
Candidate Genes for Eyelid Myoclonia with Absences, Review of the Literature

 , , and
, , and
Abstract
1. Introduction
2. Results and Discussion
2.1. SYNGAP1
2.2. KIA2022/NEXMIF
2.3. RORB
2.4. CHD2
2.5. Other Genes of Interest
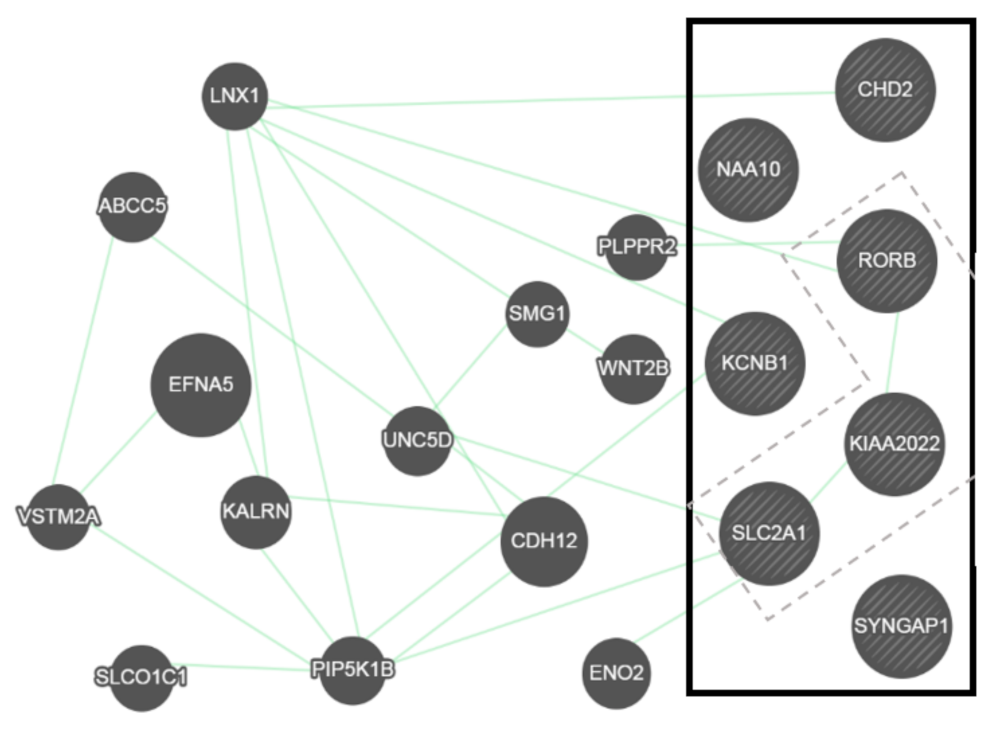
2.6. General Overview of Genetic Interactions
2.7. Animal Models
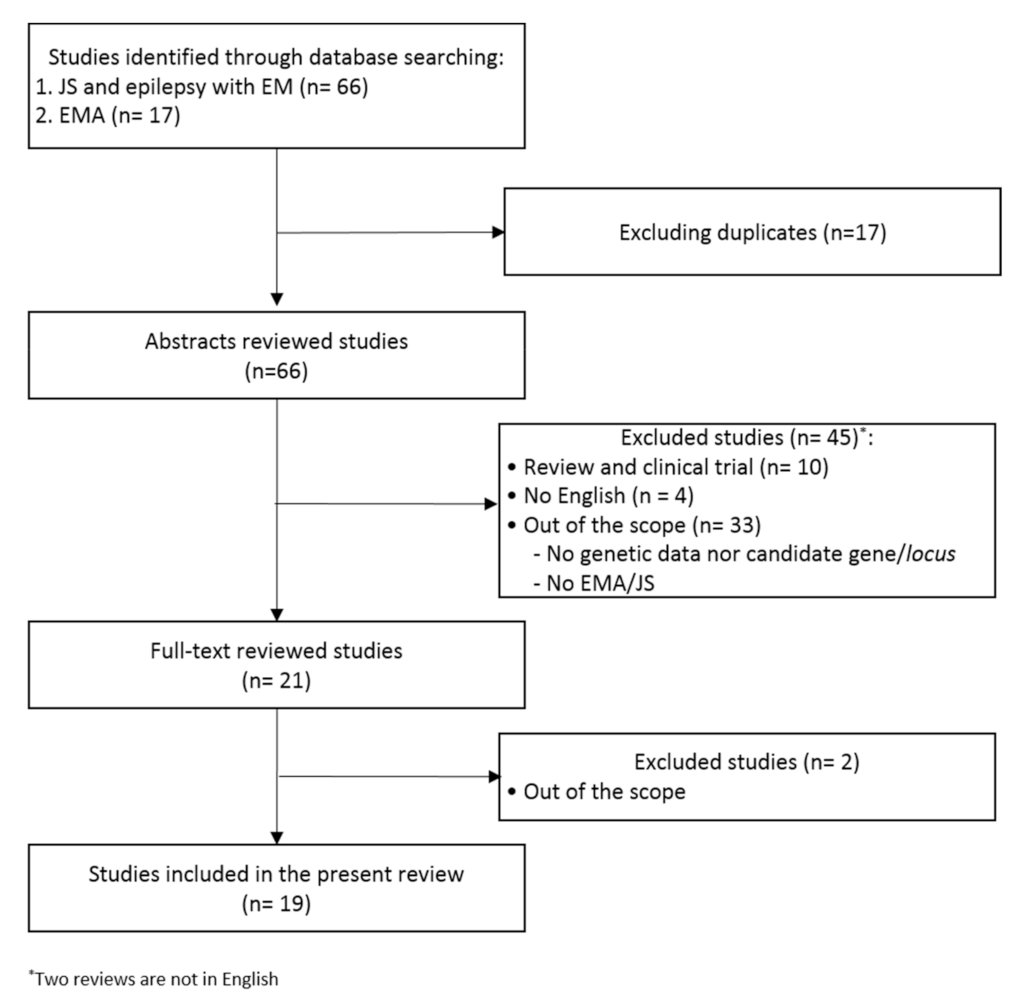
3. Methods
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
Appendix A
- ((Jeavons Syndrome) OR (epilepsy with eyelid myoclonias)) AND (genetic OR gene OR genes)
- (“Eyelid myoclonia with absences”) AND (genetic OR gene OR genes)
Abbreviations
| ADHD | Attention deficit hyperactivity disorder |
| AED | Antiepileptic drugs |
| ASD | Autism spectrum disorder |
| CHD | Chromodomain helicase DNA-binding |
| DD | Developmental delay |
| DEE | Developmental epileptic encephalopathy |
| EEOC | Childhood-onset epileptic encephalopathy |
| EEG | Electroencephalogram |
| EM | Eyelid myoclonia |
| EMA | Eyelid myoclonia with absences |
| GGE | Genetic generalized epilepsies |
| GTCS | Generalized tonic-clonic seizures |
| ID | Intellectual disability |
| ILAE | International League Against Epilepsy |
| IPS | Intermittent photic stimulation |
| JS | Jeavons syndrome |
| KO | Knock-out |
| MAE | Myoclonic-atonic epilepsy |
| MRX98 | Mental retardation X-linked 98 |
| PS | Photosensitivity |
| XCI | X-Chromosome Inactivation |
| XPN | X-linked Intellectual Disability Protein Related to Neurite Extension |
References
- Jeavons, P.M. Nosological Problems of Myoclonic Epilepsies in Childhood and Adolescence. Dev. Med. Child Neurol. 1977, 19, 3–8. [Google Scholar] [CrossRef]
- Samanta, D.; Willis, E. KIAA2022-related disorders can cause Jeavons (eyelid myoclonia with absence) syndrome. Acta Neurol. Belg. 2020, 120, 205–207. [Google Scholar] [CrossRef] [PubMed]
- Striano, S.; Capovilla, G.; Sofia, V.; Romeo, A.; Rubboli, G.; Striano, P.; Trenité, D.K.N. Eyelid myoclonia with absences (Jeavons syndrome): A well-defined idiopathic generalized epilepsy syndrome or a spectrum of photosensitive conditions? Epilepsia 2009, 50, 15–19. [Google Scholar] [CrossRef]
- Striano, S. Eyelid myoclonia with absences: An overlooked epileptic syndrome?Les myoclonies des paupières avec absences: Un syndrome épileptique sous-estimé ? Neurophysiol. Clin. Neurophysiol. 2002, 32, 287–296. [Google Scholar] [CrossRef]
- International League against Epilepsy. Epilepsy with Eyelid Myoclonias. Available online: https://www.epilepsydiagnosis.org/syndrome/emwa-overview.html (accessed on 1 April 2021).
- Parker, A.; Gardiner, R.; Panayiotopoulos, C.P.; Agathonikou, A.; Ferrie, C.D. Observations on families with eyelid myoclonia with absences. In Eyelid Myoclonia with Absences; Duncan, J.S., Panayiotopoulos, C.P., Eds.; John Libbey Ltd.: London, UK, 1996; pp. 107–115. [Google Scholar]
- Demarco, P. Eyelid Myoclonia with Absences (EMA) in Two Monovular Twins. Clin. EEG Neurosci. 1989, 20, 193–195. [Google Scholar] [CrossRef] [PubMed]
- Adachi, M.; Inoue, T.; Tsuneishi, S.; Takada, S.; Nakamura, H. Eyelid myoclonia with absences in monozygotic twins. Pediatr. Int. 2005, 47, 343–347. [Google Scholar] [CrossRef] [PubMed]
- Striano, P. Orphanet Encyclopedia Jeavons Syndrome. Available online: https://www.orpha.net/consor/cgi-bin/OC_Exp.php?lng=EN&Expert=139431#:~:text=Disease (accessed on 1 April 2021).
- Valentine, V.; Sogawa, Y.; Rajan, D.; Ortiz, D. A case of de novo NAA10 mutation presenting with eyelid myoclonias (AKA Jeavons syndrome). Seizure 2018, 60, 120–122. [Google Scholar] [CrossRef] [PubMed]
- Morea, A.; Boero, G.; Demaio, V.; Francavilla, T.; La Neve, A. Eyelid myoclonia with absences, intellectual disability and attention deficit hyperactivity disorder: A clinical phenotype of the RORB gene mutation. Neurol. Sci. 2021, 42, 2059–2062. [Google Scholar] [CrossRef] [PubMed]
- Klitten, L.L.; Møller, R.S.; Nikanorova, M.; Silahtaroglu, A.; Hjalgrim, H.; Tommerup, N. A balanced translocation disrupts SYNGAP1 in a patient with intellectual disability, speech impairment, and epilepsy with myoclonic absences (EMA). Epilepsia 2011, 52, 190–193. [Google Scholar] [CrossRef]
- Madaan, P.; Jauhari, P.; Chakrabarty, B.; Gulati, S. Jeavons syndrome in a family with GLUT1-deficiency syndrome. Seizure 2019, 71, 158–160. [Google Scholar] [CrossRef] [PubMed]
- Dragoumi, P.; Emery, J.; Chivers, F.; Brady, M.; Desurkar, A.; Cross, J.H.; Das, K.B. Crossing the lines between epilepsy syndromes: A myoclonic epilepsy variant with prominent eyelid myoclonia and atonic components. Epileptic Disord. 2018, 20, 35–41. [Google Scholar] [CrossRef] [PubMed]
- Reyhani, A.; Özkara, Ç. Pitfalls in the diagnosis of Jeavons syndrome: A study of 32 cases and review of the literature. Epileptic Disord. 2020, 22, 281–290. [Google Scholar] [CrossRef] [PubMed]
- Online Mendelian Inheritance in Man, OMIM®. Johns Hopkins University, Baltimore, M.D. SYNAPTIC RAS-GTPase-ACTIVATING PROTEIN 1; SYNGAP1. Number: * 603384. Available online: https://www.omim.org/entry/603384 (accessed on 1 April 2021).
- Gene [Internet]. Bethesda (MD): National Library of Medicine (US) National Center for Biotechnology Information. SYNGAP1 Synaptic Ras GTPase Activating Protein 1 [Homo Sapiens (Human)] Gene ID: 8831. Available online: https://www.ncbi.nlm.nih.gov/gene/8831 (accessed on 1 April 2021).
- Jeyabalan, N.; Clement, J.P. SYNGAP1: Mind the gap. Front. Cell. Neurosci. 2016, 10, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Online Mendelian Inheritance in Man, OMIM®. Johns Hopkins University, Baltimore, M.D. MENTAL RETARDATION, AUTOSOMAL DOMINANT 5; MRD5. Number: # 612621. Available online: https://www.omim.org/entry/612621 (accessed on 1 April 2021).
- Matricardi, S. Orphanet Encyclopedia SYNGAP1-Related Developmental and Epileptic Encephalopathy. Available online: https://www.orpha.net/consor/cgi-bin/Disease_Search.php?lng=EN&data_id=28070&Disease_Disease_Search_diseaseGroup=SYNGAP1&Disease_Disease_Search_diseaseType=Gen&Disease (accessed on 1 April 2021).
- Mignot, C.; von Stülpnagel, C.; Nava, C.; Ville, D.; Sanlaville, D.; Lesca, G.; Rastetter, A.; Gachet, B.; Marie, Y.; Korenke, G.C.; et al. Genetic and neurodevelopmental spectrum of SYNGAP1-associated intellectual disability and epilepsy. J. Med. Genet. 2016, 53, 511–522. [Google Scholar] [CrossRef]
- Berryer, M.H.; Hamdan, F.F.; Klitten, L.L.; Møller, R.S.; Carmant, L.; Schwartzentruber, J.; Patry, L.; Dobrzeniecka, S.; Rochefort, D.; Neugnot-Cerioli, M.; et al. Mutations in SYNGAP1 Cause Intellectual Disability, Autism, and a Specific Form of Epilepsy by Inducing Haploinsufficiency. Hum. Mutat. 2013, 34, 385–394. [Google Scholar] [CrossRef]
- Vlaskamp, D.R.M.; Shaw, B.J.; Burgess, R.; Mei, D.; Montomoli, M.; Xie, H.; Myers, C.T.; Bennett, M.F.; Xiangwei, W.; Williams, D.; et al. SYNGAP1 encephalopathy: A distinctive generalized developmental and epileptic encephalopathy. Neurology 2019, 92, E96–E107. [Google Scholar] [CrossRef]
- Lo Barco, T.; Kaminska, A.; Solazzi, R.; Cancés, C.; Barcia, G.; Chemaly, N.; Fontana, E.; Desguerre, I.; Canafoglia, L.; Hachon Le Camus, C.; et al. Syngap1-Dee: A visual sensitive epilepsy. Clin. Neurophysiol. 2021, 132, 841–850. [Google Scholar] [CrossRef]
- Kuchenbuch, M.; D’Onofrio, G.; Chemaly, N.; Barcia, G.; Teng, T.; Nabbout, R. Add-on cannabidiol significantly decreases seizures in 3 patients with SYNGAP1 developmental and epileptic encephalopathy. Epilepsia Open 2020, 5, 496–500. [Google Scholar] [CrossRef]
- Okazaki, T.; Saito, Y.; Hiraiwa, R.; Saitoh, S.; Kai, M.; Adachi, K.; Nishimura, Y.; Nanba, E.; Maegaki, Y. Pharmacoresistant epileptic eyelid twitching in a child with a mutation in SYNGAP1. Epileptic Disord. 2017, 19, 339–344. [Google Scholar] [CrossRef]
- Redin, C.; Gérard, B.; Lauer, J.; Herenger, Y.; Muller, J.; Quartier, A.; Masurel-Paulet, A.; Willems, M.; Lesca, G.; El-Chehadeh, S.; et al. Efficient strategy for the molecular diagnosis of intellectual disability using targeted high-throughput sequencing. J. Med. Genet. 2014, 51, 724–736. [Google Scholar] [CrossRef]
- von Stülpnagel, C.; Hartlieb, T.; Borggräfe, I.; Coppola, A.; Gennaro, E.; Eschermann, K.; Kiwull, L.; Kluger, F.; Krois, I.; Møller, R.S.; et al. Chewing induced reflex seizures (“eating epilepsy”) and eye closure sensitivity as a common feature in pediatric patients with SYNGAP1 mutations: Review of literature and report of 8 cases. Seizure 2019, 65, 131–137. [Google Scholar] [CrossRef] [PubMed]
- Carvill, G.L.; Heavin, S.B.; Yendle, S.C.; McMahon, J.M.; O’Roak, B.J.; Cook, J.; Khan, A.; Dorschner, M.O.; Weaver, M.; Calvert, S.; et al. Targeted resequencing in epileptic encephalopathies identifies de novo mutations in CHD2 and SYNGAP1. Nat. Genet. 2013, 45, 825–830. [Google Scholar] [CrossRef]
- Parrini, E.; Marini, C.; Mei, D.; Galuppi, A.; Cellini, E.; Chiti, L.; Rutigliano, D.; Bianchini, C.; Virdò, S.; De, D.; et al. Diagnostic Targeted Resequencing in 349 Patients with Drug-Resistant Pediatric Epilepsies Identifies Causative Mutations in 30 Different Genes. Hum. Mutat. 2017, 38, 216–225. [Google Scholar] [CrossRef] [PubMed]
- Deciphering Developmental Disorders Study. Prevalence and architecture of de novo mutations in developmental disorders. Nature 2017, 542, 433–438. [Google Scholar] [CrossRef] [PubMed]
- Online Mendelian Inheritance in Man, OMIM®. Johns Hopkins University, Baltimore, M.D. NEURITE EXTENSION AND MIGRATION FACTOR; NEXMIF. Number: * 300524. Available online: https://www.omim.org/entry/300524 (accessed on 1 April 2021).
- Cantagrel, V.; Haddad, M.R.; Ciofi, P.; Andrieu, D.; Lossi, A.M.; van Maldergem, L.; Roux, J.C.; Villard, L. Spatiotemporal expression in mouse brain of Kiaa2022, a gene disrupted in two patients with severe mental retardation. Gene Expr. Patterns 2009, 9, 423–429. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, T.; Miyata, S.; Koyama, Y.; Yoshikawa, K.; Hattori, T.; Kumamoto, N.; Shingaki, K.; Katayama, T.; Tohyama, M. Transient expression of Xpn, an XLMR protein related to neurite extension, during brain development and participation in neurite outgrowth. Neuroscience 2012, 214, 181–191. [Google Scholar] [CrossRef] [PubMed]
- Magome, T.; Hattori, T.; Taniguchi, M.; Ishikawa, T.; Miyata, S.; Yamada, K.; Takamura, H.; Matsuzaki, S.; Ito, A.; Tohyama, M.; et al. XLMR protein related to neurite extension (Xpn/KIAA2022) regulates cell-cell and cell-matrix adhesion and migration. Neurochem. Int. 2013, 63, 561–569. [Google Scholar] [CrossRef] [PubMed]
- Online Mendelian Inheritance in Man, OMIM®. Johns Hopkins University, Baltimore, M.D. MENTAL RETARDATION, X-LINKED 98; MRX98. Number: # 300912. Available online: https://www.omim.org/entry/300912 (accessed on 1 April 2021).
- Lorenzo, M.; Stolte-Dijkstra, I.; van Rheenen, P.; Smith, R.G.; Scheers, T.; Walia, J.S. Clinical spectrum of KIAA2022 pathogenic variants in males: Case report of two boys with KIAA2022 pathogenic variants and review of the literature. Am. J. Med. Genet. Part A 2018, 176, 1455–1462. [Google Scholar] [CrossRef]
- de Lange, I.M.; Helbig, K.L.; Weckhuysen, S.; Møller, R.S.; Velinov, M.; Dolzhanskaya, N.; Marsh, E.; Helbig, I.; Devinsky, O.; Tang, S.; et al. De novo mutations of KIAA2022 in females cause intellectual disability and intractable epilepsy. J. Med. Genet. 2016, 53, 850–858. [Google Scholar] [CrossRef]
- Webster, R.; Cho, M.T.; Retterer, K.; Millan, F.; Nowak, C.; Douglas, J.; Ahmad, A.; Raymond, G.V.; Johnson, M.R.; Pujol, A.; et al. De novo loss of function mutations in KIAA2022 are associated with epilepsy and neurodevelopmental delay in females. Clin. Genet. 2017, 91, 756–763. [Google Scholar] [CrossRef]
- Borlot, F.; Regan, B.M.; Bassett, A.S.; Stavropoulos, D.J.; Andrade, D.M. Prevalence of pathogenic copy number variation in adults with pediatric-onset epilepsy and intellectual disability. JAMA Neurol. 2017, 74, 1301–1311. [Google Scholar] [CrossRef] [PubMed]
- Myers, C.T.; Hollingsworth, G.; Muir, A.M.; Schneider, A.L.; Thuesmunn, Z.; Knupp, A.; King, C.; Lacroix, A.; Mehaffey, M.G.; Berkovic, S.F.; et al. Parental Mosaicism in “De Novo” Epileptic Encephalopathies. N. Engl. J. Med. 2018, 378, 1646–1648. [Google Scholar] [CrossRef] [PubMed]
- Stamberger, H.; Hammer, T.B.; Gardella, E.; Vlaskamp, D.R.M.; Bertelsen, B.; Mandelstam, S.; de Lange, I.; Zhang, J.; Myers, C.T.; Fenger, C.; et al. NEXMIF encephalopathy: An X-linked disorder with male and female phenotypic patterns. Genet. Med. 2021, 23, 363–373. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Ji, C.; Chen, Z.; Wang, K. Novel NEXMIF gene pathogenic variant in a female patient with refractory epilepsy and intellectual disability. Am. J. Med. Genet. Part A 2020, 182, 2765–2772. [Google Scholar] [CrossRef] [PubMed]
- Viravan, S.; Go, C.; Ochi, A.; Akiyama, T.; Carter Snead, O.; Otsubo, H. Jeavons syndrome existing as occipital cortex initiating generalized epilepsy. Epilepsia 2011, 52, 1273–1279. [Google Scholar] [CrossRef]
- Online Mendelian Inheritance in Man, OMIM®. Johns Hopkins University, Baltimore, M.D. RAR-RELATED ORPHAN RECEPTOR B; RORB. Number: * 601972. Available online: https://www.omim.org/entry/601972 (accessed on 1 April 2021).
- Liu, H.; Aramaki, M.; Fu, Y.; Forrest, D. Retinoid-Related Orphan Receptor β and Transcriptional Control of Neuronal Differentiation, 1st ed.; Elsevier Inc.: New York, NY, USA, 2017; Volume 125. [Google Scholar]
- Online Mendelian Inheritance in Man, OMIM®. Johns Hopkins University, Baltimore, M.D. EPILEPSY, IDIOPATHIC GENERALIZED, SUSCEPTIBILITY TO, 15; EIG15. Number: # 618357. Available online: https://www.omim.org/entry/618357 (accessed on 1 April 2021).
- Bartnik, M.; Szczepanik, E.; Derwińska, K.; Wiśniowiecka-Kowalnik, B.; Gambin, T.; Sykulski, M.; Ziemkiewicz, K.; Keogonekdzior, M.; Gos, M.; Hoffman-Zacharska, D.; et al. Application of array comparative genomic hybridization in 102 patients with epilepsy and additional neurodevelopmental disorders. Am. J. Med. Genet. Part B Neuropsychiatr. Genet. 2012, 159, 760–771. [Google Scholar] [CrossRef]
- Rudolf, G.; Lesca, G.; Mehrjouy, M.M.; Labalme, A.; Salmi, M.; Bache, I.; Bruneau, N.; Pendziwiat, M.; Fluss, J.; De Bellescize, J.; et al. Loss of function of the retinoid-related nuclear receptor (RORB) gene and epilepsy. Eur. J. Hum. Genet. 2016, 24, 1761–1770. [Google Scholar] [CrossRef]
- Sadleir, L.G.; de Valles-Ibáñez, G.; King, C.; Coleman, M.; Mossman, S.; Paterson, S.; Nguyen, J.; Berkovic, S.F.; Mullen, S.; Bahlo, M.; et al. Inherited RORB pathogenic variants: Overlap of photosensitive genetic generalized and occipital lobe epilepsy. Epilepsia 2020, 61, e23–e29. [Google Scholar] [CrossRef]
- Online Mendelian Inheritance in Man, OMIM®. Johns Hopkins University, Baltimore, M.D. CHROMODOMAIN HELICASE DNA-BINDING PROTEIN 2; CHD2. Number: * 602119. Available online: https://www.omim.org/entry/602119 (accessed on 1 April 2021).
- Wilson, M.M.; Henshall, D.C.; Byrne, S.M.; Brennan, G.P. Chd2-related cns pathologies. Int. J. Mol. Sci. 2021, 22, 588. [Google Scholar] [CrossRef] [PubMed]
- Online Mendelian Inheritance in Man, OMIM®. Johns Hopkins University, Baltimore, M.D. EPILEPTIC ENCEPHALOPATHY, CHILDHOOD-ONSET; EEOC. Number: # 615369. Available online: https://www.omim.org/entry/615369 (accessed on 1 April 2021).
- Helbig, I. Orphanet Encyclopedia Myoclonic-Astatic Epilepsy. Available online: https://www.orpha.net/consor/cgi-bin/Disease_Search.php?lng=EN&data_id=891&Disease_Disease_Search_diseaseGroup=CHD2&Disease_Disease_Search_diseaseType=Gen&Enfermedad(es) (accessed on 1 April 2021).
- Galizia, E.C.; Myers, C.T.; Leu, C.; De Kovel, C.G.F.; Afrikanova, T.; Cordero-Maldonado, M.L.; Martins, T.G.; Jacmin, M.; Drury, S.; Chinthapalli, V.K.; et al. CHD2 variants are a risk factor for photosensitivity in epilepsy. Brain 2015, 138, 1198–1207. [Google Scholar] [CrossRef]
- Thomas, R.H.; Zhang, L.M.; Carvill, G.L.; Archer, J.S.; Heavin, S.B.; Mandelstam, S.A.; Craiu, D.; Berkovic, S.F.; Gill, D.S.; Mefford, H.C.; et al. CHD2 myoclonic encephalopathy is frequently associated with self-induced seizures. Neurology 2015, 84, 951–958. [Google Scholar] [CrossRef] [PubMed]
- Mullen, S.A.; Carvill, G.L.; Bellows, S.; Bayly, M.A.; Berkovic, S.F.; Dibbens, L.M.; Scheffer, I.E.; Mefford, H.C. Copy number variants are frequent in genetic generalized epilepsy with intellectual disability. Neurology 2013, 81, 1507–1514. [Google Scholar] [CrossRef] [PubMed]
- Online Mendelian Inheritance in Man, OMIM®. Johns Hopkins University, Baltimore, M.D. SOLUTE CARRIER FAMILY 2 (FACILITATED GLUCOSE TRANSPORTER), MEMBER 1; SLC2A1. Number: * 138140. Available online: https://www.omim.org/entry/138140 (accessed on 1 April 2021).
- Online Mendelian Inheritance in Man, OMIM®. Johns Hopkins University, Baltimore, M.D. GLUT1 DEFICIENCY SYNDROME 1; GLUT1DS1. Number: # 606777. Available online: https://omim.org/entry/606777 (accessed on 1 April 2021).
- De Lonlay, P.P. Orphanet Encyclopedia Classic Glucose Transporter Type 1 Deficiency Syndrome. Available online: https://www.orpha.net/consor/cgi-bin/Disease_Search.php?lng=EN&data_id=10999&Disease_Disease_Search_diseaseGroup=GLUt1&Disease_Disease_Search_diseaseType=Pat&Disease (accessed on 1 April 2021).
- Gökben, S.; Yilmaz, S.; Klepper, J.; Serdaroǧlu, G.; Tekgül, H. Video/EEG recording of myoclonic absences in GLUT1 deficiency syndrome with a hot-spot R126C mutation in the SLC2A1 gene. Epilepsy Behav. 2011, 21, 200–202. [Google Scholar] [CrossRef] [PubMed]
- Diomedi, M.; Gan-Or, Z.; Placidi, F.; Dion, P.A.; Szuto, A.; Bengala, M.; Rouleau, G.A.; Gigli, G.L. A 23 years follow-up study identifies GLUT1 deficiency syndrome initially diagnosed as complicated hereditary spastic paraplegia. Eur. J. Med. Genet. 2016, 59, 564–568. [Google Scholar] [CrossRef]
- Altıokka-Uzun, G.; Özdemir, Ö.; Uğur-İşeri, S.; Bebek, N.; Gürses, C.; Özbek, U.; Baykan, B. Investigation of SLC2A1 gene variants in genetic generalized epilepsy patients with eyelid myoclonia. Epileptic Disord. 2018, 20, 396–400. [Google Scholar] [CrossRef]
- Online Mendelian Inheritance in Man, OMIM®. Johns Hopkins University, Baltimore, M.D. POTASSIUM CHANNEL, VOLTAGE-GATED, SHAB-RELATED SUBFAMILY, MEMBER 1; KCNB1. Number: * 600397. Available online: https://www.omim.org/entry/600397 (accessed on 1 April 2021).
- Online Mendelian Inheritance in Man, OMIM®. Johns Hopkins University, Baltimore, M.D. DEVELOPMENTAL AND EPILEPTIC ENCEPHALOPATHY 26; DEE26. Number: # 616056. Available online: https://www.omim.org/entry/616056 (accessed on 1 April 2021).
- Marini, C.; Romoli, M.; Parrini, E.; Costa, C.; Mei, D.; Mari, F.; Parmeggiani, L.; Procopio, E.; Metitieri, T.; Cellini, E.; et al. Clinical features and outcome of 6 new patients carrying de novo KCNB1 gene mutations. Neurol. Genet. 2017, 3, e206. [Google Scholar] [CrossRef]
- Saitsu, H.; Akita, T.; Tohyama, J.; Goldberg-Stern, H.; Kobayashi, Y.; Cohen, R.; Kato, M.; Ohba, C.; Miyatake, S.; Tsurusaki, Y.; et al. De novo KCNB1 mutations in infantile epilepsy inhibit repetitive neuronal firing. Sci. Rep. 2015, 5, 1–14. [Google Scholar] [CrossRef]
- Minardi, R.; Licchetta, L.; Baroni, M.C.; Pippucci, T.; Stipa, C.; Mostacci, B.; Severi, G.; Toni, F.; Bergonzini, L.; Carelli, V.; et al. Whole-exome sequencing in adult patients with developmental and epileptic encephalopathy: It is never too late. Clin. Genet. 2020, 98, 477–485. [Google Scholar] [CrossRef]
- Online Mendelian Inheritance in Man, OMIM®. Johns Hopkins University, Baltimore, M.D. N-ALPHA-ACETYLTRANSFERASE 10, NatA CATALYTIC SUBUNIT; NAA10. Number: * 300013. Available online: https://www.omim.org/entry/300013 (accessed on 1 April 2021).
- Online Mendelian Inheritance in Man, OMIM®. Johns Hopkins University, Baltimore, M.D. OGDEN SYNDROME. Number: # 300855. Available online: https://www.omim.org/entry/300855 (accessed on 1 April 2021).
- Saunier, C.; Støve, S.I.; Popp, B.; Gérard, B.; Blenski, M.; AhMew, N.; de Bie, C.; Goldenberg, P.; Isidor, B.; Keren, B.; et al. Expanding the Phenotype Associated with NAA10-Related N-Terminal Acetylation Deficiency. Hum. Mutat. 2016, 37, 755–764. [Google Scholar] [CrossRef] [PubMed]
- Popp, B.; Støve, S.I.; Endele, S.; Myklebust, L.M.; Hoyer, J.; Sticht, H.; Azzarello-Burri, S.; Rauch, A.; Arnesen, T.; Reis, A. De novo missense mutations in the NAA10 gene cause severe non-syndromic developmental delay in males and females. Eur. J. Hum. Genet. 2015, 23, 602–609. [Google Scholar] [CrossRef]
- Cantagrel, V.; Lossi, A.M.; Boulanger, S.; Depetris, D.; Mattei, M.G.; Gecz, J.; Schwartz, C.E.; Van Maldergem, L.; Villard, L. Disruption of a new X linked gene highly expressed in brain in a family with two mentally retarded males. J. Med. Genet. 2004, 41, 736–742. [Google Scholar] [CrossRef] [PubMed]
- Xiuli, G.; Meiyu, G.; Guanhua, D. Glucose transporter 1, distribution in the brain and in neural disorders: Its relationship with transport of neuroactive drugs through the blood-brain barrier. Biochem. Genet. 2005, 43, 175–187. [Google Scholar] [CrossRef] [PubMed]
- Warde-Farley, D.; Donaldson, S.L.; Comes, O.; Zuberi, K.; Badrawi, R.; Chao, P.; Franz, M.; Grouios, C.; Kazi, F.; Lopes, C.T.; et al. The GeneMANIA prediction server: Biological network integration for gene prioritization and predicting gene function. Nucleic Acids Res. 2010, 38, 214–220. [Google Scholar] [CrossRef] [PubMed]
- Clement, J.P.; Aceti, M.; Creson, T.K.; Ozkan, E.D.; Shi, Y.; Reish, N.J.; Almonte, A.G.; Miller, B.H.; Wiltgen, B.J.; Miller, C.A.; et al. Pathogenic SYNGAP1 mutations impair cognitive development by disrupting maturation of dendritic spine synapses. Cell 2012, 151, 709–723. [Google Scholar] [CrossRef]
- Ozkan, E.D.; Creson, T.K.; Kramár, E.A.; Rojas, C.; Seese, R.R.; Babyan, A.H.; Shi, Y.; Lucero, R.; Xu, X.; Noebels, J.L.; et al. Reduced cognition in Syngap1 mutants is caused by isolated damage within developing forebrain excitatory neurons. Neuron 2014, 82, 1317–1333. [Google Scholar] [CrossRef]
- Creson, T.K.; Rojas, C.; Hwaun, E.; Vaissiere, T.; Kilinc, M.; Jimenez-Gomez, A.; Holder, J.L.; Tang, J.; Colgin, L.L.; Miller, C.A.; et al. Re-expression of SynGAP protein in adulthood improves translatable measures of brain function and behavior. Elife 2019, 8, e46752. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, B.J.; Ammanuel, S.; Kipnis, P.A.; Araki, Y.; Huganir, R.L.; Kadam, S.D. Low-Dose Perampanel Rescues Cortical Gamma Dysregulation Associated With Parvalbumin Interneuron GluA2 Upregulation in Epileptic Syngap1 +/− Mice. Biol Psychiatry 2020, 87, 829–842. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, J.; O’Connor, M.; Templet, S.; Moghaddam, M.; Di Via Ioschpe, A.; Sinclair, A.; Zhu, L.Q.; Xu, W.; Man, H.Y. NExMIF/Kidlia knock-out mouse demonstrates autism-like behaviors, memory deficits, and impairments in synapse formation and function. J. Neurosci. 2020, 40, 237–254. [Google Scholar] [CrossRef] [PubMed]
- Zlotogora, J. Germ line mosaicism. Hum. Genet. 1998, 102, 381–386. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| A | ||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Reference | Klitten 2011 1 | Mingot 2016 a | Mingot 2016 | Mingot 2016 | Okazaki 2017 b | Vlaskamp 2019 1 | Vlaskamp 2019 1 | Vlaskamp 2019 1,2 | Vlaskamp 2019 1,2 | |||||||||||||||||||
| Patient | 11 | 12 | 14 | 1 | 4 | 5 | 7 | |||||||||||||||||||||
| Clinical features | gender | Male | Male | Female | Female | Male | Male | Female | Male | Male | ||||||||||||||||||
| Age | 25 yr | 3 yr | 22 yr | 8 yr | 4 yr | 3 yr 10 months | 8 yr 3 months | 11 yr 2 months | 17 | |||||||||||||||||||
| DD/ID | + (Severe) | + (Severe) | + (Severe) | + (Mild) | + | - (FSIQ: 80, low average) | + (Mild) | + (Moderate) | + (Severe) | |||||||||||||||||||
| Behavioral features | ASD traits, anxious behavior | Repetitive behaviours, stereotypies | Stereotypies | ASD, stereotypies | NR | - | ASD traits, mild tantrums, aggression, high pain thresholds, sleeping problems | - | ASD, regression severe tantrums, self-injury, aggression, high pain threshold, sleeping problems | |||||||||||||||||||
| Other parametres | Absence of language | Absence of language, truncal hypotonia, swallowing difficulties | Absence of language, mild gait ataxia, flexion deformity of left hip, hyperlordotic lumbar spine, microcephaly | Motor slowness and moderate akinesia, ataxic gait, truncal hypotonia, dystonic postures of hands and feet, plastic hypertonia | Hypotonia, hypersalivation | - | Hypotonia | - | Hypotonia, unsteady gait, reflux, obstipation, eating difficulties, benign bone tumor | |||||||||||||||||||
| Epilepsy | Age of onset | 13 months | 2 yr | 1 yr | 5 yr | 1 yr and 5 months | 16 months | 2.5 yr | 11 months | 2 yr | ||||||||||||||||||
| Abscence seizures | + (MA, AA) | NR | + (AA) | + (MA) | NR | + (MA) | NR | NR | NR | |||||||||||||||||||
| Eyelid myoclonia | + (eyelid winking) | + | + | + | NR | + | + | + | + | |||||||||||||||||||
| Photosensitivity | NR | + | NR | + | + | + | + | + | + | |||||||||||||||||||
| Other seizures | DA with MJ | FS | FS, MJ | NR | Upward eye deviation, motion arrest, loss of consciousness, and eyelid twitching. Triggered by crying and photosensivility | MS. Triggered by PS, sounds, sleep deprivation and fatigue | MS | FS, MAt, bilateral TCS. Triggered by PS, sleep deprivation and fatigue | Bilateral TCS, MS, FIAS, DA. Triggered by PS, eye closure and eating | |||||||||||||||||||
| EEG | others | Interictal: generalized synchronous 3–7 Hz (P)SW | Abnormal BG, generalized slowing, EM, and generalized seizure patterns. Triggered by photosensivility | Bursts of spikes and slow waves in the occipital region after eye closure. Triggered by FOS | Ictal: bursts of diffuse PSW with posterior predominance after eyes closer and photic stimulation. Triggered by FOS and PS | Ictal: diffuseslow or SW activity with occipitalto central predominance. Interictal: bilateral frontal spikes. Sleep: rhythmic, generalized 2–3-Hz delta activity, without visible seizures. Normal BG | Ictal: GSW (myoclonic). Interictal: GSW | BG slow. Interrictal: Frequent 2.5–4 Hz GSW, after eye closure in trains, MFD | Interictal: GPW | sleep: frequent seizures while falling asleep | ||||||||||||||||||
| Cranial MRI | NR | Normal | Normal | Normal | Normal | Normal | Normal | Normal | NR | |||||||||||||||||||
| AED Treatment | VPA, LTG, CLB | VPA | LEV, TPM | LEV, ETX | CBZ, VPA, LEV, ETX, LTG | VPA | VPA, LEV, LTG, ETX, CBD | VPA | VPA, CLZ, CBZ | |||||||||||||||||||
| Geneticinformation | Genetic test | karyotype | gene panel NGS | WES | WES | NGS panel | NR | NR | NR | NR | ||||||||||||||||||
| Genomic change (Hg19) | NR | chr6:33406650; C>C/T | chr6:33408514; C>C/T | chr6:33409458_ 33409461delAGCG | chr6:33414346; G>G/A | chr6:33391277; C>C/T | chr6:33399974; CA>CA/C | chr6:333400498_ 33400501delAAAC | chr6:33400501; C>C/T | |||||||||||||||||||
| cDNA/aa change | (truncate gene) ish, 46,XY, t(6;22)(p21.32;q11.21)dn | c.1630C>T, p.Arg544* | c.1685C>T, p.Pro562Leu | c.2214_2217delAGCG, p.Glu739Glyfs*20 | c.3583-6G>A, p.Val1195Alafs*27 | c.91C>T, p.Arg31* | c.333delA, p.Lys114Serfs*20 | c.424_ 427delAAAC, p.Lys142Glufs*31 | c.427C>T, p.Arg143* | |||||||||||||||||||
| Inheritance | de novo | de novo | de novo | de novo | (parent not tested) | de novo | de novo | de novo | de novo | |||||||||||||||||||
| Others information | FISH (probe RP11.497A24) | VUS inherited from the mother: SCN9A: c.4282G>A and c.5624G>A; ARX: c.1462A>G | - | - | Karyotype and anlysis for AS with normal results. CSF glucose normal | |||||||||||||||||||||||
| Family History | - (epilepsy) | - | - | - | - (epilepsy or ID) | - (seizures, ID or ASD) | - (seizures, ID or ASD) | - (seizures, ID or ASD) | Sister, father, and many paternal relatives with learning difficulties. Maternal cousin ASD | |||||||||||||||||||
| B | ||||||||||||||||||||||||||||
| Reference | Vlaskamp 2019 1,2/Von Stülpnagel 2019 | Vlaskamp 2019 1,2/Von Stülpnagel 2019 | Vlaskamp 2019 1,2 | Vlaskamp 2019 1,2 | Vlaskamp 2019 1,2/The DDD Study 2017 | Vlaskamp 2019 1,2 | Vlaskamp 2019 1 | Vlaskamp 2019 1 | Vlaskamp 2019 1,2 | Vlaskamp 2019 1,2 | ||||||||||||||||||
| Patient | 8/7 | 9/8 | 11 | 12 | 13/241 | 15 | 17 | 18 | 20 | 21 | ||||||||||||||||||
| Clinical features | gender | female | female | male | female | male | female | female | male | female | female | |||||||||||||||||
| Age | 8 yr | 6 yr | 6 yr | 4 yr 8 months | 7 yr 10 months | 9.5 yr | 5 yr 2 months | 5 yr 10 months | 15 yr 1 months | 10 yr 5 months | ||||||||||||||||||
| DD/ID | + (moderate-severe) | + (moderate-severe) | + (severe) | + (severe) | + (severe) | + (severe) | + (severe) | + (severe) | + (moderate) | + (moderate) | ||||||||||||||||||
| Behavioral features | regression, tantrums, self-injury, tichotillomania, high pain threshold, eating disorder, sleeping problems | ASD, echolalia, high pain threshold, eating disorder, sleeping problems | aggression, high pain threshold | regression, ASD, tantrums, self-injury, aggression. high pain threshold, sleep problems | Tantrums, aggression, high pain threshold, sleep problems | ASD traits, stereotypes, odontoprisis, high pain threshold | odontoprisis, high pain threshold | regression, obsession, self-injury, aggression, high pain threshold, sleep problems, eating disorder | ASD, self-injury, aggression, sleep problems, high pain threshold, oral hypersensitivity | regression, ASD, severe tantrums, self-injury, aggression, sleep problems, high pain threshold, oral hypersensitivity | ||||||||||||||||||
| Other parametres | hypotonia, hyperlaxity, 2 café au lait spots, small capillary hemangioma, constipation, hearing loss after infection/ataxia, problems in fine motor skills | hypotonia, hyperlaxity, hearing loss after recurrent otitis/ataxia | hypotonia, ataxia, constipation, reflux, absence of language | pes planus, strabismus, constipation, hearing loss | nystagmus | Congenital hipdyslocation, absence of language | hypotonia, poor balance and coordination, absence of language | hypotonia, hypermobility, scoliosos, constipation | congenital nystagmus, hypotonia, a few cafe au lait macules, constipation | |||||||||||||||||||
| Epilepsy | Age of onset | 8/16 months | 12–13 months | 4 yr | 23 months | 2 yr | 2 yr | 3.5 yr | 3 yr | 12–14 months | 2 yrs | |||||||||||||||||
| Abscence seizures | + | + | NR | + (AA) | NR | NR | NR | NR | + (MA) | |||||||||||||||||||
| Eyelid myoclonia | + | + | + | + | + | + | + | + | + | + | ||||||||||||||||||
| Photosensitivity | + | + | NR | NR | NR | NR | + | NR | + | + | ||||||||||||||||||
| Other seizures | MS, MAt. Triggered by touch and thinking of eating/GS, MJ, atonic drops. Reflex seizures while chewing | MS, MAt. Triggered by thinking of eating/GS, MJ, atonic drops. Reflex seizures while chewing | MS, DA. Triggered by fever and infection | MS, AS | MS, AS, bilateral TCS. Triggered by eating (chocolate), fever and fatigue | Bi- and unilateral TCS | MJ, MS | Triggered by fatigue | atonic DA, nocturnal TS. Triggered by PS and eating | MJ, bilateral TCS | ||||||||||||||||||
| EEG | others | Intal: 3 Hz GSW (EM-MAt). Interictal:G(P)SW | Intal: 2.5–3.5 Hz GSW (EM-MAt). Inerictal: 2.5–3.5 Hz GSW | BG poor. Ictal: Bilateral occipital sharps, followed by MFD (EM). Interictal: MFD | BG: slow. Ictal: GPSW (MS), 1.5–2 Hz GSW (AA). Interictal: GSW facilited by eye closure | Interictal: GSW | BG: slow. Ictal: FD (unilateral TCS). Interictal: 3–4 Hz GSW, MFD | BG: slow. Interictal: 1.5–3 Hz GSW, MFD | Interictal: 3 Hz GSW, bifrotal SW | BG: slow. Ictal: GSE (MA). Inerictal: MFD | BG: slow. Interictal: 3 Hz GSW, also following eye closure, FD | |||||||||||||||||
| MRI | Cranial | Normal | Normal | Normal | Normal | Normal | Normal | Normal | Normal | Normal (discrete hippocampal tissue loss, not progressive and without sclerosis) | Normal | |||||||||||||||||
| Treatment | AED | VPA, LEV, TPM, CLB, LTG, ETX, LCM, ZNS, CLZ, CBD, PHT | CLB, LCM, ZNS, CLZ, CBD | VPA, LEV, TPM | CLB, TPM, NZP, LTG, VPA, CLZ | VPA, CLB | VPA, CLZ, LEV, TPM | VPA, CLZ, LEV, CLB | VPA | VPA, LEV, LTG | VPA | |||||||||||||||||
| Other | KD | KD, mAD | Vitamin B6 | |||||||||||||||||||||||||
| Genetic information | Genomic change (Hg19) | chr6:33400509_33400521dup | chr6:33403058delC | chr6:33403318dupC | chr6:33403367; C>C/T | chr6:33405511_33405512insC | chr6:33406048; C>C/T | chr6:33406202delC | chr6:33406324;C>C/G | chr6:33408547G>GGCTGC | ||||||||||||||||||
| cDNA/aa change | c.435_447dup, p.Leu150Valfs*6 | c.639delC, p.Ile214Trpfs*9 | c.690dupC, p.Phe231Leufs*14 | c.739C>T, p.Gln247* | c.822_823insC, p.Lys277Glnfs*7 | c.1366C>T, p.Gln456* | c.1393delC, p.Leu465Phefs*9 | c.1515C>G, p.Tyr505* | c.1718_1719insGCTGC, p.Glu578Alafs*74 | |||||||||||||||||||
| Inheritance | de novo/mosaic parent | de novo | de novo | de novo | de novo | de novo | de novo | de novo | de novo | |||||||||||||||||||
| Family History | sisters | - (seizures, ID or ASD) | - (seizures, ID or ASD) | Maternal aunt and distant relative epilepsy, other distant relatives ASD | - (seizures, ID or ASD) | - (seizures, ID or ASD) | Maternal grandfather post-stroke epilepsy | Paternal uncle moderate ID | - (seizures, ID or ASD) | |||||||||||||||||||
| C | ||||||||||||||||||||||||||||
| Reference | Vlaskamp 2019 1,2 | Vlaskamp 2019 1,2 | Vlaskamp 2019 1 | Vlaskamp 2019 1/Carvill 2013 | Vlaskamp 2019 1 | Vlaskamp 2019 1 | Vlaskamp 2019 1,2 | Vlaskamp 2019 1 | Vlaskamp 2019 1 | Vlaskamp 2019 1 | ||||||||||||||||||
| Patient | 23 | 24 | 25 | 26/T2528 | 27 | 30 | 31 | 32 | 33 | 35 | ||||||||||||||||||
| Clinical features | gender | female | female | male | male | male | female | male | female | male | male | |||||||||||||||||
| Age | 11 yr 11 months | 7 yr | 11 yr 7 months | 30/26 yr | 6 yr | 3 yr 11 months | 11 yr 2 months | 33 yr | 15 yr 3 months | 4 yr 9 months | ||||||||||||||||||
| DD/ID | + (severe) | + (severe) | + (moderate) | + (moderate) | + (severe) | + (severe) | + (severe) | + (moderate-severe) | + (severe) | + (moderate-severe) | ||||||||||||||||||
| Behavioral features | regression, ASD, tantrums, self-injury, aggression. Sleep problems, high pain threshold, eating difficulties | regression, ASD, aggression, sleep problems, high pain threshold | ASD, self injury, aggression, sleep problems, high pain threshold | regression, OCD symptoms, tantrums, aggression | regression, ASD, high pain threshold | ASD, tantrums, self-injury, aggression, sleep problems, high pain threshold, oral hypersensibility | regression, ASD, tantrums, aggressive, sleep problems, high pain threshold, eating difficulties | regression, ASD, self-injury, aggression, poor concentration, high pain threshold | regression, ASD, aggression, sleep problems, high pain thresholds, eating difficulties | ASD, aggression, sleep problems, high pain threshold | ||||||||||||||||||
| Other parametres | hypotonia, constipation | congenital hisdysplasia, hypotonia, ataxic gait | pes planus, hypotonia, unsteady gait, constipation | mild two/three syndactyly, irregular tremor upper extremities, osteopenia | Unsteady gait | hypotyonia, unsteady gait, constipation, chronic idiopathic tromnocytopenic purpura, absence of language | microcephaly, short stature, borderline hypotonia, ataxia, Hemangioma nasal cavity | Hypotonia, coordination disoder/ataxia | right pes planus, left pes caves, hypotonia, bilateral pyramidal syndrome, unsteady gait, orthothics, hyperflexibility | mild hypotonia | ||||||||||||||||||
| Epilepsy | Age of onset | 2 yr | 6 months | 9.5 yr | 18 months | 2 yr | 18 months | 2 yr 3 months | 8 months | 18 months | 2 yr 1 month | |||||||||||||||||
| Abscence seizures | NR | NR | NR | + | NR | NR | NR | NR | NR | NR | ||||||||||||||||||
| Eyelid myoclonia | + | + | + | + | + | + | + | + | + | + | ||||||||||||||||||
| Photosensitivity | NR | NR | NR | + | NR | NR | NR | + | NR | NR | ||||||||||||||||||
| Other seizures | FS, bilateral TCS (with fiver) | bilateral TCS (with fiver), atonic DA. Triggered by fatigue and illness | Triggered by eating | FS, aura, FIAS, MJ, NCSE, bi- and unilateral TCS, MS. Triggered by PS | Triggered by hunger, self-induced with hyperventilation, fatigue and stress | TCS (with fiver) | Atonic DA. Triggered by sounds, fatigue, and drop in emperature | GTCS. Triggered by PS | - | Triggered by eating | ||||||||||||||||||
| EEG | others | IBG: Slow. Ictal: 2–3 Hz GSW with frontal maximum, (EM-AS). Interictal: MFD | Interictal: 2.5 Hz GSW | NR | BG: slow. Interictal: occipital 2 Hz GSW, occipital FD/bi-occipital ED, DS, SSW | BG: iregular. Ictal: GSW (EM). Interictal: 2–3 Hz GSW, irregular GPSW, MFD | interictal: epileptiform discharge | Ictal: irregular GSW followed by slower discharges (EM), GPSW (EM). Interictal: G(P)SW, bifrotal SW, FD | NR | BG: slow Ictal: GSW (EM). Interictal: temporo-occipital SW, 10% generalized activity in 24 hours. | BG: right occipital slowing. Ictal: eyeblink without ictal correlate. Interictal: only in sleep: right occipital slowing, focal sharp waves | |||||||||||||||||
| Cranial MRI | Normal | Normal | Normal | Normal | Patent cavus vergae | Atypical WM abnormalities | Normal | NR | Enlarged ventricles | Normal | ||||||||||||||||||
| Treatment | AED | VPA, LEV | VPA, LEV | - | VPA, LTG, CLB | VPA, LEV, ETX, ZNS, CBD, LTG | - | VPA, CLB, TPM, LTG | VPA, CBZ, TPM | VPA, LEV, LTG | - | |||||||||||||||||
| Other | KD | KD | ||||||||||||||||||||||||||
| Genetic information | Genetic test | NR | NR | NR | NGS panel | NR | NR | NR | NR | NR | NR | |||||||||||||||||
| Genomic change (Hg19) | chr6:33409006;G>G/A | chr6:33409095;C>C/T | chr6:33409095;C>C/T | chr6:33409140;C>C/T | chr6:33409419_3349422del | chr6:33411265_ 33411267delinsCA | chr6:33411735dupC | chr6:33412317;G>G/T | chr6:33414426;T>T/G | chr6:33393573;A>A/G | ||||||||||||||||||
| cDNA/aa change | c.1970G>A, p.Trp657* | c.2059C>T, p.Arg687* | c.2059C>T, p.Arg687* | c.2104C>T, p.Gln702* | c.2177_2180delGGAA, p.Arg726Thrfs*33 | c.2936_ 2938delinsCA, p.Phe979Serfs*98 | c.3406dupC, p.Gln1136Profs*17 | c.3505G>T, p.Glu1169* | c.3657T>G, p.Tyr1219* | c.190-2A>G, (splice acceptor site) | ||||||||||||||||||
| Inheritance | unknow | de novo | de novo | de novo | de novo | de novo | de novo | de novo | de novo | de novo | ||||||||||||||||||
| Family History | Distant relative ASD | Paternal grandmother GTCS 16–20 y. Distant relative ASD | - (seizures, ID or ASD) | Distant relative epilepsy | - (seizures, ID or ASD) | Maternal aunt ID. Paternal first cousin post-traumatic epilepsy | Maternal uncle ID post-meningitis. Distant relative epilepsy | Maternal and paternal first cousins learning difficulties | Distant relative ASD | - (seizures, ID or ASD) | ||||||||||||||||||
| D | ||||||||||||||||||||||||||||
| Reference | Vlaskamp 2019 1,2 | Vlaskamp 2019 1/Parrini 2017 | Vlaskamp 2019 1 | Vlaskamp 2019 1,2,c | Vlaskamp 2019 1,2 | Vlaskamp 2019 1,2 | Vlaskamp 2019 1,2,a/Berryer 2013 a | Vlaskamp 2019 1,2 | Vlaskamp 2019 1,2 | Von Stülpnagel 2019 2,c | Von Stülpnagel 2019 | |||||||||||||||||
| Patient | 36 | 39/1190N | 41 | 45 | 46 | 50 | 51/3 | 52 | 53 | 1 | 2 | |||||||||||||||||
| Clinical features | gender | female | female | female | male | male | male | female | female | female | male | male | ||||||||||||||||
| Age | 8 yr 11 months | 16.4/17 yr | 6 yr 8 months | 3 yr 2 months | 10 yr | 15 yr 1 month | 9.1/4.2 yr | 8 yr 3 months | 7 yr months | 5 yr | 14 yr | |||||||||||||||||
| DD/ID | + (mild) | + (moderate-severe) | + (moderate-severe) | + (severe) | + (moderate) | + (severe) | + (moderate/mild) | + (severe) | + (mild) | + (moderate) | + (moderate-severe) | |||||||||||||||||
| Behavioral features | regression, ASD traits, tantrums, self-injury, aggresive | regression, Aggressive, REM sleep behavioral disorder, ASD | regression, ASD, sleep problems, high pain threshold | regression, ASD traits | regression, ASD, tantrums, OCD, echolalia, high pain threshold, eating dificulties | regression, ASD, eating disorder | regression, ASD, OCD, tantrums, self-injury, sleep problems, high pain threshold, eating disorder | regression, ASD, tantrums, self-injury, sleep problems, high pain threshold | tantrums, aggressive, sleep problems | regression, ASD | ASD | |||||||||||||||||
| Other parametres | Few café au lait macules | pronated foot, coordinarition disorder, ataxic gait | hypotonia | hypospadie, 6th toe, hypotonia, nystagmus | Mild cerebral palsy, pes planus, mild musle weakness, clinodactyly toes, hypotonia, constipation, coeliac disease | obesity, ataxia with wide-based gait | Hypotonia, unsteady gait with poor balance, gross and fine motor dyspraxia | macrocephaly, knee hyperextension, pes planus, pronated feet, hypotonia, wide-based gait with poor balance, constipation | pes caves, hypotonia, balance issues | Height <3p. Tongue hypotonia and horizontal nystagmus. Postaxial hexadactylia and hypospadia | Abnormal gait; poor coordination; dysarthria. Abnormal facial shape (triangular), large anteverted, ears, wide mouth, thin lips, pointed chin | |||||||||||||||||
| Epilepsy | Age of onset | <2 yr | 6.7/5 yr | 4.5 ys | 2.5 yr | 2.8 yr | 2.5 yr | 18 months | 18 months | 2.5 yr | 2.5 yr | 20 months | ||||||||||||||||
| Abscence seizures | NR | NR | + (typical) | NR | NR | + (typical) | NR | NR | NR | |||||||||||||||||||
| Eyelid myoclonia | + | + | + | + | + | + | + | + | + | + | + | |||||||||||||||||
| Photosensitivity | NR | + | + | + | - | + | - | NR | - | - | + | |||||||||||||||||
| Other seizures | Triggered by eye closure, hunger and fatigue | Triggered by PS | - | MAt, MS, atonic DA. Triggered by eating and stress | Triggered by visual patterns | bilateral TCS (with fiver). Triggered by PS and noise | bilateral TCS, myoclonic DA. Triggered by eating | Triggered by eating, eye closure and fatigue | Triggered by illness and fatigue | Atonic head dropping | FS, GTCS, episodes characterized by loss of consciousness, backward eyeball rolling, MS; generalized with head atonia and EM; status epilepticus. | |||||||||||||||||
| EEG | others | Ictal: GD (EM-MAt). Interictal:2–3 Hz GSW, frequent GD, induced by eye closure | Ictal: G(P)SW (EM)Interictal: G(P)SW | Ictal: G(P)SW (EM) | NR | BG: slowing. Interictal: GPS, 3.5–4 GSP, FD | Interictal: GD, MFD, spikes, G(P)SW | BG slow | Interictal: GSW | BG: gegeralized slowing. Interictal: MFD | 1–3 s lasting high amplitude 3/s SW complexes with bilateral initiation and occipital predominance but never lateralized. Triggered by heat, fatigue, stress, and orofacial stimuli | slowed BG activity (theta); spikes and polyspikes over the occipital regions; abnormalities are worsened by sleep. Triggered by PS, autoinduced, and eating. | ||||||||||||||||
| MRI | Cranial | Normal | Normal | Mega cisterna magna fossa posterior | Normal | Normal | Normal | Small hyperintens subcortical WM lesions (bi-frontal, peri-ventricular), possibly post-anoxic leukopathy | Stable mild enlarged ventricles and pineal cyst | Normal | Slight frontal dilatation of the external spaces of cerebrospinal fluid and an age-appropriate myelination | Normal | ||||||||||||||||
| Treatment | AED | VPA, CLB, LTG | LZP, RUF, VPA | LEV, ZNS, RUF, VPA | VPA, LTG | VPA | Hydrocortison, VGB, NZP | VPA | VPA, LEV, ETX, CLZ, LTG, CBD | VPA, ZNS, LEV, PER, CBD | VPA, LTG | VPA, LTG, LEV, CLB | ||||||||||||||||
| Other | NR | NR | NR | NR | NR | NR | NR | NR | KD | NR | NR | |||||||||||||||||
| Genetic information | Genetic test | NR | NGS panel (95 genes) | NR | NR | NR | NR | NR | NR | NR | gene panel NGS | NR | ||||||||||||||||
| Genomic change (Hg19) | chr6:33400029;G>G/A | chr6:33400583; G>G/A | chr6:33408504_ 33408514delAGCGTGTTCCC | chr6:33405650;T>T/C | chr6:33405712; G>G/A | chr6:33406199;T>T/ TTCC | chr6:33408514;C>C/T | chr6:33408626;C>C/G | chr6:33408718;T>T/A | chr6:33405650;T>T/C | chr6:33400462;C>C/T | |||||||||||||||||
| cDNA/aa change | c.387G>A, p.Ser129Ser (splice donor site) | c.509G>A, p. Arg170Gln | c.1677-2_1685del, (splice acceptor site) | c.968T>C, p.Leu323Pro | c.1030G>A, p.Gly344Ser | c. 1390delinsTTCC, p.Leu465dup | c.1685C>T, p.Pro562Leu | c.1797C>G, p.Cys599Trp | c.1889T>A, p.Ile630Asn | c.968T>C, p.Leu323Pro | c.388C>T, p.Gln130Ter | |||||||||||||||||
| Inheritance | de novo | de novo | de novo | de novo | de novo | de novo | de novo | de novo | de novo | de novo | de novo | |||||||||||||||||
| Others information | Karyotype, aCGH, MECP2 mutation and X-fragile analysis, without relevant results | |||||||||||||||||||||||||||
| Family History | - (seizures, ID or ASD) | - (seizures, ID or ASD) | - (seizures, ID or ASD) | Paternal uncle epilepsy and behavioural problems | - (seizures, ID or ASD) | Mother FS. Paternal uncle learning difficulties | Maternal first cousin ASD | - (seizures, ID or ASD) | - (seizures, ID or ASD) | - (epilepsy or DD) | NR | |||||||||||||||||
| E | ||||||||||||||||||||||||||||
| Reference | Kuchenbuch 2020 d | Kuchenbuch 2020 e | Lo barco 2021 e | Lo barco 2021 | Lo barco 2021 | Lo barco 2021 d | Lo barco 2021 b | Lo barco 2021 | Lo barco 2021 | |||||||||||||||||||
| Patient | 1 | 3 | 2 | 4 | 5 | 6 | 8 | 9 | 10 | |||||||||||||||||||
| Clinical features | gender | male | male | male | male | female | male | male | female | female | ||||||||||||||||||
| Age | 5.6 yr | 3.5 yr | 3 yr 4 months | 11 yr 3 months | 6 yr 3 months | 5 yr 4 months | 6 yr 6 months | 14 yr | 7 yr | |||||||||||||||||||
| DD/ID | NR | NR | + (severe/moderate) | + (severe) | + (severe/moderate) | + (moderate) | + (severe) | + (severe/moderate) | + (severe/moderate) | |||||||||||||||||||
| Behavioral features | motor stereotypies, heteroaggressivity, ASD | NR | ASD, behavior disorder | ASD, behavior disorder | ASD, behavioral and sleep problems | ASD, behavior disorder | ASD, feeding problems | ASD, behavioral and sleep problems | sleep, behavior and eating disorders | |||||||||||||||||||
| Other parametres | NR | NR | NR | head growth slowdown, absence of language | growth delay, absence of language | NR | nystagmus, absence of language | head growth slowdown, absence of language | head growth slowdown, absence of language | |||||||||||||||||||
| Epilepsy | Age of onset | 3.5 yr | 8 months | 8 months | 27 months | 18 months | 36 months | 20 months | 20 months | 30 months | ||||||||||||||||||
| Abscence seizures | + (AA) | + (MA) | + (AA, AAM) | + (AA) | + (AA, AAM, AAOC, AAA) | + (AA, AAOC, MA) | + (AAM) | + (AA, AAM) | + (AAM) | |||||||||||||||||||
| Eyelid myoclonia | + | + | NR | + | + (EMA) | + (EMA) | NR | NR | + (EMA) | |||||||||||||||||||
| Photosensitivity | + | NR | NR | NR | NR | NR | NR | NR | NR | |||||||||||||||||||
| Other seizures | FS, MS, upper limb MJ, DA. Triggered by sleep and IPS | DA, upper limb MJ, reflex seizures self-induced by eye closure | MS, FS | DA | AS | NR | MS, TS | MS | NR | |||||||||||||||||||
| EEG | PPR | NR | NR | + | - | + | + | - | + | - | ||||||||||||||||||
| others | NR | 2-HZ GPSW | Sleep: Sporadic low-voltage multifocal spikes; sporadic bursts of generalized irregular polyspike or PSW in sleep. EM, AA, AAM, F. Self stimulation wirh eyes closure. | Wake: (P)SW on frontal regions; Sleep: higher frequency of generalized discharges. EM, AA. Self stimulation wirh eyes closure. | Sleep: Multifocal spikes, prominent on frontal and occipital regions; bursts of generalized irregular polyspike or PSW. EM, EMA, AA, AAOC, AS, MS, AAA. Self stimulation wirh eyes closure. | Sleep: Low-voltage centrooccipital spikes. AA, AAOC, MA | Wake: Diffuse GPSW; Sleep: PSW on frontal regions. EM, EMA, AA, AAM, MS, AS, MATS. Self stimulation wirh eyes closure. | Wake: Diffuse SW, predominant on frontal regions; Sleep: Numerous frontal SW. EMA, AAM | Wake: Temporo-parietal SW; Sleep: PSW on frontal regions | |||||||||||||||||||
| MRI | Cranial | Normal | NR | Normal | Normal | Bilateral hypersignal of WM (4 yr); cerebellar atrophy (6 yr) | Normal | Aspecific WM hypersignal | NR | Defect in frontal lobes develoment | ||||||||||||||||||
| Treatment | AED | VPA, LEV, ETX, LTG, CBD | ETX, LEV, LTG, VPA, CLB, ZNS, PER, CBD | VPA, ETX, ZNS, LTG | LEV, VPA | VPA, CLB, ETX, LTG, RUF, ZNS | LEV, VPA, LTG, ETX | LEV, VPA, TPM, CLB, CLZ, ZNS, LTG, PER | VPA, LEV, TPM | LEV, VPA, LTG, ETX, CLB, CLZ, ZNS | ||||||||||||||||||
| Other | KD | KD | KD | KD, VNS | KD | KD | ||||||||||||||||||||||
| Genetic inform. | Genomic change (Hg19) | chr6:33409002; G>G/T | chr6:33409095; C>C/T | chr6:33409095; C>C/T | chr6:33411544delA | chr6:33405604; T>T/C | chr6:33409002; G>G/T | chr6:33414346; G>G/A | chr6:33411127; A>A/G | chr6:33400531-33400532insG | ||||||||||||||||||
| cDNA/aa change | c.1966G>T, p.Glu656* | c.2059C>T, p.Arg687* | c.2059C>T, p.Arg687* | c.3215_3224del, p.Lys1072Serfs*2 | c.922T>C, p.Trp308Arg | c.1966G>T, p.Glu656* | c.3583-6G>A, p.Val1195Alafs*27 | c.2798A>G, p.His933Arg | c.456insG, p.Thr153Aspfs*15 | |||||||||||||||||||
| Inheritance | de novo | de novo | de novo | de novo | de novo | de novo | de novo | de novo | de novo | |||||||||||||||||||
| Family History | NR | Paternal grandmother with unspecified epilepsy | NR | NR | NR | NR | NR | NR | NR | |||||||||||||||||||
| A | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Reference | Samanta 2020 1 | Wu 2020 | Stamberger 2021 1,2 | Stamberger 2021 1,2 | Stamberger 2021 1,2 | Stamberger 2021 1 | Stamberger 2021 1 | Stamberger 2021 1,2/Myers 2018 2 | ||||||
| Patient | 1 (F1) | 2 (F2) | 4 (F4) | 7 (F5) | 8 (F6) | 10 (F7)/T990 (family 12) | ||||||||
| Clinical features | gender | Female | Female | Male | Male | Female | Female | Female | Female | |||||
| Age | 9 yr | 29 yr | 8 yr | 12 yr | 12 yr | 14 yr | 18 yr | 28 yr | ||||||
| DD/ID | + (mild) | + (mild) | + (severe) | + (moderate) | + (severe) | + (moderate) | + (moderate) | + (moderate-severe) | ||||||
| Behavioral features | ADHD | NR | Regression, ASD, behavioral problems | Regression, ASD, severe tantrums | Regression, ASD, ADHD | Regression, obsessive, repetitive behaviours, anxiety | Regression, stereotypes, agressive behaviour, impulsive, attention problems, anxiety | ASD traits | ||||||
| Other parametres | mild hypotonia | Minor dysmorphic features (flat nasal bridge and ocular hypertelorism), diabetes mellitus type 2 | Scaphocephaly, mild facial dysmorphisms (deep set eyes, wide spaced teetch, prominent lower lips, protruding tongue; tapering fingers), hypotonia/hupertonia, esotropia | NR | Upslanting palpebral fissures, hypoplastic eyelashes, small rounded nasal tip | ventricular septal defect; primary enuresis | NR | overweight, prominent eyebrows, hirsuitism, polycystisc ovarial syndrome | ||||||
| Epilepsy | Age of onset | 2 yr | 6 yr | 21 months | 15 months | 12–14 months | 2 yr | 3 yr | 19 months | |||||
| Abscence seizures | + | + (AAS) | + | + | + | + | + | + | ||||||
| Eyelid myoclonia | + | + (rare) | + | + | + | + | + | + | ||||||
| Photosensitivity | + | + | + | - | - | - | + | - | ||||||
| Other seizures | Rapid eye blinking with upward eye rolling associated with head bobbing | GTCS, brief blanking, behavioral arrest and states of prolonged confusion | AS, MS. Triggered by PS | MS, MAS, GTCS, NCSE. Triggered by temperature (hot) | Head nods, drop attack, AS, MS, likely NCSE. Triggered by fever, temperature, eye closure | MS, NCSE (absence status) | nocturnal MS, rare GTCS | MS, DA, GTCS, NCSE | ||||||
| EEG | PPR | + | + | NR | NR | NR | NR | NR | NR | |||||
| others | 3 Hz GSW. Eyelid jerking, generalized epileptiform discharges induced by eye cosure | Interictal: paroxysmal GPSW induced by eye closure, PS. Ictal: persistent 1.5–2.5 Hz semi-rhythmic GPSW. Epileptiform discharges induced by eye closure | Interictal: BG slowing, 2.5–3 Hz GPSW, PFA. Ictal: 2.5 Hz GPSW. Triggered by sleep abd IPS | Interictal: BG slowing, 2.5–4 Hz G(P)SW. Ictal: 2.5–4 Hz GPSW (MS, MAS, head drops). Triggered by sleep, eye closure | Interictal: BG slowing, G(P)SW. Ictal: irregular G(P)SW (eyeclosuse with EM). Triggered by eye closure, posible IPS | Interictal: excessive beta activity, G(P)SW. Ictal: irregular GSW (A-EM, MS). Triggered by eye closure, IPS | Interictal: BG slowing 2.5–5 Hz G(P)SW, MFD. Ictal: G(P)SW (EM, MS), NCSE in sleep. Triggered by sleep, eye closure, IPS | Interictal: BG slowing, G(P)SW, PFA, left rhythmic delta activity, MFD | ||||||
| MRI | Cranial | Normal | Normal | Short medulla, thin, dysmorphic CC, asymmetrical hippocampi, right incompletely rotated, delayed myelination subcortical WM and increased FL/T2 signal in posterior PVWM | Slightly small cerebellar vermis and mild tonsillar ectopia, bulky amygdalas and hippocampal heads. | Normal | Possible minimal atrophy superior cerebellar vermis | Normal | Normal | |||||
| Treatment | AED | ETX, LTG, MPH, GF, RD, LEV, VPA, RUF, TPM, ZNS | CBZ, VPA, MZN, LTG, LEV, TPM | VPA, LTG, LEV | ETX, VPA, FBM, RUF, TPM, CLB, CBZ, LEV, PLP, CLZ, VGB, LTG, GB, CS | CS, CLB, LEV, VPA. | ETX, ZNS, PB, CS, VPA, LTG, AZA, CLB, LEV, ATD, CBD | CS, NZP, LTG, CBZ, VPA, TPM, CLB, ZNS, ETX | CS, VPA, LEV, LCM, TPM, AZA, PB, ETX, LTG, PER, BRV, CBD | |||||
| Other | KD, mAD, LHID, DAP | GPO, STG | KD | KD | KD | |||||||||
| Genetic information | Genetic test | NGS panel (1148 genes) | WES | NR | NR | NR | NR | NR | NR | |||||
| Genomic change (Hg19) | chrX:73962671_73962674 del | ChrX:73963328; AG>AG/A- | chrX:73963494; C>C/A | chrX:73961747; G>G/C | chrX:73962951;G>G/A | chrX:73962417; G>G/A | chrX:73961593; G>G/T | chrX:73963428; G>G/A | ||||||
| cDNA/aa change | c.1718_1721delATCA, p.Asp573Serfs*11 | c.1063delC, p.Leu355* | c.898G>T, p.Glu300* | c.2645C>G, p.Ser882* | c.1441C>T, p.Arg481* | c.1975C>T, p.Gln659* | c.2799C>A, p.Tyr933* | c.964C>T, p.Arg322* | ||||||
| Inheritance | de novo | de novo | de novo | NR | inherited (maternal) | de novo | de novo | inherited (paternal gonadal mosaicism likely) | ||||||
| Others information | CXI 74:26 (random). CSF GLUT1 and aCGH normal | CXI 51:49 (random) | NR | ~30% mosaicism for NEXMIF alteration | CXI ~90:10 (skewed). SCN1A:p.(Met1977Val), paternal - VUS | CXI~60:40 (random) | CXI~50:50 (random) | CXI~80:20 (skewed) | ||||||
| Family History | No family history of IDnor epilepsy | Family history of GEFS+ and hypotonia: Father: seizures, hypotonia, speech/language delay, unilateral hearing loss. Sister: FS. Paternal grandfather, paternal aunt and two paternal uncles: childhood epilepsy +- FS. Paternal cousin: FS | Maternal great-great-grandmother: epilepsy | Two sisters carriers of the alteration with ID but without seizures (patient 5 and 6). Two more affected siblings not included in the study with ID without seizures. One other sister is carrier with no disease activity to date. Carrier mother has mild ID | Epileptic sister, also with MAE, carrier of the alteration (patient 9). | |||||||||
| B | ||||||||||||||
| Reference | Stamberger 2021 1,# | Stamberger 2021 1,# | Stamberger 2021 1 | Stamberger 2021 1,& | Stamberger 2021 1 | Stamberger 2021 1 | Stamberger 2021 1/Borlot 2017 1 | Stamberger 2021 1 | Stamberger 2021 1,2,& | |||||
| Patient | 13 (F10) | 15 (F12) | 16 (F13) | 18 (F15) | 23 (F20) | 33 (F30) | 34 (31)/27 | 37 (F34) | 41 (F38 | |||||
| Clinical features | gender | Female | Female | Female | Female | Female | Female | Female | Female | Female | ||||
| Age | 8 yr | 12 yr | 10 yr | 15 yr | 16 yr | 15 yr | 26/23 yr | 10 yr | 4 yr | |||||
| DD/ID | + (mild) | + (moderate) | + (moderate) | + (mild) | + (moderate) | + (moderate) | + (mild) | + (moderate) | + (mild) | |||||
| Behavioral features | Aggressive behaviour, attention problems | ADHD | ASD, Agressive behaviour | Self-abasement, ASD traits (social difficulties) | NR | Easily frustrated | Depression, anxiety | Attention deficiency and problems linked to communication difficulties during infancy, decreased satiety, tics (blinking), ASD traits. | - | |||||
| Other parametres | overweight, gastro-oesophageal reflux disease | NR | Mild facial dysmorphisms (short philtrum, low-set hairline, mild prognathism with frontal bossing) | NR | Hypotonia, hypermovility | NR | overweight, gastro-oesophageal reflux disease as infant/- | Low set backward rotated ears, protruding underlip, hypotonia | Mild hypotonia and hyperlaxity | |||||
| Epilepsy | Age of onset | 1 yr | 9 monts | 2–4 months | 2–4 yr | 18 months | 6.5 yr | 16 months | 30 months | 2 yr 10 months | ||||
| Abscence seizures | + | + | + | + | + | + | + | + | NR | |||||
| Eyelid myoclonia | + | + | + | + | + | + | + | + | + | |||||
| Photosensitivity | NR | NR | - | NR | + | - | - | - | + | |||||
| Other seizures | MS, GTCS. Triggered by sleep deprivation | Triggered by fever, eye closure | Triggered bu eye closure | GTCS | GTCS, NCSE (absences), Triguered by PS | NR | Single GTCS/BCS | MS | AS (head drops) MS (blinking). Triggered by PS | |||||
| EEGG | PPR | NR | NR | NR | NR | NR | NR | NR | NR | NR | ||||
| others | Interictal: mild BG slowing, >3 Hz G(P)SW, MFD, GPFA. Ictal: G(P)SW (Absences +-EM), GSW (MS). Triggered by sleep, IPS, hiperventilation | Interictal: normal BG, G(P)SW in sleep, MFD. Ictal: GSW (EM), Triggered by sleep, eye closure | Interictal: normal BG, G(P)SW in sleep, MFD. Ictal: 3 Hz irregular GPSW (EM). Triggered by sleep | Interictal: BG asimmetry, near continuous G(P)SW during wakefulness. Ictal: EM with impaired awareness | Interictal: Normal BG, MFD with (P)SW, multiple spikes. Triggered by hyperventilation, IPS, sleep, eye closure | Interictal: G(P)SW, PFA. IctalG(P)SW (EM, MS). Triggered by hyperventilation, IPS, eye closure, fixation of sensitivity | Interictal and ictal: sharply contoured runs of alpha activity at times/polyspike and generalized spike waves induced by eye closure | Interictal: multiple spikes and spike-wave. Ictal: quick frontal and central activity (MS). Triggered by eye closure | Interictal: BG slowing, G(P)SW, MFD, bifrontal disrythmic delta activity during sleep. Ictal: GPSW (MS). Triggered by sleep | |||||
| Cranial MRI | Normal | Normal | Normal | Normal | Normal | Normal | Normal | Normal | Normal | |||||
| Treatment | AED | ETX, LEV, VPA | LTG, CLZ, LCM, VPA, ETX, CLB, LEV | ETX, CLZ, VPA | VPA, LTG | LEV, CLZ, ZNS, VPA, ETX, OXC, LTG | VPA, CLB | TPM, CBZ, VPA | LEV, LTG, ETX, VPA | CBD, VPA, CLB, LEV | ||||
| Other | KD, VNS | vitamin B6 | ||||||||||||
| Genetic information | Genetic test | NR | NR | NR | NR | NR | NR | aCGH | NR | NR | ||||
| Genomic change (Hg19) | chrX:73962510; G>G/A | chrX:73962510; G>G/A | chrX:73961016; C>C/A | chrX:73961500; G>G/C | chrX:73964056; C>C/T | chrX:73963740; G>G/A | ChrX:73930523_74007913 del (0.08 Mb, 1 gene) | chrX:73960934dupT | chrX:73961500; G>G/C | |||||
| cDNA/aa change | c.1882C>T, p.Arg628* | c.1882C>T, p.Arg628* | c.3376G>T, p.Glu1126* | c.2892C>G, p.Tyr964* | c.336G>A, p.Trp112* | c.652C>T, p.Arg218* | Xq13.3 del (Ex 2–4) | c.3458dupA, p.Asn1153Lysfs*8 | c.2892C>G, p.Tyr964* | |||||
| Inheritance | NR | de novo | de novo | de novo | de novo | de novo | de novo | de novo | NR | |||||
| Others information | CXI~65:35 (random) | CXI~65:35 (random) | SMA: 3p24.1(30,414,405–30,878,291)x3, maternal - VUS ADGRV1: p.(Asp2942His), maternal - VUS | CXI ~60:40 (random) | GRIN2A: p.(Asn106Lys), paternal - VUS/GLUT1 deficiency (SLC2A1 sequencing) and Epilepsy panel (476 genes) normal | CXI ~50:50 (random) | ||||||||
| Family History | Maternal distant cousin: GTCS and learning disability. Distant maternal relative: absence seizures in childhood. Sister of paternal grandmother: "drop seizures" and questionable DD | Sister with normal development exhibited epilepticus status due to a fall in bicycle, attention deficiency | ||||||||||||
| Reference | Bartnik 2012 | Rudolf 2016 1 | Rudolf 2016 1 | Rudolf 2016 1 | Rudolf 2016 1 | Rudolf 2016 | Rudolf 2016 | Sadleir 2020 1 | Sadleir 2020 | Morea 2021 1 | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Patient | 12 | 4 | 13 | 14 | 20 | 9A1117 | GE0705 | Familly C II-2 | Family D II-1 | Case report | |
| Clinical features | gender | Male | Female | Female | Female | Female | Female | Female | Female | Male | Male |
| Age | NR | NR | NR | NR | NR | 25 yr | 10 yr | 40 yr | 20 yr | 21 yr | |
| DD/ID | NR | + (mild) | + (mild) | + (mild) | NR | + (mild) | + | NR | - | + (moderate) | |
| Behavioral features | ASD | NR | NR | NR | NR | NR | NR | NR | NR | ADHD | |
| Other parametres | NR | NR | NR | NR | NR | Macrocephal, overweight, and learning dificulties | convergent strabismus, hypermetropia, learning dificulties | Learning dificulties | Learning dificulties | NR | |
| Epilepsy | Age of onset | 2 yr | 13 yr | 3 yr | 9 yr | 11 yr | 5 yr 5 months | 4 yr 9 month | 3 yr | 10 yr | 4 yr |
| Abscence seizures | NR | + | + | + | + | + (TA, absence status) | + | + | + (absence status) | + | |
| Eyelid myoclonia | + | + | + | + | + | + | + | + | + | + | |
| Photosensitivity | NR | + | + | + | + | - | + | + | + | NR | |
| Other seizures | GTCS | GTCS | GTCS | GTCS | GTCS | GTCS, FS, TC | nocturnal GTCS | GTCS, occipital seizures | GTCS, occipital seizurures, | Induced by television and videogame exposure | |
| EEG | PPR | NR | + | + | + | + | NR | NR | + | + | NR |
| others | CTS | 3 Hz GSW | 3 Hz GSW | 3 HZ GSW | 3 Hz GSW | Interictal: normal BG rhythm and bilateral centrotemporal spikes. 3 Hz SW absence seizures activated by hyperventilation. Ictal Absence seizures. Interictal GSW, GPSW. Focal frontal or temporal occipital paroxysmal activity | Absence seizures ocassionally with EM triggered by IPS. 3 Hz GSW | GSW | GSW | Ictal: 3 Hz GSW. Interictal: asynchronous spikes on a physiological BG rhythm. Generalized epileptic discharges elicited by eye closure and hyperventilation | |
| Cranial MRI | NR | NR | NR | NR | NR | Normal | Normal | - | Normal | Normal | |
| Treatment | AED | NR | 3 treated with VPA, one with ETX and PB | CBZ, VPA, ETX, VGB, CLB, LTG, TPM, LEV | TEX, VPA, LTG, LEV | LTG, VPA | VPA, LEV, LTG | VPA, ETX, LEV | |||
| Other | KD | ||||||||||
| Genetic information | Genetic test | aCGH | WES | WES | sanger sequencing | WES | aCGH | aCGH | aCGH | Sanger sequencing | NGS |
| Genomic change (Hg19) | chr9:7474,400_ 77306932 del (2.57 Mb, 6 genes) | chr9:77249649; C>C/T | chr9:70984481_ 79549501 del (8.5 Mb, 47 genes) | chr9:77261322_ 77313598 del (52Kb) | Min:chr9:77249078_ 77251973del2896 Max:chr9.77248677_77251984del3308 | chr9:77286755; A>A/T | 9q21 | ||||
| cDNA/aa change | c.196C>T, p.Arg66* | 9q31.13 del (Ex 5–10) | c.96_237del141, p.Gly32_Ala79del48 | c.1162A>T, p.Ile388Phe | NR | ||||||
| Inheritance | de novo | (probably) Inherited | de novo | de novo | inherited (maternal) | inherited (paternal) | de novo | ||||
| Others information | FISH: 9q21.13 (RP 11-243A1) | aCGH array normal | Karyotype: mos 47,XX,+r [20]/46,XX,-21,+der(9)t(9;21) [5]/46,XX [25]. aGCH array: mosaic gain 9p. FISH: 9q13q21.13 del (RP11-404E6), mosaic i(9p), der(9)t(9;21) and r(9) | qPCR to validate the result | NR | NR | |||||
| Family History | Father Asperger syndrome | Family members: Patient 20; Patient 13 (Mother); Patient 14 (maternal aunt); Patient 4 (maternal grandmother). Other two carriers: Patient 23 (sister): One episode of absence seizure (probably GGE with no EEG); Patient 10 (maternal great-aunt): EEG with isolated high-amplitude spike during IPS but seizure state not confirmed. Several antecedents of PS without seizures | Maternal uncle and two of her first cousins had GTCS | No family history of seizures | Alteration inherited from her mother (normal intelligence and no seizures). Her son, with intratable DEE and severe ID, has the same microdeletion | Alteration inherited from his father, diagnosed with early onset abcense epilepsy and occipital lobe epilepsy, who also presented GTCS | - | ||||
| Reference | Galizia 2015 1 | Galizia 2015 1 | Galizia 2015 1 | Tomas 2015 1/Carvill 2013 2 | Tomas 2015 1 | Tomas 2015 1 | Tomas 2015 1/Mullen 2013 | |
|---|---|---|---|---|---|---|---|---|
| Patient | 7 | 8 | 9 | 5/T38 | 6 | 8 | 9/15 [57] | |
| Clinical features | gender | NR | NR | NR | Male | Female | Male | Female |
| Age | NR | NR | NR | 18/17 yr | 13 yr | 14 yr | 36/26 yr | |
| DD/ID | NR | NR | NR | + (moderate-severe/moderate) | + (moderate-severe) | + (mild) | + (mild) | |
| Behavioral features | ASD | NR | NR | ASD, regression, aggression/ASD, No regression | ASD, ADHD, aggresion | ADHD, regression, agression | autistic traits, regression, agression | |
| Other parametres | nephrolithiasis, migraine, scoliosis | NR | NR | Transient ataxia on valproate | Short stature | Short stature, ataxia | NR | |
| Epilepsy | Age of onset | NR | NR | NR | 12 months | 30 months | 30 months | 34 months |
| Abscence seizures | + | + | + | + (AMA, MA; TA)/+ | + (TA) | + | + | |
| Eyelid myoclonia | + | + | + | + | + | + | + | |
| Photosensitivity | + | + | + | + (Self induced with TV) | + (Self induced with TV) | +(Self induced with TV; photic stimulation) | +(Self induced with TV or light) | |
| Other seizures | NR | NR | NR | MS, FS, GTCS/AS, FS, MJ, TC | MS (self-induced with TV), TS, GTCS | AS, GTCS, MS (self-induced with TV), NCSE, CSE | GTCS, MS (self-induced with TV or light), | |
| EEG | PPR | + | + | + | - (interictal and BG) | - (interictal and BG) | + (grade 4) | NR |
| others | NR | NR | NR | 3–4-HZ GPSW during atonic myoclonic absence seizures. 9 yr: Diffusely slow BG with symmetrical theta and 3 Hz delta. 14 yr: Normal BG for brief bursts of 3–4 Hz regular rhythmic GSW/3.8 Hz GSW | 3 yr: Slow BG for age. Inter-ictal GSW GPSW, bi frontal slow spike wave (2 Hz). Eye flickering—bursts of 2 Hz bifrontal spike and wave. 4 yr: EMA, GSW, GPSW. Activated by eye closure. 7 yr: Normal BG, irregular GSW with a frontal predominance. GSW on eye closure and during eyelid myoclonia | 32 months: Normal BG, frontal predominant GSW, GPSW. 5.5 yr: Normal BG, multifocal GSW. 11 yr: Diffusely slow BG, 3 Hz GSW, bifrontal spikes, activated in sleep, 4–5 Hz posteriorly dominant GSW on eye closure. 12 yr: Continuous slowing in wake and marked generalized epileptiform activity in sleep. 14 yr: Diffuse theta slowing, active interictal GSW GPSW discharges >3 Hz increased by hyperventilation | 5 yr: Generalized epileptiform discharges every 1–2 minutes | |
| MRI | Craneal | NR | NR | NR | Normal | Generalized cerebellar atrophy with large v4 and prominent folia. The posterior corpus callosum is foreshortened and smaller posteriorly | Atrophy between scans, markedly in the cerebellum. The corpus callosum is hypoplastic posteriorly with a small splenium | Normal |
| Genetic data | Genetic test | NGS | NGS | NGS | target NGS | target NGS | target NGS | aCGH |
| Genomic change (Hg19) | chr15:93540316; A> A/- | chr15:93545442; ->-/A | chr15:93482909; C>C/T | chr15:93545504_93545507del | chr15:93557956delG | chr15:93521611; C>C/T | chr15:91027533_93477874 del | |
| cDNA/aa change | c.3725delA, p.Lys1245Asnfs*4 | c.4173dupA, p.Gln1392Thrf*17 | c.C653T, p.Pro218Leu | c.4235_4238 delAAGG, p.Glu1412Glyfs*64 | c.4720delG, p.Gly1575Valfs*17 | c.2725C>T, p.Gln909* | 15q26 del (2.4 Mb) | |
| Inheritance | NR | de novo | inherited | de novo | de novo | de novo | de novo | |
| Family History | NR | NR | Inherited from unaffected mother | NR | NR | NR | NR |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mayo, S.; Gómez-Manjón, I.; Fernández-Martínez, F.J.; Camacho, A.; Martínez, F.; Benito-León, J. Candidate Genes for Eyelid Myoclonia with Absences, Review of the Literature. Int. J. Mol. Sci. 2021, 22, 5609. https://doi.org/10.3390/ijms22115609
Mayo S, Gómez-Manjón I, Fernández-Martínez FJ, Camacho A, Martínez F, Benito-León J. Candidate Genes for Eyelid Myoclonia with Absences, Review of the Literature. International Journal of Molecular Sciences. 2021; 22(11):5609. https://doi.org/10.3390/ijms22115609
Chicago/Turabian StyleMayo, Sonia, Irene Gómez-Manjón, Fco. Javier Fernández-Martínez, Ana Camacho, Francisco Martínez, and Julián Benito-León. 2021. "Candidate Genes for Eyelid Myoclonia with Absences, Review of the Literature" International Journal of Molecular Sciences 22, no. 11: 5609. https://doi.org/10.3390/ijms22115609
APA StyleMayo, S., Gómez-Manjón, I., Fernández-Martínez, F. J., Camacho, A., Martínez, F., & Benito-León, J. (2021). Candidate Genes for Eyelid Myoclonia with Absences, Review of the Literature. International Journal of Molecular Sciences, 22(11), 5609. https://doi.org/10.3390/ijms22115609