
High Concordance of Genomic Profiles between Primary and Metastatic Colorectal Cancer


Abstract
1. Introduction
2. Results
2.1. Clinicopathologic Characteristics of Patients
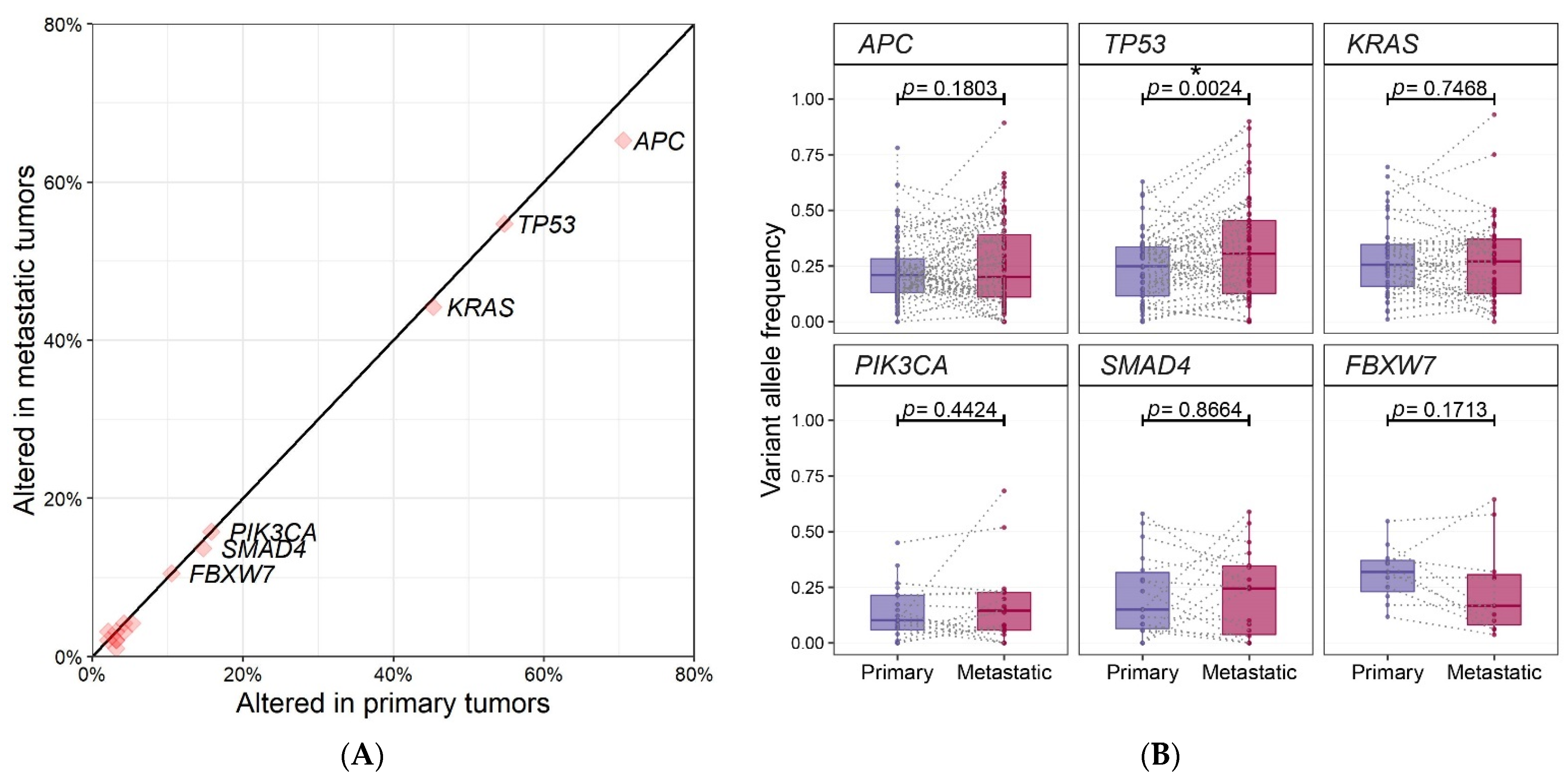
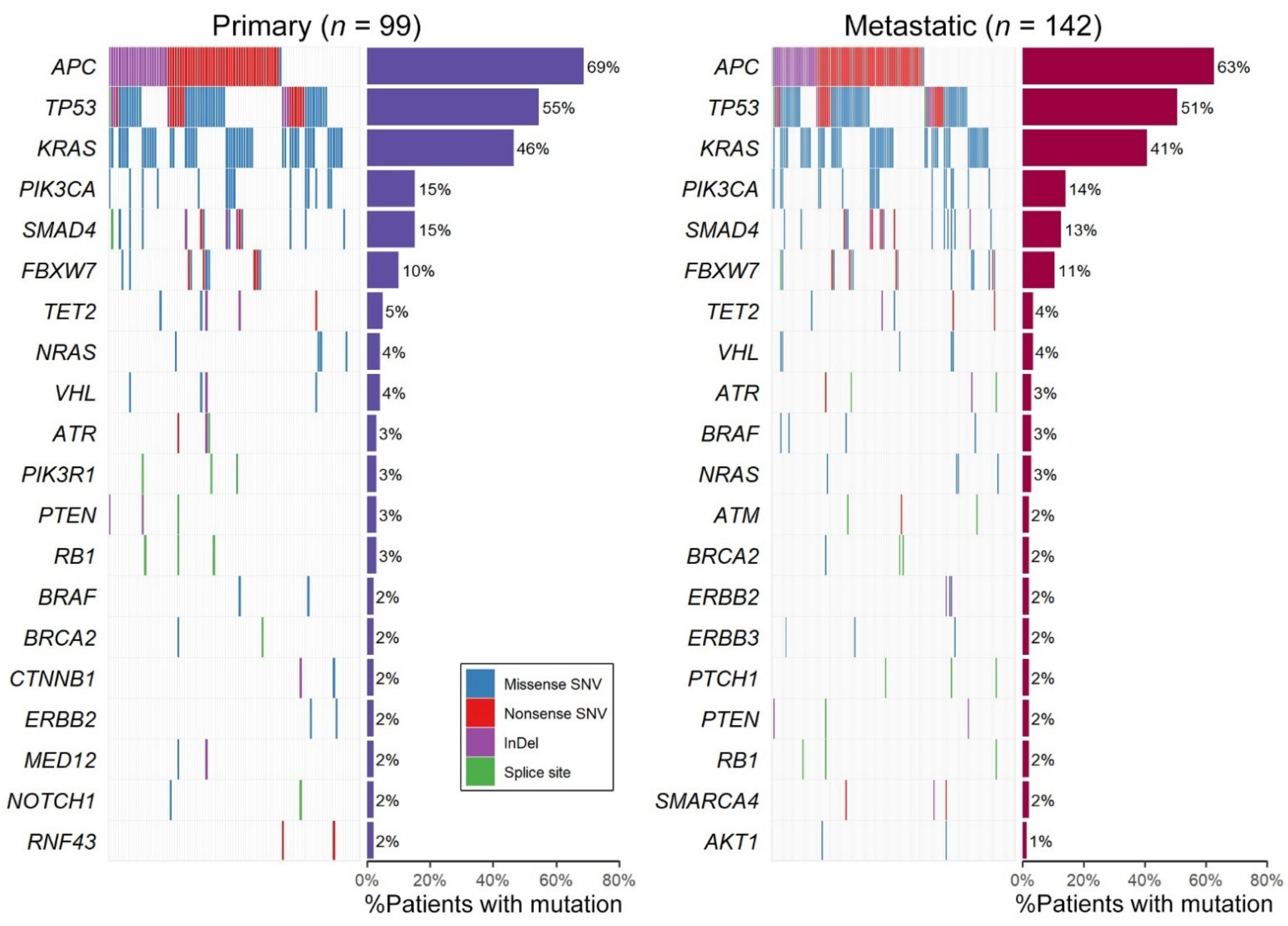
2.2. Genomic Profiling of 95 Paired Samples
2.3. Genomic Profiling of All 142 Samples Including Primary/Metastatic Singletons
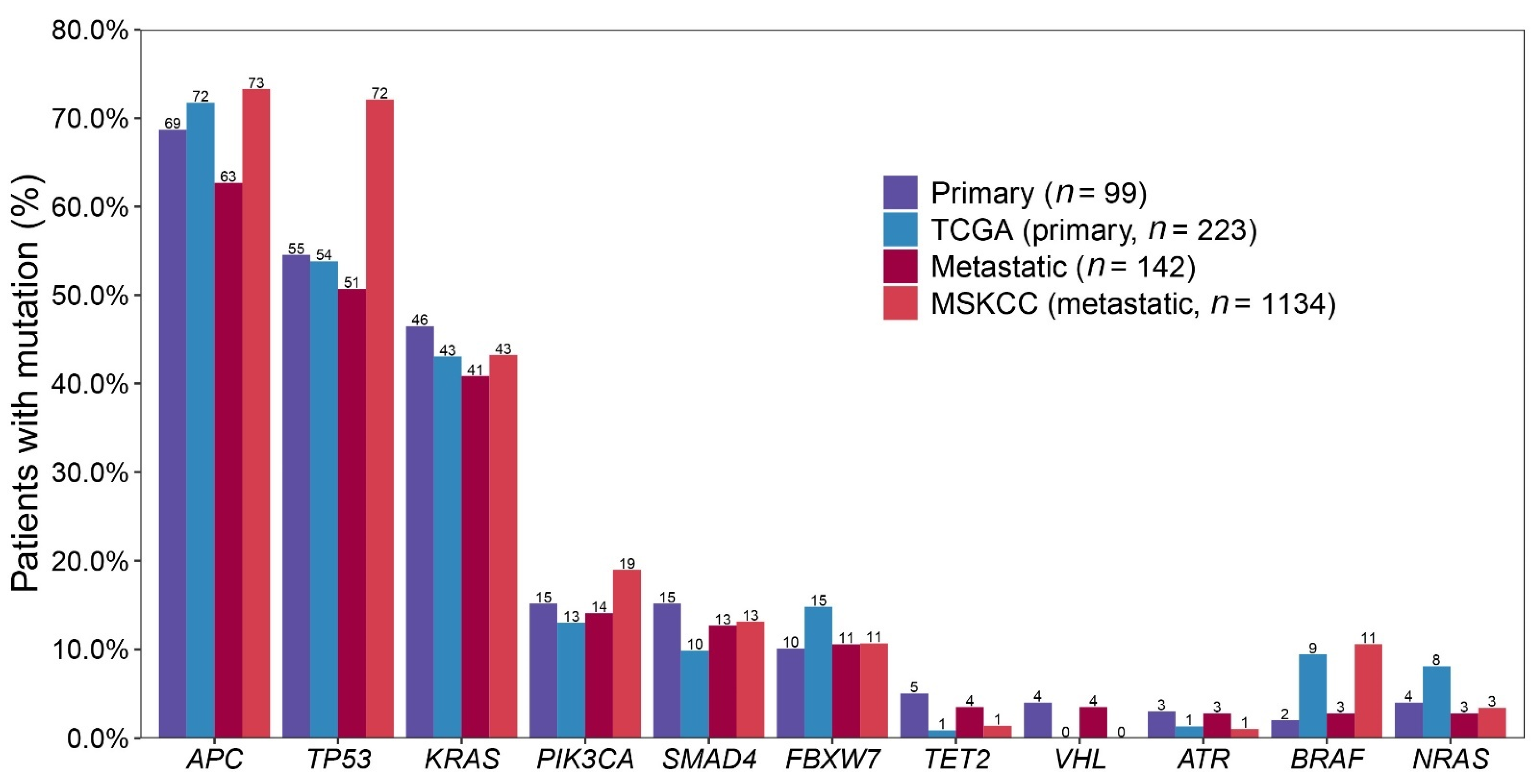
2.4. Comparing with Public Data: 99 Primary CRCs vs. TCGA and 142 Metastatic CRCs vs. MSKCC
3. Discussion
4. Materials and Methods
4.1. Patients
4.2. Targeted Sequencing & Identification of Clinically Significant Mutations
4.3. Comparing with Public Data
4.4. Molecular Findings for Microsatellite Instability (MSI)
4.5. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Vogelstein, B.; Fearon, E.R.; Hamilton, S.R.; Kern, S.E.; Preisinger, A.C.; Leppert, M.; Nakamura, Y.; White, R.; Smits, A.M.; Bos, J.L. Genetic alterations during colorectal-tumor development. N. Engl. J. Med. 1988, 319, 525–532. [Google Scholar] [CrossRef]
- Cancer Genome Atlas, N. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012, 487, 330–337. [Google Scholar] [CrossRef]
- Yaeger, R.; Chatila, W.K.; Lipsyc, M.D.; Hechtman, J.F.; Cercek, A.; Sanchez-Vega, F.; Jayakumaran, G.; Middha, S.; Zehir, A.; Donoghue, M.T.A.; et al. Clinical Sequencing Defines the Genomic Landscape of Metastatic Colorectal Cancer. Cancer Cell 2018, 33, 125–136 e3. [Google Scholar] [CrossRef]
- Jones, S.; Chen, W.D.; Parmigiani, G.; Diehl, F.; Beerenwinkel, N.; Antal, T.; Traulsen, A.; Nowak, M.A.; Siegel, C.; Velculescu, V.E.; et al. Comparative lesion sequencing provides insights into tumor evolution. Proc. Natl. Acad. Sci. USA 2008, 105, 4283–4288. [Google Scholar] [CrossRef]
- Vakiani, E.; Janakiraman, M.; Shen, R.; Sinha, R.; Zeng, Z.; Shia, J.; Cercek, A.; Kemeny, N.; D’Angelica, M.; Viale, A.; et al. Comparative genomic analysis of primary versus metastatic colorectal carcinomas. J. Clin. Oncol. 2012, 30, 2956–2962. [Google Scholar] [CrossRef] [PubMed]
- Brannon, A.R.; Vakiani, E.; Sylvester, B.E.; Scott, S.N.; McDermott, G.; Shah, R.H.; Kania, K.; Viale, A.; Oschwald, D.M.; Vacic, V.; et al. Comparative sequencing analysis reveals high genomic concordance between matched primary and metastatic colorectal cancer lesions. Genome Biol. 2014, 15, 454. [Google Scholar] [CrossRef] [PubMed]
- Lim, B.; Mun, J.; Kim, J.H.; Kim, C.W.; Roh, S.A.; Cho, D.H.; Kim, Y.S.; Kim, S.Y.; Kim, J.C. Genome-wide mutation profiles of colorectal tumors and associated liver metastases at the exome and transcriptome levels. Oncotarget 2015, 6, 22179–22190. [Google Scholar] [CrossRef]
- Williams, M.J.; Werner, B.; Barnes, C.P.; Graham, T.A.; Sottoriva, A. Identification of neutral tumor evolution across cancer types. Nat. Genet. 2016, 48, 238–244. [Google Scholar] [CrossRef]
- Saito, T.; Niida, A.; Uchi, R.; Hirata, H.; Komatsu, H.; Sakimura, S.; Hayashi, S.; Nambara, S.; Kuroda, Y.; Ito, S.; et al. A temporal shift of the evolutionary principle shaping intratumor heterogeneity in colorectal cancer. Nat. Commun. 2018, 9, 2884. [Google Scholar] [CrossRef]
- Sondka, Z.; Bamford, S.; Cole, C.G.; Ward, S.A.; Dunham, I.; Forbes, S.A. The COSMIC Cancer Gene Census: Describing genetic dysfunction across all human cancers. Nat. Rev. Cancer 2018, 18, 696–705. [Google Scholar] [CrossRef]
- Choi, J.Y.; Choi, S.; Lee, M.; Park, Y.S.; Sung, J.S.; Chang, W.J.; Kim, J.W.; Choi, Y.J.; Kim, J.; Kim, D.S.; et al. Clinical Implication of Concordant or Discordant Genomic Profiling between Primary and Matched Metastatic Tissues in Patients with Colorectal Cancer. Cancer Res. Treat. 2020, 52, 764–778. [Google Scholar] [CrossRef] [PubMed]
- Zehir, A.; Benayed, R.; Shah, R.H.; Syed, A.; Middha, S.; Kim, H.R.; Srinivasan, P.; Gao, J.; Chakravarty, D.; Devlin, S.M.; et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat. Med. 2017, 23, 703–713. [Google Scholar] [CrossRef] [PubMed]
- Priestley, P.; Baber, J.; Lolkema, M.P.; Steeghs, N.; de Bruijn, E.; Shale, C.; Duyvesteyn, K.; Haidari, S.; van Hoeck, A.; Onstenk, W.; et al. Pan-cancer whole-genome analyses of metastatic solid tumours. Nature 2019, 575, 210–216. [Google Scholar] [CrossRef]
- Vignot, S.; Lefebvre, C.; Frampton, G.M.; Meurice, G.; Yelensky, R.; Palmer, G.; Capron, F.; Lazar, V.; Hannoun, L.; Miller, V.A.; et al. Comparative analysis of primary tumour and matched metastases in colorectal cancer patients: Evaluation of concordance between genomic and transcriptional profiles. Eur. J. Cancer 2015, 51, 791–799. [Google Scholar] [CrossRef]
- Cross, W.; Graham, T.A.; Wright, N.A. New paradigms in clonal evolution: Punctuated equilibrium in cancer. J. Pathol. 2016, 240, 126–136. [Google Scholar] [CrossRef]
- Bhullar, D.S.; Barriuso, J.; Mullamitha, S.; Saunders, M.P.; O’Dwyer, S.T.; Aziz, O. Biomarker concordance between primary colorectal cancer and its metastases. EBioMedicine 2019, 40, 363–374. [Google Scholar] [CrossRef]
- Xie, Y.H.; Chen, Y.X.; Fang, J.Y. Comprehensive review of targeted therapy for colorectal cancer. Signal Transduct. Target. Ther. 2020, 5, 22. [Google Scholar] [CrossRef]
- Miyaki, M.; Iijima, T.; Konishi, M.; Sakai, K.; Ishii, A.; Yasuno, M.; Hishima, T.; Koike, M.; Shitara, N.; Iwama, T.; et al. Higher frequency of Smad4 gene mutation in human colorectal cancer with distant metastasis. Oncogene 1999, 18, 3098–3103. [Google Scholar] [CrossRef]
- Losi, L.; Luppi, G.; Benhattar, J. Assessment of K-ras, Smad4 and p53 gene alterations in colorectal metastases and their role in the metastatic process. Oncol. Rep. 2004, 12, 1221–1225. [Google Scholar] [CrossRef]
- Kemp, Z.; Rowan, A.; Chambers, W.; Wortham, N.; Halford, S.; Sieber, O.; Mortensen, N.; von Herbay, A.; Gunther, T.; Ilyas, M.; et al. CDC4 mutations occur in a subset of colorectal cancers but are not predicted to cause loss of function and are not associated with chromosomal instability. Cancer Res. 2005, 65, 11361–11366. [Google Scholar] [CrossRef]
- Akhoondi, S.; Sun, D.; von der Lehr, N.; Apostolidou, S.; Klotz, K.; Maljukova, A.; Cepeda, D.; Fiegl, H.; Dafou, D.; Marth, C.; et al. FBXW7/hCDC4 is a general tumor suppressor in human cancer. Cancer Res. 2007, 67, 9006–9012. [Google Scholar] [CrossRef]
- Malapelle, U.; Pisapia, P.; Sgariglia, R.; Vigliar, E.; Biglietto, M.; Carlomagno, C.; Giuffre, G.; Bellevicine, C.; Troncone, G. Less frequently mutated genes in colorectal cancer: Evidences from next-generation sequencing of 653 routine cases. J. Clin. Pathol. 2016, 69, 767–771. [Google Scholar] [CrossRef]
- Korphaisarn, K.; Morris, V.K.; Overman, M.J.; Fogelman, D.R.; Kee, B.K.; Raghav, K.P.S.; Manuel, S.; Shureiqi, I.; Wolff, R.A.; Eng, C.; et al. FBXW7 missense mutation: A novel negative prognostic factor in metastatic colorectal adenocarcinoma. Oncotarget 2017, 8, 39268–39279. [Google Scholar] [CrossRef]
- Kim, J.H.; Kang, G.H. Molecular and prognostic heterogeneity of microsatellite-unstable colorectal cancer. World J. Gastroenterol. 2014, 20, 4230–4243. [Google Scholar] [CrossRef]
- Koopman, M.; Kortman, G.A.; Mekenkamp, L.; Ligtenberg, M.J.; Hoogerbrugge, N.; Antonini, N.F.; Punt, C.J.; van Krieken, J.H. Deficient mismatch repair system in patients with sporadic advanced colorectal cancer. Br. J. Cancer 2009, 100, 266–273. [Google Scholar] [CrossRef]
- Lee, S.E.; Lee, H.S.; Kim, K.Y.; Park, J.H.; Roh, H.; Park, H.Y.; Kim, W.S. High prevalence of the MLH1 V384D germline mutation in patients with HER2-positive luminal B breast cancer. Sci. Rep. 2019, 9, 10966. [Google Scholar] [CrossRef]
- McLaren, W.; Gil, L.; Hunt, S.E.; Riat, H.S.; Ritchie, G.R.; Thormann, A.; Flicek, P.; Cunningham, F. The Ensembl Variant Effect Predictor. Genome Biol. 2016, 17, 122. [Google Scholar] [CrossRef]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alfoldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020, 581, 434–443. [Google Scholar] [CrossRef]
- Landrum, M.J.; Lee, J.M.; Benson, M.; Brown, G.R.; Chao, C.; Chitipiralla, S.; Gu, B.; Hart, J.; Hoffman, D.; Jang, W.; et al. ClinVar: Improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 2018, 46, D1062–D1067. [Google Scholar] [CrossRef]
- Chakravarty, D.; Gao, J.; Phillips, S.M.; Kundra, R.; Zhang, H.; Wang, J.; Rudolph, J.E.; Yaeger, R.; Soumerai, T.; Nissan, M.H.; et al. OncoKB: A Precision Oncology Knowledge Base. JCO Precis. Oncol. 2017. [Google Scholar] [CrossRef]
- Boland, C.R.; Thibodeau, S.N.; Hamilton, S.R.; Sidransky, D.; Eshleman, J.R.; Burt, R.W.; Meltzer, S.J.; Rodriguez-Bigas, M.A.; Fodde, R.; Ranzani, G.N.; et al. A National Cancer Institute Workshop on Microsatellite Instability for cancer detection and familial predisposition: Development of international criteria for the determination of microsatellite instability in colorectal cancer. Cancer Res. 1998, 58, 5248–5257. [Google Scholar] [PubMed]
- Umar, A.; Boland, C.R.; Terdiman, J.P.; Syngal, S.; de la Chapelle, A.; Ruschoff, J.; Fishel, R.; Lindor, N.M.; Burgart, L.J.; Hamelin, R.; et al. Revised Bethesda Guidelines for hereditary nonpolyposis colorectal cancer (Lynch syndrome) and microsatellite instability. J. Natl. Cancer Inst. 2004, 96, 261–268. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variable | Category | Total | |
|---|---|---|---|
| (n = 142) | |||
| Age | |||
| Mean ± SD | 60.51 ± 11.17 | ||
| Median | 61 | ||
| Range (min–max) | 34–89 | ||
| Gender | |||
| Male | 95 | 66.90% | |
| Female | 47 | 33.10% | |
| Site of metastasis | |||
| Liver | 77 | 54.23% | |
| Lung | 35 | 24.65% | |
| Abdominopelvic cavity | 18 | 12.68% | |
| Soft tissue | 6 | 4.23% | |
| Brain | 2 | 1.41% | |
| Lymph node | 2 | 1.41% | |
| Bone | 1 | 0.70% | |
| Skin | 1 | 0.70% | |
| Location of primary cancer | |||
| Ascending colon | 16 | 11.30% | |
| Hepatic flexure | 6 | 4.20% | |
| Transverse colon | 5 | 3.50% | |
| Splenic flexure | 2 | 1.40% | |
| Descending colon | 6 | 4.20% | |
| Rectosigmoid | 107 | 75.40% | |
| Patterns of metastasis | |||
| Synchronous | 78 | 54.90% | |
| Metachronous | 64 | 45.10% | |
| T stage | |||
| 1 | 3 | 2.11% | |
| 2 | 7 | 4.93% | |
| 3 | 101 | 71.13% | |
| 4 | 26 | 18.31% | |
| NA | 5 | 3.52% | |
| N stage | |||
| 0 | 39 | 27.46% | |
| 1 | 48 | 33.80% | |
| 2 | 48 | 33.80% | |
| NA | 7 | 4.93% | |
| Grade | |||
| 1 | 2 | 1.41% | |
| 2 | 132 | 92.96% | |
| 3 | 4 | 2.82% | |
| NA | 4 | 2.82% | |
| Vascular invasion | |||
| Absent | 117 | 82.39% | |
| Present | 20 | 14.08% | |
| NA | 5 | 3.52% | |
| Lymphatic invasion | |||
| Absent | 72 | 50.70% | |
| Present | 65 | 45.77% | |
| NA | 5 | 3.52% | |
| Perineural invasion | |||
| Absent | 97 | 68.31% | |
| Present | 40 | 28.17% | |
| NA | 5 | 3.52% | |
| MSI status | |||
| MSI-H | 7 | 4.93% | |
| MSS | 107 | 75.35% | |
| NA | 21 | 14.79% |
| Genes | No. of variants | VAF of Primary Tumors (Mean ± SD) | VAF of Metastatic Tumors (Mean ± SD) | p-Value |
|---|---|---|---|---|
| APC | 87 | 0.23 ± 0.14 | 0.26 ± 0.19 | 0.1803 |
| TP53 | 55 | 0.24 ± 0.15 | 0.33 ± 0.22 | 0.0024 |
| KRAS | 43 | 0.28 ± 0.16 | 0.27 ± 0.19 | 0.7468 |
| PIK3CA | 20 | 0.14 ± 0.12 | 0.17 ± 0.17 | 0.4424 |
| SMAD4 | 18 | 0.21 ± 0.19 | 0.22 ± 0.20 | 0.8664 |
| FBXW7 | 11 | 0.31 ± 0.12 | 0.24 ± 0.21 | 0.1713 |
| Mutational Status | Total | MSI Status | p Value | Total | Location of Primary Cancer | p Value | Total | Patterns of Metastasis | p Value | |||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Patients | MSI-H | MSS | Patients | Liver | Others | Patients | Synchronous | Metachronous | ||||
| (n = 118) | (n = 7) | (n = 111) | (n = 142) | (n = 77) | (n = 65) | (n = 142) | (n = 78) | (n = 64) | ||||
| APC | ||||||||||||
| Wild type (%) | 47 (39.83) | 3 (42.86) | 44 (39.64) | >0.99 | 53 (37.32) | 24 (31.17) | 29 (44.62) | 0.14 | 53 (37.32) | 32 (41.03) | 21 (32.81) | 0.41 |
| Mutated (%) | 71 (60.17) | 4 (57.14) | 67 (60.36) | 89 (62.68) | 53 (68.83) | 36 (55.38) | 89 (62.68) | 46 (58.97) | 43 (67.19) | |||
| TP53 | ||||||||||||
| Wild type (%) | 58 (49.15) | 5 (71.43) | 53 (47.75) | 0.27 | 70 (49.3) | 33 (42.86) | 37 (56.92) | 0.13 | 70 (49.3) | 32 (41.03) | 38 (59.38) | 0.04 |
| Mutated (%) | 60 (50.85) | 2 (28.57) | 58 (52.25) | 72 (50.7) | 44 (57.14) | 28 (43.08) | 72 (50.7) | 46 (58.97) | 26 (40.62) | |||
| KRAS | ||||||||||||
| Wild type (%) | 69 (58.47) | 3 (42.86) | 66 (59.46) | 0.45 | 84 (59.15) | 49 (63.64) | 35 (53.85) | 0.31 | 84 (59.15) | 52 (66.67) | 32 (50) | 0.07 |
| Mutated (%) | 49 (41.53) | 4 (57.14) | 45 (40.54) | 58 (40.85) | 28 (36.36) | 30 (46.15) | 58 (40.85) | 26 (33.33) | 32 (50) | |||
| PIK3CA | ||||||||||||
| Wild type (%) | 99 (83.9) | 6 (85.71) | 93 (83.78) | >0.99 | 122 (85.92) | 64 (83.12) | 58 (89.23) | 0.42 | 122 (85.92) | 68 (87.18) | 54 (84.38) | 0.81 |
| Mutated (%) | 19 (16.1) | 1 (14.29) | 18 (16.22) | 20 (14.08) | 13 (16.88) | 7 (10.77) | 20 (14.08) | 10 (12.82) | 10 (15.62) | |||
| SMAD4 | ||||||||||||
| Wild type (%) | 102 (86.44) | 3 (42.86) | 99 (89.19) | 0.01 | 124 (87.32) | 66 (85.71) | 58 (89.23) | 0.71 | 124 (87.32) | 67 (85.9) | 57 (89.06) | 0.76 |
| Mutated (%) | 16 (13.56) | 4 (57.14) | 12 (10.81) | 18 (12.68) | 11 (14.29) | 7 (10.77) | 18 (12.68) | 11 (14.1) | 7 (10.94) | |||
| FBXW7 | ||||||||||||
| Wild type (%) | 106 (89.83) | 7 (100) | 99 (89.19) | >0.99 | 127 (89.44) | 66 (85.71) | 61 (93.85) | 0.19 | 127 (89.44) | 68 (87.18) | 59 (92.19) | 0.49 |
| Mutated (%) | 12 (10.17) | 0 (0) | 12 (10.81) | 15 (10.56) | 11 (14.29) | 4 (6.15) | 15 (10.56) | 10 (12.82) | 5 (7.81) | |||
| Primary (n = 99) | Metastatic (n = 142) | TCGA (n = 223) | MSKCC (n = 1134) | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Order | Gene | # Mutated Patients | % Mutation | Order | Gene | # Mutated Patients | % Mutation | Order | Gene | # Mutated Patients | % Mutation | Order | Gene | # Mutated Patients | % Mutation |
| 1 | APC | 68 | 69% | 1 | APC | 89 | 63% | 1 | APC | 160 | 72% | 1 | APC | 831 | 73% |
| 2 | TP53 | 54 | 55% | 2 | TP53 | 72 | 51% | 2 | TP53 | 120 | 54% | 2 | TP53 | 818 | 72% |
| 3 | KRAS | 46 | 46% | 3 | KRAS | 58 | 41% | 3 | KRAS | 96 | 43% | 3 | KRAS | 490 | 43% |
| 4 | PIK3CA | 15 | 15% | 4 | PIK3CA | 20 | 14% | 4 | FBXW7 | 33 | 15% | 4 | PIK3CA | 215 | 19% |
| 5 | SMAD4 | 15 | 15% | 5 | SMAD4 | 18 | 13% | 5 | PIK3CA | 29 | 13% | 5 | SMAD4 | 149 | 13% |
| 6 | FBXW7 | 10 | 10% | 6 | FBXW7 | 15 | 11% | 6 | SMAD4 | 22 | 10% | 6 | FBXW7 | 121 | 11% |
| 7 | TET2 | 5 | 5% | 7 | TET2 | 5 | 4% | 7 | BRAF | 21 | 9% | 7 | BRAF | 120 | 11% |
| 8 | NRAS | 4 | 4% | 8 | VHL | 5 | 4% | 8 | AMER1 | 19 | 9% | 8 | SOX9 | 99 | 9% |
| 9 | VHL | 4 | 4% | 9 | ATR | 4 | 3% | 9 | NRAS | 18 | 8% | 9 | ARID1A | 83 | 7% |
| 10 | ATR | 3 | 3% | 10 | BRAF | 4 | 3% | 10 | ARID1A | 12 | 5% | 10 | TCF7L2 | 77 | 7% |
| 11 | PIK3R1 | 3 | 3% | 11 | NRAS | 4 | 3% | 11 | ATM | 10 | 4% | 11 | PTEN | 68 | 6% |
| 12 | PTEN | 3 | 3% | 12 | ATM | 3 | 2% | 12 | ERBB3 | 10 | 4% | 12 | RNF43 | 67 | 6% |
| 13 | RB1 | 3 | 3% | 13 | BRCA2 | 3 | 2% | 13 | SOX9 | 10 | 4% | 13 | AMER1 | 49 | 4% |
| 14 | BRAF | 2 | 2% | 14 | ERBB2 | 3 | 2% | 14 | SMAD2 | 9 | 4% | 14 | ATM | 45 | 4% |
| 15 | BRCA2 | 2 | 2% | 15 | ERBB3 | 3 | 2% | 15 | PIK3R1 | 8 | 4% | 15 | PIK3R1 | 40 | 4% |
| 16 | CTNNB1 | 2 | 2% | 16 | PTCH1 | 3 | 2% | 16 | CREBBP | 7 | 3% | 16 | NRAS | 39 | 3% |
| 17 | ERBB2 | 2 | 2% | 17 | PTEN | 3 | 2% | 17 | FBN1 | 7 | 3% | 17 | B2M | 37 | 3% |
| 18 | MED12 | 2 | 2% | 18 | RB1 | 3 | 2% | 18 | PTEN | 7 | 3% | 18 | BRCA2 | 35 | 3% |
| 19 | NOTCH1 | 2 | 2% | 19 | SMARCA4 | 3 | 2% | 19 | BCOR | 6 | 3% | 19 | SMAD3 | 32 | 3% |
| 20 | RNF43 | 2 | 2% | 20 | AKT1 | 2 | 1% | 20 | ELF3 | 6 | 3% | 20 | TGFBR2 | 30 | 3% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, S.E.; Park, H.Y.; Hwang, D.-Y.; Han, H.S. High Concordance of Genomic Profiles between Primary and Metastatic Colorectal Cancer. Int. J. Mol. Sci. 2021, 22, 5561. https://doi.org/10.3390/ijms22115561
Lee SE, Park HY, Hwang D-Y, Han HS. High Concordance of Genomic Profiles between Primary and Metastatic Colorectal Cancer. International Journal of Molecular Sciences. 2021; 22(11):5561. https://doi.org/10.3390/ijms22115561
Chicago/Turabian StyleLee, Seung Eun, Ha Young Park, Dae-Yong Hwang, and Hye Seung Han. 2021. "High Concordance of Genomic Profiles between Primary and Metastatic Colorectal Cancer" International Journal of Molecular Sciences 22, no. 11: 5561. https://doi.org/10.3390/ijms22115561
APA StyleLee, S. E., Park, H. Y., Hwang, D.-Y., & Han, H. S. (2021). High Concordance of Genomic Profiles between Primary and Metastatic Colorectal Cancer. International Journal of Molecular Sciences, 22(11), 5561. https://doi.org/10.3390/ijms22115561