantonio, B.J.; Carberry, M.; Wissel, P.; Jacobs-Small, M.; O’Dwyer, P.J.; et al. A phase II trial of gemcitabine, irinotecan and panitumumab in advanced cholangiocarcinoma. Ann. Oncol. 2013, 24, 3061–3065. [Google Scholar] [CrossRef] [PubMed]
- Leone, F.; Marino, D.; Cereda, S.; Filippi, R.; Belli, C.; Spadi, R.; Nasti, G.; Montano, M.; Amatu, A.; Aprile, G.; et al. Panitumumab in combination with gemcitabine and oxaliplatin does not prolong survival in wild-typeKRASadvanced biliary tract cancer: A randomized phase 2 trial (Vecti-BIL study). Cancer 2016, 122, 574–581. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Park, S.H.; Chang, H.-M.; Kim, J.S.; Choi, H.J.; Lee, M.A.; Chang, J.S.; Jeung, H.C.; Kang, J.H.; Lee, H.W.; et al. Gemcitabine and oxaliplatin with or without erlotinib in advanced biliary-tract cancer: A multicentre, open-label, randomised, phase 3 study. Lancet Oncol. 2012, 13, 181–188. [Google Scholar] [CrossRef]
- Kim, S.T.; Jang, K.-T.; Lee, S.J.; Jang, H.-L.; Lee, J.; Park, S.H.; Park, Y.S.; Lim, H.Y.; Kang, W.K.; Park, J.O. Tumour shrinkage at 6 weeks predicts favorable clinical outcomes in a phase III study of gemcitabine and oxaliplatin with or without erlotinib for advanced biliary tract cancer. BMC Cancer 2015, 15, 530. [Google Scholar] [CrossRef]
- Corti, F.; Nichetti, F.; Raimondi, A.; Niger, M.; Prinzi, N.; Torchio, M.; Tamborini, E.; Perrone, F.; Pruneri, G.; Di Bartolomeo, M.; et al. Targeting the PI3K/AKT/mTOR pathway in biliary tract cancers: A review of current evidences and future perspectives. Cancer Treat. Rev. 2019, 72, 45–55. [Google Scholar] [CrossRef]
- Pierre-Jean, M.; Deleuze, J.-F.; Le Floch, E.; Mauger, F. Clustering and variable selection evaluation of 13 unsupervised methods for multi-omics data integration. Briefings Bioinform. 2020, 21, 2011–2030. [Google Scholar] [CrossRef]
- Peck, J.; Wei, L.; Zalupski, M.; O’Neil, B.; Calero, M.V.; Bekaii-Saab, T. HER2/neu May Not Be an Interesting Target in Biliary Cancers: Results of an Early Phase II Study with Lapatinib. Oncology 2012, 82, 175–179. [Google Scholar] [CrossRef] [PubMed]
- Dang, L.; White, D.W.; Gross, S.; Bennett, B.D.; Bittinger, M.A.; Driggers, E.M.; Fantin, V.R.; Jang, H.G.; Jin, S.; Keenan, M.C.; et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 2010, 465, 966. [Google Scholar] [CrossRef]
- Ray-Coquard, I.; Pautier, P.; Pignata, S.; Pérol, D.; González-Martín, A.; Berger, R.; Fujiwara, K.; Vergote, I.; Colombo, N.; Mäenpää, J.; et al. Olaparib plus Bevacizumab as First-Line Maintenance in Ovarian Cancer. N. Engl. J. Med. 2019, 381, 2416–2428. [Google Scholar] [CrossRef]
- Marquez, J.; Flores, J.; Kim, A.H.; Nyamaa, B.; Nguyen, A.T.T.; Park, N.; Han, J. Rescue of TCA Cycle Dysfunction for Cancer Therapy. J. Clin. Med. 2019, 8, 2161. [Google Scholar] [CrossRef]
- Molenaar, R.J.; Coelen, R.J.S.; Khurshed, M.; Roos, E.; Caan, A.M.W.; Van Linde, M.E.; Kouwenhoven, M.; Bramer, J.A.M.; Bovée, J.V.M.G.; Mathôt, A.R.; et al. Study protocol of a phase IB/II clinical trial of metformin and chloroquine in patients withIDH1-mutated orIDH2-mutated solid tumours. BMJ Open 2017, 7, e014961. [Google Scholar] [CrossRef]
- Abou-Alfa, G.K.; Sahai, V.; Hollebecque, A.; Vaccaro, G.; Melisi, D.; Al-Rajabi, R.; Paulson, A.S.; Borad, M.J.; Gallinson, D.; Murphy, A.G.; et al. Pemigatinib for previously treated, locally advanced or metastatic cholangiocarcinoma: A multicentre, open-label, phase 2 study. Lancet Oncol. 2020, 21, 671–684. [Google Scholar] [CrossRef]
- Bekaii-Saab, T.S.; Valle, J.W.; Van Cutsem, E.; Rimassa, L.; Furuse, J.; Ioka, T.; Melisi, D.; Macarulla, T.; Bridgewater, J.; Wasan, H.; et al. FIGHT-302: First-line pemigatinib vs gemcitabine plus cisplatin for advanced cholangiocarcinoma with FGFR2 rearrangements. Futur. Oncol. 2020, 16, 2385–2399. [Google Scholar] [CrossRef]
- Krook, M.A.; Bonneville, R.; Chen, H.-Z.; Reeser, J.W.; Wing, M.R.; Martin, D.M.; Smith, A.M.; Dao, T.; Samorodnitsky, E.; Paruchuri, A.; et al. Tumor heterogeneity and acquired drug resistance in FGFR2-fusion-positive cholangiocarcinoma through rapid research autopsy. Mol. Case Stud. 2019, 5, 8. [Google Scholar] [CrossRef]
- Cleary, J.M.; Raghavan, S.; Wu, Q.; Li, Y.Y.; Spurr, L.F.; Gupta, H.V.; Rubinson, D.A.; Fetter, I.J.; Hornick, J.L.; Nowak, J.A.; et al. FGFR2 Extracellular Domain In-Frame Deletions are Therapeutically Targetable Genomic Alterations that Function as Oncogenic Drivers in Cholangiocarcinoma. Cancer Discov. 2021. [Google Scholar] [CrossRef]
- Rizzo, A.; Ricci, A.D.; Brandi, G. Futibatinib, an investigational agent for the treatment of intrahepatic cholangiocarcinoma: Evidence to date and future perspectives. Expert Opin. Investig. Drugs 2021, 30, 317–324. [Google Scholar] [CrossRef]
- Goyal, L.; Saha, S.K.; Liu, L.Y.; Siravegna, G.; Leshchiner, I.; Ahronian, L.G.; Lennerz, J.K.; Vu, P.; Deshpande, V.; Kambadakone, A.; et al. Polyclonal Secondary FGFR2 Mutations Drive Acquired Resistance to FGFR Inhibition in Patients with FGFR2 Fusion–Positive Cholangiocarcinoma. Cancer Discov. 2017, 7, 252–263. [Google Scholar] [CrossRef]
- Yoshikawa, D.; Ojima, H.; Iwasaki, M.; Hiraoka, N.; Kosuge, T.; Kasai, S.; Hirohashi, S.; Shibata, T. Clinicopathological and prognostic significance of EGFR, VEGF, and HER2 expression in cholangiocarcinoma. Br. J. Cancer 2007, 98, 418–425. [Google Scholar] [CrossRef]
- Cavalloni, G.; Peraldo-Neia, C.; Varamo, C.; Chiorino, G.; Sassi, F.; Aglietta, M.; Leone, F. Preclinical activity of EGFR and MEK1/2 inhibitors in the treatment of biliary tract carcinoma. Oncotarget 2016, 7, 52354–52363. [Google Scholar] [CrossRef]
- Bang, Y.-J.; Van Cutsem, E.; Feyereislova, A.; Chung, H.C.; Shen, L.; Sawaki, A.; Lordick, F.; Ohtsu, A.; Omuro, Y.; Satoh, T.; et al. Trastuzumab in combination with chemotherapy versus chemotherapy alone for treatment of HER2-positive advanced gastric or gastro-oesophageal junction cancer (ToGA): A phase 3, open-label, randomised controlled trial. Lancet 2010, 376, 687–697. [Google Scholar] [CrossRef]
- Denlinger, C.S.; Meropol, N.J.; Li, T.; Lewis, N.L.; Engstrom, P.F.; Weiner, L.M.; Cheng, J.D.; Alpaugh, R.K.; Cooper, H.; Wright, J.J.; et al. A Phase II Trial of the Proteasome Inhibitor Bortezomib in Patients with Advanced Biliary Tract Cancers. Clin. Color. Cancer 2014, 13, 81–86. [Google Scholar] [CrossRef]
- Cadamuro, M.; Brivio, S.; Spirli, C.; Joplin, R.E.; Strazzabosco, M.; Fabris, L. Autocrine and Paracrine Mechanisms Promoting Chemoresistance in Cholangiocarcinoma. Int. J. Mol. Sci. 2017, 18, 149. [Google Scholar] [CrossRef]
- Raggi, C.; Correnti, M.; Sica, A.; Andersen, J.B.; Cardinale, V.; Alvaro, D.; Chiorino, G.; Forti, E.; Glaser, S.; Alpini, G.; et al. Cholangiocarcinoma stem-like subset shapes tumor-initiating niche by educating associated macrophages. J. Hepatol. 2017, 66, 102–115. [Google Scholar] [CrossRef]
- Job, S.; Rapoud, D.; Dos Santos, A.; Gonzalez, P.; Desterke, C.; Pascal, G.; Elarouci, N.; Ayadi, M.; Adam, R.; Azoulay, D.; et al. Identification of Four Immune Subtypes Characterized by Distinct Composition and Functions of Tumor Microenvironment in Intrahepatic Cholangiocarcinoma. Hepatology 2020, 72, 965–981. [Google Scholar] [CrossRef]
- Marabelle, A.; Le, D.T.; Ascierto, P.A.; Di Giacomo, A.M.; De Jesus-Acosta, A.; Delord, J.-P.; Geva, R.; Gottfried, M.; Penel, N.; Hansen, A.R.; et al. Efficacy of Pembrolizumab in Patients with Noncolorectal High Microsatellite Instability/Mismatch Repair–Deficient Cancer: Results From the Phase II KEYNOTE-158 Study. J. Clin. Oncol. 2020, 38, 1–10. [Google Scholar] [CrossRef]
- Le, D.T.; Uram, J.N.; Wang, H.; Bartlett, B.R.; Kemberling, H.; Eyring, A.D.; Skora, A.D.; Luber, B.S.; Azad, N.S.; Laheru, D.; et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N. Engl. J. Med. 2015, 372, 2509–2520. [Google Scholar] [CrossRef]
- Winkelmann, R.; Schneider, M.; Hartmann, S.; Schnitzbauer, A.A.; Zeuzem, S.; Peveling-Oberhag, J.; Hansmann, M.L.; Walter, D. Microsatellite Instability Occurs Rarely in Patients with Cholangiocarcinoma: A Retrospective Study from a German Tertiary Care Hospital. Int. J. Mol. Sci. 2018, 19, 1421. [Google Scholar] [CrossRef]
- Alvaro, D.; Hassan, C.; Cardinale, V.; Carpino, G.; Fabris, L.; Gringeri, E.; Granata, V.; Mutignani, M.; Morement, H.; Giuliante, F.; et al. Italian Clinical Practice Guidelines on Cholangiocarcinoma—Part I: Classification, diagnosis and staging. Dig. Liver Dis. 2020, 52, 1282–1293. [Google Scholar] [CrossRef]
- Taghizadeh, H.; Müllauer, L.; Mader, R.; Prager, G.W. Applied precision cancer medicine in metastatic biliary tract cancer. Hepatol. Int. 2020, 14, 288–295. [Google Scholar] [CrossRef]
- Lowery, M.A.; Ptashkin, R.N.; Jordan, E.J.; Berger, M.F.; Zehir, A.; Capanu, M.; Kemeny, N.E.; O’Reilly, E.M.; El-Dika, I.; Jarnagin, W.R.; et al. Comprehensive Molecular Profiling of Intrahepatic and Extrahepatic Cholangiocarcinomas: Potential Targets for Intervention. Clin. Cancer Res. 2018, 24, 4154–4161. [Google Scholar] [CrossRef] [PubMed]


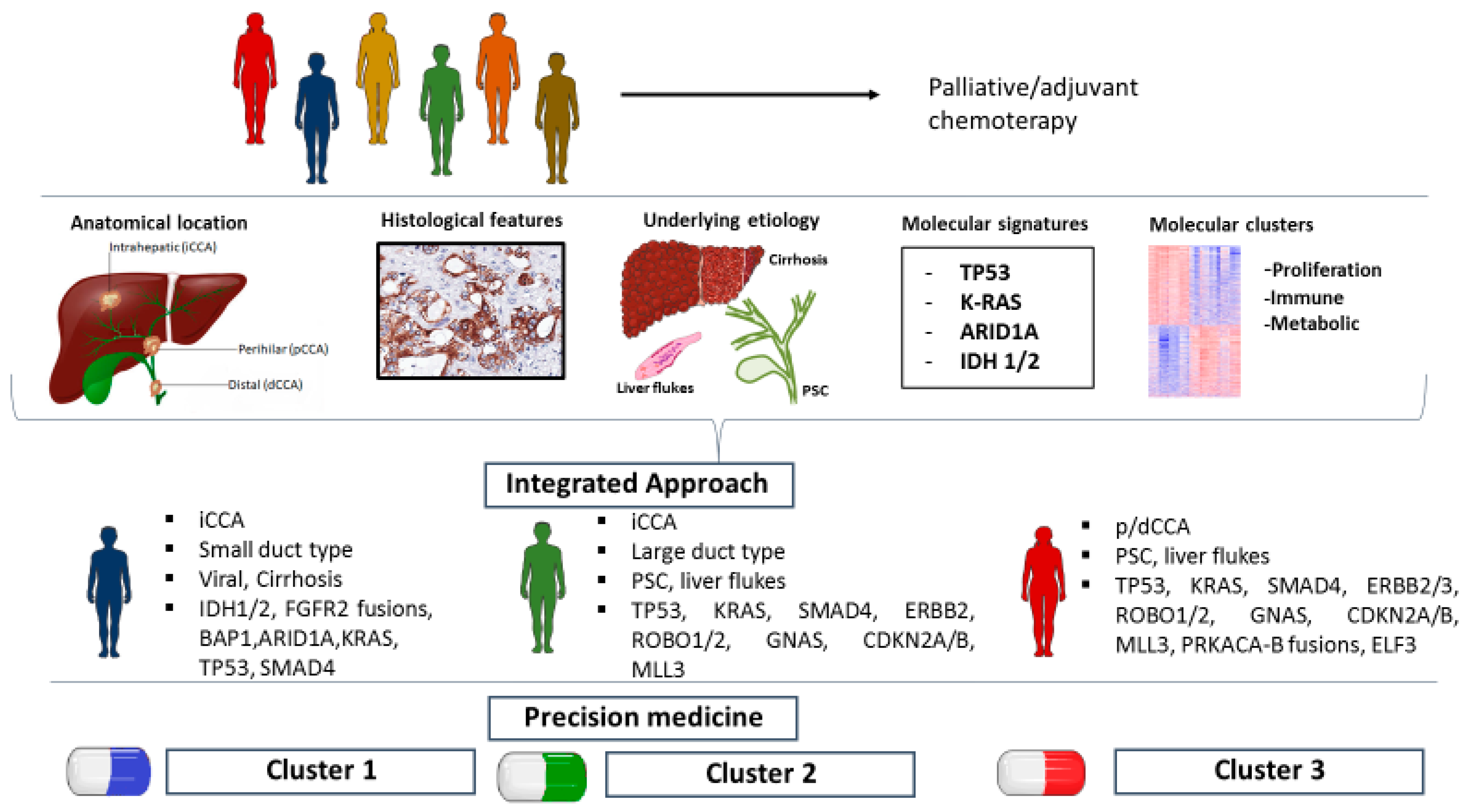{kind=link}
| Study | Nakamura et al., 2015 [23] | Wardell et al., 2018 [24] | Sia et al., 2013 [26] | Nepal et al., 2018 [29] | Montal et al., 2020 [27] | Jusakul et al., 2017 [36] |
|---|---|---|---|---|---|---|
| Samples features | 145 iCCA, 86 p/dCCA, 29 GBC; surgical specimens; Japanese cohort; fluke- | 136 iCCA, 109 pCCA, 101 dCCA, and 66 GBC/CDC; surgical specimens; Japanese and Italian cohort | 149 iCCA; surgical specimens; American, Italian and Spanish cohort genes; poorest prognosis | 496 iCCA | 189 p/dCCA; surgical specimens; American, Spanish and Swiss cohort | 489 samples from Brazil, China, France, Italy, Japan, Romania, Singapore, South Korea, Taiwan, Thailand 133 Fluke+, 39 HBV-HCV+, 5 PSC+ |
| High throughput technique | Whole-exome and transcriptome sequencing | Whole-exome sequencing (n = 107), whole-genome sequencing (n = 39), and targeted sequencing (n = 266) | Whole-genome gene expression microarray profiles, SNP array, and mutation analyses | WES (277), TES (150), genome-wide DNA methylation profiling (69), RNA sequencing (135) | Whole-genome expression profiling, targeted DNA sequencing | WGS (71), targeted sequencing (188), published exome sequencing (200), SNP arrays (175), DNA methylation arrays (138), gene expression arrays (118) |
| Driver genes mutations 1 | TP53 (23% iCCA, 26% p/dCCA) KRAS (25% iCCA, 12% p/d CCA), ARID1A (15% iCCA, 7% p/dCCA), SMAD4 (10% iCCA, 10% p/dCCA), BAP1 (12% iCCA, 3% p/dCCA), PIK3CA(8% iCCA, 4% p/dCCA), ARID2 (4% iCCA, 5% p/dCCA) | TP53 (26%), KRAS (17%), SMAD4 (8%), NF1 (6%), ARID1A (6%), PBRM1 (6%), KMT2D (6%), ATR (6%) | IDH1 (14%), TP53 (13%) and KRAS (12%) | KRAS (36.7%), TP53 (34.7%), ARID1A (14%), SMAD4 (10.7%). Main altered pathways: RTK-RAS-PI3K (53%), TP53- RB (47%), histone modification (22%) and TGFβ (18%) | TP53 (32%) ARID1A (17.4%) KRAS (16.5%) SMAD4 (13%) BAP1 (8.5%) APC (7.1%) PBRM1 (6.5%) ELF3 (6.3%) STK11 (5%) | |
| Molecular clusters | Cluster 1: mostly p/dCCA, negative enrichment of RAS and MAPK activation signatures, best prognosis Cluster 3: mostly iCCA, BAP1, IDH1 and NRAS mutations and FGFR2 fusions Cluster 4: enrichment of immune system and antiapoptotic genes; poorest prognosis | Signature A (5-methylcytosine deamination), Signature B (AID/APOBEC deaminases), Signature C (nucleotide excision repair deficiency) | Proliferation (62%): enrichment of EGFR, RAS, MAPK, AKT, MET, VEGF, PDGF, and HDAC1 pathways; poorest prognosis Inflammation (38%): overexpression of Th2 cytokines (IL4, IL10) and pSTAT3, and downregulation of Th1 cytokines | IDH group: BCLAF1, ARID1A, BAP1, enriched for metabolic pathways, including glutathione metabolism and citrate cycle. KRAS group: SMAD4; EGFR, VEGF and actin cytoskeleton rearrangement TP53 group: PTEN, RB1, LATS2, MAPK, WNT Udt group: KDM6B, mTOR signaling | Metabolic (18.7%): HNF4A = key regulator, HDAC6. Proliferation (22.5%): overexpression of CSNK2A1, MYC targets, activation of cell cycle signaling and DNA repair pathways, enrichment of Ras/MAPK and AKT/mTOR pathways; ERBB2 alterations. Mesenchymal (47.3%): TGF-β1 = key regulator; TNFα signaling, and periostin. Immune (11.5%): IFN-γ = key regulator; over-expression of PD-1 and PD-L1 | Cluster 1: enrichment of ARID1A and BRCA1/2 mutations, TP53 mutations and ERBB2 amplifications Cluster 2 upregulated CTNNB1, WNT5B and AKT expression, downregulation of genes involving EIF translation initiation factors; TP53 mutations and ERBB2 amplifications Cluster 3 Upregulation PD-1, PD-L2 and BTLA Cluster 4 BAP1, IDH1 mutations, FGFR alterations, PI3K pathway signatures |
| Candidate targeted therapy | Ivosidenib [12] Enasidinib NCT02273739 Pemigatinib [38,39] Derazatinib NCT03230318 Infigratinib NCT03773302 Futibatinib NCT04093362 Pembrolizumab NCT04003636 Durvalumab NCT03875235 Bintrafusp alfa NCT04066491 mTOR pathway modulators [37,40] | Ceralasertib NCT03878095 | Sorafenib [20] Regorafenib [41] Cetuximab [19] Panitumumab [42,43] Erlotinib [44,45] mTOR pathway modulators [46,47] Bevacizumab [18] | Ivosidenib [12] Enasidinib NCT02273739 Cetuximab [19] Panitumumab [42,43] Erlotinib [44,45] Bevacizumab [18] Microtubule-targeting drugs [29] mTOR pathway modulators [46,47] | mTOR pathway modulators [46,47] Trastuzumab NCT03613168 Lapatinib [48] Varlitinib NCT03093870 Pembrolizumab NCT04003636 Durvalumab NCT03875235 Bintrafusp alfa NCT04066491 Nivolumab NCT02834013 | Trastuzumab NCT03613168 Lapatinib [48] Varlitinib NCT03093870 mTOR pathway modulators [46,47] Nivolumab NCT02834013 Ivosidenib [12] Enasidinib NCT02273739 Pemigatinib [38,39] Derazatinib NCT03230318 Infigratinib NCT03773302 Futibatinib NCT0409336 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Casadio, M.; Biancaniello, F.; Overi, D.; Venere, R.; Carpino, G.; Gaudio, E.; Alvaro, D.; Cardinale, V. Molecular Landscape and Therapeutic Strategies in Cholangiocarcinoma: An Integrated Translational Approach towards Precision Medicine. Int. J. Mol. Sci. 2021, 22, 5613. https://doi.org/10.3390/ijms22115613
Casadio M, Biancaniello F, Overi D, Venere R, Carpino G, Gaudio E, Alvaro D, Cardinale V. Molecular Landscape and Therapeutic Strategies in Cholangiocarcinoma: An Integrated Translational Approach towards Precision Medicine. International Journal of Molecular Sciences. 2021; 22(11):5613. https://doi.org/10.3390/ijms22115613
Chicago/Turabian StyleCasadio, Marco, Francesca Biancaniello, Diletta Overi, Rosanna Venere, Guido Carpino, Eugenio Gaudio, Domenico Alvaro, and Vincenzo Cardinale. 2021. "Molecular Landscape and Therapeutic Strategies in Cholangiocarcinoma: An Integrated Translational Approach towards Precision Medicine" International Journal of Molecular Sciences 22, no. 11: 5613. https://doi.org/10.3390/ijms22115613
APA StyleCasadio, M., Biancaniello, F., Overi, D., Venere, R., Carpino, G., Gaudio, E., Alvaro, D., & Cardinale, V. (2021). Molecular Landscape and Therapeutic Strategies in Cholangiocarcinoma: An Integrated Translational Approach towards Precision Medicine. International Journal of Molecular Sciences, 22(11), 5613. https://doi.org/10.3390/ijms22115613