
Latency Reversing Agents: Kick and Kill of HTLV-1?


Abstract
1. Introduction
1.1. HTLV-1, a Persistent Human Tumorvirus
1.2. The HTLV-1 Viral Reservoir
1.3. Immune Responses Targeting Tax
1.4. Kick and Kill, Shock and Kill, or Gene Activation Therapy to Achieve Latency Reversal
1.5. Brief Categorization of Latency Reversing Agents (LRAs)
2. Manipulation of Histone Modification
2.1. Histone Modification
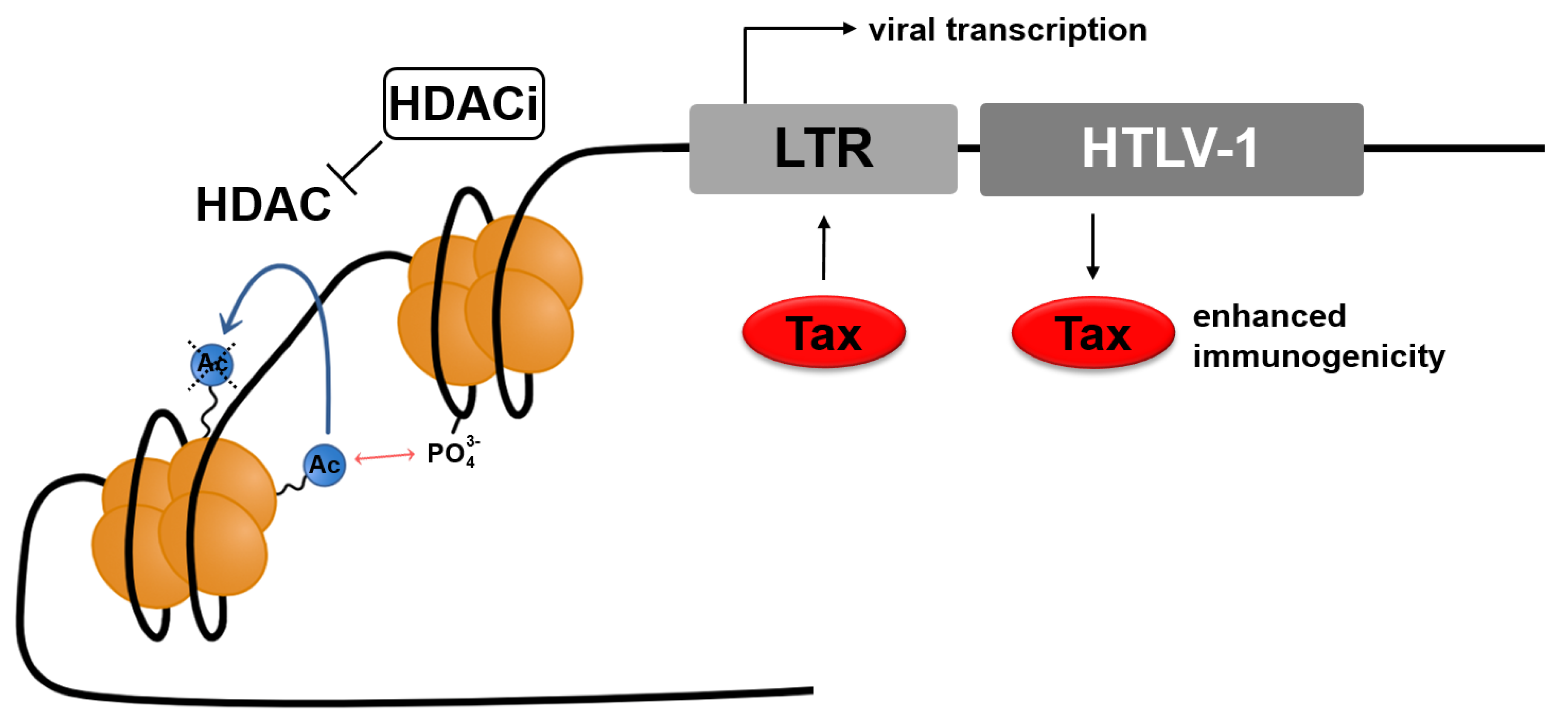
2.1.1. Histone Modification: Histone Deacetylases (HDACs)
2.1.2. HDACs at the HTLV-1 Promoter
2.2. Histone Deacetylase Inhibitors (HDACi)
2.2.1. HDACi as the Anticancer Drugs
2.2.2. HDACi: Mode of Action
2.2.3. HDACi: Immune Modulation
2.3. Classes of Histone Deacetylase Inhibitors (HDACi)
2.3.1. HDACi: Overview of Classes
2.3.2. Hydroxamates
2.3.3. Benzamides
2.3.4. Short-Chain Fatty Acids
2.3.5. Cyclic Peptides
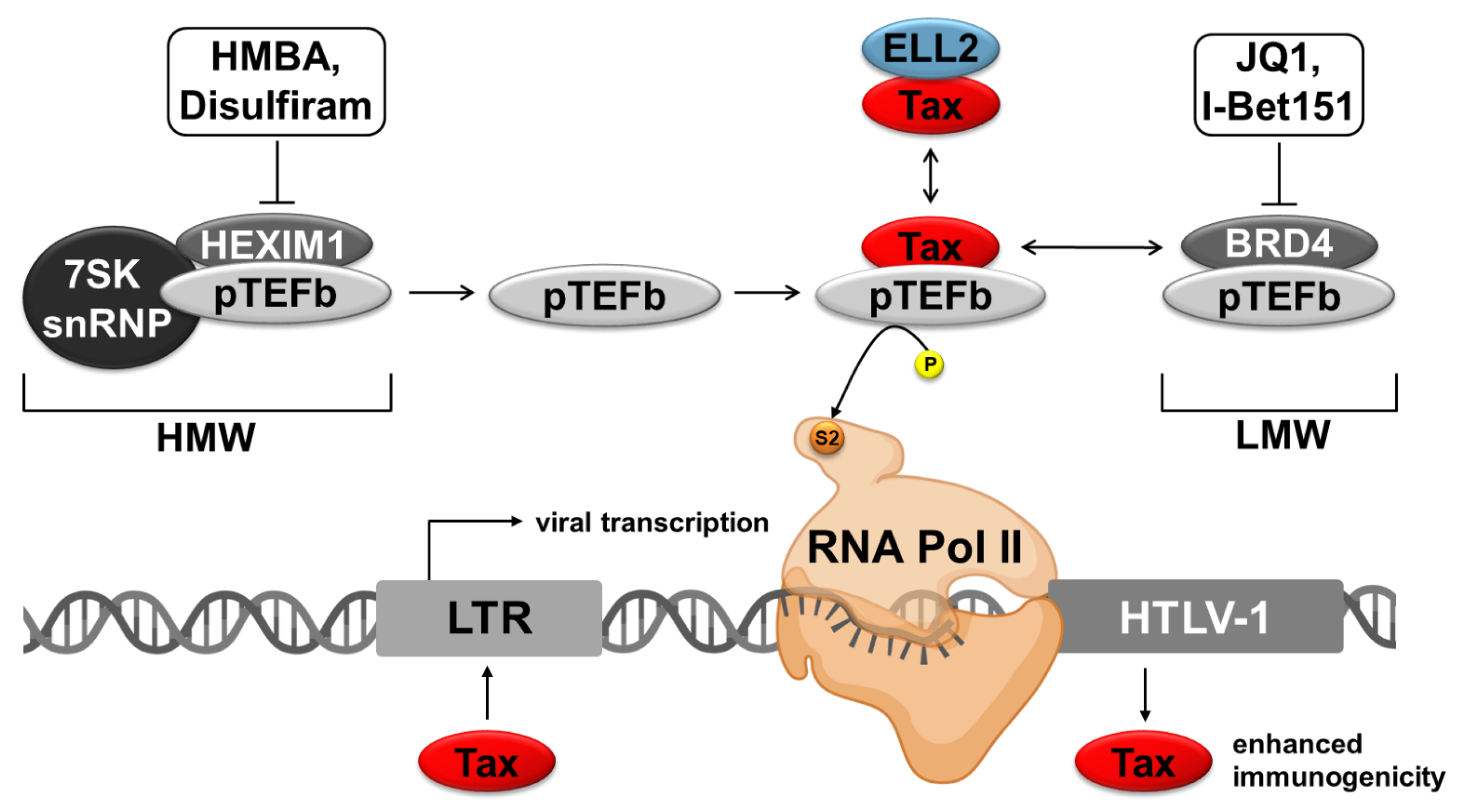
3. Agents for Targeting P-TEFb Complexes
3.1. Regulation of the RNA Polymerase II
3.2. Different P-TEFb Modulating Agents
3.2.1. Agents Interfering with the LMW-Complex
3.2.2. Agents Interfering with the HMW
4. Other Strategies to Reactivate HTLV-1 from Latency
4.1. Classification of Other Stimuli
4.2. Mitogens
4.3. Sirtuin Inhibitors
4.4. Extracellular Factors and Circumstances
5. Caveats of the Kick and Kill Approach and Open Questions
- The treatment of HTLV-1-infected patients with HDACi or related compounds does not specifically target virus-infected cells. Rather, other cells like CTLs themselves may also be affected by the treatment and may be functionally impaired. Thus, individual compounds have to be monitored more closely.
- Enhanced viral transcription may result in increased viral replication and worsening of the phenotype if the CTL response does not work properly. Since viral reactivation also poses the risk that a yet repressed but harmful HTLV-1-infected T-cell clone proliferates upon treatment, close monitoring of patients would be required.
- It is unclear whether the combination of antiretroviral therapy with different compounds used for the Kick and Kill strategy could improve clinical parameters comparable to the approaches used in HIV reactivation.
- The outcome of viral reactivation may differ depending on the viral integration sites in the human genome.
- Viral reservoirs are largely unknown for HTLV-1. Most studies performed thus far monitored the impact of LRAs on peripheral blood mononuclear cells. However, whether and how LRAs may impact viral gene expression in other cell types and tissues and how accessible these viral reservoirs are, is not understood.
- The impact of LRAs on HBZ expression and HBZ-specific CTLs remains to be elucidated in more detail.
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Poiesz, B.J.; Ruscetti, F.W.; Gazdar, A.F.; Bunn, P.A.; Minna, J.D.; Gallo, R.C. Detection and isolation of type C retrovirus particles from fresh and cultured lymphocytes of a patient with cutaneous T-cell lymphoma. Proc. Natl. Acad. Sci. USA 1980, 77, 7415–7419. [Google Scholar] [CrossRef]
- Yoshida, M.; Miyoshi, I.; Hinuma, Y. Isolation and characterization of retrovirus from cell lines of human adult T-cell leukemia and its implication in the disease. Proc. Natl. Acad. Sci. USA 1982, 79, 2031–2035. [Google Scholar] [CrossRef]
- Osame, M.; Matsumoto, M.; Usuku, K.; Izumo, S.; Ijichi, N.; Amitani, H.; Tara, M.; Igata, A. Chronic progressive myelopathy associated with elevated antibodies to human T-lymphotropic virus type I and adult T-cell leukemialike cells. Ann. Neurol. 1987, 21, 117–122. [Google Scholar] [CrossRef]
- Gessain, A.; Barin, F.; Vernant, J.C.; Gout, O.; Maurs, L.; Calender, A.; de Thé, G. Antibodies to human T-lymphotropic virus type-I in patients with tropical spastic paraparesis. Lancet 1985, 2, 407–410. [Google Scholar] [CrossRef]
- Martin, F.; Tagaya, Y.; Gallo, R. Time to eradicate HTLV-1: An open letter to WHO. Lancet 2018, 391, 1893–1894. [Google Scholar] [CrossRef]
- Gessain, A.; Cassar, O. Epidemiological Aspects and World Distribution of HTLV-1 Infection. Front. Microbiol. 2012, 3, 388. [Google Scholar] [CrossRef]
- Bangham, C.R.M. Human T Cell Leukemia Virus Type 1: Persistence and Pathogenesis. Annu. Rev. Immunol. 2018, 36, 43–71. [Google Scholar] [CrossRef]
- Tagaya, Y.; Matsuoka, M.; Gallo, R. 40 years of the human T-cell leukemia virus: Past, present, and future. F1000Research 2019, 8, 228. [Google Scholar] [CrossRef]
- Martin, F.; Taylor, G.P.; Jacobson, S. Inflammatory manifestations of HTLV-1 and their therapeutic options. Expert Rev. Clin. Immunol. 2014, 10, 1531–1546. [Google Scholar] [CrossRef] [PubMed]
- Kamoi, K.; Mochizuki, M. HTLV-1 uveitis. Front. Microbiol. 2012, 3, 270. [Google Scholar] [CrossRef]
- Mochizuki, M.; Watanabe, T.; Yamaguchi, K.; Yoshimura, K.; Nakashima, S.; Shirao, M.; Araki, S.; Takatsuki, K.; Mori, S.; Miyata, N. Uveitis associated with human T-cell lymphotropic virus type I. Am. J. Ophthalmol. 1992, 114, 123–129. [Google Scholar] [CrossRef]
- Iwanaga, M.; Watanabe, T.; Yamaguchi, K. Adult T-cell leukemia: A review of epidemiological evidence. Front. Microbiol. 2012, 3, 322. [Google Scholar] [CrossRef] [PubMed]
- Olindo, S.; Jeannin, S.; Saint-Vil, M.; Signate, A.; Edimonana-Kaptue, M.; Joux, J.; Merle, H.; Richard, P.; Granjeaud, S.; Cabre, P.; et al. Temporal trends in Human T-Lymphotropic virus 1 (HTLV-1) associated myelopathy/tropical spastic paraparesis (HAM/TSP) incidence in Martinique over 25 years (1986–2010). PLoS Negl. Trop. Dis. 2018, 12, e0006304. [Google Scholar] [CrossRef]
- Shimoyama, M. Diagnostic criteria and classification of clinical subtypes of adult T-cell leukaemia-lymphoma. A report from the Lymphoma Study Group (1984–1987). Br. J. Haematol. 1991, 79, 428–437. [Google Scholar] [CrossRef] [PubMed]
- Hermine, O.; Ramos, J.C.; Tobinai, K. A Review of New Findings in Adult T-cell Leukemia-Lymphoma: A Focus on Current and Emerging Treatment Strategies. Adv. Ther. 2018, 35, 135–152. [Google Scholar] [CrossRef] [PubMed]
- Cook, L.B.; Fuji, S.; Hermine, O.; Bazarbachi, A.; Ramos, J.C.; Ratner, L.; Horwitz, S.; Fields, P.; Tanase, A.; Bumbea, H.; et al. Revised Adult T-Cell Leukemia-Lymphoma International Consensus Meeting Report. J. Clin. Oncol. 2019, 37, 677–687. [Google Scholar] [CrossRef] [PubMed]
- Cook, L.B.; Phillips, A.A. How I treat adult T-cell leukemia/lymphoma. Blood 2021, 137, 459–470. [Google Scholar] [CrossRef]
- El Hajj, H.; Tsukasaki, K.; Cheminant, M.; Bazarbachi, A.; Watanabe, T.; Hermine, O. Novel Treatments of Adult T Cell Leukemia Lymphoma. Front. Microbiol. 2020, 11, 1062. [Google Scholar] [CrossRef]
- Sato, T.; Coler-Reilly, A.L.G.; Yagishita, N.; Araya, N.; Inoue, E.; Furuta, R.; Watanabe, T.; Uchimaru, K.; Matsuoka, M.; Matsumoto, N.; et al. Mogamulizumab (Anti-CCR4) in HTLV-1-Associated Myelopathy. N. Engl. J. Med. 2018, 378, 529–538. [Google Scholar] [CrossRef]
- Phillips, A.A.; Fields, P.A.; Hermine, O.; Ramos, J.C.; Beltran, B.E.; Pereira, J.; Wandroo, F.; Feldman, T.; Taylor, G.P.; Sawas, A.; et al. Mogamulizumab versus investigator’s choice of chemotherapy regimen in relapsed/refractory adult T-cell leukemia/lymphoma. Haematologica 2019, 104, 993–1003. [Google Scholar] [CrossRef]
- Willems, L.; Hasegawa, H.; Accolla, R.; Bangham, C.; Bazarbachi, A.; Bertazzoni, U.; Carneiro-Proietti, A.B.; Cheng, H.; Chieco-Bianchi, L.; Ciminale, V.; et al. Reducing the global burden of HTLV-1 infection: An agenda for research and action. Antivir. Res. 2017, 137, 41–48. [Google Scholar] [CrossRef]
- Kirk, P.D.; Huvet, M.; Melamed, A.; Maertens, G.N.; Bangham, C.R. Retroviruses integrate into a shared, non-palindromic DNA motif. Nat. Microbiol. 2016, 2, 16212. [Google Scholar] [CrossRef]
- Currer, R.; Van Duyne, R.; Jaworski, E.; Guendel, I.; Sampey, G.; Das, R.; Narayanan, A.; Kashanchi, F. HTLV tax: A fascinating multifunctional co-regulator of viral and cellular pathways. Front. Microbiol. 2012, 3, 406. [Google Scholar] [CrossRef] [PubMed]
- Bangham, C.R.M.; Miura, M.; Kulkarni, A.; Matsuoka, M. Regulation of Latency in the Human T Cell Leukemia Virus, HTLV-1. Annu. Rev. Virol. 2019, 6, 365–385. [Google Scholar] [CrossRef]
- Laverdure, S.; Polakowski, N.; Hoang, K.; Lemasson, I. Permissive Sense and Antisense Transcription from the 5’ and 3’ Long Terminal Repeats of Human T-Cell Leukemia Virus Type 1. J. Virol. 2016, 90, 3600–3610. [Google Scholar] [CrossRef]
- Nyborg, J.K.; Egan, D.; Sharma, N. The HTLV-1 Tax protein: Revealing mechanisms of transcriptional activation through histone acetylation and nucleosome disassembly. Biochim. Biophys. Acta 2010, 1799, 266–274. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.J.; Giam, C.Z. Human T-cell lymphotropic virus type I (HTLV-I) transcriptional activator, Tax, enhances CREB binding to HTLV-I 21-base-pair repeats by protein-protein interaction. Proc. Natl. Acad. Sci. USA 1992, 89, 7070–7074. [Google Scholar] [CrossRef]
- Adya, N.; Giam, C.Z. Distinct regions in human T-cell lymphotropic virus type I tax mediate interactions with activator protein CREB and basal transcription factors. J. Virol. 1995, 69, 1834–1841. [Google Scholar] [CrossRef]
- Cho, W.K.; Jang, M.K.; Huang, K.; Pise-Masison, C.A.; Brady, J.N. Human T-lymphotropic virus type 1 Tax protein complexes with P-TEFb and competes for Brd4 and 7SK snRNP/HEXIM1 binding. J. Virol. 2010, 84, 12801–12809. [Google Scholar] [CrossRef]
- Zaborowska, J.; Isa, N.F.; Murphy, S. P-TEFb goes viral. Inside Cell 2016, 1, 106–116. [Google Scholar] [CrossRef]
- Melamed, A.; Laydon, D.J.; Al Khatib, H.; Rowan, A.G.; Taylor, G.P.; Bangham, C.R. HTLV-1 drives vigorous clonal expansion of infected CD8+ T cells in natural infection. Retrovirology 2015, 12, 91. [Google Scholar] [CrossRef] [PubMed]
- De Castro-Amarante, M.F.; Pise-Masison, C.A.; McKinnon, K.; Washington Parks, R.; Galli, V.; Omsland, M.; Andresen, V.; Massoud, R.; Brunetto, G.; Caruso, B.; et al. Human T Cell Leukemia Virus Type 1 Infection of the Three Monocyte Subsets Contributes to Viral Burden in Humans. J. Virol. 2015, 90, 2195–2207. [Google Scholar] [CrossRef]
- Macatonia, S.E.; Cruickshank, J.K.; Rudge, P.; Knight, S.C. Dendritic cells from patients with tropical spastic paraparesis are infected with HTLV-1 and stimulate autologous lymphocyte proliferation. AIDS Res. Hum. Retrovir. 1992, 8, 1699–1706. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, H.; Nishikata, I.; Shiraga, T.; Akamatsu, E.; Fukami, T.; Hidaka, T.; Kubuki, Y.; Okayama, A.; Hamada, K.; Okabe, H.; et al. Overexpression of a cell adhesion molecule, TSLC1, as a possible molecular marker for acute-type adult T-cell leukemia. Blood 2005, 105, 1204–1213. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, A.; Matsuoka, M. HTLV-1 Alters T Cells for Viral Persistence and Transmission. Front. Microbiol. 2018, 9, 461. [Google Scholar] [CrossRef] [PubMed]
- Manivannan, K.; Rowan, A.G.; Tanaka, Y.; Taylor, G.P.; Bangham, C.R. CADM1/TSLC1 Identifies HTLV-1-Infected Cells and Determines Their Susceptibility to CTL-Mediated Lysis. PLoS Pathog. 2016, 12, e1005560. [Google Scholar] [CrossRef] [PubMed]
- Furuta, R.; Yasunaga, J.I.; Miura, M.; Sugata, K.; Saito, A.; Akari, H.; Ueno, T.; Takenouchi, N.; Fujisawa, J.I.; Koh, K.R.; et al. Human T-cell leukemia virus type 1 infects multiple lineage hematopoietic cells in vivo. PLoS Pathog. 2017, 13, e1006722. [Google Scholar] [CrossRef]
- Lewin, S.R.; Rasmussen, T.A. Kick and kill for HIV latency. Lancet 2020, 395, 844–846. [Google Scholar] [CrossRef]
- Matsuoka, M.; Jeang, K.T. Human T-cell leukaemia virus type 1 (HTLV-1) infectivity and cellular transformation. Nat. Rev. Cancer 2007, 7, 270–280. [Google Scholar] [CrossRef]
- Goon, P.K.; Igakura, T.; Hanon, E.; Mosley, A.J.; Barfield, A.; Barnard, A.L.; Kaftantzi, L.; Tanaka, Y.; Taylor, G.P.; Weber, J.N.; et al. Human T cell lymphotropic virus type I (HTLV-I)-specific CD4+ T cells: Immunodominance hierarchy and preferential infection with HTLV-I. J. Immunol. 2004, 172, 1735–1743. [Google Scholar] [CrossRef]
- Kannagi, M.; Harada, S.; Maruyama, I.; Inoko, H.; Igarashi, H.; Kuwashima, G.; Sato, S.; Morita, M.; Kidokoro, M.; Sugimoto, M.; et al. Predominant recognition of human T cell leukemia virus type I (HTLV-I) pX gene products by human CD8+ cytotoxic T cells directed against HTLV-l-infected cells. Int. Immunol. 1991, 3, 761–767. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, S.; Shidat, H.; McFarlin, D.E.; Faucit, A.S.; Koenig, S. Circulating CD8+ cytotoxic T lymphocytes specific for HTL V-1 pX in patients with HTLV-1 associated neurological disease. Nature 1990, 215, 245–248. [Google Scholar] [CrossRef] [PubMed]
- Goon, P.K.; Biancardi, A.; Fast, N.; Igakura, T.; Hanon, E.; Mosley, A.J.; Asquith, B.; Gould, K.G.; Marshall, S.; Taylor, G.P.; et al. Human T cell lymphotropic virus (HTLV) type-1-specific CD8+ T cells: Frequency and immunodominance hierarchy. J. Infect. Dis. 2004, 189, 2294–2298. [Google Scholar] [CrossRef] [PubMed]
- Bangham, C.R.M.; Meekings, K.; Toulza, F.; Nejmeddine, M.; Majorovits, E.; Asquith, B.; Taylor, G.P. The immune control of HTLV-1 infection: Selection forces and dynamics. Front. Biosci. 2009, 14, 2889–2903. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, A.; Bangham, C.R.M. HTLV-1: Regulating the Balance Between Proviral Latency and Reactivation. Front. Microbiol. 2018, 9, 449. [Google Scholar] [CrossRef] [PubMed]
- Furukawa, Y.; Kubota, R.; Tara, M.; Izumo, S.; Osame, M. Existence of escape mutant in HTLV-I tax during the development of adult T-cell leukemia. Blood 2001, 97, 987–993. [Google Scholar] [CrossRef]
- Tsukasa Koiwa, A.H.-U.; Ishida, T.; Okayama, A.; Yamaguchi, K.; Kamihira, S.; Watanabe, T. 5 -Long Terminal Repeat-Selective CpG Methylation of Latent HumanT-Cell Leukemia Virus Type 1 Provirus In Vitro and In Vivo. J. Virol. 2002, 76, 9389–9397. [Google Scholar] [CrossRef]
- Takeda, S.; Maeda, M.; Morikawa, S.; Taniguchi, Y.; Yasunaga, J.; Nosaka, K.; Tanaka, Y.; Matsuoka, M. Genetic and epigenetic inactivation of tax gene in adult T-cell leukemia cells. Int. J. Cancer 2004, 109, 559–567. [Google Scholar] [CrossRef]
- Shah, U.A.; Chung, E.Y.; Giricz, O.; Pradhan, K.; Kataoka, K.; Gordon-Mitchell, S.; Bhagat, T.D.; Mai, Y.; Wei, Y.Q.; Ishida, E.; et al. North American ATLL has a distinct mutational and transcriptional profile and responds to epigenetic therapies. Blood 2018, 132, 1507–1518. [Google Scholar] [CrossRef]
- Harrod, R. Silencers of HTLV-1 and HTLV-2: The pX-encoded latency-maintenance factors. Retrovirology 2019, 16, 25. [Google Scholar] [CrossRef]
- Kattan, T.; MacNamara, A.; Rowan, A.G.; Nose, H.; Mosley, A.J.; Tanaka, Y.; Taylor, G.P.; Asquith, B.; Bangham, C.R. The avidity and lytic efficiency of the CTL response to HTLV-1. J. Immunol. 2009, 182, 5723–5729. [Google Scholar] [CrossRef]
- Mosley, A.J.; Asquith, B.; Bangham, C.R. Cell-mediated immune response to human T-lymphotropic virus type I. Viral Immunol. 2005, 18, 293–305. [Google Scholar] [CrossRef] [PubMed]
- Cook, L.B.; Elemans, M.; Rowan, A.G.; Asquith, B. HTLV-1: Persistence and pathogenesis. Virology 2013, 435, 131–140. [Google Scholar] [CrossRef] [PubMed]
- Macnamara, A.; Rowan, A.; Hilburn, S.; Kadolsky, U.; Fujiwara, H.; Suemori, K.; Yasukawa, M.; Taylor, G.; Bangham, C.R.; Asquith, B. HLA class I binding of HBZ determines outcome in HTLV-1 infection. PLoS Pathog. 2010, 6, e1001117. [Google Scholar] [CrossRef] [PubMed]
- Rende, F.; Cavallari, I.; Corradin, A.; Silic-Benussi, M.; Toulza, F.; Toffolo, G.M.; Tanaka, Y.; Jacobson, S.; Taylor, G.P.; D’Agostino, D.M.; et al. Kinetics and intracellular compartmentalization of HTLV-1 gene expression: Nuclear retention of HBZ mRNAs. Blood 2011, 117, 4855–4859. [Google Scholar] [CrossRef]
- Ma, G.; Yasunaga, J.I.; Shimura, K.; Takemoto, K.; Watanabe, M.; Amano, M.; Nakata, H.; Liu, B.; Zuo, X.; Matsuoka, M. Human retroviral antisense mRNAs are retained in the nuclei of infected cells for viral persistence. Proc. Natl. Acad. Sci. USA 2021, 118, e2014783118. [Google Scholar] [CrossRef] [PubMed]
- Lezin, A.; Gillet, N.; Olindo, S.; Signate, A.; Grandvaux, N.; Verlaeten, O.; Belrose, G.; de Carvalho Bittencourt, M.; Hiscott, J.; Asquith, B.; et al. Histone deacetylase mediated transcriptional activation reduces proviral loads in HTLV-1 associated myelopathy/tropical spastic paraparesis patients. Blood 2007, 110, 3722–3728. [Google Scholar] [CrossRef]
- Margolis, D.M.; Archin, N.M.; Cohen, M.S.; Eron, J.J.; Ferrari, G.; Garcia, J.V.; Gay, C.L.; Goonetilleke, N.; Joseph, S.B.; Swanstrom, R.; et al. Curing HIV: Seeking to Target and Clear Persistent Infection. Cell 2020, 181, 189–206. [Google Scholar] [CrossRef]
- Barre-Sinoussi, F.; Chermann, J.C.; Rey, F.; Nugeyre, M.T.; Chamaret, S.; Gruest, J.; Dauguet, C.; Axler-Blin, C.; Vezinet-Brun, F.; Rouzioux, C.; et al. Isolation of a T-lymphotropic retrovirus from a patient at risk for acquired immune deficiency syndrome (AIDS). Science 1983, 220, 868–871. [Google Scholar] [CrossRef]
- Margolis, D.M.; Garcia, J.V.; Hazuda, D.J.; Haynes, B.F. Latency reversal and viral clearance to cure HIV-1. Science 2016, 353, aaf6517. [Google Scholar] [CrossRef]
- Deeks, S.G. HIV: Shock and kill. Nature 2012, 487, 439–440. [Google Scholar] [CrossRef] [PubMed]
- Thorlund, K.; Horwitz, M.S.; Fife, B.T.; Lester, R.; Cameron, D.W. Landscape review of current HIV ’kick and kill’ cure research—Some kicking, not enough killing. BMC Infect. Dis. 2017, 17, 595. [Google Scholar] [CrossRef]
- Sengupta, S.; Siliciano, R.F. Targeting the Latent Reservoir for HIV-1. Immunity 2018, 48, 872–895. [Google Scholar] [CrossRef] [PubMed]
- Archin, N.M.; Liberty, A.L.; Kashuba, A.D.; Choudhary, S.K.; Kuruc, J.D.; Crooks, A.M.; Parker, D.C.; Anderson, E.M.; Kearney, M.F.; Strain, M.C.; et al. Administration of vorinostat disrupts HIV-1 latency in patients on antiretroviral therapy. Nature 2012, 487, 482–485. [Google Scholar] [CrossRef] [PubMed]
- Jones, R.B.; Mueller, S.; O’Connor, R.; Rimpel, K.; Sloan, D.D.; Karel, D.; Wong, H.C.; Jeng, E.K.; Thomas, A.S.; Whitney, J.B.; et al. A Subset of Latency-Reversing Agents Expose HIV-Infected Resting CD4+ T-Cells to Recognition by Cytotoxic T-Lymphocytes. PLoS Pathog. 2016, 12, e1005545. [Google Scholar] [CrossRef] [PubMed]
- Lehrman, G.; Hogue, I.B.; Palmer, S.; Jennings, C.; Spina, C.A.; Wiegand, A.; Landay, A.L.; Coombs, R.W.; Richman, D.D.; Mellors, J.W.; et al. Depletion of latent HIV-1 infection in vivo: A proof-of-concept study. Lancet 2005, 366, 549–555. [Google Scholar] [CrossRef]
- Damaskos, C.; Garmpis, N.; Karatzas, T.; Nikolidakis, L.; Kostakis, I.D.; Garmpi, A.; Karamaroudis, S.; Boutsikos, G.; Damaskou, Z.; Kostakis, A.; et al. Histone Deacetylase (HDAC) Inhibitors: Current Evidence for Therapeutic Activities in Pancreatic Cancer. Anticancer Res. 2015, 35, 3129–3135. [Google Scholar]
- Monneret, C. Histone deacetylase inhibitors for epigenetic therapy of cancer. Anticancer Drugs 2007, 18, 363–370. [Google Scholar] [CrossRef]
- Lemasson, I.; Polakowski, N.J.; Laybourn, P.J.; Nyborg, J.K. Transcription regulatory complexes bind the human T-cell leukemia virus 5′ and 3′ long terminal repeats to control gene expression. Mol. Cell. Biol. 2004, 24, 6117–6126. [Google Scholar] [CrossRef] [PubMed]
- Lemasson, I.; Polakowski, N.J.; Laybourn, P.J.; Nyborg, J.K. Transcription factor binding and histone modifications on the integrated proviral promoter in human T-cell leukemia virus-I-infected T-cells. J. Biol. Chem. 2002, 277, 49459–49465. [Google Scholar] [CrossRef]
- Contreras, X.; Barboric, M.; Lenasi, T.; Peterlin, B.M. HMBA releases P-TEFb from HEXIM1 and 7SK snRNA via PI3K/Akt and activates HIV transcription. PLoS Pathog. 2007, 3, 1459–1469. [Google Scholar] [CrossRef] [PubMed]
- Boehm, D.; Calvanese, V.; Dar, R.D.; Xing, S.; Schroeder, S.; Martins, L.; Aull, K.; Li, P.C.; Planelles, V.; Bradner, J.E.; et al. BET bromodomain-targeting compounds reactivate HIV from latency via a Tat-independent mechanism. Cell Cycle 2013, 12, 452–462. [Google Scholar] [CrossRef] [PubMed]
- Luger, K.; Richmond, T.J. The histone tails of the nucleosome. Curr. Opin. Genet. Dev. 1998, 8, 140–146. [Google Scholar] [CrossRef]
- Luger, K.; Mä Der, A.W.; Richmond, R.K.; Sargent, D.F.; Richmond, T.J. Crystal structure of the nucleosome core particle at 2.8 A° resolution. Nature 1997, 389, 251–260. [Google Scholar] [CrossRef] [PubMed]
- Strahl, B.D.; Allis, C.D. The language of covalent histone modifications. Nature 2000, 403, 41–45. [Google Scholar] [CrossRef]
- Jenuwein, T.; Allis, C.D. Translating the histone code. Science 2001, 293, 1074–1080. [Google Scholar] [CrossRef]
- Thiagalingam, S.; Cheng, K.H.; Lee, H.J.; Mineva, N.; Thiagalingam, A.; Ponte, J.F. Histone deacetylases: Unique players in shaping the epigenetic histone code. Ann. N. Y. Acad. Sci. 2003, 983, 84–100. [Google Scholar] [CrossRef] [PubMed]
- Filippakopoulos, P.; Qi, J.; Picaud, S.; Shen, Y.; Smith, W.B.; Fedorov, O.; Morse, E.M.; Keates, T.; Hickman, T.T.; Felletar, I.; et al. Selective inhibition of BET bromodomains. Nature 2010, 468, 1067–1073. [Google Scholar] [CrossRef]
- Dey, A.; Nishiyama, A.; Karpova, T.; McNally, J.; Ozato, K. Brd4 marks select genes on mitotic chromatin and directs postmitotic transcription. Mol. Biol. Cell 2009, 20, 4899–4909. [Google Scholar] [CrossRef]
- Allfrey, V.G. Structural modifications of histones and their possible role in the regulation of ribonucleic acid synthesis. Proc. Can. Cancer Conf. 1966, 6, 313–335. [Google Scholar]
- Seto, E.; Yoshida, M. Erasers of histone acetylation: The histone deacetylase enzymes. Cold Spring Harb. Perspect. Biol. 2014, 6, a018713. [Google Scholar] [CrossRef]
- Miller, T.A.; Witter, D.J.; Belvedere, S. Histone deacetylase inhibitors. J. Med. Chem. 2003, 46, 5097–5116. [Google Scholar] [CrossRef] [PubMed]
- De Ruijter, A.J.; van Gennip, A.H.; Caron, H.N.; Kemp, S.; van Kuilenburg, A.B. Histone deacetylases (HDACs): Characterization of the classical HDAC family. Biochem. J. 2003, 370, 737–749. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Seto, E. HDACs and HDAC Inhibitors in Cancer Development and Therapy. Cold Spring Harb. Perspect. Med. 2016, 6, a026831. [Google Scholar] [CrossRef] [PubMed]
- Minucci, S.; Pelicci, P.G. Histone deacetylase inhibitors and the promise of epigenetic (and more) treatments for cancer. Nat. Rev. Cancer 2006, 6, 38–51. [Google Scholar] [CrossRef] [PubMed]
- Ego, T.; Ariumi, Y.; Shimotohno, K. The interaction of HTLV-1 Tax with HDAC1 negatively regulates the viral gene expression. Oncogene 2002, 21, 7241–7246. [Google Scholar] [CrossRef]
- Lu, H.; Pise-Masison, C.A.; Linton, R.; Park, H.U.; Schiltz, R.L.; Sartorelli, V.; Brady, J.N. Tax relieves transcriptional repression by promoting histone deacetylase 1 release from the human T-cell leukemia virus type 1 long terminal repeat. J. Virol. 2004, 78, 6735–6743. [Google Scholar] [CrossRef]
- McClure, J.J.; Li, X.; Chou, C.J. Advances and Challenges of HDAC Inhibitors in Cancer Therapeutics. Adv. Cancer Res. 2018, 138, 183–211. [Google Scholar]
- Jones, P.A.; Baylin, S.B. The fundamental role of epigenetic events in cancer. Nat. Rev. Genet. 2002, 3, 415–428. [Google Scholar] [CrossRef]
- Timp, W.; Feinberg, A.P. Cancer as a dysregulated epigenome allowing cellular growth advantage at the expense of the host. Nat. Rev. Cancer 2013, 13, 497–510. [Google Scholar] [CrossRef]
- Benedetti, R.; Conte, M.; Altucci, L. Targeting Histone Deacetylases in Diseases: Where Are We? Antioxid. Redox Signal. 2015, 23, 99–126. [Google Scholar] [CrossRef] [PubMed]
- Hull, E.E.; Montgomery, M.R.; Leyva, K.J. HDAC Inhibitors as Epigenetic Regulators of the Immune System: Impacts on Cancer Therapy and Inflammatory Diseases. Biomed. Res. Int. 2016, 2016, 8797206. [Google Scholar] [CrossRef] [PubMed]
- Chueh, A.C.; Tse, J.W.; Togel, L.; Mariadason, J.M. Mechanisms of Histone Deacetylase Inhibitor-Regulated Gene Expression in Cancer Cells. Antioxid. Redox Signal. 2015, 23, 66–84. [Google Scholar] [CrossRef] [PubMed]
- Drummond, D.C.; Noble, C.O.; Kirpotin, D.B.; Guo, Z.; Scott, G.K.; Benz, C.C. Clinical development of histone deacetylase inhibitors as anticancer agents. Annu. Rev. Pharmacol. Toxicol. 2005, 45, 495–528. [Google Scholar] [CrossRef]
- Mottamal, M.; Zheng, S.; Huang, T.L.; Wang, G. Histone deacetylase inhibitors in clinical studies as templates for new anticancer agents. Molecules 2015, 20, 3898–3941. [Google Scholar] [CrossRef] [PubMed]
- Cameron, E.E.; Bachman, K.E.; Myohanen, S.; Herman, J.G.; Baylin, S.B. Synergy of demethylation and histone deacetylase inhibition in the re-expression of genes silenced in cancer. Nat. Genet. 1999, 21, 103–107. [Google Scholar] [CrossRef] [PubMed]
- Cusack, M.; King, H.W.; Spingardi, P.; Kessler, B.M.; Klose, R.J.; Kriaucionis, S. Distinct Contribution of DNA Methylation and Histone Acetylation to the Genomic Occupancy of Transcription Factors. Genome Res. 2020, 30, 1393–1406. [Google Scholar] [CrossRef]
- Chen, R.; Yik, J.H.; Lew, Q.J.; Chao, S.H. Brd4 and HEXIM1: Multiple roles in P-TEFb regulation and cancer. Biomed. Res. Int. 2014, 2014, 232870. [Google Scholar] [CrossRef]
- Elliott, J.H.; Wightman, F.; Solomon, A.; Ghneim, K.; Ahlers, J.; Cameron, M.J.; Smith, M.Z.; Spelman, T.; McMahon, J.; Velayudham, P.; et al. Activation of HIV transcription with short-course vorinostat in HIV-infected patients on suppressive antiretroviral therapy. PLoS Pathog. 2014, 10, e1004473. [Google Scholar] [CrossRef]
- Olindo, S.; Belrose, G.; Gillet, N.; Rodriguez, S.; Boxus, M.; Verlaeten, O.; Asquith, B.; Bangham, C.; Signate, A.; Smadja, D.; et al. Safety of long-term treatment of HAM/TSP patients with valproic acid. Blood 2011, 118, 6306–6309. [Google Scholar] [CrossRef]
- Afonso, P.V.; Mekaouche, M.; Mortreux, F.; Toulza, F.; Moriceau, A.; Wattel, E.; Gessain, A.; Bangham, C.R.; Dubreuil, G.; Plumelle, Y.; et al. Highly active antiretroviral treatment against STLV-1 infection combining reverse transcriptase and HDAC inhibitors. Blood 2010, 116, 3802–3808. [Google Scholar] [CrossRef] [PubMed]
- Ning, Z.Q.; Li, Z.B.; Newman, M.J.; Shan, S.; Wang, X.H.; Pan, D.S.; Zhang, J.; Dong, M.; Du, X.; Lu, X.P. Chidamide (CS055/HBI-8000): A new histone deacetylase inhibitor of the benzamide class with antitumor activity and the ability to enhance immune cell-mediated tumor cell cytotoxicity. Cancer Chemother. Pharmacol. 2012, 69, 901–909. [Google Scholar] [CrossRef] [PubMed]
- Noureen, N.; Rashid, H.; Kalsoom, S. Identification of type-specific anticancer histone deacetylase inhibitors: Road to success. Cancer Chemother. Pharmacol. 2010, 66, 625–633. [Google Scholar] [CrossRef] [PubMed]
- Mwakwari, S.C.; Patil, V.; Guerrant, W.; Oyelere, A.K. Macrocyclic histone deacetylase inhibitors. Curr. Top. Med. Chem. 2010, 10, 1423–1440. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Zhang, J.; Jiang, Q.; Zhang, L.; Song, W. Zinc binding groups for histone deacetylase inhibitors. J. Enzym. Inhib. Med. Chem. 2018, 33, 714–721. [Google Scholar] [CrossRef]
- Marks, P.; Rifkind, R.A.; Richon, V.M.; Breslow, R.; Miller, T.; Kelly, W.K. Histone deacetylases and cancer: Causes and therapies. Nat. Rev. Cancer 2001, 1, 194–202. [Google Scholar] [CrossRef]
- Freese, K.; Seitz, T.; Dietrich, P.; Lee, S.M.L.; Thasler, W.E.; Bosserhoff, A.; Hellerbrand, C. Histone Deacetylase Expressions in Hepatocellular Carcinoma and Functional Effects of Histone Deacetylase Inhibitors on Liver Cancer Cells In Vitro. Cancers 2019, 11, 1587. [Google Scholar] [CrossRef]
- Matalon, S.; Rasmussen, T.A.; Dinarello, C.A. Histone deacetylase inhibitors for purging HIV-1 from the latent reservoir. Mol. Med. 2011, 17, 466–472. [Google Scholar] [CrossRef]
- Merezak, C.; Reichert, M.; Van Lint, C.; Kerkhofs, P.; Portetelle, D.; Willems, L.; Kettmann, R. Inhibition of histone deacetylases induces bovine leukemia virus expression in vitro and in vivo. J. Virol. 2002, 76, 5034–5042. [Google Scholar] [CrossRef]
- Mahgoub, M.; Yasunaga, J.I.; Iwami, S.; Nakaoka, S.; Koizumi, Y.; Shimura, K.; Matsuoka, M. Sporadic on/off switching of HTLV-1 Tax expression is crucial to maintain the whole population of virus-induced leukemic cells. Proc. Natl. Acad. Sci. USA 2018, 115, E1269–E1278. [Google Scholar] [CrossRef]
- Rasmussen, T.A.; Schmeltz Sogaard, O.; Brinkmann, C.; Wightman, F.; Lewin, S.R.; Melchjorsen, J.; Dinarello, C.; Ostergaard, L.; Tolstrup, M. Comparison of HDAC inhibitors in clinical development: Effect on HIV production in latently infected cells and T-cell activation. Hum. Vaccin. Immunother. 2013, 9, 993–1001. [Google Scholar] [CrossRef]
- Nishioka, C.; Ikezoe, T.; Yang, J.; Komatsu, N.; Bandobashi, K.; Taniguchi, A.; Kuwayama, Y.; Togitani, K.; Koeffler, H.P.; Taguchi, H. Histone deacetylase inhibitors induce growth arrest and apoptosis of HTLV-1-infected T-cells via blockade of signaling by nuclear factor kappaB. Leuk. Res. 2008, 32, 287–296. [Google Scholar] [CrossRef]
- Wightman, F.; Lu, H.K.; Solomon, A.E.; Saleh, S.; Harman, A.N.; Cunningham, A.L.; Gray, L.; Churchill, M.; Cameron, P.U.; Dear, A.E.; et al. Entinostat is a histone deacetylase inhibitor selective for class 1 histone deacetylases and activates HIV production from latently infected primary T cells. AIDS 2013, 27, 2853–2862. [Google Scholar] [CrossRef]
- Li, J.H.; Ma, J.; Kang, W.; Wang, C.F.; Bai, F.; Zhao, K.; Yao, N.; Liu, Q.; Dang, B.L.; Wang, B.W.; et al. The histone deacetylase inhibitor chidamide induces intermittent viraemia in HIV-infected patients on suppressive antiretroviral therapy. HIV Med. 2020, 21, 747–757. [Google Scholar] [CrossRef]
- Yang, W.; Sun, Z.; Hua, C.; Wang, Q.; Xu, W.; Deng, Q.; Pan, Y.; Lu, L.; Jiang, S. Chidamide, a histone deacetylase inhibitor-based anticancer drug, effectively reactivates latent HIV-1 provirus. Microbes Infect. 2018, 20, 626–634. [Google Scholar] [CrossRef]
- Davie, J.R. Inhibition of histone deacetylase activity by butyrate. J. Nutr. 2003, 133, 2485S–2493S. [Google Scholar] [CrossRef]
- Lin, H.C.; Dezzutti, C.S.; Lal, R.B.; Rabson, A.B. Activation of human T-cell leukemia virus type 1 tax gene expression in chronically infected T cells. J. Virol. 1998, 72, 6264–6270. [Google Scholar] [CrossRef]
- Bilen, M.A.; Fu, S.; Falchook, G.S.; Ng, C.S.; Wheler, J.J.; Abdelrahim, M.; Erguvan-Dogan, B.; Hong, D.S.; Tsimberidou, A.M.; Kurzrock, R.; et al. Phase I trial of valproic acid and lenalidomide in patients with advanced cancer. Cancer Chemother. Pharmacol. 2015, 75, 869–874. [Google Scholar] [CrossRef]
- Achachi, A.; Florins, A.; Gillet, N.; Debacq, C.; Urbain, P.; Foutsop, G.M.; Vandermeers, F.; Jasik, A.; Reichert, M.; Kerkhofs, P.; et al. Valproate activates bovine leukemia virus gene expression, triggers apoptosis, and induces leukemia/lymphoma regression in vivo. Proc. Natl. Acad. Sci. USA 2005, 102, 10309–10314. [Google Scholar] [CrossRef]
- Belrose, G.; Gross, A.; Olindo, S.; Lezin, A.; Dueymes, M.; Komla-Soukha, I.; Smadja, D.; Tanaka, Y.; Willems, L.; Mesnard, J.M.; et al. Effects of valproate on Tax and HBZ expression in HTLV-1 and HAM/TSP T lymphocytes. Blood 2011, 118, 2483–2491. [Google Scholar] [CrossRef]
- Chen, J.; Zhang, M.; Ju, W.; Waldmann, T.A. Effective treatment of a murine model of adult T-cell leukemia using depsipeptide and its combination with unmodified daclizumab directed toward CD25. Blood 2009, 113, 1287–1293. [Google Scholar] [CrossRef] [PubMed]
- Sogaard, O.S.; Graversen, M.E.; Leth, S.; Olesen, R.; Brinkmann, C.R.; Nissen, S.K.; Kjaer, A.S.; Schleimann, M.H.; Denton, P.W.; Hey-Cunningham, W.J.; et al. The Depsipeptide Romidepsin Reverses HIV-1 Latency In Vivo. PLoS Pathog. 2015, 11, e1005142. [Google Scholar] [CrossRef]
- Yoshida, M.; Kijima, M.; Akita, M.; Beppu, T. Potent and specific inhibition of mammalian histone deacetylase both in vivo and in vitro by trichostatin A. J. Biol. Chem. 1990, 265, 17174–17179. [Google Scholar] [CrossRef]
- Arnold, J.; Zimmerman, B.; Li, M.; Lairmore, M.D.; Green, P.L. Human T-cell leukemia virus type-1 antisense-encoded gene, Hbz, promotes T-lymphocyte proliferation. Blood 2008, 112, 3788–3797. [Google Scholar] [CrossRef] [PubMed]
- Florins, A.; de Brogniez, A.; Elemans, M.; Bouzar, A.B.; Francois, C.; Reichert, M.; Asquith, B.; Willems, L. Viral expression directs the fate of B cells in bovine leukemia virus-infected sheep. J. Virol. 2012, 86, 621–624. [Google Scholar] [CrossRef] [PubMed]
- Siddiqi, T.; Frankel, P.; Beumer, J.H.; Kiesel, B.F.; Christner, S.; Ruel, C.; Song, J.Y.; Chen, R.; Kelly, K.R.; Ailawadhi, S.; et al. Phase 1 study of the Aurora kinase A inhibitor alisertib (MLN8237) combined with the histone deacetylase inhibitor vorinostat in lymphoid malignancies. Leuk. Lymphoma 2020, 61, 309–317. [Google Scholar] [CrossRef]
- Olsen, E.A.; Kim, Y.H.; Kuzel, T.M.; Pacheco, T.R.; Foss, F.M.; Parker, S.; Frankel, S.R.; Chen, C.; Ricker, J.L.; Arduino, J.M.; et al. Phase IIb multicenter trial of vorinostat in patients with persistent, progressive, or treatment refractory cutaneous T-cell lymphoma. J. Clin. Oncol. 2007, 25, 3109–3115. [Google Scholar] [CrossRef]
- Mann, B.S.; Johnson, J.R.; Cohen, M.H.; Justice, R.; Pazdur, R. FDA approval summary: Vorinostat for treatment of advanced primary cutaneous T-cell lymphoma. Oncologist 2007, 12, 1247–1252. [Google Scholar] [CrossRef]
- Spina, C.A.; Anderson, J.; Archin, N.M.; Bosque, A.; Chan, J.; Famiglietti, M.; Greene, W.C.; Kashuba, A.; Lewin, S.R.; Margolis, D.M.; et al. An in-depth comparison of latent HIV-1 reactivation in multiple cell model systems and resting CD4+ T cells from aviremic patients. PLoS Pathog. 2013, 9, e1003834. [Google Scholar] [CrossRef]
- Archin, N.M.; Kirchherr, J.L.; Sung, J.A.; Clutton, G.; Sholtis, K.; Xu, Y.; Allard, B.; Stuelke, E.; Kashuba, A.D.; Kuruc, J.D.; et al. Interval dosing with the HDAC inhibitor vorinostat effectively reverses HIV latency. J. Clin. Investig. 2017, 127, 3126–3135. [Google Scholar] [CrossRef]
- Blazkova, J.; Chun, T.W.; Belay, B.W.; Murray, D.; Justement, J.S.; Funk, E.K.; Nelson, A.; Hallahan, C.W.; Moir, S.; Wender, P.A.; et al. Effect of histone deacetylase inhibitors on HIV production in latently infected, resting CD4+ T cells from infected individuals receiving effective antiretroviral therapy. J. Infect. Dis. 2012, 206, 765–769. [Google Scholar] [CrossRef] [PubMed]
- Mohammadi, P.; di Iulio, J.; Munoz, M.; Martinez, R.; Bartha, I.; Cavassini, M.; Thorball, C.; Fellay, J.; Beerenwinkel, N.; Ciuffi, A.; et al. Dynamics of HIV latency and reactivation in a primary CD4+ T cell model. PLoS Pathog. 2014, 10, e1004156. [Google Scholar] [CrossRef]
- Chen, I.C.; Sethy, B.; Liou, J.P. Recent Update of HDAC Inhibitors in Lymphoma. Front. Cell Dev. Biol. 2020, 8, 576391. [Google Scholar] [CrossRef]
- Androutsopoulos, V.P.; Spandidos, D.A. Antiproliferative effects of TSA, PXD101 and MS275 in A2780 and MCF7 cells: Acetylated histone H4 and acetylated tubulin as markers for HDACi potency and selectivity. Oncol. Rep. 2017, 38, 3412–3418. [Google Scholar] [PubMed]
- Acharya, M.R.; Sparreboom, A.; Venitz, J.; Figg, W.D. Rational development of histone deacetylase inhibitors as anticancer agents: A review. Mol. Pharmacol. 2005, 68, 917–932. [Google Scholar] [CrossRef]
- Zhang, Q.; Wang, S.; Chen, J.; Yu, Z. Histone Deacetylases (HDACs) Guided Novel Therapies for T-cell lymphomas. Int. J. Med. Sci. 2019, 16, 424–442. [Google Scholar] [CrossRef]
- Khan, N.; Jeffers, M.; Kumar, S.; Hackett, C.; Boldog, F.; Khramtsov, N.; Qian, X.; Mills, E.; Berghs, S.C.; Carey, N.; et al. Determination of the class and isoform selectivity of small-molecule histone deacetylase inhibitors. Biochem. J. 2008, 409, 581–589. [Google Scholar] [CrossRef]
- Yadav, R.; Mishra, P.; Yadav, D. Histone Deacetylase Inhibitors: A Prospect in Drug Discovery. Turk J. Pharm. Sci. 2019, 16, 101–114. [Google Scholar] [CrossRef]
- Anne, M.; Sammartino, D.; Barginear, M.F.; Budman, D. Profile of panobinostat and its potential for treatment in solid tumors: An update. Onco Targets Ther. 2013, 6, 1613–1624. [Google Scholar] [CrossRef]
- Atadja, P. Development of the pan-DAC inhibitor panobinostat (LBH589): Successes and challenges. Cancer Lett. 2009, 280, 233–241. [Google Scholar] [CrossRef]
- Rasmussen, T.A.; Tolstrup, M.; Brinkmann, C.R.; Olesen, R.; Erikstrup, C.; Solomon, A.; Winckelmann, A.; Palmer, S.; Dinarello, C.; Buzon, M.; et al. Panobinostat, a histone deacetylase inhibitor, for latent-virus reactivation in HIV-infected patients on suppressive antiretroviral therapy: A phase 1/2, single group, clinical trial. Lancet HIV 2014, 1, e13–e21. [Google Scholar] [CrossRef]
- Hasegawa, H.; Yamada, Y.; Tsukasaki, K.; Mori, N.; Tsuruda, K.; Sasaki, D.; Usui, T.; Osaka, A.; Atogami, S.; Ishikawa, C.; et al. LBH589, a deacetylase inhibitor, induces apoptosis in adult T-cell leukemia/lymphoma cells via activation of a novel RAIDD-caspase-2 pathway. Leukemia 2011, 25, 575–587. [Google Scholar] [CrossRef]
- Moradei, O.; Vaisburg, A.; Martell, R.E. Histone deacetylase inhibitors in cancer therapy: New compounds and clinical update of benzamide-type agents. Curr. Top. Med. Chem. 2008, 8, 841–858. [Google Scholar] [CrossRef]
- Hasegawa, H.; Bissonnette, R.P.; Gillings, M.; Sasaki, D.; Taniguchi, H.; Kitanosono, H.; Tsuruda, K.; Kosai, K.; Uno, N.; Morinaga, Y.; et al. Induction of apoptosis by HBI-8000 in adult T-cell leukemia/lymphoma is associated with activation of Bim and NLRP3. Cancer Sci. 2016, 107, 1124–1133. [Google Scholar] [CrossRef]
- Johnstone, R.W. Histone-deacetylase inhibitors: Novel drugs for the treatment of cancer. Nat. Rev. Drug Discov. 2002, 1, 287–299. [Google Scholar] [CrossRef]
- Newmark, H.L.; Lupton, J.R.; Young, C.W. Butyrate as a Differentiating Agent—Pharmacokinetics, Analogs and Current Status. Cancer Lett. 1994, 78, 1–5. [Google Scholar] [CrossRef]
- Göttlicher, M.; Minucci, S.; Zhu, P.; Kramer, O.H.; Schimpf, A.; Giavara, S.; Sleeman, J.P.; Lo Coco, F.; Nervi, C.; Pelicci, P.G.; et al. Valproic acid defines a novel class of HDAC inhibitors inducing differentiation of transformed cells. EMBO J. 2001, 20, 6969–6978. [Google Scholar] [CrossRef]
- Phiel, C.J.; Zhang, F.; Huang, E.Y.; Guenther, M.G.; Lazar, M.A.; Klein, P.S. Histone Deacetylase Is a Direct Target of Valproic Acid, a Potent Anticonvulsant, Mood Stabilizer, and Teratogen. J. Biol. Chem. 2001, 276, 36734–36741. [Google Scholar] [CrossRef]
- Gillet, N.; Vandermeers, F.; de Brogniez, A.; Florins, A.; Nigro, A.; Francois, C.; Bouzar, A.B.; Verlaeten, O.; Stern, E.; Lambert, D.M.; et al. Chemoresistance to Valproate Treatment of Bovine Leukemia Virus-Infected Sheep; Identification of Improved HDAC Inhibitors. Pathogens 2012, 1, 65–82. [Google Scholar] [CrossRef]
- Whittaker, S.J.; Demierre, M.F.; Kim, E.J.; Rook, A.H.; Lerner, A.; Duvic, M.; Scarisbrick, J.; Reddy, S.; Robak, T.; Becker, J.C.; et al. Final results from a multicenter, international, pivotal study of romidepsin in refractory cutaneous T-cell lymphoma. J. Clin. Oncol. 2010, 28, 4485–4491. [Google Scholar] [CrossRef]
- Suraweera, A.; O’Byrne, K.J.; Richard, D.J. Combination Therapy With Histone Deacetylase Inhibitors (HDACi) for the Treatment of Cancer: Achieving the Full Therapeutic Potential of HDACi. Front. Oncol. 2018, 8, 92. [Google Scholar] [CrossRef]
- Nakajima, H.; Kim, Y.B.; Terano, H.; Yoshida, M.; Horinouchi, S. FR901228, a potent antitumor antibiotic, is a novel histone deacetylase inhibitor. Exp. Cell Res. 1998, 241, 126–133. [Google Scholar] [CrossRef]
- Moskowitz, A.J.; Horwitz, S.M. Targeting histone deacetylases in T-cell lymphoma. Leuk. Lymphoma 2017, 58, 1306–1319. [Google Scholar] [CrossRef] [PubMed]
- Furumai, R.; Matsuyama, A.; Kobashi, N.; Lee, K.-H.; Nishiyama, M.; Nakajima, H.; Tanaka, A.; Komatsu, Y.; Nishino, N.; Yoshida, M.; et al. FK228 (Depsipeptide) as a Natural Prodrug That Inhibits Class I Histone Deacetylases 1. Cancer Res. 2002, 62, 4916–4921. [Google Scholar]
- Yu, P.; Petrus, M.N.; Ju, W.; Zhang, M.; Conlon, K.C.; Nakagawa, M.; Maeda, M.; Bamford, R.N.; Waldmann, T.A. Augmented efficacy with the combination of blockade of the Notch-1 pathway, bortezomib and romidepsin in a murine MT-1 adult T-cell leukemia model. Leukemia 2015, 29, 556–566. [Google Scholar] [CrossRef]
- Shilatifard, A.; Conaway, R.C.; Conaway, J.W. The RNA polymerase II elongation complex. Annu. Rev. Biochem. 2003, 72, 693–715. [Google Scholar] [CrossRef]
- Kornberg, R.D. The molecular basis of eukaryotic transcription. Proc. Natl. Acad. Sci. USA 2007, 104, 12955–12961. [Google Scholar] [CrossRef]
- Missra, A.; Gilmour, D.S. Interactions between DSIF (DRB sensitivity inducing factor), NELF (negative elongation factor), and the Drosophila RNA polymerase II transcription elongation complex. Proc. Natl. Acad. Sci. USA 2010, 107, 11301–11306. [Google Scholar] [CrossRef]
- Marshall, N.F.; Price, D.H. Purification of P-Tefb, a Transcription Factor Required for the Transition into Productive Elongation. J. Biol. Chem. 1995, 270, 12335–12338. [Google Scholar] [CrossRef]
- Wada, T.; Takagi, T.; Yamaguchi, Y.; Ferdous, A.; Imai, T.; Hirose, S.; Sugimoto, S.; Yano, K.; Hartzog, G.A.; Winston, F.; et al. DSIF, a novel transcription elongation factor that regulates RNA polymerase II processivity, is composed of human Spt4 and Spt5 homologs. Genes Dev. 1998, 12, 343–356. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, Y.; Takagi, T.; Wada, T.; Yano, K.; Furuya, A.; Sugimoto, S.; Hasegawa, J.; Handa, H. NELF, a multisubunit complex containing RD, cooperates with DSIF to repress RNA polymerase II elongation. Cell 1999, 97, 41–51. [Google Scholar] [CrossRef]
- Bres, V.; Yoh, S.M.; Jones, K.A. The multi-tasking P-TEFb complex. Curr. Opin. Cell Biol. 2008, 20, 334–340. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Erickson, B.; Luo, W.; Seward, D.; Graber, J.H.; Pollock, D.D.; Megee, P.C.; Bentley, D.L. Gene-specific RNA polymerase II phosphorylation and the CTD code. Nat. Struct. Mol. Biol. 2010, 17, 1279–1286. [Google Scholar] [CrossRef] [PubMed]
- Chapman, R.D.; Heidemann, M.; Albert, T.K.; Mailhammer, R.; Flatley, A.; Meisterernst, M.; Kremmer, E.; Eick, D. Transcribing RNA polymerase II is phosphorylated at CTD residue serine-7. Science 2007, 318, 1780–1782. [Google Scholar] [CrossRef]
- Egloff, S.; O’Reilly, D.; Chapman, R.D.; Taylor, A.; Tanzhaus, K.; Pitts, L.; Eick, D.; Murphy, S. Serine-7 of the RNA polymerase II CTD is specifically required for snRNA gene expression. Science 2007, 318, 1777–1779. [Google Scholar] [CrossRef] [PubMed]
- Buratowski, S. Progression through the RNA Polymerase II CTD Cycle. Mol. Cell 2009, 36, 541–546. [Google Scholar] [CrossRef]
- Margaritis, T.; Holstege, F.C. Poised RNA polymerase II gives pause for thought. Cell 2008, 133, 581–584. [Google Scholar] [CrossRef]
- Cho, E.J.; Kobor, M.S.; Kim, M.; Greenblatt, J.; Buratowski, S. Opposing effects of Ctk1 kinase and Fcp1 phosphatase at Ser 2 of the RNA polymerase II C-terminal domain. Genes Dev. 2001, 15, 3319–3329. [Google Scholar] [CrossRef]
- Liu, P.; Xiang, Y.; Fujinaga, K.; Bartholomeeusen, K.; Nilson, K.A.; Price, D.H.; Peterlin, B.M. Release of positive transcription elongation factor b (P-TEFb) from 7SK small nuclear ribonucleoprotein (snRNP) activates hexamethylene bisacetamide-inducible protein (HEXIM1) transcription. J. Biol. Chem. 2014, 289, 9918–9925. [Google Scholar] [CrossRef]
- Qiu, H.; Hu, C.; Hinnebusch, A.G. Phosphorylation of the Pol II CTD by KIN28 enhances BUR1/BUR2 recruitment and Ser2 CTD phosphorylation near promoters. Mol. Cell 2009, 33, 752–762. [Google Scholar] [CrossRef]
- Zhou, K.; Kuo, W.H.; Fillingham, J.; Greenblatt, J.F. Control of transcriptional elongation and cotranscriptional histone modification by the yeast BUR kinase substrate Spt5. Proc. Natl. Acad. Sci. USA 2009, 106, 6956–6961. [Google Scholar] [CrossRef]
- Ott, M.; Geyer, M.; Zhou, Q. The control of HIV transcription: Keeping RNA polymerase II on track. Cell Host Microbe 2011, 10, 426–435. [Google Scholar] [CrossRef] [PubMed]
- Peterlin, B.M.; Price, D.H. Controlling the elongation phase of transcription with P-TEFb. Mol. Cell 2006, 23, 297–305. [Google Scholar] [CrossRef] [PubMed]
- Core, L.J.; Lis, J.T. Transcription regulation through promoter-proximal pausing of RNA polymerase II. Science 2008, 319, 1791–1792. [Google Scholar] [CrossRef]
- Levine, M. Paused RNA Polymerase II as a Developmental Checkpoint. Cell 2011, 145, 502–511. [Google Scholar] [CrossRef] [PubMed]
- Smith, E.; Lin, C.; Shilatifard, A. The super elongation complex (SEC) and MLL in development and disease. Genes Dev. 2011, 25, 661–672. [Google Scholar] [CrossRef]
- Fujinaga, K.; Irwin, D.; Huang, Y.; Taube, R.; Kurosu, T.; Peterlin, B.M. Dynamics of human immunodeficiency virus transcription: P-TEFb phosphorylates RD and dissociates negative effectors from the transactivation response element. Mol. Cell. Biol. 2004, 24, 787–795. [Google Scholar] [CrossRef]
- Garriga, J.; Grana, X. Cellular control of gene expression by T-type cyclin/CDK9 complexes. Gene 2004, 337, 15–23. [Google Scholar] [CrossRef]
- Gilmour, D.S. Promoter proximal pausing on genes in metazoans. Chromosoma 2009, 118, 1–10. [Google Scholar] [CrossRef]
- Ivanov, D.; Kwak, Y.T.; Guo, J.; Gaynor, R.B. Domains in the SPT5 protein that modulate its transcriptional regulatory properties. Mol. Cell. Biol. 2000, 20, 2970–2983. [Google Scholar] [CrossRef]
- Kim, J.B.; Sharp, P.A. Positive transcription elongation factor B phosphorylates hSPT5 and RNA polymerase II carboxyl-terminal domain independently of cyclin-dependent kinase-activating kinase. J. Biol. Chem. 2001, 276, 12317–12323. [Google Scholar] [CrossRef] [PubMed]
- Renner, D.B.; Yamaguchi, Y.; Wada, T.; Handa, H.; Price, D.H. A highly purified RNA polymerase II elongation control system. J. Biol. Chem. 2001, 276, 42601–42609. [Google Scholar] [CrossRef] [PubMed]
- Wada, T.; Takagi, T.; Yamaguchi, Y.; Watanabe, D.; Handa, H. Evidence that P-TEFb alleviates the negative effect of DSIF on RNA polymerase II-dependent transcription in vitro. EMBO J. 1998, 17, 7395–7403. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.R.; Peery, T.; Peng, T.M.; Ramanathan, Y.; Marshall, N.; Marshall, T.; Amendt, B.; Mathews, M.B.; Price, D.H. Transcription elongation factor P-TEFb is required for HIV-1 Tat transactivation in vitro. Genes Dev. 1997, 11, 2622–2632. [Google Scholar] [CrossRef]
- Fujinaga, K.; Luo, Z.; Schaufele, F.; Peterlin, B.M. Visualization of positive transcription elongation factor b (P-TEFb) activation in living cells. J. Biol. Chem. 2015, 290, 1829–1836. [Google Scholar] [CrossRef]
- He, N.; Liu, M.; Hsu, J.; Xue, Y.; Chou, S.; Burlingame, A.; Krogan, N.J.; Alber, T.; Zhou, Q. HIV-1 Tat and host AFF4 recruit two transcription elongation factors into a bifunctional complex for coordinated activation of HIV-1 transcription. Mol. Cell 2010, 38, 428–438. [Google Scholar] [CrossRef] [PubMed]
- Kiss, T.; Michels, A.A.; Bensaude, O. 7SK small nuclear RNA binds to and inhibits the activity of CDK9/cyclin T complexes. Nature 2001, 414, 322–325. [Google Scholar]
- Michels, A.A.; Nguyen, V.T.; Fraldi, A.; Labas, V.; Edwards, M.; Bonnet, F.; Lania, L.; Bensaude, O. MAQ1 and 7SK RNA interact with CDK9/cyclin T complexes in a transcription-dependent manner. Mol. Cell. Biol. 2003, 23, 4859–4869. [Google Scholar] [CrossRef]
- Jang, M.K.; Mochizuki, K.; Zhou, M.S.; Jeong, H.S.; Brady, J.N.; Ozato, K. The bromodomain protein Brd4 is a positive regulatory component of P-TEFb and stimulates RNA polymerase II-dependent transcription. Mol. Cell 2005, 19, 523–534. [Google Scholar] [CrossRef]
- Yang, Z.; Yik, J.H.; Chen, R.; He, N.; Jang, M.K.; Ozato, K.; Zhou, Q. Recruitment of P-TEFb for stimulation of transcriptional elongation by the bromodomain protein Brd4. Mol. Cell 2005, 19, 535–545. [Google Scholar] [CrossRef]
- Sobhian, B.; Laguette, N.; Yatim, A.; Nakamura, M.; Levy, Y.; Kiernan, R.; Benkirane, M. HIV-1 Tat assembles a multifunctional transcription elongation complex and stably associates with the 7SK snRNP. Mol. Cell 2010, 38, 439–451. [Google Scholar] [CrossRef]
- Mann, M.C.; Strobel, S.; Fleckenstein, B.; Kress, A.K. The transcription elongation factor ELL2 is specifically upregulated in HTLV-1-infected T-cells and is dependent on the viral oncoprotein Tax. Virology 2014, 464, 98–110. [Google Scholar] [CrossRef][Green Version]
- Dey, A.; Ellenberg, J.; Farina, A.; Coleman, A.E.; Maruyama, T.; Sciortino, S.; Lippincott-Schwartz, J.; Ozato, K. A bromodomain protein, MCAP, associates with mitotic chromosomes and affects G2-to-M transition. Mol. Cell. Biol. 2000, 20, 6537–6549. [Google Scholar] [CrossRef] [PubMed]
- Kanno, T.; Kanno, Y.; Siegel, R.M.; Jang, M.K.; Lenardo, M.J.; Ozato, K. Selective recognition of acetylated histones by bromodomain proteins visualized in living cells. Mol. Cell 2004, 13, 33–43. [Google Scholar] [CrossRef]
- Ladurner, A.G.; Inouye, C.; Jain, R.; Tjian, R. Bromodomains mediate an acetyl-histone encoded antisilencing function at heterochromatin boundaries. Mol. Cell 2003, 11, 365–376. [Google Scholar] [CrossRef]
- Jeanmougin, F.; Wurtz, J.M.; Le Douarin, B.; Chambon, P.; Losson, R. The bromodomain revisited. Trends Biochem. Sci. 1997, 22, 151–153. [Google Scholar] [CrossRef]
- Cho, W.K.; Zhou, M.; Jang, M.K.; Huang, K.; Jeong, S.J.; Ozato, K.; Brady, J.N. Modulation of the Brd4/P-TEFb interaction by the human T-lymphotropic virus type 1 tax protein. J. Virol. 2007, 81, 11179–11186. [Google Scholar] [CrossRef]
- Darcis, G.; Kula, A.; Bouchat, S.; Fujinaga, K.; Corazza, F.; Ait-Ammar, A.; Delacourt, N.; Melard, A.; Kabeya, K.; Vanhulle, C.; et al. An In-Depth Comparison of Latency-Reversing Agent Combinations in Various In Vitro and Ex Vivo HIV-1 Latency Models Identified Bryostatin-1+JQ1 and Ingenol-B+JQ1 to Potently Reactivate Viral Gene Expression. PLoS Pathog. 2015, 11, e1005063. [Google Scholar] [CrossRef]
- Fowler, T.; Ghatak, P.; Price, D.H.; Conaway, R.; Conaway, J.; Chiang, C.M.; Bradner, J.E.; Shilatifard, A.; Roy, A.L. Regulation of MYC expression and differential JQ1 sensitivity in cancer cells. PLoS ONE 2014, 9, e87003. [Google Scholar] [CrossRef]
- Lockwood, W.W.; Zejnullahu, K.; Bradner, J.E.; Varmus, H. Sensitivity of human lung adenocarcinoma cell lines to targeted inhibition of BET epigenetic signaling proteins. Proc. Natl. Acad. Sci. USA 2012, 109, 19408–19413. [Google Scholar] [CrossRef]
- Mertz, J.A.; Conery, A.R.; Bryant, B.M.; Sandy, P.; Balasubramanian, S.; Mele, D.A.; Bergeron, L.; Sims, R.J., 3rd. Targeting MYC dependence in cancer by inhibiting BET bromodomains. Proc. Natl. Acad. Sci. USA 2011, 108, 16669–16674. [Google Scholar] [CrossRef]
- Zuber, J.; Shi, J.W.; Wang, E.; Rappaport, A.R.; Herrmann, H.; Sison, E.A.; Magoon, D.; Qi, J.; Blatt, K.; Wunderlich, M.; et al. RNAi screen identifies Brd4 as a therapeutic target in acute myeloid leukaemia. Nature 2011, 478, 524–528. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, C.; Archin, N.; Michaels, D.; Belkina, A.C.; Denis, G.V.; Bradner, J.; Sebastiani, P.; Margolis, D.M.; Montano, M. BET bromodomain inhibition as a novel strategy for reactivation of HIV-1. J. Leukoc. Biol. 2012, 92, 1147–1154. [Google Scholar] [CrossRef]
- Zhu, J.; Gaiha, G.D.; John, S.P.; Pertel, T.; Chin, C.R.; Gao, G.; Qu, H.; Walker, B.D.; Elledge, S.J.; Brass, A.L. Reactivation of latent HIV-1 by inhibition of BRD4. Cell Rep. 2012, 2, 807–816. [Google Scholar] [CrossRef] [PubMed]
- Jiang, G.; Mendes, E.A.; Kaiser, P.; Wong, D.P.; Tang, Y.; Cai, I.; Fenton, A.; Melcher, G.P.; Hildreth, J.E.; Thompson, G.R.; et al. Synergistic Reactivation of Latent HIV Expression by Ingenol-3-Angelate, PEP005, Targeted NF-kB Signaling in Combination with JQ1 Induced p-TEFb Activation. PLoS Pathog. 2015, 11, e1005066. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Qi, J.; Bradner, J.E.; Xiao, G.; Chen, L.F. Bromodomain and extraterminal (BET) protein inhibition suppresses human T cell leukemia virus 1 (HTLV-1) Tax protein-mediated tumorigenesis by inhibiting nuclear factor kappaB (NF-kappaB) signaling. J. Biol. Chem. 2013, 288, 36094–36105. [Google Scholar] [CrossRef]
- Li, G.; Zhang, Z.; Reszka-Blanco, N.; Li, F.; Chi, L.; Ma, J.; Jeffrey, J.; Cheng, L.; Su, L. Specific Activation In Vivo of HIV-1 by a Bromodomain Inhibitor from Monocytic Cells in Humanized Mice under Antiretroviral Therapy. J. Virol. 2019, 93, e00233-19. [Google Scholar] [CrossRef] [PubMed]
- Coude, M.M.; Braun, T.; Berrou, J.; Dupont, M.; Bertrand, S.; Masse, A.; Raffoux, E.; Itzykson, R.; Delord, M.; Riveiro, M.E.; et al. BET inhibitor OTX015 targets BRD2 and BRD4 and decreases c-MYC in acute leukemia cells. Oncotarget 2015, 6, 17698–17712. [Google Scholar] [CrossRef]
- Lu, P.P.; Qu, X.Y.; Shen, Y.Z.; Jiang, Z.T.; Wang, P.F.; Zeng, H.X.; Ji, H.Y.; Deng, J.X.; Yang, X.Y.; Li, X.; et al. The BET inhibitor OTX015 reactivates latent HIV-1 through P-TEFb. Sci. Rep. 2016, 6, 1–13. [Google Scholar] [CrossRef]
- Huang, H.; Liu, S.; Jean, M.; Simpson, S.; Huang, H.; Merkley, M.; Hayashi, T.; Kong, W.; Rodriguez-Sanchez, I.; Zhang, X.; et al. A Novel Bromodomain Inhibitor Reverses HIV-1 Latency through Specific Binding with BRD4 to Promote Tat and P-TEFb Association. Front. Microbiol. 2017, 8, 1035. [Google Scholar] [CrossRef] [PubMed]
- Bartholomeeusen, K.; Fujinaga, K.; Xiang, Y.H.; Peterlin, B.M. Histone Deacetylase Inhibitors (HDACis) That Release the Positive Transcription Elongation Factor b (P-TEFb) from Its Inhibitory Complex Also Activate HIV Transcription. J. Biol. Chem. 2013, 288, 14400–14407. [Google Scholar] [CrossRef] [PubMed]
- Quaresma, A.; Bugai, A.; Barboric, M. Cracking the control of RNA polymerase II elongation by 7SK snRNP and P-TEFb. Nucleic Acids Res. 2016, 44, 7527–7539. [Google Scholar] [CrossRef] [PubMed]
- Jamaluddin, M.S.; Hu, P.W.; Jan, Y.; Siwak, E.B.; Rice, A.P. Short Communication: The Broad-Spectrum Histone Deacetylase Inhibitors Vorinostat and Panobinostat Activate Latent HIV in CD4+ T Cells In Part Through Phosphorylation of the T-Loop of the CDK9 Subunit of P-TEFb. AIDS Res. Hum. Retrovir. 2016, 32, 169–173. [Google Scholar] [CrossRef] [PubMed]
- Ramakrishnan, R.; Liu, H.; Rice, A.P. Short communication: SAHA (vorinostat) induces CDK9 Thr-186 (T-loop) phosphorylation in resting CD4+ T cells: Implications for reactivation of latent HIV. AIDS Res. Hum. Retrovir. 2015, 31, 137–141. [Google Scholar] [CrossRef] [PubMed]
- Elliott, J.H.; McMahon, J.H.; Chang, C.C.; Lee, S.A.; Hartogensis, W.; Bumpus, N.; Savic, R.; Roney, J.; Hoh, R.; Solomon, A.; et al. Short-term administration of disulfiram for reversal of latent HIV infection: A phase 2 dose-escalation study. Lancet HIV 2015, 2, e520–e529. [Google Scholar] [CrossRef]
- Knights, H.D. A critical review of the evidence concerning the HIV latency reversing effect of disulfiram, the possible explanations for its inability to reduce the size of the latent reservoir in vivo, and the caveats associated with its use in practice. AIDS Res. Treat. 2017, 2017, 8239428. [Google Scholar] [CrossRef]
- Xing, S.; Bullen, C.K.; Shroff, N.S.; Shan, L.; Yang, H.C.; Manucci, J.L.; Bhat, S.; Zhang, H.; Margolick, J.B.; Quinn, T.C.; et al. Disulfiram reactivates latent HIV-1 in a Bcl-2-transduced primary CD4+ T cell model without inducing global T cell activation. J. Virol. 2011, 85, 6060–6064. [Google Scholar] [CrossRef]
- Cho, S.Y.; Schroeder, S.; Kaehlcke, K.; Kwon, H.S.; Pedal, A.; Herker, E.; Schnoelzer, M.; Ott, M. Acetylation of cyclin T1 regulates the equilibrium between active and inactive P-TEFb in cells. EMBO J. 2009, 28, 1407–1417. [Google Scholar] [CrossRef]
- Zhao, M.; De Crignis, E.; Rokx, C.; Verbon, A.; van Gelder, T.; Mahmoudi, T.; Katsikis, P.D.; Mueller, Y.M. T cell toxicity of HIV latency reversing agents. Pharmacol. Res. 2019, 139, 524–534. [Google Scholar] [CrossRef]
- Laird, G.M.; Bullen, C.K.; Rosenbloom, D.I.; Martin, A.R.; Hill, A.L.; Durand, C.M.; Siliciano, J.D.; Siliciano, R.F. Ex vivo analysis identifies effective HIV-1 latency-reversing drug combinations. J. Clin. Investig. 2015, 125, 1901–1912. [Google Scholar] [CrossRef]
- Li, Q.T.; Price, J.P.; Byers, S.A.; Cheng, D.M.; Peng, J.M.; Price, D.H. Analysis of the large inactive P-TEFb complex indicates that it contains one 7SK molecule, a dimer of HEXIM1 or HEXIM2, and two P-TEFb molecules containing Cdk9 phosphorylated at threonine 186. J. Biol. Chem. 2005, 280, 28819–28826. [Google Scholar] [CrossRef]
- Yang, Z.; Zhu, Q.; Luo, K.; Zhou, Q. The 7SK small nuclear RNA inhibits the CDK9/cyclin T1 kinase to control transcription. Nature 2001, 414, 317–322. [Google Scholar] [CrossRef]
- Yik, J.H.; Chen, R.; Nishimura, R.; Jennings, J.L.; Link, A.J.; Zhou, Q. Inhibition of P-TEFb (CDK9/Cyclin T) kinase and RNA polymerase II transcription by the coordinated actions of HEXIM1 and 7SK snRNA. Mol. Cell 2003, 12, 971–982. [Google Scholar] [CrossRef]
- Zhou, M.; Lu, H.; Park, H.; Wilson-Chiru, J.; Linton, R.; Brady, J.N. Tax interacts with P-TEFb in a novel manner to stimulate human T-lymphotropic virus type 1 transcription. J. Virol. 2006, 80, 4781–4791. [Google Scholar] [CrossRef]
- Fibach, E.; Reuben, R.C.; Rifkind, R.A.; Marks, P.A. Effect of hexamethylene bisacetamide on the commitment to differentiation of murine erythroleukemia cells. Cancer Res. 1977, 37, 440–444. [Google Scholar] [PubMed]
- Richon, V.M.; Webb, Y.; Merger, R.; Sheppard, T.; Jursic, B.; Ngo, L.; Civoli, F.; Breslow, R.; Rifkind, R.A.; Marks, P.A. Second generation hybrid polar compounds are potent inducers of transformed cell differentiation. Proc. Natl. Acad. Sci. USA 1996, 93, 5705–5708. [Google Scholar] [CrossRef] [PubMed]
- Siegel, D.S.; Zhang, X.; Feinman, R.; Teitz, T.; Zelenetz, A.; Richon, V.M.; Rifkind, R.A.; Marks, P.A.; Michaeli, J. Hexamethylene bisacetamide induces programmed cell death (apoptosis) and down-regulates BCL-2 expression in human myeloma cells. Proc. Natl. Acad. Sci. USA 1998, 95, 162–166. [Google Scholar] [CrossRef]
- Kusuhara, M.; Nagasaki, K.; Kimura, K.; Maass, N.; Manabe, T.; Ishikawa, S.; Aikawa, M.; Miyazaki, K.; Yamaguchi, K. Cloning of hexamethylene-bis-acetamide-inducible transcript, HEXIM1, in human vascular smooth muscle cells. Biomed. Res. 1999, 20, 273–279. [Google Scholar] [CrossRef]
- Ouchida, R.; Kusuhara, M.; Shimizu, N.; Hisada, T.; Makino, Y.; Morimoto, C.; Handa, H.; Ohsuzu, F.; Tanaka, H. Suppression of NF-kappaB-dependent gene expression by a hexamethylene bisacetamide-inducible protein HEXIM1 in human vascular smooth muscle cells. Genes Cells 2003, 8, 95–107. [Google Scholar] [CrossRef] [PubMed]
- Bartholomeeusen, K.; Xiang, Y.; Fujinaga, K.; Peterlin, B.M. Bromodomain and extra-terminal (BET) bromodomain inhibition activate transcription via transient release of positive transcription elongation factor b (P-TEFb) from 7SK small nuclear ribonucleoprotein. J. Biol. Chem. 2012, 287, 36609–36616. [Google Scholar] [CrossRef]
- Liu, Y.; Denlinger, C.E.; Rundall, B.K.; Smith, P.W.; Jones, D.R. Suberoylanilide hydroxamic acid induces Akt-mediated phosphorylation of p300, which promotes acetylation and transcriptional activation of RelA/p65. J. Biol. Chem. 2006, 281, 31359–31368. [Google Scholar] [CrossRef]
- Richon, V.M.; Emiliani, S.; Verdin, E.; Webb, Y.; Breslow, R.; Rifkind, R.A.; Marks, P.A. A class of hybrid polar inducers of transformed cell differentiation inhibits histone deacetylases. Proc. Natl. Acad. Sci. USA 1998, 95, 3003–3007. [Google Scholar] [CrossRef] [PubMed]
- Chick, J.; Gough, K.; Falkowski, W.; Kershaw, P.; Hore, B.; Mehta, B.; Ritson, B.; Ropner, R.; Torley, D. Disulfiram treatment of alcoholism. Br. J. Psychiatry 1992, 161, 84–89. [Google Scholar] [CrossRef]
- Fuller, R.K.; Branchey, L.; Brightwell, D.R.; Derman, R.M.; Emrick, C.D.; Iber, F.L.; James, K.E.; Lacoursiere, R.B.; Lee, K.K.; Lowenstam, I. Disulfiram treatment of alcoholism: A Veterans Administration cooperative study. JAMA 1986, 256, 1449–1455. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Warfield, L.; Zhang, C.; Luo, J.; Allen, J.; Lang, W.H.; Ranish, J.; Shokat, K.M.; Hahn, S. Phosphorylation of the transcription elongation factor Spt5 by yeast Bur1 kinase stimulates recruitment of the PAF complex. Mol. Cell. Biol. 2009, 29, 4852–4863. [Google Scholar] [CrossRef]
- Christman, J.K. 5-Azacytidine and 5-aza-2′-deoxycytidine as inhibitors of DNA methylation: Mechanistic studies and their implications for cancer therapy. Oncogene 2002, 21, 5483–5495. [Google Scholar] [CrossRef]
- Fernandez, G.; Zeichner, S.L. Cell line-dependent variability in HIV activation employing DNMT inhibitors. Virol. J. 2010, 7, 266. [Google Scholar] [CrossRef]
- Jeeninga, R.E.; Westerhout, E.M.; van Gerven, M.L.; Berkhout, B. HIV-1 latency in actively dividing human T cell lines. Retrovirology 2008, 5, 37. [Google Scholar] [CrossRef]
- Kauder, S.E.; Bosque, A.; Lindqvist, A.; Planelles, V.; Verdin, E. Epigenetic regulation of HIV-1 latency by cytosine methylation. PLoS Pathog. 2009, 5, e1000495. [Google Scholar] [CrossRef]
- Pillat, M.M.; Correa, B.L.; da Rocha, C.F.; Muller, G.C.; Lopes, R.P.; Lampert, S.S.; Teixeira, A.L.; Menna-Barreto, M.; Bauer, M.E. Changes in T cell phenotype and activated MAPKs are correlated to impaired cellular responses to antigens and glucocorticoids during HTLV-I infection. J. Neuroimmunol. 2009, 216, 76–84. [Google Scholar] [CrossRef]
- Radonovich, M.; Jeangt, K.-T. Activation of the Human T-Cell Leukemia Virus Type I Long Terminal Repeat by 12-O-Tetradecanoylphorbol-13-Acetate and by Tax (p4Ox) Occurs through Similar but Functionally Distinct Target Sequences. J. Virol. 1989, 63, 2987–2994. [Google Scholar] [CrossRef]
- Abou-Kandil, A.; Chamias, R.; Huleihel, M.; Godbey, W.T.; Aboud, M. Differential role of PKC-induced c-Jun in HTLV-1 LTR activation by 12-O-tetradecanoylphorbol-13-acetate in different human T-cell lines. PLoS ONE 2012, 7, e29934. [Google Scholar] [CrossRef] [PubMed]
- Jabareen, A.; Suleman, M.; Abu-Jaafar, A.; Huleihel, M. Different molecular mechanisms of HTLV-1 and HIV LTR activation by TPA. Biochem. Biophys. Res. Commun. 2018, 500, 538–543. [Google Scholar] [CrossRef] [PubMed]
- Roth, D.; Krammer, P.H.; Gulow, K. Dynamin related protein 1-dependent mitochondrial fission regulates oxidative signalling in T cells. FEBS Lett. 2014, 588, 1749–1754. [Google Scholar] [CrossRef] [PubMed]
- Torgeman, A.; Ben-Aroya, Z.; Grunspan, A.; Zelin, E.; Butovsky, E.; Hallak, M.; Lochelt, M.; Flugel, R.M.; Livneh, E.; Wolfson, M.; et al. Activation of HTLV-I long terminal repeat by stress-inducing agents and protection of HTLV-I-infected T-cells from apoptosis by the viral tax protein. Exp. Cell Res. 2001, 271, 169–179. [Google Scholar] [CrossRef] [PubMed]
- Chatila, T.; Silverman, L.; Miller, R.; Geha, R. Mechanisms of T cell activation by the calcium ionophore ionomycin. J. Immunol. 1989, 143, 1283–1289. [Google Scholar] [PubMed]
- Spivak, A.M.; Planelles, V. Novel Latency Reversal Agents for HIV-1 Cure. Annu. Rev. Med. 2018, 69, 421–436. [Google Scholar] [CrossRef] [PubMed]
- Pichler, K.; Kattan, T.; Gentzsch, J.; Kress, A.K.; Taylor, G.P.; Bangham, C.R.; Grassmann, R. Strong induction of 4-1BB, a growth and survival promoting costimulatory receptor, in HTLV-1-infected cultured and patients’ T cells by the viral Tax oncoprotein. Blood 2008, 111, 4741–4751. [Google Scholar] [CrossRef]
- Wang, Y.; He, J.; Liao, M.; Hu, M.; Li, W.; Ouyang, H.; Wang, X.; Ye, T.; Zhang, Y.; Ouyang, L. An overview of Sirtuins as potential therapeutic target: Structure, function and modulators. Eur. J. Med. Chem. 2019, 161, 48–77. [Google Scholar] [CrossRef]
- Kozako, T. New Strategy of Adult T-cell Leukemia Treatment Targeted for Anti-tumor Immunity and a Longevity Gene-encoded Protein. Yakugaku Zasshi 2011, 131, 1061–1072. [Google Scholar] [CrossRef]
- Kozako, T.; Aikawa, A.; Shoji, T.; Fujimoto, T.; Yoshimitsu, M.; Shirasawa, S.; Tanaka, H.; Honda, S.; Shimeno, H.; Arima, N.; et al. High expression of the longevity gene product SIRT1 and apoptosis induction by sirtinol in adult T-cell leukemia cells. Int. J. Cancer 2012, 131, 2044–2055. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.M.; Gao, W.W.; Chan, C.P.; Cheng, Y.; Deng, J.J.; Yuen, K.S.; Iha, H.; Jin, D.Y. SIRT1 Suppresses Human T-Cell Leukemia Virus Type 1 Transcription. J. Virol. 2015, 89, 8623–8631. [Google Scholar] [CrossRef] [PubMed]
- Kozako, T.; Suzuki, T.; Yoshimitsu, M.; Uchida, Y.; Kuroki, A.; Aikawa, A.; Honda, S.; Arima, N.; Soeda, S. Novel small-molecule SIRT1 inhibitors induce cell death in adult T-cell leukaemia cells. Sci. Rep. 2015, 5, 11345. [Google Scholar] [CrossRef]
- Kulkarni, A.; Taylor, G.P.; Klose, R.J.; Schofield, C.J.; Bangham, C.R. Histone H2A monoubiquitylation and p38-MAPKs regulate immediate-early gene-like reactivation of latent retrovirus HTLV-1. JCI Insight 2018, 3, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, A.; Mateus, M.; Thinnes, C.C.; McCullagh, J.S.; Schofield, C.J.; Taylor, G.P.; Bangham, C.R.M. Glucose Metabolism and Oxygen Availability Govern Reactivation of the Latent Human Retrovirus HTLV-1. Cell Chem. Biol. 2017, 24, 1377–1387. [Google Scholar] [CrossRef]
- Miura, M.; Dey, S.; Ramanayake, S.; Singh, A.; Rueda, D.S.; Bangham, C.R.M. Kinetics of HTLV-1 reactivation from latency quantified by single-molecule RNA FISH and stochastic modelling. PLoS Pathog. 2019, 15, e1008164. [Google Scholar] [CrossRef]
- Bahrami, S.; Drablos, F. Gene regulation in the immediate-early response process. Adv. Biol. Regul. 2016, 62, 37–49. [Google Scholar] [CrossRef]
- Mohyeldin, A.; Garzon-Muvdi, T.; Quinones-Hinojosa, A. Oxygen in stem cell biology: A critical component of the stem cell niche. Cell Stem Cell 2010, 7, 150–161. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| HDACi | HDAC Selectivity | Use in Latency Reversal or Retroviral Infection | Result | Ref. | |
|---|---|---|---|---|---|
| HIV-1 | HTLV-1 or Related Viruses | ||||
| Hydroxamates | |||||
| Trichostatin A (TSA) | pan-HDACi | 🗸 | HTLV-1 pos. cell lines (MT-2, SLB-1), PBMCs from BLV-infected sheep |
| [69,70,107,108,109] |
| Vorinostat (Suberoylanilide hydroxamic acid, SAHA, Zolinza) | pan-HDACi | 🗸 | MT-1-GFP+ reporter cells |
| [105,108,110] |
| Belinostat (PXD-101, Beleodaq) | pan-HDACi | 🗸 | X |
| [84,105,111] |
| Panobinostat (LBH589, Farydak) | pan-HDACi | 🗸 | MT-1-GFP+ reporter cells |
| [105,110,111] |
| Benzamides | |||||
| Entinostat (MS-275) | class I | 🗸 | HTLV-1 pos. cell lines (MT-1, -2, -4) |
| [95,112,113] |
| Chidamide (HBI-8000, Epidaza) | class I, class IIb | 🗸 | ATLL-derived cells lines |
| [84,102,114,115] |
| Short-chain fatty acids | |||||
| Butyrates | class I, class IIa | 🗸 | HTLV-1 pos. cell line(SLB-1, HUT 102) |
| [70,108,116,117] |
| Valproate (Valproic acid) | class I, class IIa | 🗸 | cell and animal models (BLV, STLV), HAM/TSP patients |
| [57,66,101,118,119,120] |
| Cyclic peptides | |||||
| Romidepsin (FK228, Depsipeptide, Istodax) | class I | 🗸 | HTLV-1 pos. cell lines (HUT 102, MT-2) |
| [84,121,122] |
| Mode of Action | Use in Latency Reversal or Retroviral Infection | Result | Ref. | ||
|---|---|---|---|---|---|
| HIV-1 | HTLV | ||||
| LMW-complex | |||||
| JQ1 | BRD4 inhibitor | HIV-1 pos. cell lines (A2, A72, J-Lat clone 10.6) | 🗸 |
| [72,110,203,204,206] |
| PEP005 | BRD4 inhibitor | cell line (ART patients) | X |
| [205] |
| iBET151 (GSK1210151A) | BRD4 inhibitor | HIV-1 pos. cell lines | X |
| [72,207] |
| Birabresib (OTX-015) | BRD4 inhibitor | AML and ALL cell lines, cell line (ART patients) | X |
| [208,209] |
| UMB-136 | BRD4 inhibitor | HIV pos. cell lines harboring latent proviruses | X |
| [210] |
| HMW-complex | |||||
| HMBA | PI3K/Akt pathway activation/ | chronically infected cells (U1 ACH-2, and JΔK) | X |
| [71,169,211,212] |
| Vorinostat (SAHA) | CDK9 T-loop phosphorylation | resting CD4+ T cells (aviremic patients) | X |
| [130,169,211,213,214] |
| Disulfiram | PI3K/Akt pathway activation/ | cell line (HAART patients) dose-escalation study | X |
| [215,216,217] |
| Trichostatin (TSA) | reporter in 293T cells | X |
| [169,218] | |
| 5-Azacytidine (AzaC) | analog of cytidine | HeLa cells 293T cells | 🗸 |
| [185,47] |
| Stimulus | Mode of Action | Use in HTLV-1 Infection | Result | Ref. |
|---|---|---|---|---|
| Mitogens | ||||
| Phytohemagglutinin-P (PHA) | T-cell activation | HTLV-1 pos. cell line (FS) |
| [117] |
| Phorbol 12-myristate 13-acetate (PMA) | PKC activation | HTLV-1 pos. cell lines (HUT 102, FS, 1996, 1657) |
| [117] |
| SIRT inhibitors | ||||
| Sirtinol | SIRT 1 inhibition | PBMCs from ATLL patients, HTLV-1 pos. cell lines |
| [251,252] |
| Ex527 | SIRT 1 inhibition | HTLV-1 pos. cell line (C8166) |
| [252] |
| Extracellular factors and circumstances | ||||
| Oxidative stress | p38 MAPK- activation | HTLV-1 pos. cell line (MT-1-GFP+ reporter cells) |
| [110] |
| Physiological hypoxia | p38 MAPK- activation | PBMCs from HTLV-1 infected patients |
| [254,255] |
| Glucose metabolism | link to Tax protein pos. feedback loop | PBMCs from HTLV-1 infected patients |
| [24,254,255] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schnell, A.P.; Kohrt, S.; Thoma-Kress, A.K. Latency Reversing Agents: Kick and Kill of HTLV-1? Int. J. Mol. Sci. 2021, 22, 5545. https://doi.org/10.3390/ijms22115545
Schnell AP, Kohrt S, Thoma-Kress AK. Latency Reversing Agents: Kick and Kill of HTLV-1? International Journal of Molecular Sciences. 2021; 22(11):5545. https://doi.org/10.3390/ijms22115545
Chicago/Turabian StyleSchnell, Annika P., Stephan Kohrt, and Andrea K. Thoma-Kress. 2021. "Latency Reversing Agents: Kick and Kill of HTLV-1?" International Journal of Molecular Sciences 22, no. 11: 5545. https://doi.org/10.3390/ijms22115545
APA StyleSchnell, A. P., Kohrt, S., & Thoma-Kress, A. K. (2021). Latency Reversing Agents: Kick and Kill of HTLV-1? International Journal of Molecular Sciences, 22(11), 5545. https://doi.org/10.3390/ijms22115545