
C3 Deficiency Leads to Increased Angiogenesis and Elevated Pro-Angiogenic Leukocyte Recruitment in Ischemic Muscle Tissue


 ,
, 
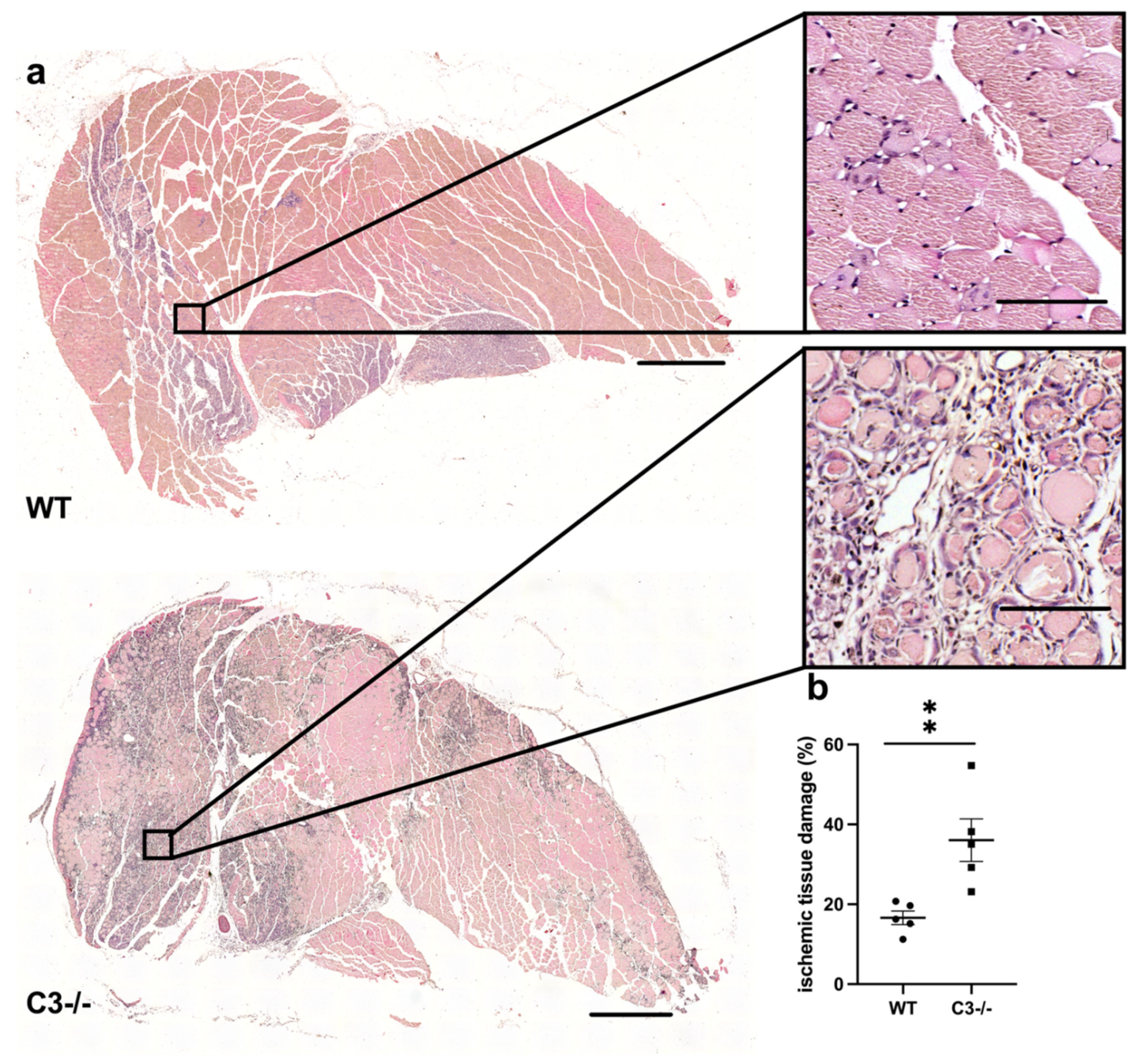{kind=link}
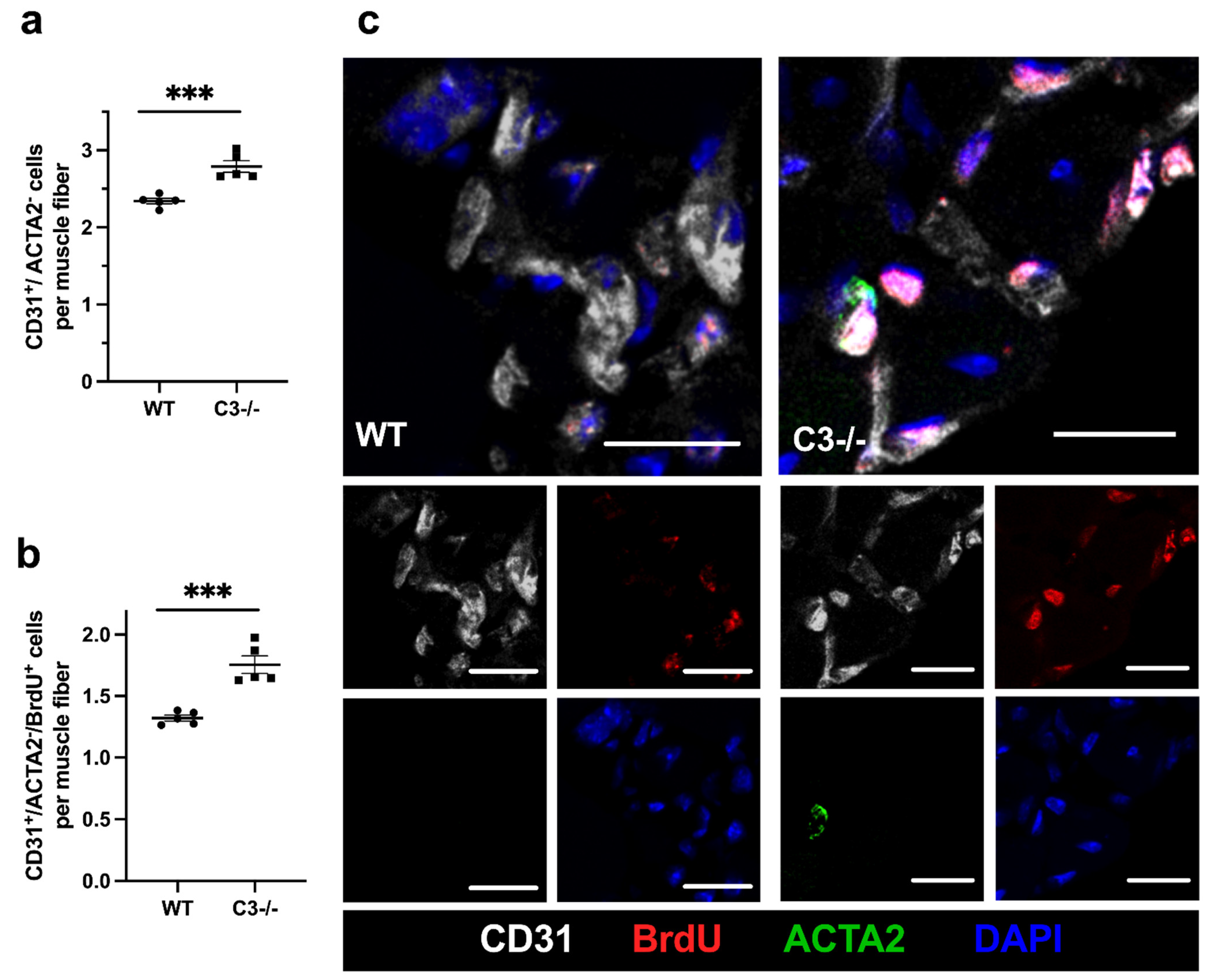{kind=link}
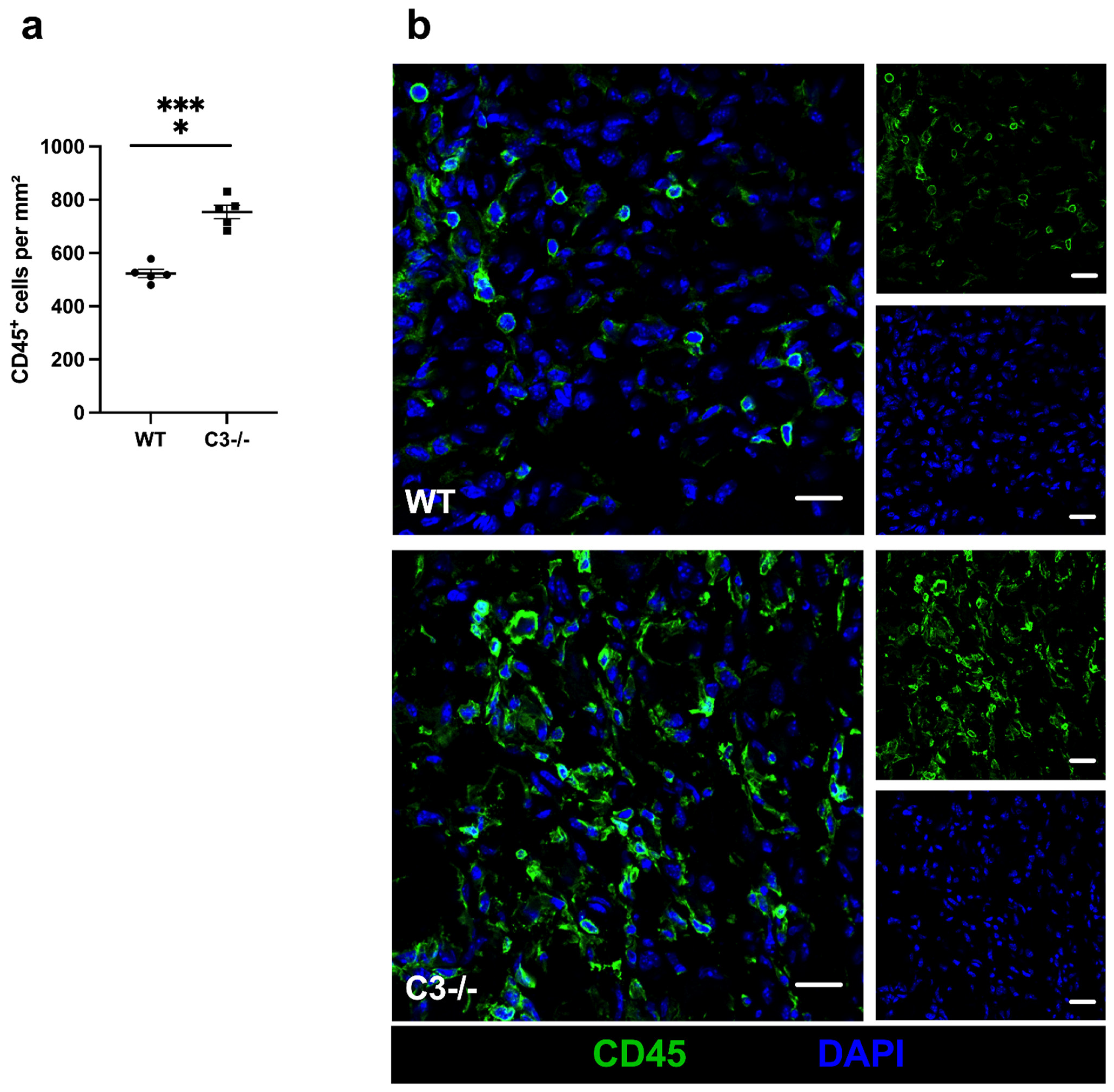{kind=link}
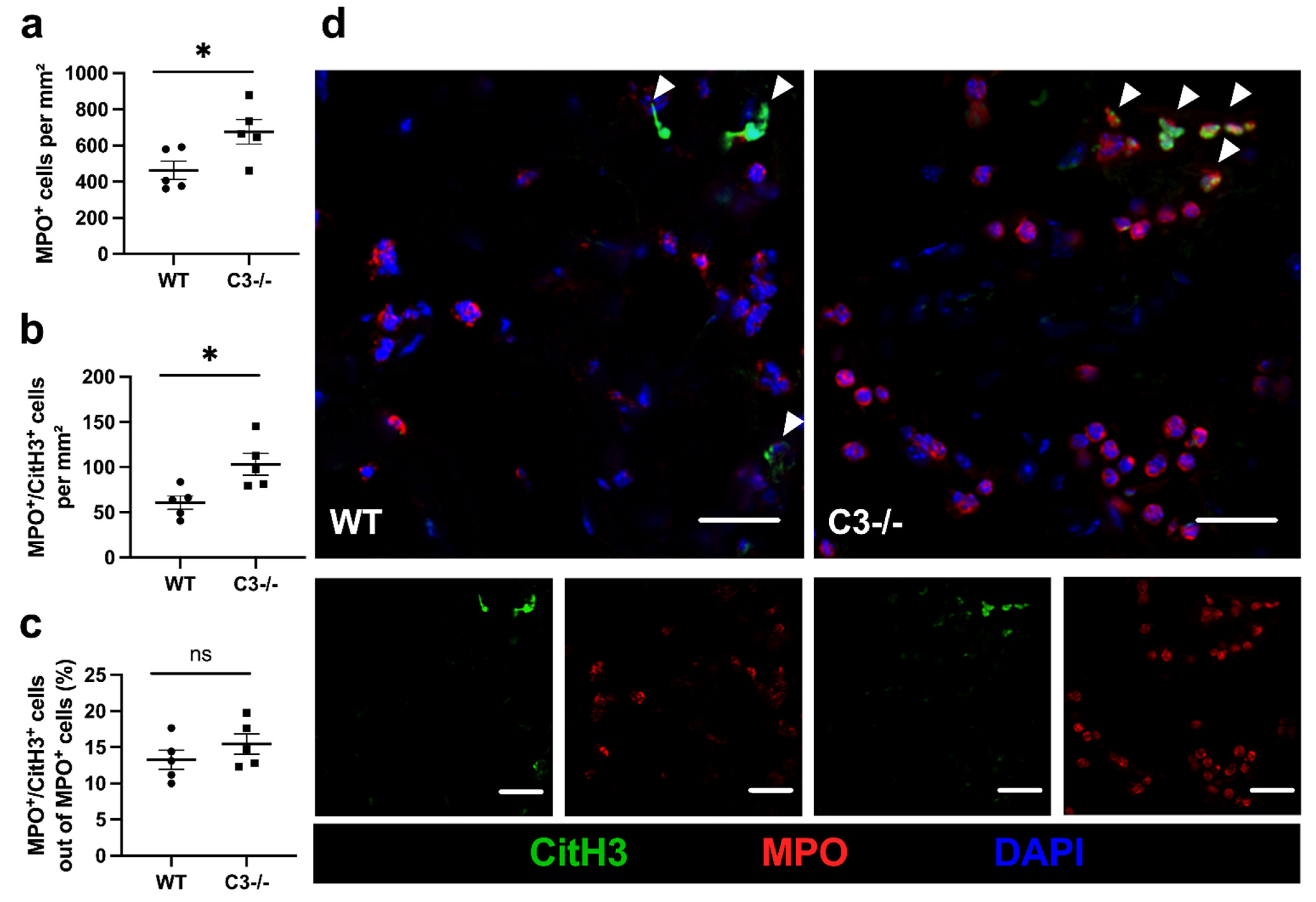{kind=link}
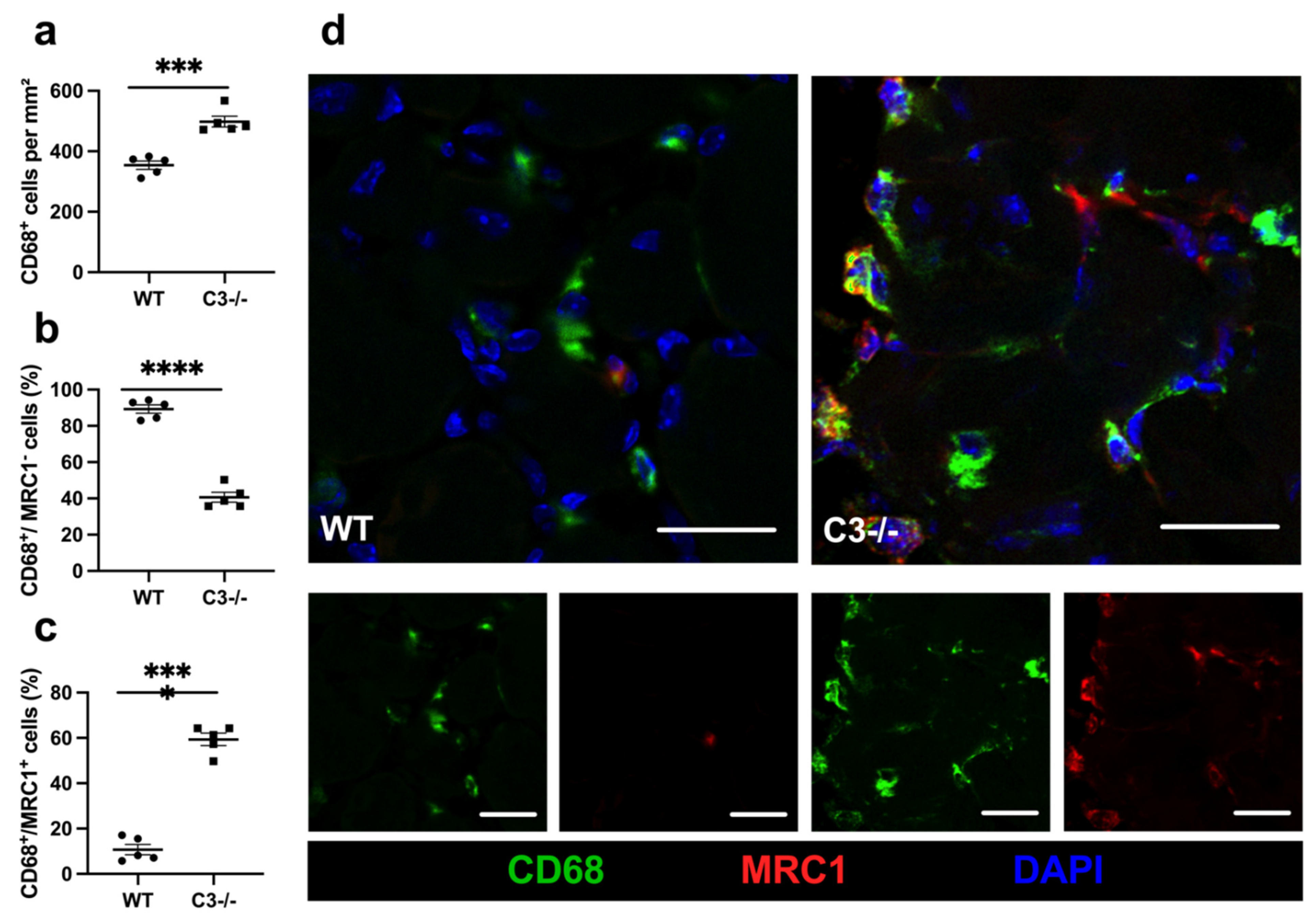{kind=link}
Abstract
1. Introduction
2. Results
3. Discussion
4. Materials and Methods
4.1. Animals and Experimental Procedures
4.2. Histological and Immunofluorescence Analysis
4.3. Statistical Analyses
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Adams, R.H.; Alitalo, K. Molecular regulation of angiogenesis and lymphangiogenesis. Nat. Rev. Mol. Cell Biol. 2007, 8, 464–478. [Google Scholar] [CrossRef]
- Carmeliet, P. Mechanisms of angiogenesis and arteriogenesis. Nat. Med. 2000, 6, 389–395. [Google Scholar] [CrossRef]
- Egginton, S.; Zhou, A.-L.; Brown, M.D.; Hudlická, O. Unorthodox angiogenesis in skeletal muscle. Cardiovasc. Res. 2001, 49, 634–646. [Google Scholar] [CrossRef]
- Carmeliet, P. Angiogenesis in health and disease. Nat. Med. 2003, 9, 653–660. [Google Scholar] [CrossRef] [PubMed]
- Weckbach, L.T.; Preissner, K.T.; Deindl, E. The Role of Midkine in Arteriogenesis, Involving Mechanosensing, Endothelial Cell Proliferation, and Vasodilation. Int. J. Mol. Sci. 2018, 19, 2559. [Google Scholar] [CrossRef]
- Deindl, E.; Zamba, M.; Brunner, S.; Huber, B.; Mehl, U.; Assmann, G.; Hoefer, I.E.; Mueller-Hoecker, J.; Franz, W. G-CSF administration after myocardial infarction in mice attenuates late ischemic cardiomyopathy by enhanced arteriogenesis. FASEB J. 2006, 20, 956–958. [Google Scholar] [CrossRef]
- Fukumura, D.; Xu, L.; Chen, Y.; Gohongi, T.; Seed, B.; Jain, R.K. Hypoxia and acidosis independently up-regulate vascular endothelial growth factor transcription in brain tumors in vivo. Cancer Res. 2001, 61, 6020–6024. [Google Scholar]
- Shima, D.T.; Adamis, A.P.; Ferrara, N.; Yeo, K.-T.; Yeo, T.-K.; Allende, R.; Folkman, J.; D Amore, P.A. Hypoxic induction of endothelial cell growth factors in retinal cells: Identification and characterization of vascular endothelial growth factor (VEGF) as the mitogen. Mol. Med. 1995, 1, 182–193. [Google Scholar] [CrossRef]
- Scapini, P.; Morini, M.; Tecchio, C.; Minghelli, S.; DI Carlo, E.; Tanghetti, E.; Albini, A.; Lowell, C.; Berton, G.; Noonan, D.M.; et al. CXCL1/Macrophage Inflammatory Protein-2-Induced Angiogenesis In Vivo Is Mediated by Neutrophil-Derived Vascular Endothelial Growth Factor-A. J. Immunol. 2004, 172, 5034–5040. [Google Scholar] [CrossRef]
- Gaudry, M.; Brégerie, O.; Andrieu, V.; El Benna, J.; Pocidalo, M.A.; Hakim, J. Intracellular pool of vascular endothelial growth factor in human neutrophils. Blood 1997, 90, 4153–4161. [Google Scholar] [CrossRef]
- Wang, J. Neutrophils in tissue injury and repair. Cell Tissue Res. 2018, 371, 531–539. [Google Scholar] [CrossRef] [PubMed]
- Du Cheyne, C.; Tay, H.; De Spiegelaere, W. The complex TIE between macrophages and angiogenesis. Anat. Histol. Embryol. 2019, 49, 585–596. [Google Scholar] [CrossRef] [PubMed]
- Vestweber, D. How leukocytes cross the vascular endothelium. Nat. Rev. Immunol. 2015, 15, 692–704. [Google Scholar] [CrossRef] [PubMed]
- Qiu, D.; Nikita, D.; Zhang, L.; Deng, J.; Xia, Z.; Zhan, J.; Huang, J.; Liu, L.; Liu, F.; Duan, J.; et al. ICAM-1 deletion delays the repair process in aging diabetic mice. Metabolism 2020, 114, 154412. [Google Scholar] [CrossRef]
- Benedicto, A.; Herrero, A.; Romayor, I.; Marquez, J.; Smedsrød, B.; Olaso, E.; Arteta, B. Liver sinusoidal endothelial cell ICAM-1 mediated tumor/endothelial crosstalk drives the development of liver metastasis by initiating inflammatory and angiogenic responses. Sci. Rep. 2019, 9, 1–12. [Google Scholar] [CrossRef]
- Hoefer, I.E.; van Royen, N.; Rectenwald, J.E.; Deindl, E.; Hua, J.; Jost, M.; Grundmann, S.; Voskuil, M.; Ozaki, C.K.; Piek, J.J.; et al. Arteriogenesis Proceeds via ICAM-1/Mac-1- Mediated Mechanisms. Circ. Res. 2004, 94, 1179–1185. [Google Scholar] [CrossRef]
- Kaufmann, S.H. Immunology’s foundation: The 100-year anniversary of the Nobel Prize to Paul Ehrlich and Elie Metchnikoff. Nat. Immunol. 2008, 9, 705–712. [Google Scholar] [CrossRef]
- Merle, N.S.; Noé, R.; Halbwachs-Mecarelli, L.; Fremeaux-Bacchi, V.; Roumenina, L.T. Complement System Part II: Role in Immunity. Front. Immunol. 2015, 6, 257. [Google Scholar] [CrossRef]
- Sjöberg, A.; Trouw, L.; Blom, A. Complement activation and inhibition: A delicate balance. Trends Immunol. 2009, 30, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Ricklin, D.; Hajishengallis, G.; Yang, K.; Lambris, J.D. Complement: A key system for immune surveillance and homeostasis. Nat. Immunol. 2010, 11, 785–797. [Google Scholar] [CrossRef]
- Merle, N.S.; Church, S.E.; Fremeaux-Bacchi, V.; Roumenina, L.T. Complement System Part I–Molecular Mechanisms of Activation and Regulation. Front. Immunol. 2015, 6, 262. [Google Scholar] [CrossRef]
- Wu, M.C.L.; Brennan, F.H.; Lynch, J.P.L.; Mantovani, S.; Phipps, S.; Wetsel, R.A.; Ruitenberg, M.J.; Taylor, S.M.; Woodruff, T.M. The receptor for complement component C3a mediates protection from intestinal ischemia-reperfusion injuries by inhibiting neutrophil mobilization. Proc. Natl. Acad. Sci. USA 2013, 110, 9439–9444. [Google Scholar] [CrossRef]
- Peng, Q.; Li, K.; Patel, H.; Sacks, S.; Zhou, W. Dendritic Cell Synthesis of C3 Is Required for Full T Cell Activation and Development of a Th1 Phenotype. J. Immunol. 2006, 176, 3330–3341. [Google Scholar] [CrossRef] [PubMed]
- Strainic, M.G.; Liu, J.; Huang, D.; An, F.; Lalli, P.N.; Muqim, N.; Shapiro, V.S.; Dubyak, G.; Heeger, P.S.; Medof, M.E. Locally Produced Complement Fragments C5a and C3a Provide Both Costimulatory and Survival Signals to Naive CD4+ T Cells. Immunity 2008, 28, 425–435. [Google Scholar] [CrossRef] [PubMed]
- Nozaki, M.; Raisler, B.J.; Sakurai, E.; Sarma, J.V.; Barnum, S.R.; Lambris, J.D.; Chen, Y.; Zhang, K.; Ambati, B.K.; Baffi, J.Z.; et al. Drusen complement components C3a and C5a promote choroidal neovascularization. Proc. Natl. Acad. Sci. USA 2006, 103, 2328–2333. [Google Scholar] [CrossRef]
- Fischetti, F.; Tedesco, F. Cross-talk between the complement system and endothelial cells in physiologic conditions and in vascular diseases. Autoimmunity 2006, 39, 417–428. [Google Scholar] [CrossRef]
- Riedl, M.; Thorner, P.; Licht, C. C3 Glomerulopathy. Pediatr. Nephrol. 2016, 32, 43–57. [Google Scholar] [CrossRef] [PubMed]
- Seifert, P.S.; Hugo, F.; Hansson, G.K.; Bhakdi, S. Prelesional complement activation in experimental atherosclerosis. Terminal C5b-9 complement deposition coincides with cholesterol accumulation in the aortic intima of hypercholesterolemic rabbits. Lab. Investig. 1989, 60, 747–754. [Google Scholar]
- Torzewski, M.; Klouche, M.; Hock, J.; Meßner, M.; Dorweiler, B.; Torzewski, J.; Gabbert, H.E.; Bhakdi, S. Immunohistochemical Demonstration of Enzymatically Modified Human LDL and Its Colocalization With the Terminal Complement Complex in the Early Atherosclerotic Lesion. Arter. Thromb. Vasc. Biol. 1998, 18, 369–378. [Google Scholar] [CrossRef]
- Lin, Z.; Lin, H.; Li, W.; Huang, Y.; Dai, H. Complement Component C3 Promotes Cerebral Ischemia/Reperfusion Injury Mediated by TLR2/NFκB Activation in Diabetic Mice. Neurochem. Res. 2018, 43, 1599–1607. [Google Scholar] [CrossRef]
- Wei, L.-L.; Ma, N.; Wu, K.-Y.; Wang, J.-X.; Diao, T.-Y.; Zhao, S.-J.; Bai, L.; Liu, E.; Li, Z.-F.; Zhou, W.; et al. Protective Role of C3aR (C3a Anaphylatoxin Receptor) Against Atherosclerosis in Atherosclerosis-Prone Mice. Arter. Thromb. Vasc. Biol. 2020, 40, 2070–2083. [Google Scholar] [CrossRef]
- Bora, P.S.; Sohn, J.-H.; Cruz, J.M.C.; Jha, P.; Nishihori, H.; Wang, Y.; Kaliappan, S.; Kaplan, H.J.; Bora, N.S. Role of Complement and Complement Membrane Attack Complex in Laser-Induced Choroidal Neovascularization. J. Immunol. 2004, 174, 491–497. [Google Scholar] [CrossRef] [PubMed]
- Langer, H.F.; Chung, K.-J.; Orlova, V.V.; Choi, E.Y.; Kaul, S.; Kruhlak, M.J.; Alatsatianos, M.; DeAngelis, R.A.; Roche, P.; Magotti, P.; et al. Complement-mediated inhibition of neovascularization reveals a point of convergence between innate immunity and angiogenesis. Blood 2010, 116, 4395–4403. [Google Scholar] [CrossRef] [PubMed]
- Collard, C.D.; Väkevä, A.; Morrissey, M.A.; Agah, A.; Rollins, S.A.; Reenstra, W.R.; Buras, J.A.; Meri, S.; Stahl, G.L. Complement activation after oxidative stress: Role of the lectin complement pathway. Am. J. Pathol. 2000, 2000. 156, 1549–1556. [Google Scholar] [CrossRef]
- Väkevä, A.; Meri, S. Complement activation and regulator expression after anoxic injury of human endothelial cells. APMIS 1998, 106, 1149–1156. [Google Scholar] [CrossRef]
- Vercellotti, G.M.; Dalmasso, A.P.; Schaid, T.R., Jr.; Nguyen, J.; Chen, C.; Ericson, M.E.; Fuad, A.; Killeen, T.; Lindorfer, M.A.; Taylor, R.P.; et al. Critical role of C5a in sickle cell disease. Am. J. Hematol. 2019, 94, 327–337. [Google Scholar] [CrossRef] [PubMed]
- Limbourg, A.; Korff, T.; Napp, L.C.; Schaper, W.; Drexler, H.; Limbourg, F. Evaluation of postnatal arteriogenesis and angiogenesis in a mouse model of hind-limb ischemia. Nat. Protoc. 2009, 4, 1737–1748. [Google Scholar] [CrossRef]
- Chillo, O.; Kleinert, E.C.; Lautz, T.; Lasch, M.; Pagel, J.-I.; Heun, Y.; Troidl, K.; Fischer, S.; Caballero-Martinez, A.; Mauer, A.; et al. Perivascular Mast Cells Govern Shear Stress-Induced Arteriogenesis by Orchestrating Leukocyte Function. Cell Rep. 2016, 16, 2197–2207. [Google Scholar] [CrossRef]
- Heil, M.; Eitenmüller, I.; Schmitz-Rixen, T.; Schaper, W. Arteriogenesis versus angiogenesis: Similarities and differences. J. Cell. Mol. Med. 2006, 10, 45–55. [Google Scholar] [CrossRef]
- Deindl, E.; Buschmann, I.; Hoefer, I.E.; Podzuweit, T.; Boengler, K.; Vogel, S.; Van Royen, N.; Fernandez, B.; Schaper, W. Role of Ischemia and of Hypoxia-Inducible Genes in Arteriogenesis After Femoral Artery Occlusion in the Rabbit. Circ. Res. 2001, 89, 779–786. [Google Scholar] [CrossRef]
- Pipp, F.; Boehm, S.; Cai, W.-J.; Adili, F.; Ziegler, B.; Karanovic, G.; Ritter, R.; Balzer, J.; Scheler, C.; Schaper, W.; et al. Elevated Fluid Shear Stress Enhances Postocclusive Collateral Artery Growth and Gene Expression in the Pig Hind Limb. Arter. Thromb. Vasc. Biol. 2004, 24, 1664–1668. [Google Scholar] [CrossRef] [PubMed]
- Vogel, C.-W. The Role of Complement in Myocardial Infarction Reperfusion Injury: An Underappreciated Therapeutic Target. Front. Cell Dev. Biol. 2020, 8, 606407. [Google Scholar] [CrossRef] [PubMed]
- Duehrkop, C.; Rieben, R. Ischemia/reperfusion injury: Effect of simultaneous inhibition of plasma cascade systems versus specific complement inhibition. Biochem. Pharmacol. 2014, 88, 12–22. [Google Scholar] [CrossRef]
- Zhou, W.; Farrar, C.A.; Abe, K.; Pratt, J.R.; Marsh, J.E.; Wang, Y.; Stahl, G.; Sacks, S.H. Predominant role for C5b-9 in renal ischemia/reperfusion injury. J. Clin. Investig. 2000, 105, 1363–1371. [Google Scholar] [CrossRef]
- Gong, Y.; Koh, D.-R. Neutrophils promote inflammatory angiogenesis via release of preformed VEGF in an in vivo corneal model. Cell Tissue Res. 2009, 339, 437–448. [Google Scholar] [CrossRef]
- Hong, H.; Tian, X.Y. The Role of Macrophages in Vascular Repair and Regeneration after Ischemic Injury. Int. J. Mol. Sci. 2020, 21, 6328. [Google Scholar] [CrossRef] [PubMed]
- Heijink, I.H.; Pouwels, S.D.; Leijendekker, C.; De Bruin, H.G.; Zijlstra, G.J.; Van Der Vaart, H.; Hacken, N.H.T.T.; Van Oosterhout, A.J.M.; Nawijn, M.C.; Van Der Toorn, M. Cigarette Smoke–Induced Damage-Associated Molecular Pattern Release from Necrotic Neutrophils Triggers Proinflammatory Mediator Release. Am. J. Respir. Cell Mol. Biol. 2015, 52, 554–562. [Google Scholar] [CrossRef]
- Pittman, K.; Kubes, P. Damage-Associated Molecular Patterns Control Neutrophil Recruitment. J. Innate Immun. 2013, 5, 315–323. [Google Scholar] [CrossRef]
- Wolf, D.; Ley, K. Immunity and Inflammation in Atherosclerosis. Circ. Res. 2019, 124, 315–327. [Google Scholar] [CrossRef]
- Capitão, M.; Soares, R. Angiogenesis and Inflammation Crosstalk in Diabetic Retinopathy. J. Cell. Biochem. 2016, 117, 2443–2453. [Google Scholar] [CrossRef] [PubMed]
- Frangogiannis, N.G. The inflammatory response in myocardial injury, repair, and remodelling. Nat. Rev. Cardiol. 2014, 11, 255–265. [Google Scholar] [CrossRef]
- Huber-Lang, M.; Sarma, J.V.; Zetoune, F.S.; Rittirsch, D.; Neff, T.A.; McGuire, S.R.; Lambris, J.D.; Warner, R.L.; Flierl, M.A.; Hoesel, L.M.; et al. Generation of C5a in the absence of C3: A new complement activation pathway. Nat. Med. 2006, 12, 682–687. [Google Scholar] [CrossRef]
- Mustafa, A.; Nityanand, S.; Berglund, L.; Lithell, H.; Lefvert, A.K. Circulating Immune Complexes in 50-Year-Old Men as a Strong and Independent Risk Factor for Myocardial Infarction. Circulation 2000, 102, 2576–2581. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Oksjoki, R.; Kovanen, P.T.; Pentikäinen, M.O. Role of complement activation in atherosclerosis. Curr. Opin. Lipidol. 2003, 14, 477–482. [Google Scholar] [CrossRef] [PubMed]
- Buono, C.; Come, C.E.; Witztum, J.L.; Maguire, G.F.; Connelly, P.W.; Carroll, M.; Lichtman, A.H. Influence of C3 Deficiency on Atherosclerosis. Circulation 2002, 105, 3025–3031. [Google Scholar] [CrossRef] [PubMed]
- Niculescu, F.; Rus, H. The Role of Complement Activation in Atherosclerosis. Immunol. Res. 2004, 30, 73–80. [Google Scholar] [CrossRef]
- Christoffersson, G.; Henriksnäs, J.; Johansson, L.; Rolny, C.; Ahlström, H.; Caballero-Corbalan, J.; Segersvärd, R.; Permert, J.; Korsgren, O.; Carlsson, P.-O.; et al. Clinical and experimental pancreatic islet transplantation to striated muscle: Establishment of a vascular system similar to that in native islets. Diabetes 2010, 59, 2569–2578. [Google Scholar] [CrossRef]
- Mittal, M.; Siddiqui, M.R.; Tran, K.; Reddy, S.P.; Malik, A.B. Reactive Oxygen Species in Inflammation and Tissue Injury. Antioxid. Redox Signal. 2014, 20, 1126–1167. [Google Scholar] [CrossRef] [PubMed]
- Yipp, B.G.; Petri, B.; Salina, D.; Jenne, C.N.; Scott, B.N.V.; Zbytnuik, L.D.; Pittman, K.; Asaduzzaman, M.; Wu, K.; Meijndert, H.C.; et al. Infection-induced NETosis is a dynamic process involving neutrophil multitasking in vivo. Nat. Med. 2012, 18, 1386–1393. [Google Scholar] [CrossRef]
- Denning, N.-L.; Aziz, M.; Gurien, S.D.; Wang, P. DAMPs and NETs in Sepsis. Front. Immunol. 2019, 10, 2536. [Google Scholar] [CrossRef]
- Lefrançais, E.; Mallavia, B.; Zhuo, H.; Calfee, C.S.; Looney, M.R. Maladaptive role of neutrophil extracellular traps in pathogen-induced lung injury. JCI Insight 2018, 3. [Google Scholar] [CrossRef]
- Mutua, V.; Gershwin, L.J. A Review of Neutrophil Extracellular Traps (NETs) in Disease: Potential Anti-NETs Therapeutics. Clin. Rev. Allergy Immunol. 2020, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Aldabbous, L.; Abdul-Salam, V.; McKinnon, T.; Duluc, L.; Pepke-Zaba, J.; Southwood, M.; Ainscough, A.J.; Hadinnapola, C.; Wilkins, M.R.; Toshner, M.; et al. Neutrophil Extracellular Traps Promote Angiogenesis: Evidence From Vascular Pathology in Pulmonary Hypertension. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 2078–2087. [Google Scholar] [CrossRef] [PubMed]
- Binet, F.; Cagnone, G.; Crespo-Garcia, S.; Hata, M.; Neault, M.; Dejda, A.; Wilson, A.M.; Buscarlet, M.; Mawambo, G.T.; Howard, J.P.; et al. Neutrophil extracellular traps target senescent vasculature for tissue remodeling in retinopathy. Science 2020, 369, eaay5356. [Google Scholar] [CrossRef] [PubMed]
- Barnett, F.H.; Rosenfeld, M.; Wood, M.; Kiosses, W.B.; Usui, Y.; Marchetti, V.; Aguilar, E.; Friedlander, M. Macrophages form functional vascular mimicry channels in vivo. Sci. Rep. 2016, 6, 36659. [Google Scholar] [CrossRef]
- Lertkiatmongkol, P.; Liao, D.; Mei, H.; Hu, Y.; Newman, P.J. Endothelial functions of platelet/endothelial cell adhesion molecule-1 (CD31). Curr. Opin. Hematol. 2016, 23, 253–259. [Google Scholar] [CrossRef]
- Kübler, M.; Beck, S.; Fischer, S.; Götz, P.; Kumaraswami, K.; Ishikawa-Ankerhold, H.; Lasch, M.; Deindl, E. Absence of Cold-Inducible RNA-Binding Protein (CIRP) Promotes Angiogenesis and Regeneration of Ischemic Tissue by Inducing M2-Like Macrophage Polarization. Biomedicines 2021, 9, 395. [Google Scholar] [CrossRef]
- Moore, E.M.; West, J.L. Harnessing Macrophages for Vascularization in Tissue Engineering. Ann. Biomed. Eng. 2018, 47, 354–365. [Google Scholar] [CrossRef]
- Murray, P.J. Macrophage Polarization. Annu. Rev. Physiol. 2017, 79, 541–566. [Google Scholar] [CrossRef]
- Wynn, T.A.; Vannella, K.M. Macrophages in Tissue Repair, Regeneration, and Fibrosis. Immunity 2016, 44, 450–462. [Google Scholar] [CrossRef]
- Gurevich, D.B.; Severn, C.E.; Twomey, C.; Greenhough, A.; Cash, J.; Toye, A.M.; Mellor, H.; Martin, P. Live imaging of wound angiogenesis reveals macrophage orchestrated vessel sprouting and regression. EMBO J. 2018, 37. [Google Scholar] [CrossRef]
- Zajac, E.; Schweighofer, B.; Kupriyanova, T.A.; Juncker-Jensen, A.; Minder, P.; Quigley, J.P.; Deryugina, E.I. Angiogenic capacity of M1- and M2-polarized macrophages is determined by the levels of TIMP-1 complexed with their secreted proMMP-9. Blood 2013, 122, 4054–4067. [Google Scholar] [CrossRef] [PubMed]
- Dort, J.; Fabre, P.; Molina, T.; Dumont, N.A. Macrophages Are Key Regulators of Stem Cells during Skeletal Muscle Regeneration and Diseases. Stem Cells Int. 2019, 2019, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Asgari, E.; Le Friec, G.; Yamamoto, H.; Perucha, E.; Sacks, S.S.; Köhl, J.; Cook, H.T.; Kemper, C. C3a modulates IL-1beta secretion in human monocytes by regulating ATP efflux and subsequent NLRP3 inflammasome activation. Blood 2013, 122, 3473–3481. [Google Scholar] [CrossRef]
- Takabayashi, T.; Vannier, E.; Clark, B.D.; Margolis, N.H.; Dinarello, C.A.; Burke, J.F.; Gelfand, J.A. A new biologic role for C3a and C3a desArg: Regulation of TNF-alpha and IL-1 beta synthesis. J. Immunol. 1996, 156, 3455–3460. [Google Scholar] [PubMed]
- Olfert, I.M.; Baum, O.; Hellsten, Y.; Egginton, S. Advances and challenges in skeletal muscle angiogenesis. Am. J. Physiol. Circ. Physiol. 2016, 310, H326–H336. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Götz, P.; Braumandl, A.; Kübler, M.; Kumaraswami, K.; Ishikawa-Ankerhold, H.; Lasch, M.; Deindl, E. C3 Deficiency Leads to Increased Angiogenesis and Elevated Pro-Angiogenic Leukocyte Recruitment in Ischemic Muscle Tissue. Int. J. Mol. Sci. 2021, 22, 5800. https://doi.org/10.3390/ijms22115800
Götz P, Braumandl A, Kübler M, Kumaraswami K, Ishikawa-Ankerhold H, Lasch M, Deindl E. C3 Deficiency Leads to Increased Angiogenesis and Elevated Pro-Angiogenic Leukocyte Recruitment in Ischemic Muscle Tissue. International Journal of Molecular Sciences. 2021; 22(11):5800. https://doi.org/10.3390/ijms22115800
Chicago/Turabian StyleGötz, Philipp, Anna Braumandl, Matthias Kübler, Konda Kumaraswami, Hellen Ishikawa-Ankerhold, Manuel Lasch, and Elisabeth Deindl. 2021. "C3 Deficiency Leads to Increased Angiogenesis and Elevated Pro-Angiogenic Leukocyte Recruitment in Ischemic Muscle Tissue" International Journal of Molecular Sciences 22, no. 11: 5800. https://doi.org/10.3390/ijms22115800
APA StyleGötz, P., Braumandl, A., Kübler, M., Kumaraswami, K., Ishikawa-Ankerhold, H., Lasch, M., & Deindl, E. (2021). C3 Deficiency Leads to Increased Angiogenesis and Elevated Pro-Angiogenic Leukocyte Recruitment in Ischemic Muscle Tissue. International Journal of Molecular Sciences, 22(11), 5800. https://doi.org/10.3390/ijms22115800